

Abstract
Radioimmunotherapy (RIT) is a therapeutic modality that allows delivering of ionizing radiation directly to targeted cancer cells. Conventional RIT uses β-emitting radioisotopes, but recently, a growing interest has emerged for the clinical development of α particles. α emitters are ideal for killing isolated or small clusters of tumor cells, thanks to their specific characteristics (high linear energy transfer and short path in the tissue), and their effect is less dependent on dose rate, tissue oxygenation, or cell cycle status than γ and X rays. Several studies have been performed to describe α emitter radiobiology and cell death mechanisms induced after α irradiation. But so far, no investigation has been undertaken to analyze the impact of α particles on the immune system, when several studies have shown that external irradiation, using γ and X rays, can foster an antitumor immune response. Therefore, we decided to evaluate the immunogenicity of murine adenocarcinoma MC-38 after bismuth-213 (213Bi) irradiation using a vaccination approach. In vivo studies performed in immunocompetent C57Bl/6 mice induced a protective antitumor response that is mediated by tumor-specific T cells. The molecular mechanisms potentially involved in the activation of adaptative immunity were also investigated by in vitro studies. We observed that 213Bi-treated MC-38 cells release “danger signals” and activate dendritic cells. Our results demonstrate that α irradiation can stimulate adaptive immunity, elicits an efficient antitumor protection, and therefore is an immunogenic cell death inducer, which provides an attractive complement to its direct cytolytic effect on tumor cells.
Abbreviations: BMDC, bone marrow-derived dendritic cell; CTL, cytotoxic T lymphocyte; DAMP, danger-associated molecular pattern; RIT, radioimmunotherapy
Introduction
Radiotherapy is one of the most common treatments of cancer and has been efficiently used for decades. Ionizing radiation is used to eradicate cancer cells through direct cytotoxicity potentially associated with a bystander and other nontargeted effects [1–3]. Numerous radiotherapy modalities have been developed, among which radioimmunotherapy (RIT) is one of the most promising for the treatment of disseminated cancers. RIT is an internal form of radiotherapy using radiolabeled vectors to target antigens expressed on tumor cells [4]. This therapeutic approach has significantly progressed for the past 20 years with the development of new vectors, improvement of labeling efficiencies, and availability of new radionuclides [5]. Among those, α particles are very attractive for clinical development. Indeed, the physical and biologic characteristics of those radioisotopes appear to be especially suited for targeting isolated cancer cells, small clusters of tumor cells, or micrometastasis. α particles exhibit a high linear energy transfer (~ 100 keV/μm) with an energy comprised in between 5 and 9 MeV and a short path of 50 to 90 μm in the tissues. Like other high linear energy transfer particles, α emitters induce more DNA double-strand breaks than γ or X rays and provoke a cell cycle arrest in the G2 phase that is more marked than with γ rays [6,7]. It has been shown that irradiation of the nucleus with a few α particles is sufficient to result in cell death [8] and only a few dozens are needed when the membrane is targeted, whereas several thousands of β emitters are required for the same effect [9]. Furthermore, radiobiologic effects associated to α radionuclides are advantageously less sensitive to dose rate, hypoxia, and cell cycle distribution than β particles or γ rays [10]. Other aspects of α emitter radiobiology have been less investigated. For instance, few studies have analyzed cell death mechanisms, and those are sometimes conflicting. Some groups showed that cells undergo apoptosis following exposure to α particles [11,12] when others observed cell death independent from apoptosis [13,14].
