

Abstract
Background
The estuarine tapertail anchovy (Coilia nasus) is widely distributed in the Yangtze River, the coastal waters of China, Korea, and the Ariake Sound of Japan. It is a commercially important species owing to its nutritional value and delicate flavor. However, Coilia nasus is strongly responsive to stress, this often results in death, which causes huge losses. In this study, we used next-generation sequencing technologies to study changes in gene expression in response to loading stress and the mechanism of death caused by loading stress in Coilia nasus.
Results
Using next-generation RNA-seq technologies on an Illumina HiSeq 2000 platform, we assembled a de novo transcriptome and tested for differential expression in response to stress. A total of 65,129 unigenes were generated, the mean unigene size and N50 were 607 bp and 813 bp, respectively. Of the assembled unigenes, we identified 2,990 genes that were significantly up-regulated, while 3,416 genes were significantly down-regulated in response to loading stress. Pathway enrichment analysis based on loading stress-responsive unigenes identified significantly stress related pathways. “Metabolism” and “immunity” were the two most frequently represented categories. In the “metabolism” category, “glucose metabolism” and “lipid metabolism” were major subclasses. The transcriptional expression of rate-limiting enzymes in “glucose metabolism” and “lipid metabolism” was detected by RT-qPCR, all were significantly increased after stress. Apoptosis associated proteins tumor necrosis factor alpha (TNF-α), caspase 9, cytochrome c and caspase 3 in the stress group were significantly elevated, moreover, liver injury indicators (Alanine aminotransferase, ALT, and aspartate transaminase, AST) were also significantly elevated, which indicates that loading stress induced liver injury.
Conclusion
This study provided abundant unigenes that could contribute greatly to the discovery of novel genes in fish. The alterations in predicted gene expression patterns reflected possible responses to stress. Loading stress may induce liver injury through the mitochondrial apoptosis pathway, which was activated by TNF-α. Taken together, our data not only provide information that will aid the identification of novel genes from fish, but also shed new light on the understanding of mechanisms by which physical stressors cause death in fish.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-558) contains supplementary material, which is available to authorized users.
Keywords: Gene expression, Coilia nasus, Stress, Liver injury, Apoptosis
Background
Stress exists in all aspects of aquaculture, daily management such as netting, loading and transporting can lead to a stress response. Stress may result in inhibition of growth, reproductive failure, and reduced resistance to pathogens [1–5]. The estuarine tapertail anchovy (Coilia nasus, junior synonym C. ectenes) is widely distributed in the Yangtze River, the coastal waters of China, Korea, and the Ariake Sound of Japan [6]. It is a commercially important species owing to its nutritional value and delicate flavor. However, Coilia nasus is strongly responsive to stress, this often results in death, which causes huge losses. Currently, little is known of the mechanism by which loading stress causes death in fish. In this study, we used next-generation sequencing technologies to study gene expression changes in response to loading stress and the mechanism of death caused by loading stress in Coilia nasus.
Recently, several species-specific cDNA microarrays have been developed for teleosts, and they are being used increasingly to reveal global gene expression patterns in response to stressor exposure and/or hormone treatment [7–10]. These studies in fish have revealed that the majority of changes, inspite of the use of different arrays and different types of acute stressor, can be broadly categorized functionally into genes encoding proteins involved in metabolism, immune function and reproduction. Although these studies have identified several genes that were previously not known to be stress responsive, the significance of these observed transcript changes to overall stress adaptation is far from clear.
While the previous studies that used various microarray platforms in fish were limited by large representation of expressed sequence tags (EST), which made gene-specific interpretation of the data difficult, the advent of next-generation sequencing methods, including pyrosequencing, has effectively overcome this limitation [11, 12]. Rapid progress in next-generation sequencing technologies has allowed large-scale efficient and economical production of ESTs. Transcriptome sequencing facilitates functional genomic studies, including global gene expression, novel gene discovery, and assembly of full-length genes [13–16]. To date, available molecular information on Coilia nasus remains sparse, which limits research on the mechanism by which loading stress causes death. The powerful new technologies provide a new opportunity for studies of species without genome reference databases, and non-model organisms.
Methods
Experimental animals
Coilia nasus (average weight, 9.6 g) were adapted to the conditions in a 7.0 × 5.0 × 1.0 m3 aquarium with a water temperature of 24.5 ± 1.0°C, pH 7.2, and dissolved oxygen concentration of 9.2 ± 0.5 mg O2/L dechlorinated and aerated water. The fish were fed twice daily, at 7:00 AM and 5:00 PM. At the onset of the experiments, all fish appeared healthy.
Stressing experiment
In March 2012, three 7.0 × 5.0 × 1.0 m3 ponds were stocked with 120 juvenile Coilia nasus each. The fish were acclimated to the ponds for approximately 14 months before the experiment, at which time the fish were 15 months old. Excess fish were stocked in the ponds in order to ensure subsequent access to the intended numbers. In detail, Coilia nasus prefer to swim towards lighted areas in dark situations. To take advantage of this phototaxis, a sifter was put into the water and lit, following which the fish moved into the sifter. The fish were euthanized immediately with 70 mg/L buffered tricaine methanesulfonate (MS-222). Using this method, five fish were removed from each pond, these 15 fish were the non-stressed controls and were processed immediately (see tissue sampling below). Subsequently, another five fish were netted from each pond and loaded into 75 × 55 × 33 cm3 tanks. After 0.5 h, the fish were euthanized as described above. Compared with the stressed fish, they did not experience the netting and handling used for the control fish. The mean length was 136.98 mm ± 9.26 SEM and mass was 8.86 g ± 1.76 SEM for all fish (n = 30) sampled in this experiment.
Tissue sampling
During the experiments, euthanized fish were submerged immediately in crushed ice to retard degradation of RNA. Blood was collected into ammonium-heparinized capillary tubes after severance of the caudal fin. All fish appeared healthy during dissection and their livers were removed and placed in liquid nitrogen. Plasma was separated by centrifugation. Plasma and liver samples were stored at -80°C until later analysis.
