

Abstract
Despite evidence that kinesin family member 14 (KIF14) can serve as a prognostic biomarker in various solid tumors, how it contributes to tumorigenesis remains unclear. We observed that experimental decrease in KIF14 expression increases docetaxel chemosensitivity in estrogen receptor–negative/progesterone receptor–negative/human epidermal growth factor receptor 2-negative, “triple-negative” breast cancers (TNBC). To investigate the oncogenic role of KIF14, we used noncancerous human mammary epithelial cells and ectopically expressed KIF14 and found increased proliferative capacity, increased anchorage-independent grown in vitro, and increased resistance to docetaxel but not to doxorubicin, carboplatin, or gemcitabine. Seventeen benign breast biopsies of BRCA1 or BRCA2 mutation carriers showed increased KIF14 mRNA expression by fluorescence in situ hybridization compared to controls with no known mutations in BRCA1 or BRCA2, suggesting increased KIF14 expression as a biomarker of high-risk breast tissue. Evaluation of 34 cases of locally advanced TNBC showed that KIF14 expression significantly correlates with chemotherapy-resistant breast cancer. KIF14 knockdown also correlates with decreased AKT phosphorylation and activity. Live-cell imaging confirmed an insulin-induced temporal colocalization of KIF14 and AKT at the plasma membrane, suggesting a potential role of KIF14 in promoting activation of AKT. An experimental small-molecule inhibitor of KIF14 was then used to evaluate the potential anticancer benefits of downregulating KIF14 activity. Inhibition of KIF14 shows a chemosensitizing effect and correlates with decreasing activation of AKT. Together, these findings show an early and critical role for KIF14 in the tumorigenic potential of TNBC, and therapeutic targeting of KIF14 is feasible and effective for TNBC.
Abbreviations: TNBC, triple-negative breast cancerestrogen-receptor negative/progesterone-receptor negative/Her2 negative; siRNA, small-interfering RNA; LC50, median lethal concentration
Introduction
Triple-negative breast cancer (TNBC) is clinically defined as approximately 20% of all breast cancers that lack the expression of estrogen receptor and progesterone receptor and overexpression of human epidermal growth factor receptor 2 or HER2. TNBC is increasingly recognized as a very heterogeneous disease with various molecular, genetic, and clinical subgroups [1–3]. The problem of molecularly targeting TNBC is trying to find oncogenic dependence across the heterogeneity of the disease. We previously performed a targeted RNA interference screen of the known somatic mutations found in breast cancer in a basal, claudin-low, TNBC cell line, MDA-MB-231 [4], representing possibly the most aggressive TNBC with characteristics of breast cancer–initiating or stem cells and with the poorest response to treatment [1,5,6]. We found that kinesin family member 14 (KIF14) stands out as a gene that, when knocked down, significantly increases sensitivity of multiple TNBC cell lines to docetaxel, one of the most commonly used chemotherapies for breast cancer. KIF14 is specifically expressed in 92% of all TNBCs, a much higher fraction than in the other clinical subtypes found in the Cancer Genome Atlas [4].
Kinesins are molecular motors important for intracellular transport [7,8]. KIF14 was initially found to be involved in cytokinesis by its localization at the central spindle and midbody along with its interaction with citron kinase and protein-regulating cytokinesis 1 [9,10]. KIF14 was first noted to be located in a region of genomic gain in multiple cancers [11] and has subsequently been found to be a significant prognostic biomarker and a likely oncogene in breast cancer, lung cancer, ovarian cancer, retinoblastomas, and gliomas [12–15]. As an ATPase, it also has potential as a therapeutic target [16]. KIF14 can act as a scaffold protein tethering the Rap1 effector Radil to microtubules to regulate integrin signaling [17]. In the present study, we show that increased KIF14 expression correlates with resistance to neoadjuvant chemotherapy in patients with locally advanced TNBC and that decreased expression of KIF14 leads to chemosensitization that may be due to decreased prosurvival pathways as a consequence of decreased AKT activity. We demonstrate that increased KIF14 expression is detected in benign breast tissue of high-risk BRCA mutation carriers and may be an early biomarker for tumor progression. We further show that KIF14 is a druggable target that could be exploited to achieve therapeutic efficacy.
Materials and Methods
Plasmids and Transfection
Myristoylated pCMV6 AKT1 was gift from M. White [18] (Department of Cell Biology, University of Texas Southwestern Medical Center, Dallas, TX). pEGFP-KIF14 was a gift from F. Barr [9]. pDsRed2-N1 was obtained from Clontech Laboratories (Mountain View, CA), and pDsRed2-Akt1 was cloned from pCMV6 AKT1 with BamH1/EcoR1 (New England Biolabs, Ipswich, MA) restriction sites. cDNA transfections were performed with Lipofectamine LTX Reagent (Invitrogen, Grand Island, NY) as per manufacturer's protocol.
Human Tissues
Benign, deidentified breast biopsies were obtained from the Komen Tissue Bank (Indianapolis, IN) and University of Texas Southwestern Tissue Repository (Dallas, TX). Primary human breast tumors were deidentified and obtained from Parkland Hospital (Dallas, TX) and used with approval from Parkland Hospital and University of Texas Southwestern Medical Center Institutional Review Boards. All primary human breast tumors examined in this study underwent treatment with docetaxel or paclitaxel-containing chemotherapy regimen given with doxorubicin.