However, it has become increasingly clear that direct cytotoxicity is not the only factor accounting for tumor destruction by ionizing radiation in vivo. Indirect effects such as radiation-induced biologic bystander effects significantly contribute to the effectiveness of irradiation [15], and numerous studies have demonstrated that external treatment with γ and X rays can have a beneficial effect at distance from the field of irradiation. This phenomenon is called abscopal effect and has been shown to be mediated by the immune system [16,17]. Accumulating evidence also shows that the immune response may play an important role in patient response to radiation [18]. Several mechanisms have been proposed to explain the implementation of such an antitumor response after radiotherapy. First, irradiation induces local inflammation of tumor sites and microenvironment that favors the recruitment of immune cells, in particular dendritic cells (DCs). Additionally, DCs are capable of cross-presenting antigens from the tumor cells killed by irradiation to stimulate a specific T cell response. Finally, the stress induced by ionizing radiation provides the immune system with signals, called “danger signals” or danger-associated molecular patterns (DAMPs), needed for activation of antigen-presenting cells (APCs) such as DCs [19]. These results, obtained in animals after external irradiation, underline the importance of studying the impact of ionizing radiation on immune cells and their potential in stimulating an immune response that could complement the direct effect of irradiation and establish a long-term antitumor response. Nevertheless, the influence of α radiation on immunity has not been investigated so far.
Therefore, our study aims to investigate the potential of bismuth-213 (213Bi), an α emitter generated from an actinium-225/213Bi generator, in stimulating immune cells. We used MC-38 tumor cells, a murine adenocarcinoma, which has been reported to be weakly immunogenic and a good model for immunotherapy studies [20,21]. To study the impact of the radioelement on the tumor cells only, without irradiating the microenvironment and without any vectorization that could also act on the tumor cells, we chose a vaccination approach in immunocompetent C57Bl/6 mice. Additional in vitro studies were conducted to investigate the molecular mechanisms involved after MC-38 irradiation on the activation of adaptative immunity, in particular DCs and T cells, and the establishment of long-term protection toward tumor cells. Here, we report for the first time that tumor cells irradiated with an α particle emitter lead to the development of a long-lasting antitumor immune response mediated by specific T cells in vivo. We also demonstrate in vitro that irradiation of MC-38 cells with 213Bi induces the release of DAMPs [i.e., heat shock protein 70 (Hsp70) and homeostatic group box protein 1 (HMGB1)] and triggers the activation of DCs.
Materials and Methods
Cell Culture
MC-38 murine colon carcinoma (established by Rosenberg's laboratory, National Cancer Institute, Bethesda, MD [20] and kindly provided by Dr Pèlegrin, CRLC Val d'Aurelle-Paul Lamarque, Montpellier, France) and B16-F10 murine melanoma (ATCC®: CRL-6475, LGC Standards, Molsheim, France) were maintained in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal calf serum (PAA Laboratories, Velizy-Villacoublay, France), 2 mM glutamine (Invitrogen, Cergy Pontoise, France), 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco) at 37°C and 5% CO2.
213Bi Irradiation
Cyclohexyl diethylene triamine penta-acetic acid (Macrocyclics, Dallas, Texas) was conjugated to BSA as previously described [22] and controlled by indium labeling. For labeling with 213Bi, conjugated BSA was incubated with 213Bi eluted from a actinium-225/213Bi generator (Institute for Transuranium Elements, Karlsruhe, Germany) for 10 minutes at 37°C in 0.6 M sodium acetate (pH 5.3) and 0.01% ascorbic acid. The resulting 213Bi-BSA conjugate was purified from unbound 213Bi by size exclusion chromatography using a PD-10 column (GE Healthcare, Velizy-Villacoublay, France). Radiochemical purity was > 95%, as determined by Instant Thin Layer Chromatography - Silica Gel using 0.1 M citrate buffer (pH 4.5). A solution containing 213Bi-BSA diluted in culture media was then added to the cells at a final activity of 2.22 MBq/ml 213Bi for vaccination and 0.74 MBq/ml for in vitro studies. 213Bi-BSA was removed after 6 hours, by centrifugation and washing the cells with fresh medium, when used for vaccination.