Animal welfare and experimental procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China, 2006), and were approved by the animal ethics committee of the Chinese Academy of Fishery Sciences.
Cortisol and glucose
Blood glucose measurement was performed by glucose-oxidase-superoxide enzyme endpoint colorimetry, the test kit was obtained from Shanghai Biological Product Research Institute, Ministry of Public Health, China. Cortisol was measured by radioimmunoassay in accordance with the method described by Pickering and Pottinger [17], the test kit was purchased from Beijing Beifang Biotech Research Institute, China. Plasma samples were measured using a Beckman Cx-4 spectrophotometer (Beckman Coulter, Fullerton, CA, USA). Student’s t test was used to analyze differences among all treatments (P < 0.05).
RNA sequencing, assembly and annotation
Transcriptome sequencing was carried out on an Illumina HiSeq 2000 platform that generated approximately 100-bp paired-end (PE) raw reads (BGI, Shenzhen, China). After removing adaptor sequences, ambiguous ‘N’ nucleotides (with the ratio of ‘N’ greater than 5%) and low quality sequences (with quality score less than 10), the remaining clean reads were assembled using trinity software [18] as described for de novo transcriptome assembly without a reference genome. For homology annotation, non-redundant sequences were subjected to public databases, including NCBI (http://www.ncbi.nlm.nih.gov/) non-redundant protein (Nr) and non-redundant nucleotide (Nt), Swiss-Prot (http://www.ebi.ac.uk/uniprot/), Gene Ontology (GO, http://www.geneontology.org/), Clusters of Orthologous Groups (COG, http://www.ncbi.nlm.nih.gov/COG/) and the Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg/). If the results from different databases conflicted, a priority order of alignments from Nr, Nt, KEGG, Swiss-Prot, GO and COG databases was followed. Comparison with the Nr, Nt and Swiss-Prot databases was carried out using the BlastX algorithm with an E-value cut-off of 0.00001; GO terms at the 2nd level were used to perform GO annotation; COG and KEGG classification were performed using BlastX with an E-value cut-off of 0.00001.
Analysis of differentially expressed genes
To analyze stress-responsive, differentially expressed genes in Coilia nasus, the number of reads for each of the contigs from the two samples was converted to reads per kilo base per million (RPKM) [19]. Following this, the MA-plot-based method with Random Sampling model (MARS) in the DEGseq package was used to calculate the expression abundance of each contig among the analyzed samples. We used an FDR (false discovery rate) to determine the P-value threshold. An FDR < 0.001 was considered to indicate significant expression abundance.
Pathway enrichment analysis identifies significantly enriched metabolic pathways or signal transduction pathways in differentially expressed genes by comparing them with the whole genome background. Bonferroni adjustments [20] were used to estimate levels of significance. After correction for multiple testing, we chose pathways with a Q-value ≤ 0.05 to represent those significantly enriched in differentially expressed genes. The Q-value is defined to be the FDR analog of the P-value. The Q-value of an individual hypothesis test is the minimum FDR at which the test maybe reported as significant.
Gene expression validation
Genes identified in this transcriptome sequencing analysis were validated and quantified by real-time PCR (RT-qPCR). The primers (Additional file 1: Table S1) were designed according to Illumina sequencing data with Primer Premier 5. The prepared total RNA used in RT-PCR analysis was isolated from the same sample as that used for Illumina sequencing. The RT-qPCR was performed on the ABI 7500 real-time PCR system (ABI, USA) using 2× SYBR green real-time PCR mix (Takara, Japan). The PCR amplification was performed in triplicate, using the following cycling parameters: 94°C for 2 min, followed by 40 cycles of 15 s at 94°C, 15 s at 60°C, and 34 s at 72°C. All samples were analyzed in triplicate and the expression of target genes was calculated as relative fold values using the 2-△△CT method.
Analysis of liver lipid peroxides, tumor necrosis factor alpha and apoptosis-associated proteins
Hepatocyte damage following stress was assessed by measuring alanine aminotransferase (ALT) and aspartate transaminase (AST) activities in plasma, using corresponding detection kits (Jusbio, Shanghai, China) according to the manufacturer’s instructions. TNF-α, cytochrome c, caspase-9, and caspase-3 in hepatic tissue were analyzed using an enzyme-linked immunosorbent assay kit (Zhaorui, Shanghai, China), as described by the manufacturer. Lipid peroxides (LPO) were detected by colorimetric methods, using a kit (Jiancheng, Nanjing, China) according to the manufacturer’s instructions.
Statistical analysis
Student’s t test or ANOVA, where appropriate, were used to identify significant differences among the treatments at p = 0.05. All data in this study were expressed as mean ± standard (S.D.).
Results
Plasma cortisol and glucose
Plasma cortisol was significantly elevated (P < 0.01) in the stressed fish (407.72 ng/mL ± 9.50 SEM, n = 15) compared with the control fish (187.76 ng/mL ± 9.07, n = 15) (Figure 1A). There were no significant differences between control replicates (P > 0.05).
Figure 1.