Fluorescence In Situ Hybridization
Stellaris mRNA FISH probes against KIF14 and CAL Fluor Red 610 were obtained from Biosearch Technologies (Novato, CA), and hybridization was performed as per manufacturer's protocol. Images were obtained with Deltavision (Applied Precision, Issaquah, WA) and quantification with ImageJ (National Institute of Health, Bethesda, MD). Specificity of the KIF14 mRNA fluorescence in situ hybridization (FISH) probes was performed with MDA-MB-231 cells with knocked-down KIF14 compared to parental cells (data not shown). Four commercially available KIF14 antibodies tested were No. A300-912A from Bethyl Laboratories (Montgomery, TX), No. ABT46 from Millipore (Billerica, MA), and No. 48562 and No. 365553 from Santa Cruz Biotechnology (Dallas, TX).
Expression Array Analysis
Expression array data from FACS-sorted mammary populations were obtained from Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/) (National Center for Biotechnology Information, Bethesda, MD) with Accession No. GSE16997 [19]. Hierarchical clustering was used to segregate samples, and all analyses were performed with R/Bioconductor (Fred Hutchinson Cancer Research Center, Seattle, WA).
Western Blot Analysis and Immunoprecipitation
Cell lysates were prepared by harvesting cells in Laemmli sodium dodecyl sulfate reducing buffer, resolved on an 8% to 10% polyacrylamide gel, and transferred to a polyvinylidine difluoride membrane. The following antibodies were from Cell Signaling Technology (Beverly, MA): Glyceraldehyde 3-phosphate dehydrogenase (2118), phospho-AKT pathway antibody sampler (9916), p21 (2947), p62 (8025), and pS6K (9234). KIF14 antibody was from Bethyl Laboratories. Detection of peroxidase activity from HRP-conjugated antibodies was performed with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, Rockford, IL). Images were captured with the G:BOX-F3 with GeneSys software (Syngene, Frederick, MD). Cells for immunoprecipitation were harvested with nondenaturing cell lysis buffer (No. 9803; Cell Signaling Technology) supplemented with protease inhibitors (Millipore, Darmstadt, Germany) and further lysed with 22-gauge needle and syringe. Cell lysates were incubated at 4°C overnight with pan-AKT antibody at 1:50 (Cell Signaling Technology) or immunoglobulin control. Protein A/G agarose beads (Santa Cruz Biotechnology) were incubated with cell lysates for 4 hours at 4°C and washed with phosphate-buffered saline–Tween(R)20 before sodium dodecyl sulfate–polyacrylamide gel electrophoresis and subsequent immunoblot analysis.
Anchorage-Independent Colony Formation Assay
Cultured cells were seeded in 0.375% Noble agar (Difco, Sparks, MD) in a 24-well plate on top of 0.5% presolidified agar. Each density was seeded in triplicates, and each assay was performed two separate times. After 21 days, wells were imaged with SteREO scope (Carl Zeiss, Thornwood, NY) and scored for macroscopically visible colonies > 1mm. GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA) was used to plot data and perform two-tailed Student's t tests.
Cell Lines and RNA Interference
All cell lines, culture conditions, and the generation of stable KIF14 knockdowns are as described previously [4]. The two KIF14 No. 1 and No. 2 small-interfering RNAs used are J-003319-05 and J-003319-06, respectively, from Dharmacon/Thermo Scientific. Unless specified, siKIF14 refers to J-003319-06.
Kinase Assay
Evaluation of KIF14 inhibitor (KIF14i) activity against phosphoinositide 3-kinase (PI3K) (p110α/p85α) and AKT1 was performed by SignalChem Lifesciences Corp (Richmond, British Colombia). Profiling of KIF14i activity in duplicate was performed at four concentrations (10 nM, 100 nM, 1000 nM, and 10,000 nM). Staurosporine and wortmanin were used as positive controls against AKT1 and PI3K, respectively. 33P-ATP was used for AKT1 radioisotope assay, whereas the ADP-Glo assay kit from Promega (Madison, WI) was used for the PI3K assay.
Viability Assays
Chemicals or DMSO as control was added to cells at 60% confluency, and cell viability was determined 72 hours later with CellTiter-Glo or CytoTox-Glo (Promega) as per manufacturer's protocols. Whereas viability is directly inferred from luminescent signal in the CellTiter-Glo assay, viability in the CytoTox-Glo assay is calculated by subtracting the luminescent signal resulting from experimental agent–induced cell death from total luminescent values after addition of lysis reagent. Data are means from two independent experiments performed in triplicate. Reported median lethal concentration (LC50) values are calculated from dose-response curves obtained with CellTiter-Glo assays. Direct comparisons of the two assays on experiments performed in parallel are shown in Figure W1. Unless otherwise specified, all viability assays in the figures are performed with CellTiter-Glo assays.
Chemicals
Insulin (Sigma, St Louis, MO) was used at 100 nM for 30 minutes unless otherwise specified. Docetaxel, doxorubicin, gemcitabine, and carboplatin were obtained from Sigma. MK2206 was obtained from Selleck Chemicals (Houston, TX).