Annexin V and 4′, 6-Diamidino-2-phenylindol (DAPI) Staining
Staining was performed according to manufacturer's instructions (Annexin V-APC Apoptosis detection kit, BD, Le Pont de Claix, France). Briefly, cells were washed in phosphate-buffered saline (PBS) and resuspended at 1 × 106 cells per milliliter in 1 × Annexin binding buffer [10 mM Hepes/NaOH (pH 7.4), 140 mM NaCl, and 2.5 mM CaCl2]. A population of 105 cells was then stained with 5 μl of Annexin V-APC and/or 1 μl of DAPI for 2 minutes at room temperature in the dark. At least 10,000 events were analyzed using BD FACSCanto II flow cytometer and FlowJo software (TreeStar, Ashland, OR).
Caspase-3 Assay
Caspase-3 assay was performed using the PE Active Caspase-3 Apoptosis kit (BD Pharmingen, Le Pont de Claix, France) according to the manufacturer's instructions. Briefly, cells were washed once in PBS and resuspended in BD Cytofix/Cytoperm on ice for 20 minutes. After two washes in BD Perm/Wash, cells were stained with the PE antiactive caspase-3 antibody for 30 minutes at room temperature. At least 50,000 events were analyzed using BD FACSCalibur flow cytometer and FlowJo software.
Mice and Vaccination
Wild-type C57Bl/6 (H-2b) mice aged between 11 and 17 weeks and 14-week-old nude mice (RjOrl:NMRI-Foxn1nu/Foxn1nu; Janvier Labs, Le Genest-Saint-Isle, France) were immunized with 3 × 106 irradiated MC-38 injected subcutaneously in the left flank. The injection was performed after 6 hours of incubation with the radioconjugate to allow for complete radioactive decay of 213Bi before injection, thus limiting irradiation of the mice. Seven days later, the mice were challenged with 2 × 105 live MC-38 s.c. in the right flank. Tumor progression was then assessed regularly using calipers, and mice were killed when tumor volume reached 2500 mm3 or when signs of tumor necrosis were observed. Experiments performed in this study were approved by the local veterinary committee (License No. CEEA.2013.2).
T Cell Preparation
Mice were killed 5 days after their last injection, and axillary, inguinal, popliteal, and mesenteric lymph nodes were recovered. T cells were then purified using Pan T Cell Isolation Kit II (Miltenyi Biotec, Bergish Gladbach, Germany) and stimulated in vitro with 5 μg/ml anti-CD3ε (clone 145-2C11; eBioscience) and 2 μg/ml anti-CD28 (clone 37.51; eBioscience). T cells were then cultured until they went back to a resting state (no proliferation).
Cytotoxicity Assay
Cytotoxic activity was tested using a standard 51Cr release assay. Briefly, MC-38 or B16-F10 autologous tumor cell lines were used as a target and labeled with 2.77 MBq of Na251CrO4 for 1 hour at 37°C, washed five times, and then plated onto 96-well U-bottom plates. Effector-to-target ratios (E:T ratios) were 20:1, 10:1, 5:1, and 2.5:1. After 16 hours of incubation at 37°C, 25 μl of supernatant from each well was removed, mixed with 100 μl of Betaplate Scint (PerkinElmer, Waltham, MA), and read using 1450 MicroBeta Plus counter (Wallac, Gaithersburg, MD). Each test was performed in triplicate. Results were expressed as a percentage of lysis according to the following formula: (experimental release − spontaneous release)/(maximal release − spontaneous release) × 100, where experimental release represents mean cpm release from target cells in the presence of effector cells, spontaneous release represents that from targets incubated without effectors, and maximum release represents that from target cells incubated with 1% triton.
DAMP Detection in Culture Media
DAMP release in cell culture was assessed using ELISA directed against Hsp70 (R&D Systems, Abingdon, UK) and HMGB1 (IBL International, Hamburg, Germany) following the manufacturers' protocols.