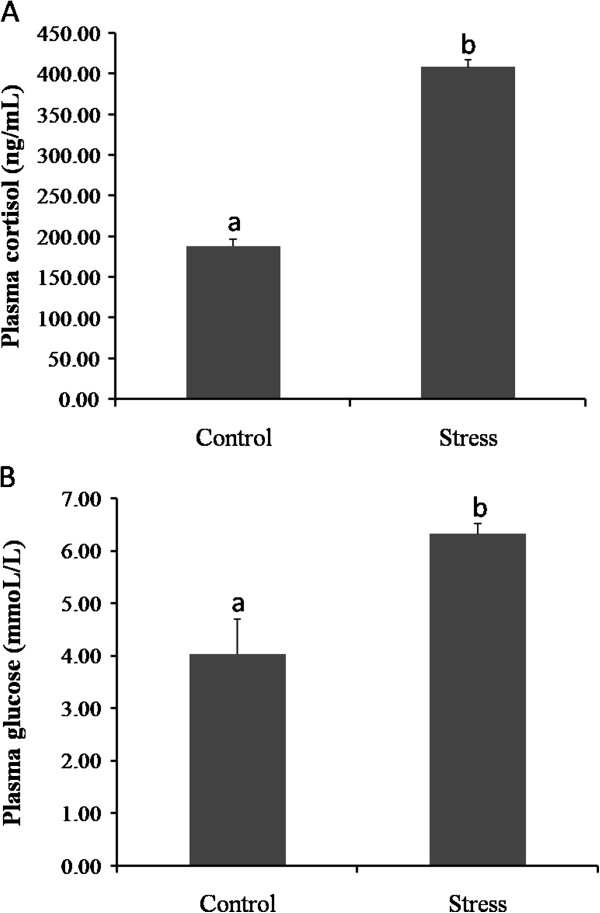
Plasma cortisol and glucose concentrations in control and loading stress groups. Bars represent the mean for replicates. Values with different letters are significantly different at P < 0.05. (A). Plasma cortisol concentrations for control and stressed groups (n = 15). (B). Plasma glucose levels for the two groups.
Plasma glucose concentrations (Figure 1B) in the stressed group (6.31 mmol/L ± 0.20, n = 15) were statistically higher than in the controls (4.03 mmol/L ± 0.66, P < 0.05, n = 15).
Generation and de novo assembly of Coilia nasus transcriptome data
In this study, we performed transcriptome sequencing of two libraries from liver samples in Coilia nasus via an Illumina HiSeq 2000 platform sequencer: 64.8 and 67.8 million reads were obtained from the two libraries. After removing low-quality reads, short reads and reads belonging to mitochondria, a total of 111,053,176 clean reads corresponding to mRNAs were obtained, these reads covered a total of 9,994,785,840 bases (Table 1).
Table 1.
Summary of sequence data generated for the Coilia nasus transcriptome, and quality filtering
| Sample | Total reads | Clean reads | Clean nucleotides (nt) | Q20 percentage (%) |
|---|---|---|---|---|
| Control | 64,827,364 | 55,526,588 | 4,997,392,920 | 98.20 |
| Stress | 67,782,560 | 55,526,588 | 4,997,392,920 | 98.28 |
| Total | 132,609,924 | 111,053,176 | 9,994,785,840 | 98.24 |
Using the Trinity assembly program, we generated a total of 65,129 unigenes (Table 2). The length distribution of unigenes larger than 200 bp is shown in Figure 2. The mean unigene size and N50 were 607 bp and 813 bp, respectively. About half of the unigenes (30,582; 47.0%) were ≥ 500 bp and 648 unigenes were > 3,000 bp in length. The largest unigene was 10,911 bp in length (Table 2).
Table 2.
Assembly statistics of reads
| Parameter | Numbers |
|---|---|
| Number of Unigene | 65,129 |
| Total bases of Unigene (bp) | 39,474,010 |
| Unigene mean lengths (bp) | 606 |
| Number of Unigene ≥500 bp | 30,582 |
| N50 | 835 |
| Max length (bp) | 10,911 |
Figure 2.
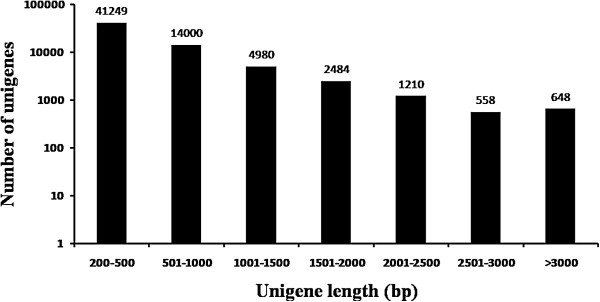
Length distribution of unigenes.
Functional annotation and pathway assignment
According to Gene Ontology (GO), an internationally standardized gene functional classification system, 21,688 non-redundant unigenes were classified into three major functional categories (biological process, cellular component and molecular function) and 49 subcategories (Table 3, Figure 3). In the category of cellular components, “cell part” (15,015, 69.2%) were the most commonly represented, followed by “organelle” (11,679, 53.9%) and “organelle part” (7,270, 33.5%). Among the molecular function terms, a significant proportion of clusters were assigned to “binding” (14,201, 65.5%) and “catalytic activity” (9,099, 50.0%). Of sequences categorized as biological processes, dominant subcategories were “cellular process” (15,851,73.1%) and “metabolic process” (13,020, 60.0%). However, within each of the three categories, few genes were assigned to the subcategories of “growth”, “cell junction” and “receptor regulator activity”.
Table 3.
Blast analysis of non-redundant unigenes against public databases
| Datebase | Number of annotated unigenes | Percentage of annoted unigenes (%) |
|---|---|---|
| Nr | 33,723 | 51.8 |
| Nt | 31,224 | 47.9 |
| Swiss-prot | 30,476 | 46.8 |
| KEGG | 25,188 | 38.7 |
| GO | 21,688 | 33.3 |
| COG | 10,631 | 16.3 |
Figure 3.
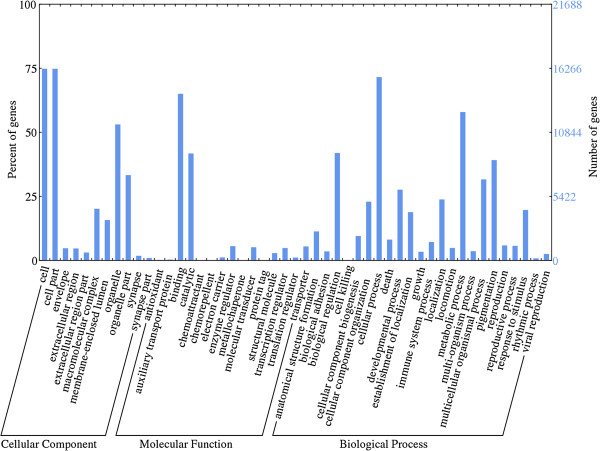
GO categorization of non-redundant unigenes.