Synthesis of KIF14i
(E)-2-(4-isopropyl-3-nitrobenzylidene)hydrazinecarbothioamide [20]
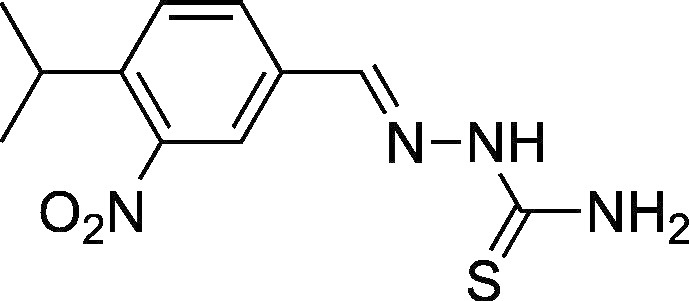
On the basis of a published method [21], 4-isopropyl-3-nitrobenzaldehyde (100 mg, 0.52 mmol, 1 eq) and thiosemicarbazide (47.2 mg, 0.52 mmol, 1 eq) were combined with MeOH (10 ml, 0.05 M). The mixture (which was poorly soluble at room temperature) was heated to 70°C in a capped 1-dram vial for 6 hours. The mixture was then concentrated and purified by column chromatography (155%-100% EtOAc/hexanes) to give a cream-colored solid (116.1 mg, 0.436 mmol, 84%). 1H nuclear magnetic resonance (500 MHz, DMSO-d6) spectrum showed δ 11.56 (s, 1H), 8.32 (d, J = 1.7 Hz, 1H), 8.28 (s, 1H), 8.21 (s, 1H), 8.05 (s, 1H), 7.97 (dd, J = 8.2, 1.7 Hz, 1H), 7.63 (d, J = 8.2 Hz, 1H), 3.12 (hept, J = 6.6 Hz, 1H), and 1.25 (d, J = 6.8 Hz, 6H). 13C nuclear magnetic resonance (126 MHz, DMSO) spectrum showed δ 178.24, 150.07, 141.60, 139.47, 133.61, 131.22, 127.98, 120.75, 28.28, and 23.10.
Statistical Analysis
All experiments were performed at least three times except for viability assays that were performed twice with data as means from triplicates. Means were compared using Student's t test except analysis of covariance was used to compute the P value for the survival curves. All statistical tests were two sided, with P < .05 taken as significance threshold.
Live-cell Imaging and Image Processing
Cells were seeded onto chambered cover glass (Thermo Scientific) at 70% confluency. Images were obtained with Deltavision (Applied Precision) with 60 × oil immersion lens, whereas cells were in enclosure supplied with 5% CO2 and kept at 37°C. Forty-micron sections were obtained at 10 to 15 minutes apart for up to 2 hours. Images were then generated into .lsm files and were processed through the Imaris software (Bitplane AG, Zurich, Switzerland) for 3-dimensional reconstruction.
Results
KIF14 Overexpression is Correlated with Increased Proliferation and Anchorage-Independent Growth In Vitro
To investigate if increased KIF14 expression is sufficient to confer an oncogenic phenotype to benign cells, we generated stably transfected pEGFP-KIF14 in benign, normal, diploid, telomerase reverse transcriptase-immortalized noncancerous human mammary epithelial cells (HME1) (Figure 1A). We found that KIF14 modestly overexpressing cells proliferate significantly faster compared to vector-transfected cells (doubling time for vector-control and pEGFP KIF14 cells are 2.8 days vs 3.1 days, respectively; P = .0021 by Student's t test for values at 8 days after seeding in equal numbers, Figure 1B). Increased KIF14 expression in HME1 cells is sufficient to increase anchorage-independent growth (Figure 1, C and D). Decreased KIF14 expression correlates with decreased migratory ability of breast cancer cell lines [17]. However, increased KIF14 expression in HME1 cells is not sufficient to increase the ability of these cells to migrate through extracellular matrices such as Matrigel™ (BD Biosciences, San Jose, CA) or to increase tumorigenicity of HME1 cells in mammary fat pads of immunocompromised mice (data not shown).
Figure 1.

Functional assays of KIF14 overexpression in benign breast epithelial cells and expression of KIF14 in benign breast biopsies of BRCA mutation carriers. (A) Immunoblot of HME1 cells stably transfected with pEGFP vector and pEGFP KIF14. (B) Proliferation of HME1 cells when transfected with vector only versus pEGFP KIF14. P value from two-sided Student's t test compares the manually counted number of cells at 8 days between the two groups. (C) Representative images of soft agar colony formation. (D) Quantification of soft agar colony formation of HME1 cells when transfected with vector only versus pEGFP KIF14. Results in B and D represent triplicates performed two independent times. (E) Representative KIF14 mRNA FISH images from a BRCA1 mutation carrier and age-matched control (red denotes KIF14 mRNA foci; green denotes nuclei). Scale bar, 20 μm. (F) Summary quantification of KIF14 mRNA FISH between BRCA mutation carrier and control subjects with no known mutations. All P values are from unpaired two-sided Student's t test. All error bars represent SEM. (G) Hierarchical clustering of GSE16997 showing KIF14 expression among subpopulations of benign mammary epithelial cells. Each GEO Sample number refers to a microarray. Each microarray was performed on a subpopulation of benign breast cells from one of three human subjects that were FACS sorted into four populations (luminal progenitor, mammary stem-cell enriched, mature luminal, and stromal). The luminal progenitor subpopulation from all three human subjects expressed higher KIF14 expression than any other subpopulation from each subject.
KIF14 Genetic Alteration is an Early Event in Breast Cancer Pathogenesis
Recent whole-genome sequencing efforts have shown that multiple mutations are acquired during breast carcinogenesis [2,22–25]. KIF14 is located at chromosome 1q, gain of which has been found to be one of the earliest genetic alterations seen in breast cancer [23]. KIF14 is found to be altered in 75 of 825 (9%) of breast cancer cases in The Cancer Genome Atlas, with 40 of 75 (53%) of KIF14 alterations being copy number amplification (cbioportal.org). Because BRCA1 or BRCA2 mutation carriers have 60% to 80% lifetime risk of developing breast cancer [26,27], we hypothesized that early events that drive tumorigenesis may be present before overt cancer is detected. We examined KIF14 expression in 35 benign breast cases, from the Komen Tissue Bank and the University of Texas Southwestern Medical Center Tissue Repository. Of these, 16 had BRCA1 or BRCA2 mutations, whereas 19 of them had no known BRCA1 or BRCA2 mutations. No commercially available KIF14 antibodies for immunohistochemistry were reliable (see Materials and Methods section for the four antibodies we tested). We found that KIF14 expression by mRNA FISH (i.e., the average number of cytoplasmic mRNA foci per 50 cells) was significantly increased in BRCA mutation carriers (Figure 1, E and F). Interestingly, KIF14 mRNA expression was seen in the luminal epithelial layer, where aberrant luminal progenitors for basal tumor development in BRCA1 mutation carriers have been found [19]. We evaluated KIF14 expression in expression arrays of subpopulations of cells within dissociated mammary glands [19] by clustering analysis and found that KIF14 expression is highest among luminal progenitors (Figure 1G).