Bone Marrow-Derived Dendritic Cell Production
Bone marrow cells were flushed with RPMI medium from C57Bl/6 thighbones and filtered through a 70-μm cell strainer. Adherent cells were cultured in RPMI 1640 (Gibco) supplemented with 10% fetal calf serum (PAA Laboratories), 2 mM glutamine (Gibco), 100 U/ml penicillin, 100 μg/ml streptomycin (Gibco), 100 μM 2-mercaptoethanol (Carl Roth, Karlsruhe, Germany), minimum essential medium nonessential amino acids (Gibco), and murine GM-CSF (purified from the supernatant of a transgenic GM-CSF (Granulocyte macrophage colony-stimulating factor)–producing cell line [23]) to induce bone marrow-derived dendritic cell (BMDC) differentiation. On day 3, fresh medium was added; on days 6 and 8, half of the medium was removed and replaced by fresh medium; and BMDCs were used on day 10.
Coculture Assays
For BMDC coculture assay with tumor cells, 1 × 106 murine BMDCs were plated onto six-well dishes with 5 × 106 MC-38 and incubated 24 hours at 37°C. Maturation was then analyzed by immunofluorescence phenotyping. For BMDC maturation with tumor cell supernatant, 1 ml of tumor cell supernatant was added to 1 × 106 BMDCs and plated onto 12-well dishes. BMDC phenotype was analyzed by immunofluorescence after 24 hours of incubation.
Immunofluorescence Analysis
Cells were washed once in PBS-BSA (0.1%) and then stained for 1 hour at 4°C with primary antibody. When secondary antibody was needed, cells were washed three times in PBS-BSA (0.1%) before incubation with secondary antibody. After staining, cells were washed twice in PBS-BSA (0.1%) and once in PBS before acquisition in flow cytometer. The following antibodies and their respective control isotypes were used in this study: Alexa 647 anti-mouse CD11c (N418; eBioscience), fluorescein isothiocyanate (FITC) anti-mouse CD11b (M1/70; BD), FITC anti-mouse CD34 (RAM34; BD), FITC anti-mouse CD80 (16-10A1; BD), APC anti-mouse CD86 (GL1; BD), and APC anti-mouse CD40 (1C10; eBioscience). Immunofluorescence analyses were performed using BD FACSCalibur flow cytometer and analyzed with FlowJo software.
Data Analysis
Data are expressed as the means ± SD. Survival data were analyzed using the log-rank test and Kaplan-Meier method. Comparisons of continuous variables were done using nonparametric Mann-Whitney tests or two-way analysis of variance (GraphPad Prism version 5.0, La Jolla, CA). P values of less than or equal to .05 are considered significant.
Results
Vaccination of Mice with Irradiated Cells Induces a Protective Antitumor Response In Vivo
To determine whether α-irradiated cells could foster an antitumor response in vivo, we conducted vaccination studies on syngeneic C57Bl/6 mice. Mice were vaccinated 7 days before MC-38 engraftment by s.c. injection of 3 × 106 213Bi-treated MC-38 (without any adjuvant), in the left flank. Irradiation was performed using 213Bi-BSA conjugates; however, BSA does not target tumor cells. This approach was chosen to avoid any interaction that specific vectors such as antibodies could have on tumor cells as well as with the immune response. This way, we only studied the effects of the radioelement and not the effects of the vector. Cells were incubated for 6 hours with the radioconjugate and then washed with fresh medium before injection. This allowed for complete radioactive decay and elimination of 213Bi, thus limiting irradiation of the mice. At the time of injection, viability of 213Bi-treated MC-38 was controlled by Annexin V/DAPI double staining and detection of caspase-3 activation. Both analyses showed that irradiation had no impact yet on death mechanisms because percentages of the live cells were identical in 213Bi-treated MC-38 and nonirradiated MC-38. However, irradiation induced > 95% cell killing on the long term, as determined by clonogenic assays (data not shown). On day 0, mice were engrafted with 2 × 105 live MC-38 injected s.c. in the opposite flank to make sure that any response observed would have to be systemic and not just due to local control. Among nonvaccinated controls, 21 of the 25 mice rapidly developed a tumor and reached end-point value within 40 days (Figure 1A). However, only 3 of 25 vaccinated mice developed a tumor (Figure 1B). Median survival was 37 days for controls and was not reached for the vaccinated group after more than 100 days of monitoring. Overall survival at 73 days after live tumor cell engraftment was 88% for the vaccinated mice compared to 16% in control group (Figure 1C). This demonstrates that 213Bi-irradiated tumor cells are highly immunogenic and can elicit a strong antitumor response in vivo.