To classify orthologous gene products, 10,631 (16.3%) non-redundant unigenes (Table 3) were subdivided into 25 COG classifications. Among them, the cluster of “general function prediction only” (3,830, 36.0%) represented the largest group, followed by “translation, ribosomal structure and biogenesis” (2,260, 21.3%), “transcription” (1,893, 17.8%), “replication, recombination and repair” (1,858, 17.5%), “cell cycle control, cell division, chromosome partitioning” (1,608, 15.1%), “post-translational modification, protein turnover, chaperon” (1,522, 14.3%) and “function unknown” (1,506, 14.2%), “nuclear structure” (4, 0.03%) was the smallest group (Figure 4).
Figure 4.
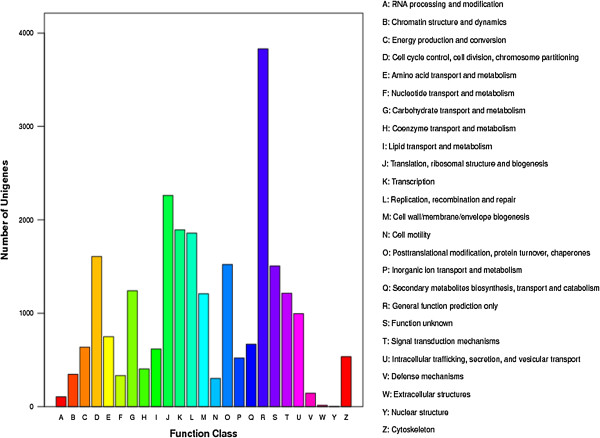
COG annotation of putative proteins.
The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed that diverse pathways were represented in our transcriptome dataset: 25,188 unigenes (Additional file 2: Table S2) were assigned to 259 specific pathways. Among them, “metabolism pathways”, “regulation of actin cytoskeleton”, “pathways in cancer”, “focal adhesion”, and “endocytosis” were the five most frequently represented pathways (Additional file 2: Table S2). Some important pathways involved in signal transduction were also identified, including “MAPK signaling pathway”, “calcium signaling pathway” and “Jak-STAT signal pathway” (Additional file 2: Table S2).
Loading stress-responsive unigenes in Coilia nasus
Unigene expression was estimated by the FPKM method, and differentially expressed genes were identified by referenceto Audic [21]. We found that 2,990 genes were significantly up-regulated, while 3,416 genes were significantly down-regulated in response to loading stress (Figure 5). The up-regulated and down-regulated genes are listed in Additional file 3: Table S3. The indicated genes included metabolic genes, enzymes, and other immune-related genes, such as the GTPase gene, threonine-protein kinase and MHC class I heavy chain gene. These genes showed different expression patterns after stress, which implies that they may play an important role in physiological processes associated with stress.
Figure 5.
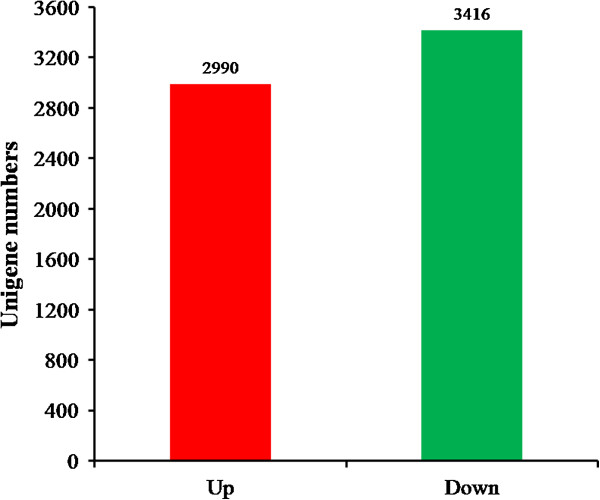
Genes differentially expressed following stress.
Pathway enrichment analysis based on loading stress-responsive unigenes identified significantly stress-related pathways. Among these pathways, “metabolism” and “immunity” were the two most commonly represented categories. In the “metabolism” category, “glucose metabolism” and “lipid metabolism” were major subclasses, and included “glycolysis/gluconeogenesis”, “starch and sucrose metabolism”, “glycerophospholipid metabolism” and “fat digestion and absorption” pathways. In the “immunity” category, “phagosome”, “HTLV-I infection” and “amoebiasis” were the most frequently represented pathways (Table 4).
Table 4.