KIF14 Overexpression Correlates with Chemoresistance
Another key oncogenic phenotype is resistance to apoptosis. We evaluated if KIF14 expression correlates with response to neoadjuvant chemotherapy in TNBC. We examined 68 locally advanced TNBCs that had undergone neoadjuvant chemotherapy from January 2008 to January 2012 from a single large community hospital in Dallas, TX, and found that 18 cases (26.5%) had pathologic complete response (pCR), whereas 10 (14.7%) had resistant disease despite chemotherapy (tumor at the time of surgical resection was the same or larger size as measured by ultrasound at the time of diagnosis). KIF14 mRNA FISH up-regulation correlated with chemoresistant tumors, whereas low KIF14 expression correlated with pCR (Figure 2, A and B).
Figure 2.

KIF14 expression correlates with response to chemotherapy. (A) KIF14 mRNA FISH (red denotes KIF14 mRNA foci; green denotes nuclei) on representative breast tissues from patients with locally advanced TNBC with known chemosensitive (i.e., pCR) versus chemoresistant disease. Scale bar, 25 μM. (B) Summary scatterplot of KIF14 mRNA expression among patients with pCR and resistant disease. P values are from paired two-sided Student's t test. Error bars represent SEM. Relative viability of HME1 cells transfected with vector only versus pEGFP KIF14 when given indicated dosages of docetaxel (C) or doxorubicin (D) for 72 hours. Relative viability of MDA-MB-231 (E), HCC38 (F), or Htb126 (G) with si-control versus si-KIF14 (J-003319-06; see Materials and Methods section) when given indicated dosages of docetaxel. P < .0001 by analysis of covariance for C, E, F, and G.
To further evaluate if KIF14 expression modulates chemoresistance, we evaluated if increased KIF14 expression in HME1 is sufficient to significantly increase resistance to docetaxel, doxorubicin, carboplatin, or gemcitabine, a few of the most commonly used chemotherapies given for breast cancer, in cell viability assays (see Materials and Methods section). We found that when we overexpressed KIF14 in HME1, cells were significantly more resistant to docetaxel but not to doxorubicin (Figure 2, C and D, and Figure W1, A and B), nor carboplatin or gemcitabine (data not shown) (docetaxel LC50 for vector-control and pEGFP KIF14 were 0.002 and 0.013 μM, respectively; P < .0001 by analysis of covariance). Although KIF14 overexpression may lead to increased proliferative capacity (Figure 1B), because viability assays are performed within 3 days of drug administration, increased proliferation rate alone (~ 10%) is unlikely to account for increased docetaxel chemoresistance seen in KIF14-overexpressing HME1 cells. In addition, we decreased KIF14 expression by siRNA in TNBC cell lines (MDA-MB-231, HCC38, and Htb126) and found significant chemosensitization when cells were exposed to docetaxel (Figure 2, E–G, and Figure W1, C–E).
Because AKT activation is antiapoptotic and is known to correlate with chemoresistance [28,29], we evaluated the expression of pAKT in various TNBC cell lines by immunoblot analysis (Figure 3A). We used insulin stimulation to identify the cell lines that we could use to study the temporal activation of AKT because most TNBC cell lines shown have high basal expression of pAKT (Figure 3A). We found that phosphorylation of AKT is a rapid event with significant phosphorylation occurring in less than 30 minutes in MDA-MB-231 cells (Figure 3B). To evaluate the potential role of KIF14 in growth factor–inducible AKT phosphorylation, we cotransfected pEGFP-KIF14 with pDsRed2-Akt1 in TNBC cells (Figure 3, C and D) and found that colocalization (denoted by yellow signal) of KIF14 (green) and AKT1 (red) is most prominent at the plasma membrane (Figure 3C) where phosphorylation of AKT1 takes place by PI3 kinase [30]. We further performed immunoprecipitation using total AKT antibody under nondenaturing conditions and found that there is endogenous association of AKT with KIF14 (Figure 3E).
Figure 3.

Insulin-induced phosphorylation of AKT and KIF14 localization. (A) Immunoblot of pAKT in various TNBC cell lines and their response to insulin treatment. (B) Immunoblot shows time course of AKT phosphorylation in MDA-MB-231 cells. (C) Images from live-cell imaging of HCC38 (top panels) and MDA-MB-231 (bottom panels) at indicated time points after insulin treatment. Arrows in top panels indicate a marked signal (yellow, indicating colocalization of red and green signals; green, KIF14; red, AKT). Scale bars in top and bottom panels, 15 μm and 10 μm, respectively. (D) Immunoblot of KIF14 and AKT showing specific signals for KIF14 and AKT from transfected vectors used in C. (E) Immunoblot of KIF14 after MDA-MB-231 cell lysates were immunoprecipitated with pan-AKT.