Figure 1.

Tumor cells irradiated with 213Bi give rise to antitumor protection in vivo. (A and B) Tumor volume progression for control (A) and immunized mice (B) after MC-38 engraftment. Volume was calculated as (length × width × height × π)/6. (C) Kaplan-Meier analysis of mice immunized with 213Bi-treated cells (square) versus control C57Bl/6 (circles) after subcutaneous challenge with 2 × 105 live MC-38. Median survival was 37 days for controls and was not reached for the immunized group. P value determined by the log-rank test was extremely significant (P < .0001). (D) Tumor-free fraction of mice after a second challenge with live MC38. P < .0001.
To follow the induced immune response, 17 of the 22 mice that rejected MC-38 on live tumor cell injection were challenged a second time on day 73. Remarkably, 100% of the animals showed full protection by rejecting that second engraftment (Figure 1D). This result demonstrates that the antitumor response was long lasting, hence most likely driven by the adaptive immunity.
An additional group of 10 mice was included in the study, receiving the vaccination but not the first challenge with live MC-38 cells on day 0. Vaccinated mice did not grow any tumor, showing that irradiated cells could not participate in tumor growth (Figure 2A). Those mice were then challenged 84 days after vaccination. Interestingly, among that group, 90% of the animals rejected the live tumor cell graft (Figure 2B). Those data confirm that α-irradiated tumor cells are highly immunogenic and that a long-lasting immune response has been elicited without any boost or adjuvant.
Figure 2.
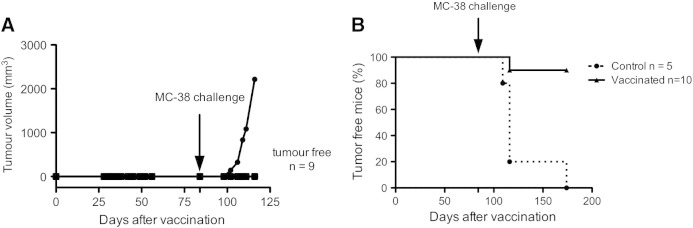
Tumor cells irradiated with 213Bi do not give rise to tumors and allow for long-term antitumor protection. (A and B) Tumor volume progression for vaccinated mice (A), which did not receive live MC-38 engraftment before day 84. Kaplan-Meier analysis of mice immunized with 213Bi-treated cells (triangles) versus control C57Bl/6 (circles) after subcutaneous challenge with 2 × 105 live MC-38. P value determined by the log-rank test was highly significant (P < .0001).
The Antitumor Response Induced In Vivo Is T Cell Mediated
To investigate the cell subsets involved in the antitumor response induced through α-irradiated MC-38, we performed vaccination studies on immunodeficient athymic mice (nude) (n = 5), which lack T cells. We observed no protection against tumor challenge after vaccination. Kinetics of tumor development appeared very similar in all the mice (Figure 3, A and B), and median survival was 13 days for both the control and vaccinated groups (Figure 3C), indicating that T cells are essential for the antitumor effect obtained through vaccination.
Figure 3.

Immunization with α-irradiated MC38 fails in nude mice. (A and B) Tumor volume progression for control (n = 5) (A) and immunized nude mice (n = 5) (B) after MC38 engraftment. (C) Kaplan-Meier analysis of mice immunized with 213Bi-treated cells (square) versus control nude mice (circles) after subcutaneous challenge with 2 × 105 live MC-38. Median survival was 13 days for both the control and immunized groups. P = .5716.