KEGG pathways of differentially expressed genes
| Category | Pathway | Pathway ID | Gene number | Q-value |
|---|---|---|---|---|
| Carbohydrate metabolism | Glycolysis/Gluconeogenesis | ko00010 | 38 | 2.09E-03 |
| Starch and sucrose metabolism | ko00500 | 32 | 2.67E-03 | |
| Pyruvate metabolism | ko00620 | 32 | 2.89E-03 | |
| Type I diabetes mellitus | ko04940 | 31 | 1.39E-08 | |
| Glyoxylate and dicarboxylate metabolism | ko00630 | 24 | 1.34E-04 | |
| Butanoate metabolism | ko00650 | 17 | 4.16E-03 | |
| D-Glutamine and D-glutamate metabolism | ko00471 | 5 | 2.12E-02 | |
| Lipid metabolism | Glycerophospholipid metabolism | ko00564 | 36 | 4.43E-02 |
| Fat digestion and absorption | ko04975 | 28 | 3.61E-03 | |
| Steroid hormone biosynthesis | ko00140 | 24 | 3.40E-02 | |
| Steroid biosynthesis | ko00100 | 23 | 1.22E-12 | |
| Fatty acid biosynthesis | ko00061 | 12 | 9.34E-04 | |
| Ether lipid metabolism | ko00565 | 16 | 3.56E-02 | |
| Other metabolism | Protein processing in endoplasmic reticulum | ko04141 | 92 | 3.53E-03 |
| Protein digestion and absorption | ko04974 | 60 | 1.53E-04 | |
| Metabolic pathways | ko01100 | 448 | 2.46E-09 | |
| Glutathione metabolism | ko00480 | 26 | 2.67E-03 | |
| Caffeine metabolism | ko00232 | 8 | 3.56E-03 | |
| PPAR signaling pathway | ko03320 | 64 | 2.52E-06 | |
| Arachidonic acid metabolism | ko00590 | 38 | 6.51E-04 | |
| Vitamin digestion and absorption | ko04977 | 24 | 1.34E-03 | |
| Propanoate metabolism | ko00640 | 21 | 1.24E-02 | |
| Terpenoid backbone biosynthesis | ko00900 | 16 | 5.07E-06 | |
| alpha-Linolenic acid metabolism | ko00592 | 14 | 7.18E-03 | |
| Valine, leucine and isoleucine biosynthesis | ko00290 | 8 | 2.67E-03 | |
| Immunity | Phagosome | ko04145 | 114 | 1.09E-08 |
| HTLV-I infection | ko05166 | 107 | 2.67E-02 | |
| Amoebiasis | ko05146 | 104 | 2.07E-02 | |
| Epstein-Barr virus infection | ko05169 | 100 | 2.42E-02 | |
| Herpes simplex infection | ko05168 | 100 | 3.40E-02 | |
| Tuberculosis | ko05152 | 89 | 7.47E-03 | |
| Viral myocarditis | ko05416 | 73 | 2.46E-03 | |
| Antigen processing and presentation | ko04612 | 72 | 1.17E-13 | |
| Systemic lupus erythematosus | ko05322 | 68 | 2.59E-09 | |
| Pertussis | ko05133 | 63 | 1.38E-02 | |
| Complement and coagulation cascades | ko04610 | 60 | 3.61E-03 | |
| Natural killer cell mediated cytotoxicity | ko04650 | 59 | 1.50E-03 | |
| Staphylococcus aureus infection | ko05150 | 54 | 4.71E-04 | |
| Rheumatoid arthritis | ko05323 | 51 | 8.59E-06 | |
| Legionellosis | ko05134 | 49 | 2.67E-03 | |
| Leishmaniasis | ko05140 | 48 | 2.84E-04 | |
| Chagas disease (American trypanosomiasis) | ko05142 | 48 | 4.65E-02 | |
| Autoimmune thyroid disease | ko05320 | 41 | 3.43E-11 | |
| Allograft rejection | ko05330 | 40 | 3.43E-11 | |
| Prion diseases | ko05020 | 40 | 2.67E-02 | |
| Graft-versus-host disease | ko05332 | 27 | 5.09E-07 | |
| Primary immunodeficiency | ko05340 | 27 | 3.53E-03 | |
| Intestinal immune network for IgA production | ko04672 | 24 | 2.32E-04 | |
| African trypanosomiasis | ko05143 | 23 | 1.34E-03 | |
| Asthma | ko05310 | 22 | 8.22E-05 | |
| Others | Spliceosome | ko03040 | 113 | 3.29E-02 |
| Insulin signaling pathway | ko04910 | 91 | 1.34E-04 | |
| Alzheimer’s disease | ko05010 | 65 | 3.57E-02 | |
| Pancreatic secretion | ko04972 | 56 | 1.05E-05 | |
| Ribosome | ko03010 | 55 | 1.72E-06 | |
| Calcium signaling pathway | ko04020 | 55 | 1.38E-02 | |
| Adipocytokine signaling pathway | ko04920 | 45 | 1.09E-02 | |
| Gastric acid secretion | ko04971 | 42 | 1.18E-03 | |
| Circadian rhythm - mammal | ko04710 | 31 | 1.28E-06 | |
| DNA replication | ko03030 | 27 | 5.09E-07 | |
| Olfactory transduction | ko04740 | 23 | 2.95E-02 | |
| Phototransduction - fly | ko04745 | 19 | 3.40E-02 | |
| Proximal tubule bicarbonate reclamation | ko04964 | 18 | 3.48E-03 | |
| Mismatch repair | ko03430 | 16 | 8.76E-04 | |
| Circadian rhythm - fly | ko04711 | 15 | 4.32E-03 | |
| Protein export | ko03060 | 10 | 3.21E-02 |
Loading stress-induced liver injury
Glucose-6-phosphataseand glucokinase are rate-limiting enzymes in gluconeogenesis and glycolysis. Liver glucose-6-phosphatase and glucokinase revealed 13.2- and 1.4-fold increased expression after stressing, respectively (Figure 6; P < 0.05). Hormone-sensitive lipase and carnitine actyltransferase I are rate-limiting enzymes for adipokinetic action and fatty acid beta-oxidation. The RT-qPCR results also revealed 11.3- and 6.0-fold increased expression for liver hormone-sensitive lipase and carnitine actyltransferase I in stressed Coilia nasus (Figure 6; P < 0.05).
Figure 6.
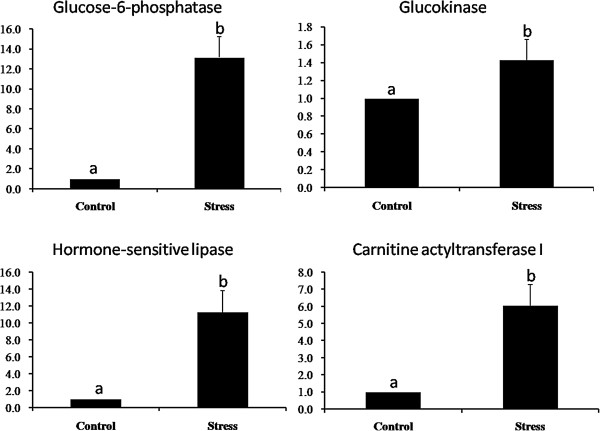
Glucose-6-phosphatase, glucokinase, hormone-sensitive lipase and carnitine actyltransferase I mRNA expression levels in liver in response to stress. The vertical axis shows the relative gene expression levels (means ± SD), bars represent the mean for each replicate ± SD. Replicates or treatments with different letters are significantly different at P < 0.05.