Chemosensitization with KIF14 Knockdown is Associated with Decreased pAKT
We previously observed that multiple TNBC cell lines with KIF14 knockdown were significantly more sensitive to docetaxel compared to control parental cell lines [4]. This suggests a prosurvival advantage that KIF14 may confer on these cell lines when under cytotoxic stress, consistent with our finding of increased docetaxel resistance when KIF14 is overexpressed (Figure 2C). The PI3K/Akt pathway is a well-known prosurvival pathway in breast cancer with 57% of 507 sequenced breast tumors in The Cancer Genome Atlas containing at least one mutation in AKT1, phosphatase and tensin homolog, or phosphatidylinositol-4,5-bisphosphate 3-kinase [31]. Because KIF14 is highly overexpressed in TNBC [4], we tested if KIF14 expression confers a survival advantage through activation of AKT. We chose MDA-MB-231 cells to study the effect of shKIF14 on PI3K/Akt pathway activation because this cell line does not have any mutations in phosphatidylinositol-4,5-bisphosphate 3-kinase, AKT1, or phosphatase and tensin homolog [32]. We examined a small panel of genes involved in the PI3K/Akt signaling pathway [33-35] in MDA-MB-231 cells with KIF14 knockdown using two distinct siRNAs and re-expression (rescue) using pEGFP KIF14 and found that decreased KIF14 correlates with decreased levels of phosphorylated or activated AKT1 (at S473 and T308), phospho-glycogen synthase kinase-3beta (S9), and pS6K (T389) along with decreased levels of p62 and p21 (Figure 4A). However, expression of phospho-Phosphoinositide-dependent kinase-1 (S241) remained the same (Figure 4A) indicating that KIF14 expression correlates more with activation of AKT, not PI3K. Although decreased KIF14 expression was associated with decreased pAKT, we also investigated if decreased activation of AKT per se is responsible for chemosensitization with KIF14 knockdown. We tested if activated AKT can rescue the chemosensitization seen in siKIF14 cells and found that constitutively active myristoylated Akt1 reverses docetaxel chemosensitivity seen in siKIF14 cells as effectively as reconstituted KIF14 (LC50, in μM, are as follows: si-control = 9.912e-3, siKIF1 = 1.846e-3, siKIF14+pEGFP KIF14 = 6.851e-3, siKIF14 + myristoylated plasmid-Cytomegalovirus-6 (pCMV6) Akt1 = 1.484e-2) (Figure 4B).
Figure 4.

KIF14 expression is correlated with pAKT in MDA-MB-231. (A) Immunoblot analysis of various proteins associated with the PI3K/AKT pathway in MDA-MB-231 cells with knockdown of KIF14 and reconstituted KIF14 expression. (B) Relative viability of MDA-MB-231 cells transfected with si-control, siKIF14 No. 2, pEGFP KIF14, or myristoylated pCMV6 Akt1 when given indicated concentrations of docetaxel for 72 hours. Results represent triplicates performed two independent times. Error bars represent SEM.
KIF14 Chemical Inhibition is Associated with Decreased AKT Activation and Chemosensitization
A putative KIF14i, (E)-2-(4-isopropyl-3-nitrobenzylidene)hydrazinecarbothioamide, was identified in a screen for small molecules that selectively inhibit the ATPase activity of KIF14 [20]. This molecule has an inhibitory concentration at 50% for KIF14 ATPase activity in vitro of 54 nM [20]. We synthesized this compound (Figure 5A; see Materials and Methods section) and showed that as a monotherapy, KIF14i decreases viability of MDA-MB-231, HCC38, and Htb126 cells (− LC50 = 11.14, 1.998, and 3.196 μM, respectively; Figure 5B and Figure W1F).
Figure 5.

Chemical inhibition of KIF14 results in decreased pAKT levels. (A) Chemical structure of KIF14i. (B) Relative viability of MDA-MB-231, HCC38, and Htb126 cells when given indicated dosages of KIF14i for 72 hours. Data are from triplicates performed two independent times. Error bars represent SEM. (C) Immunoblot analysis of MDA-MB-231 cells treated with indicated doses of KIF14i in the presence or absence of docetaxel for 24 hours. Relative viability of MDA-MB-231 (D), HCC38 (E), or Htb126 (F) with si-control versus si-KIF14 (J-003319-06; see Materials and Methods section) or when si-control cells are treated also with 5-μM KIF14i or 0.25-μM MK2206 when given indicated dosages of docetaxel.
KIF14i has a concentration-dependent effect on AKT phosphorylation and the amount of cleaved caspase 3 (as an indicator of apoptosis) in MDA-MB-231 cells (Figure 5C). KIF14i has no activity against AKT1 (see Materials and Methods section) and only minimal activity against PI3K (p110α/p85α) (12% activity decrease at 1 μM and 27% decrease at 10 μM, respectively; see Materials and Methods section). We also evaluated the effects of MK-2206 (an AKT inhibitor currently in clinical trials for breast cancer) on TNBC cells in comparison with KIF14i. MK2206 has single-agent activity (Figure W2) and is also effective when given with chemotherapy [36]. We found that there is a comparable degree of chemosensitization when 0.25-μM MK-2206 or 5-μM KIFi is given with docetaxel in MDA-MB-231, HCC38, and Htb126 and that these chemical inhibitors mimic the effects of KIF14 knockdown by siRNA (Figure 5, D–F).
Discussion
There is an increasing number of genes found mutated in breast cancer with unclear mechanistic underpinnings. We found KIF14 to be significantly overexpressed in TNBC and that this expression correlated with poor prognosis. Importantly, we determined that decreased KIF14 expression sensitized breast cancer cells to docetaxel [4]. We now provide evidence that KIF14 may be a driver gene in carcinogenesis, modulated through the PI3K/AKT pathway, and is a potential therapeutic target for treatment of chemoresistant breast cancers.