Cytotoxic T lymphocytes (CTL) are known as potent effectors in anticancer treatments; therefore, we investigated the presence of CTL specifically targeted toward MC-38 following immunization. T cells were purified from the mice lymph nodes and tested through a conventional chromium release assay (Figure 4). T cells from naïve mice exhibited a weak cytotoxic efficiency toward MC-38 cells, varying from 6.7% to 11.7% independently from the E:T ratio (Figure 4A). Similarly, cytotoxic efficiency toward another syngeneic, but nonrelated, tumor cell line (B16-F10 melanoma) was weak and did not exceed 3.4 ± 3.8% at any E:T ratio tested (Figure 4A). After vaccination with 213Bi-irradiated MC-38 and subsequent tumor challenge a week later, a significant increase of cytotoxic efficiency toward MC-38 was observed, ranging from 40.0 ± 9.7% for an E:T ratio of 2.5:1 to 74.0 ± 24.7% for an E:T ratio of 20:1 (Figure 4B), indicating that an antitumor T cell response had been raised. On the contrary, cytotoxic efficiency toward B16-F10 melanoma did not increase and was only 1.0 ± 1.8% at the highest E:T ratio, indicating that the T cell response was specific to MC-38. Comparison of naïve, challenged, vaccinated, and vaccinated + challenged mice showed that CTL activity was only significantly increased in vaccinated + challenged mice at the E:T ratio of 10:1 with 55.9% cytotoxicity compared to 7.9% in naïve animals (Figure 4C). Together with our data on the vaccination of nude mice, these results suggest that CTL is the main effector of the antitumor response raised after vaccination with α-irradiated tumor cells.
Figure 4.

Antitumor response is dependent on cytotoxic T cells. (A and B) 51Cr release assay against MC-38 or the irrelevant cell line B16-F10 was performed on T cells purified from naïve mice (A) or mice that had been immunized with 213Bi-treated MC-38 and subsequently challenged with live MC-38 (B). Assays were performed at different E:T ratios ; data points represent means ± SD of triplicate measurements for three naïve mice (A) and four vaccinated + challenged mice (B). (C) Scatterplot of mean percentage of T cell cytotoxicity against MC-38 at the E:T ratio of 10:1 for different groups of mice. In parentheses, number of mice in each group. P < .05 between naïve mice and mice that have been immunized and challenged. P value was determined by nonparametric Mann-Whitney test. Mean cytotoxicity was 7.87% for naïve mice, 19.15% for challenged mice, 16.2% for vaccinated mice, and 55.85% for vaccinated + challenged mice.
Tumor Cells Treated with 213Bi Activate DCs In Vitro
To further explore the mechanisms supporting the antitumor immune response to α particles, we analyzed the DC phenotype after in vitro irradiation of MC-38 cells. DCs express costimulatory molecules such as CD40, CD80, and CD86 on their cell surface, which are needed for activation of naïve T cells. Activation of immature DCs results in increased expression of those costimulatory molecules. Immature BMDCs derived from syngeneic C57Bl/6 mice have been incubated for 48 hours with conditioned media from control or 213Bi -treated MC-38. The media of irradiated tumor cells elicited a significant increase of 32% in CD40 expression [ratio of fluorescence intensity (RFI) increased from 4.5 to 5.9] and 44.8% in CD86 expression (22.7 to 32.6) and a slight increase, however not significant, of 4.4% in CD80 expression (3.3 to 3.4) on BMDCs (Figure 5, A–C). Besides, we observed that BMDCs aggregated in clumps (Figure 5, D and E) when incubated in culture inserts with irradiated MC-38, which is a phenotypic characteristic of DC activation [24]. No activation was observed when immature BMDCs were exposed to irradiated culture medium only (data not shown). These results suggest that 213Bi induces the release of soluble agents from MC-38 capable of activating DCs in vitro.