Lipid peroxides (LPO) are produced in the fatty acid beta-oxidation process, and are cytotoxic. The LPO content in the stressed group was significantly higher than that in the control group (P < 0.05; Table 5). We measured hepatic TNF-α levels in both control and stressed groups. As shown in Table 5, stress markedly increased hepatic TNF-α levels. Apoptosis-associated proteins caspase 9, cytochrome c and caspase 3 were significantly elevated in the stressed group (38.45 ± 1.39, 232.50 ± 3.54, 81.45 ± 3.23) compared with those in control group (34.34 ± 0.43, 187.22 ± 5.36, 68.28 ± 4.06; P < 0.05).
Table 5.
Levels of lipid peroxides and apoptosis-associated proteins
| Control | Stress | |
|---|---|---|
| LPO (nmol/mg) | 0.32 ± 0.03 | 0.44 ± 0.09a |
| TNFα (ng/L) | 4800.00 ± 494.97 | 7075 ± 106.07a |
| Caspase 9 (IU/L) | 34.34 ± 0.43 | 38.45 ± 1.39a |
| Cytochrome c (nmol/L) | 187.22 ± 5.36 | 232.50 ± 3.54a |
| Caspase 3 (IU/L) | 68.28 ± 4.06 | 81.45 ± 3.23a |
athe value in the stressed group is significantly different from that in the control group, P < 0.05.
Plasma aminotransferases were assayed to evaluate hepatotoxicity in Coilia nasusu. The ALT and AST concentrations in the control group were 4.10 ± 0.85 and 41.45 ± 7.79 IU/L, while in the stressed group the concentrations were 8.76 ± 1.08 and 202.12 ± 10.01 IU/L, respectively. The results indicate that significantly increasing plasma levels of ALT and AST developed following stress (Figure 7).
Figure 7.
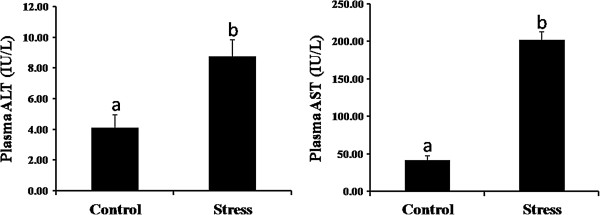
Liver injury induced by stress in Coilia nasus ( n = 15). Plasma samples were collected 0.5 h after challenge, and plasma transaminase (ALT and AST) concentrations were determined. Bars represent the mean for each replicate or treatment ± SEM. Replicates or treatments with different letters are significantly different at P < 0.05.
Discussion
The physiological and biochemical response to exposure to stressors has been studied widely in teleosts. One of the evolutionarily conserved stress responses is the rapid elevation in plasma cortisol and plasma glucose levels in response to stressor challenges [22]. In teleosts, the production of cortisol is under the control of the hypothalamus–pituitary–adrenal (HPA) axis [23, 24]. Cortisol is released from the adrenal tissue, resulting in up-regulation of the plasma glucose level. Therefore, plasma cortisol and plasma glucose can be used as indicators of the stress response. In our experiment, these parameters were significantly elevated in the stressed fish when compared with the control fish (Figure 1), this suggested that our sample could be used for transcriptome sequencing.
We found that 2,990 genes were significantly up-regulated, while 3,416 genes were significantly down-regulated in response to loading stress (Figure 5). On pathway enrichment analysis, “metabolism” and “immunity” were the two most frequently represented categories. In the “metabolism” category, “carbohydrate metabolism” and “lipid metabolism” were major subclasses, and included the “glycolysis/gluconeogenesis”, “starch and sucrose metabolism”, “glycerophospholipid metabolism” and “fat digestion and absorption” pathways. These carbohydrate metabolic pathways are mainly involved in blood glucose elevation (gluconeogenesis) and glucose utilization (glycolysis). The transcriptional expression of the rate-limiting enzymes (glucose-6-phosphatase and glucokinase) in this process was detected by RT-qPCR, both enzymes were significantly increased following stress (Figure 6). In addition, plasma glucose levels were significantly elevated after stress (Figure 1B), which provided further confirmation of the findings. These results are in agreement with studies that have reported higher activities of glycolytic enzymes after exposure to an acute stressor in fish, this may be necessary to cope with the increased energy demand of the liver, including enhanced gluconeogenesis, that is required to re-establish homeostasis [24]. Those two lipid metabolism pathways are involved mainly in fat digestion, absorption and oxidation. Transcript levels for the rate-limiting enzymes (hormone-sensitive lipase and carnitine actyltransferase I) in adipokinetic activity and the fatty acid beta-oxidation process were also significantly increased after stress.
Taken together, these results suggest that molecular regulation of enzymes critical for energy substrate mobilization and utilization is a key mechanism involved in acute stress adaptation in fish. However, problems may be associated with these processes, for example, the fatty acid beta-oxidation process may produce lipid peroxides (LPO) [25]. High levels of LPO or plasma glucose may induce an increase in TNF-α [26]. It is now well accepted that trimerization of the respective receptor by TNF leads to the assembly of the death-initiating signaling complex (disc) [27]. The disc consists of the death domain of the receptor, one or several associated proteins, and procaspase-8. Upon assembly of the disc, procaspase-8 is autocatalytically cleaved and activated [27]. The active caspase-8 can either process downstream effector caspases such as caspase-3, -6, and -7 directly [28, 29] or activate mitochondria as an amplification mechanism [30]. If mitochondria are involved, caspase-8 cleaves bid, a protein of the Bcl-2 family [31]. The C-terminal portion of the bid molecule inserts into the outer mitochondrial membrane and induces the release of cytochrome c into the cytosol [31, 32]. Cytochrome c, together with dATP, apoptosis activating factor-1 (Apaf-1) and procaspase-9, forms the apoptosome, which leads to the activation of caspase-9 and subsequently caspase-3 [33]. Caspase-3 and other effector caspases cleave death substrates, leading to apoptosis [34]. In our study, the levels of TNF-α, caspase 9, cytochrome c and caspase 3 in the liver were significantly elevated in the stressed fish compared with the control fish (Table 5). ALT and AST are important indicators of liver injury, and were also significantly elevated (Figure 7). These lines of evidence suggested that loading stress induces liver cell apoptosis mediated by TNF-α, which causes liver injury.