KIF14 has been implicated in various studies as oncogenic, but its relative significance as a driver gene in breast cancer pathogenesis remained unclear [11–13,17]. In the present studies, we demonstrate that increased KIF14 expression is sufficient to increase proliferative capacity and increase anchorage-independent growth of noncancerous mammary epithelial cells in vitro. BRCA1 or BRCA2 mutations are associated with genetic instability and gross chromosomal abnormalities [37–40]. We show that increased KIF14 expression can be seen in benign, luminal breast tissue of high-risk BRCA mutation carriers before overt cancer is detected and thus may be a novel biomarker for high-risk breast tissue. It remains to be determined whether the population of luminal cells that overexpress KIF14 correspond to the expanded luminal progenitor population that may be preneoplastic in BRCA mutation carriers [19]. Our finding of higher KIF14 expression among luminal progenitors suggests that KIF14 may act as an oncogene in luminal progenitors in BRCA carriers. This is important for future studies where cancer progression can be studied in a FACS-isolatable and increasingly well-defined subpopulation of breast epithelial cells rather than a heterogeneous mix of cells from benign mammary gland.
Drug resistance, either intrinsic or acquired, is a major hurdle in cancer treatment. KIF14 is a biomarker of chemotherapy resistance, and the study of molecular pathways associated with KIF14 chemoresistance is important for further understanding its clinical implication in the treatment of TNBC. KIF14 is a known microtubule-binding kinesin motor. We have shown that KIF14 expression specifically affects chemosensitivity to docetaxel, a microtubule-targeting agent. In addition, the activation of the PI3K/AKT pathway initially occurs at the plasma membrane where PI3K is localized. We show by live-cell imaging that AKT movement toward the plasma membrane under insulin stimulation is rapid and colocalizes with KIF14. Whether KIF14 interacts directly with AKT is undetermined although our findings support that insulin-stimulated Akt1 phosphorylation may be coupled to KIF14. An oncogenic role of KIF14 at the plasma membrane has been suggested where KIF14 was found to negatively regulate Rap1a-Radil signaling during breast cancer progression [17].
Multiple components of the PI3K/AKT pathway have been implicated in chemotherapy resistance, and direct targeting of PI3K, AKT1, and mammalian target of rapamycin is promising in the treatment of breast cancer [29,41,42]. We show here that Akt1 phosphorylation is affected by KIF14 knockdown, suggesting a significant role for KIF14 in the activation of the PI3K/AKT pathway in subsets of breast cancer. Because constitutively active Akt1 (myristoylated Akt1) reverses chemosensitization of shKIF14 in MDA-MB-231 cells, activation of Akt1 appears to be a potential molecular mechanism for chemoresistance seen with KIF14 overexpression.
The complexity of cellular functions and regulations of Akt1 may render it a challenging molecular target. In contrast, scaffolding proteins such as molecular motors that tether signaling components and help to localize signaling components in the cell may be potential targets that can disrupt multiple aberrant cellular processes simultaneously. Of note, small-molecule inhibitors of dynein, another motor, have been described with potential utility in drug-resistant forms of medulloblastoma and basal cell carcinoma [43]. Here, we demonstrate that chemical inhibition of KIF14 is feasible and targeting KIF14 may increase chemosensitivity in TNBC. KIF14 is expressed at only low levels in normal human mammary epithelial cells and postnatal tissues [11], suggesting that a specific KIF14 inhibitor may provide a high therapeutic index. We show here that high KIF14 expression in primary tumors correlates with chemoresistance. The disease-associated up-regulation of KIF14 and its apparent association with activation of AKT render it an attractive therapeutic target.
Acknowledgments
We thank the Komen Foundation and the Harold Simmons Cancer Center Tissue Repository for tissue materials. Normal breast tissue samples from the Susan G. Komen Tissue Bank at the Indiana University Simon Cancer Center were used in this study. We thank contributors, including Indiana University who collected samples used in this study, as well as donors and their families, whose help and participation made this work possible. We also thank Yi-Hung Ou, Michael White, and Melanie Cobb (Department of Cell Biology, Department of Pharmacology, University of Texas, Southwestern Medical Center) for helpful discussions.
Footnotes
This work was supported by the Simmons Cancer Center Support grant (5P30 CA 142543–03), T32CA136515 from the National Cancer Institute, and Susan G. Komen Foundation Postdoctoral Fellowship (to S.M.S.), an Alcon Research Institute Young Investigator Award (to T.W.C.), and The Southland Financial Corporation Distinguished Chair in Geriatric Research (to J.W.S). L.L. was supported by Welch (I-1665) and National Institute of Health (R01CA168761). This work was performed in laboratories constructed with support from National Institutes of Health grantC06 RR30414. This work was also supported in part by the Indiana Clinical and Translational Sciences Institute funded, in part, by the National Institutes of Health, National Center for Advancing Translational Sciences, and Clinical and Translational Sciences Awards (TR000163; to T.W.C.). Conflict of interest: The authors declare no conflict of interest.
This article refers to supplementary materials, which are designated by Figures W1 and W2 and are available online at www.neoplasia.com.
Appendix A. Supplementary materials
Figure W1.

Relative viability of HME1 cells transfected with vector only versus pEGFP KIF14 when given indicated dosages of docetaxel (A) or doxorubicin (B) for 72 hours. Relative viability of MDA-MB-231 (C), HCC38 (D), or Htb126 (E) with si-control versus si-KIF14 (J-003319-06; see Materials and Methods section) when given indicated dosages of docetaxel. Relative viability of MDA-MB-231 cells when given indicated dosages of KIF14i for 72 hours. Data are from triplicates performed two independent times. Error bars represent SEM. Solid lines indicate data from CellTiter-Glo, and dashed lines indicate data from CytoTox-Glo assays.