Conclusions
In the present study, using Illumina sequencing and bioinformatics analysis we analyzed the liver transcriptome of Coilia nasus that were stressed by loading. The main objective of this study was to annotate genes from this transcriptome analysis and identify potential stress gene and signaling pathways. On the basis of the transcriptome date, we explored the mechanism by which loading stress causes death in Coilia nasus. A total of 65,129 unigenes were generated, the mean unigene size and N50 were 607 bp and 813 bp, respectively. Of the assembled unigenes, we identified 2,990 genes that were significantly up-regulated, while 3,416 genes were significantly down-regulated in response to loading stress. Pathway enrichment analysis based on loading stress-responsive unigenes identified significantly stress-related pathways. “Metabolism” and “immunity” were the two most frequently represented categories. In the “metabolism” category, “glucose metabolism” and “lipid metabolism” were the major subclasses. The transcriptional expression of rate-limiting enzymes in “glucose metabolism” and “lipid metabolism” was investigated by RT-qPCR, all were significantly increased following stress. Apoptosis-associated proteins TNF-α, caspase 9, cytochrome c and caspase 3 were significantly elevated in the stressed group, indicators of liver injury (ALT and AST) were also significantly elevated, which indicates that loading stress induces liver injury in this fish.
Availability of supporting data
Raw sequencing data is available through the NCBI Sequence Read Archive under Project Accession SRP034828 (http://www.ncbi.nlm.nih.gov/). All samples were sequenced as 90 bp paired-end reads on an Illumina HiSeq 2000 sequencer.
Electronic supplementary material
Additional file 1: Table S1: Genes and specific primers used for real-time PCR. (DOCX 12 KB)
Additional file 2: Table S2: KEGG pathway analysis for all unigenes. (XLSX 21 MB)
Additional file 3: Table S3: List of the genes up-regulated or down-regulated in response to stress. (XLSX 2 MB)
Acknowledgments
This work was supported by a grant from the National Key Technology R&D Program (2012BAD26B05), the Three New Projects of Agricultural Aquaculture Program of Jiangsu Province (DZ2012-1, Y2013-33), and the National Special Research Fund for Non-Profit Sector (201203065).
Abbreviations
- RNA-Seq
RNA sequencing
- GO
Gene ontology
- RPKM
Reads/Kb/Million
- ALT
Alanine aminotransferase
- AST
Aspartate transaminase
- TNF-α
Tumor necrosis factor alpha
- Cyt c
Cytochrome c.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
FKD performed the bioinformatics analysis, qRT-PCR validation and drafted the manuscript. GCX and ZJN participated in RNA extraction, primer design, and bioinformatics analysis. RBG and PX contributed to the experimental design and manuscript editing. All authors read and approved the final manuscript.
Contributor Information
Fukuan Du, Email: adublg@126.com.
Gangchun Xu, Email: xugc@ffrc.cn.
Zhijuan Nie, Email: niezj@ffrc.cn.
Pao Xu, Email: xup@ffrc.cn.
Ruobo Gu, Email: gurb@ffrc.cn.
References
- 1.Barton BA, Iwama GK. Physiological changes in fish from stress in aquaculture with emphasis on the response and effects of corticosteroids. Ann Rev Fish Dis. 1991;1:3–26. doi: 10.1016/0959-8030(91)90019-G. [DOI] [Google Scholar]
- 2.Goede RW, Barton BA. Organismic indices and an autopsy-based assessment as indicators of health and condition of fish. Bethesda: American Fisheries Society; 1990. [Google Scholar]
- 3.Campbell PM, Pottinger TG, Sumpter JP. Preliminary evidence that chronic confinement stress reduces the quality of gametes produced by brown and rainbow trout. Aquaculture. 1994;120(1–2):151–169. doi: 10.1016/0044-8486(94)90230-5. [DOI] [Google Scholar]
- 4.Schreck CB, Contreras-Sanchez W, Fitzpatrick MS. Effects of stress on fish reproduction, gamete quality, and progeny. Aquaculture. 2001;197(1–4):3–24. doi: 10.1016/S0044-8486(01)00580-4. [DOI] [Google Scholar]
- 5.Alford PB, Tomasso JR, Bodine AB, Kendall C. Apoptotic death of Peripheral Leukocytes in Channel Catfish: effect of confinement-induced stress. J Aquat Anim Health. 1994;6(1):64–69. doi: 10.1577/1548-8667(1994)006<0064:ADOPLI>2.3.CO;2. [DOI] [Google Scholar]
- 6.Jiang T, Yang J, Liu H, Shen X-q. Life history of Coilia nasus from the Yellow Sea inferred from otolith Sr:Ca ratios. Environ Biol Fishes. 2012;95(4):503–508. doi: 10.1007/s10641-012-0066-6. [DOI] [Google Scholar]
- 7.Wiseman S, Osachoff H, Bassett E, Malhotra J, Bruno J, Vanaggelen G, Mommsen TP, Vijayan MM. Gene expression pattern in the liver during recovery from an acute stressor in rainbow trout. Comp Biochem Physiol Part D Genomics Proteomics. 2007;2(3):234–244. doi: 10.1016/j.cbd.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Aluru N, Vijayan MM. Hepatic transcriptome response to glucocorticoid receptor activation in rainbow trout. Physiol Genomics. 2007;31(3):483–491. doi: 10.1152/physiolgenomics.00118.2007. [DOI] [PubMed] [Google Scholar]
- 9.Moran PW, Aluru N, Black RW, Vijayan MM. Tissue contaminants and associated transcriptional response in trout liver from high elevation lakes of Washington. Environ Sci Technol. 2007;41(18):6591–6597. doi: 10.1021/es070550y. [DOI] [PubMed] [Google Scholar]