Figure W2.

Relative viability of MDA-MB-231, HCC38, and Htb126 cells when given indicated dosages of MK2206 for 72 hours. Dose-response curves were generated with data from CellTiter-Glo. Data are from triplicates performed two independent times. Error bars represent SEM.
References
- 1.Lehmann B.D., Bauer J.A., Chen X., Sanders M.E., Chakravarthy A.B., Shyr Y., Pietenpol J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shah S.P., Roth A., Goya R., Oloumi A., Ha G., Zhao Y., Turashvili G., Ding J., Tse K., Haffari G. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu K.D., Zhu R., Zhan M., Rodriguez A.A., Yang W., Wong S., Makris A., Lehmann B.D., Chen X., Mayer I. Identification of prognosis-relevant subgroups in patients with chemoresistant triple-negative breast cancer. Clin Cancer Res. 2013;19:2723–2733. doi: 10.1158/1078-0432.CCR-12-2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singel S.M., Cornelius C., Batten K., Fasciani G., Wright W.E., Lum L., Shay J.W. A targeted RNAi screen of the breast cancer genome identifies KIF14 and TLN1 as genes that modulate docetaxel chemosensitivity in triple-negative breast cancer. Clin Cancer Res. 2013;19:2061–2070. doi: 10.1158/1078-0432.CCR-13-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Creighton C.J., Li X., Landis M., Dixon J.M., Neumeister V.M., Sjolund A., Rimm D.L., Wong H., Rodriguez A., Herschkowitz J.I. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106:13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prat A., Parker J.S., Karginova O., Fan C., Livasy C., Herschkowitz J.I., He X., Perou C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirokawa N., Noda Y., Tanaka Y., Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- 8.Verhey K.J., Hammond J.W. Traffic control: regulation of kinesin motors. Nat Rev Mol Cell Biol. 2009;10:765–777. doi: 10.1038/nrm2782. [DOI] [PubMed] [Google Scholar]
- 9.Gruneberg U., Neef R., Li X., Chan E.H., Chalamalasetty R.B., Nigg E.A., Barr F.A. KIF14 and citron kinase act together to promote efficient cytokinesis. J Cell Biol. 2006;172:363–372. doi: 10.1083/jcb.200511061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carleton M., Mao M., Biery M., Warrener P., Kim S., Buser C., Marshall C.G., Fernandes C., Annis J., Linsley P.S. RNA interference-mediated silencing of mitotic kinesin KIF14 disrupts cell cycle progression and induces cytokinesis failure. Mol Cell Biol. 2006;26:3853–3863. doi: 10.1128/MCB.26.10.3853-3863.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corson T.W., Huang A., Tsao M.S., Gallie B.L. KIF14 is a candidate oncogene in the 1q minimal region of genomic gain in multiple cancers. Oncogene. 2005;24:4741–4753. doi: 10.1038/sj.onc.1208641. [DOI] [PubMed] [Google Scholar]
- 12.Corson T.W., Gallie B.L. KIF14 mRNA expression is a predictor of grade and outcome in breast cancer. Int J Cancer. 2006;119:1088–1094. doi: 10.1002/ijc.21954. [DOI] [PubMed] [Google Scholar]
- 13.Corson T.W., Zhu C.Q., Lau S.K., Shepherd F.A., Tsao M.S., Gallie B.L. KIF14 messenger RNA expression is independently prognostic for outcome in lung cancer. Clin Cancer Res. 2007;13:3229–3234. doi: 10.1158/1078-0432.CCR-07-0393. [DOI] [PubMed] [Google Scholar]
- 14.Thériault B.L., Pajovic S., Bernardini M.Q., Shaw P.A., Gallie B.L. Kinesin family member 14: an independent prognostic marker and potential therapeutic target for ovarian cancer. Int J Cancer. 2012;130:1844–1854. doi: 10.1002/ijc.26189. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q., Wang L., Li D., Deng J., Zhao Z., He S., Zhang Y., Tu Y. Kinesin family member 14 is a candidate prognostic marker for outcome of glioma patients. Cancer Epidemiol. 2013;37:79–84. doi: 10.1016/j.canep.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 16.Basavarajappa H.D., Corson T.W. KIF14 as an oncogene in retinoblastoma: a target for novel therapeutics? Future Med Chem. 2012;4:2149–2152. doi: 10.4155/fmc.12.158. [DOI] [PubMed] [Google Scholar]
- 17.Ahmed S.M., Thériault B.L., Uppalapati M., Chiu C.W., Gallie B.L., Sidhu S.S., Angers S. KIF14 negatively regulates Rap1a–Radil signaling during breast cancer progression. J Cell Biol. 2012;199:951–967. doi: 10.1083/jcb.201206051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ou Y.H., Torres M., Ram R., Formstecher E., Roland C., Cheng T., Brekken R., Wurz R., Tasker A., Polverino T. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell. 2011;41:458–470. doi: 10.1016/j.molcel.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim E., Vaillant F., Wu D., Forrest N.C., Pal B., Hart A.H., Asselin-Labat M.L., Gyorki D.E., Ward T., Partanen A. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 20.Mao M, Linsley PS, Buser CA, Marshall CG, Kim AS, inventors. Methods for identifying modulators of kinesin activity. Patent WO 2004/109290 A2. 2004.
- 21.Du X., Guo C., Hansell E., Doyle P.S., Caffrey C.R., Holler T.P., McKerrow J.H., Cohen F.E. Synthesis and structure-activity relationship study of potent trypanocidal thio semicarbazone inhibitors of the trypanosomal cysteine protease cruzain. J Med Chem. 2002;45:2695–2707. doi: 10.1021/jm010459j. [DOI] [PubMed] [Google Scholar]
- 22.Wood L.D., Parsons D.W., Jones S., Lin J., Sjöblom T., Leary R.J., Shen D., Boca S.M., Barber T., Ptak J. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 23.Nik-Zainal S., Van Loo P., Wedge D.C., Alexandrov L.B., Greenman C.D., Lau K.W., Raine K., Jones D., Marshall J., Ramakrishna M. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stephens P.J., Tarpey P.S., Davies H., Van Loo P., Greenman C., Wedge D.C., Nik-Zainal S., Martin S., Varela I., Bignell G.R. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curtis C., Shah S.P., Chin S.F., Turashvili G., Rueda O.M., Dunning M.J., Speed D., Lynch A.G., Samarajiwa S., Yuan Y. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi H., Ohno S., Sasaki Y., Matsuura M. Hereditary breast and ovarian cancer susceptibility genes (review) Oncol Rep. 2013;30:1019–1029. doi: 10.3892/or.2013.2541. [DOI] [PubMed] [Google Scholar]
- 27.Kramer J.L., Velazquez I.A., Chen B.E., Rosenberg P.S., Struewing J.P., Greene M.H. Prophylactic oophorectomy reduces breast cancer penetrance during prospective, long-term follow-up of BRCA1 mutation carriers. J Clin Oncol. 2005;23:8629–8635. doi: 10.1200/JCO.2005.02.9199. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt M., Hovelmann S., Beckers T.L. A novel form of constitutively active farnesylated Akt1 prevents mammary epithelial cells from anoikis and suppresses chemotherapy-induced apoptosis. Br J Cancer. 2002;87:924–932. doi: 10.1038/sj.bjc.6600566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knuefermann C., Lu Y., Liu B., Jin W., Liang K., Wu L., Schmidt M., Mills G.B., Mendelsohn J., Fan Z. HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene. 2003;22:3205–3212. doi: 10.1038/sj.onc.1206394. [DOI] [PubMed] [Google Scholar]
- 30.Franke T.F., Yang S.I., Chan T.O., Datta K., Kazlauskas A., Morrison D.K., Kaplan D.R., Tsichlis P.N. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 31.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forbes S.A., Bindal N., Bamford S., Cole C., Kok C.Y., Beare D., Jia M., Shepherd R., Leung K., Menzies A. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou B.P., Liao Y., Xia W., Spohn B., Lee M.H., Hung M.C. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001;3:245–252. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- 34.Wang R.C., Wei Y., An Z., Zou Z., Xiao G., Bhagat G., White M., Reichelt J., Levine B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338:956–959. doi: 10.1126/science.1225967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saini K.S., Loi S., de Azambuja E., Metzger-Filho O., Saini M.L., Ignatiadis M., Dancey J.E., Piccart-Gebhart M.J. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat Rev. 2013;39:935–946. doi: 10.1016/j.ctrv.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 36.Sangai T., Akcakanat A., Chen H., Tarco E., Wu Y., Do K.A., Miller T.W., Arteaga C.L., Mills G.B., Gonzalez-Angulo A.M. Biomarkers of response to Akt inhibitor MK-2206 in breast cancer. Clin Cancer Res. 2012;18:5816–5828. doi: 10.1158/1078-0432.CCR-12-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu X., Weaver Z., Linke S.P., Li C., Gotay J., Wang X.W., Harris C.C., Ried T., Deng C.X. Centrosome amplification and a defective G2–M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform–deficient cells. Mol Cell. 1999;3:389–395. doi: 10.1016/s1097-2765(00)80466-9. [DOI] [PubMed] [Google Scholar]
- 38.Chene G., Tchirkov A., Pierre-Eymard E., Dauplat J., Raoelfils I., Cayre A., Watkin E., Vago P., Penault-Llorca F. Early telomere shortening and genomic instability in tubo-ovarian preneoplastic lesions. Clin Cancer Res. 2013;19:2873–2882. doi: 10.1158/1078-0432.CCR-12-3947. [DOI] [PubMed] [Google Scholar]
- 39.Pradhan M., Risberg B.A., Tropé C.G., van de Rijn M., Gilks C.B., Lee C.H. Gross genomic alterations and gene expression profiles of high- grade serous carcinoma of the ovary with and without BRCA1 inactivation. BMC Cancer. 2010;10:493. doi: 10.1186/1471-2407-10-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Popova T., Manié E., Rieunier G., Caux-Moncoutier V., Tirapo C., Dubois T., Delattre O., Sigal-Zafrani B., Bollet M., Longy M. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72:5454–5462. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 41.Mondesire W.H., Jian W., Zhang H., Ensor J., Hung M.C., Mills G.B., Meric-Bernstam F. Targeting mammalian target of rapamycin synergistically enhances chemotherapy-induced cytotoxicity in breast cancer cells. Clin Cancer Res. 2004;10:7031–7042. doi: 10.1158/1078-0432.CCR-04-0361. [DOI] [PubMed] [Google Scholar]
- 42.Xu S., Li S., Guo Z., Luo J., Elllis M.J., Ma C.X. Combined targeting of mTOR and AKT is an effective strategy for basal-like breast cancer in patient-derived xenograft models. Mol Cancer Ther. 2013;12:1665–1675. doi: 10.1158/1535-7163.MCT-13-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Firestone A.J., Weinger J.S., Maldonado M., Barlan K., Langston L.D., O'Donnell M., Gelfand V.I., Kapoor T.M., Chen J.K. Small-molecule inhibitors of the AAA + ATPase motor cytoplasmic dynein. Nature. 2012;484:125–129. doi: 10.1038/nature10936. [DOI] [PMC free article] [PubMed] [Google Scholar]