- 10.Villeneuve DL, Knoebl I, Larkin P, Miracle AL, Carter BJ, Denslow ND, Ankley GT. Altered gene expression in the brain and liver of female fathead minnows Pimephales promelas Rafinesque exposed to fadrozole. J Fish Biol. 2008;72(9):2281–2340. doi: 10.1111/j.1095-8649.2008.01897.x. [DOI] [Google Scholar]
- 11.Garcia-Reyero N, Griffitt RJ, Liu L, Kroll KJ, Farmerie WG, Barber DS, Denslow ND. Construction of a robust microarray from a non-model species largemouth bass, Micropterus salmoides (Lacèpede), using pyrosequencing technology. J Fish Biol. 2008;72(9):2354–2376. doi: 10.1111/j.1095-8649.2008.01904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quinn NL, Levenkova N, Chow W, Bouffard P, Boroevich KA, Knight JR, Jarvie TP, Lubieniecki KP, Desany BA, Koop BF, Harkins TT, Davidson WS. Assessing the feasibility of GS FLX Pyrosequencing for sequencing the Atlantic salmon genome. BMC Genomics. 2008;9:404. doi: 10.1186/1471-2164-9-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Y, Huang X, Yan Y, Cai J, Ouyang Z, Cui H, Wang P, Qin Q. Transcriptome analysis of orange-spotted grouper (Epinephelus coioides) spleen in response to Singapore grouper iridovirus. BMC Genomics. 2011;12:556. doi: 10.1186/1471-2164-12-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vera JC, Wheat CW, Fescemyer HW, Frilander MJ, Crawford DL, Hanski I, Marden JH. Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Mol Ecol. 2008;17(7):1636–1647. doi: 10.1111/j.1365-294X.2008.03666.x. [DOI] [PubMed] [Google Scholar]
- 15.Alagna F, D’Agostino N, Torchia L, Servili M, Rao R, Pietrella M, Giuliano G, Chiusano ML, Baldoni L, Perrotta G. Comparative 454 pyrosequencing of transcripts from two olive genotypes during fruit development. BMC Genomics. 2009;10:399. doi: 10.1186/1471-2164-10-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salem M, Rexroad CE, 3rd, Wang J, Thorgaard GH, Yao J. Characterization of the rainbow trout transcriptome using Sanger and 454-pyrosequencing approaches. BMC Genomics. 2010;11:564. doi: 10.1186/1471-2164-11-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pickering AD, Pottinger TG, Sumpter JP. On the use of dexamethasone to block the pituitary-interrenal axis in the brown trout, Salmo trutta L. Gen Comp Endocrinol. 1987;65(3):346–353. doi: 10.1016/0016-6480(87)90119-5. [DOI] [PubMed] [Google Scholar]
- 18.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Palma F, Birren BW, Nusbaum C, Lindblad T, Friedman N, Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Meth. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 20.Cabral HJ. Multiple comparisons procedures. Circulation. 2008;117(5):698–701. doi: 10.1161/CIRCULATIONAHA.107.700971. [DOI] [PubMed] [Google Scholar]
- 21.Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7(10):986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- 22.Barton BA, Morgan JD, Vijayan MM, Adams SM. Physiological and condition-related indicators of environmental stress in fish. Bethesda: American Fisheries Society; 2002. [Google Scholar]
- 23.Wendelaar Bonga SE. The stress response in fish. Physiol Rev. 1997;77(3):591–625. doi: 10.1152/physrev.1997.77.3.591. [DOI] [PubMed] [Google Scholar]
- 24.Mommsen T, Vijayan M, Moon T. Cortisol in teleosts: dynamics, mechanisms of action, and metabolic regulation. Rev Fish Biol Fisheries. 1999;9(3):211–268. doi: 10.1023/A:1008924418720. [DOI] [Google Scholar]
- 25.Yagi K. Lipid peroxides in biology and medicine. Tokyo: Academic Press; 1982. [Google Scholar]
- 26.Day CP, James OFW. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114(4):842–845. doi: 10.1016/S0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 27.Peter ME, Krammer PH. Mechanisms of CD95 (APO-1/Fas)-mediated apoptosis. Curr Opin Immunol. 1998;10(5):545–551. doi: 10.1016/S0952-7915(98)80222-7. [DOI] [PubMed] [Google Scholar]
- 28.Fernandes-Alnemri T, Armstrong RC, Krebs J, Srinivasula SM, Wang L, Bullrich F, Fritz LC, Trapani JA, Tomaselli KJ, Litwack G, Alnemri ES. In vitro activation of CPP32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc Natl Acad Sci U S A. 1996;93(15):7464–7469. doi: 10.1073/pnas.93.15.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Enari M, Talanian RV, Wong WW, Nagata S. Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature. 1996;380(6576):723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- 30.Kuwana T, Smith JJ, Muzio M, Dixit V, Newmeyer DD, Kornbluth S. Apoptosis induction by caspase-8 is amplified through the mitochondrial release of cytochrome c. J Biol Chem. 1998;273(26):16589–16594. doi: 10.1074/jbc.273.26.16589. [DOI] [PubMed] [Google Scholar]
- 31.Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem. 1999;274(2):1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- 32.Bossy-Wetzel E, Green DR. Caspases induce cytochrome c release from mitochondria by activating cytosolic factors. J Biol Chem. 1999;274(25):17484–17490. doi: 10.1074/jbc.274.25.17484. [DOI] [PubMed] [Google Scholar]
- 33.Jiang X, Wang X. Cytochrome c promotes Caspase-9 activation by inducing Nucleotide binding to Apaf-1. J Biol Chem. 2000;275(40):31199–31203. doi: 10.1074/jbc.C000405200. [DOI] [PubMed] [Google Scholar]
- 34.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326(Pt 1):1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1: Genes and specific primers used for real-time PCR. (DOCX 12 KB)
Additional file 2: Table S2: KEGG pathway analysis for all unigenes. (XLSX 21 MB)
Additional file 3: Table S3: List of the genes up-regulated or down-regulated in response to stress. (XLSX 2 MB)