Figure 5.

213Bi-irradiated MC-38 elicit DC maturation. (A–C) Triplicate analysis of RFI for CD40 (A), CD80 (B), and CD86 (C). RFI is calculated as geometric mean of fluorescence intensity of the specific antibody divided by that of the control isotype. (D and E) Snapshot of BMDC after 24 hours in culture with 5 × 106 control MC38 (D) or 5 × 106 irradiated MC-38 (E). *P ≤ .05 determined using Mann-Whitney nonparametric test.
213Bi Irradiation Causes Release of DAMPs from MC-38 Cells
Then, we tested the conditioned media of 213Bi-treated MC-38 for the presence of DAMPs. DAMPs such as Hsp70 or HMGB1 are self-molecules normally expressed intracellularly, which can be released in the extracellular space upon cell stress or damage as a danger signal to the immune system [25,26]. Those molecules are known to activate DCs and have been implicated in the establishment of efficient antitumor response [27]. ELISA on MC-38 conditioned media showed a significant increase of HMGB1 and Hsp70 levels following 213Bi irradiation (Figure 6). This increase in both DAMP concentrations started 24 and 48 hours post-irradiation for Hsp70 and HMGB1, respectively. Altogether, these data show that 213Bi induces the release of DAMPs from irradiated tumor cells, which may contribute to the antitumor response by activating DCs.
Figure 6.

DAMP release after irradiation. (A and B) Evolution of Hsp70 (A) and HMGB1 (B) concentrations determined by ELISA on MC-38 conditioned media after irradiation with 213Bi (squares) and on controls (circles). Data points represent means ± SD of triplicate measurements. ***P < .001 as determined by two-way analysis of variance and Bonferonni posttests. Results are representative of two independent experiments.
Discussion
Radiotherapy is traditionally used for its cytotoxic effect on cancer cells. There is however growing evidence showing that direct cytotoxicity is not the only process through which radiation may contribute to tumor elimination. Here, we demonstrate that 213Bi-irradiated tumor cells are highly immunogenic and can elicit a strong antitumor response in vivo that protects immunocompetent hosts against further tumor challenge with the same tumor. Indeed, 88% of the vaccinated animals survived to the injection of live MC-38 tumor cells. α particles therefore fulfill the first requirement defined by Kroemer's group to be defined as an immunogenic cell death inducer [28]. Our results show that irradiation of tumor cells with 213Bi can lead to the elimination of other tumor cells in a distant site, even long after the irradiation. The protective effect of vaccination was indeed long lasting (at least 2 months) and must therefore involve memory cells. Strikingly, this immune protection was observed in all the animals subjected to a second challenge with live MC-38 cells. The response was T cell mediated as demonstrated by the presence of specific cytotoxic T cells and by the lack of protection in nude mice. This kind of specific, systemic, and long-lasting response would be highly desirable for anticancer therapy because it should help in eliminating distant metastases and preventing relapse. However, T cells may not be the only cells needed for an efficient antitumor effect. Nude mice have functional Natural Killer (NK) and B cells that could participate in the immune response. Further experiments will help to clarify the role of each immune subset in the antitumor response.
Interestingly, tumors grew much faster in nude mice with a median survival of 13 versus 37 days in C57Bl/6 immunocompetent mice. Moreover, 4 of the 25 challenged mice did not develop a tumor, and the rest of the challenged animals showed a slight increase in cytotoxicity against MC-38 compared to naïve mice (however not significant). These data suggest that live tumor cells exhibit some level of immunogenicity, probably related to the fact that the MC-38 cells we used express high levels of major histocompatibility complex class I molecules (data not shown). Although this immune response slows down tumor growth to some extent, it is not sufficient for tumor control in 84% of the animals. This could mean two things: either tumor cells multiply too quickly for the immune response to cope with it, or the immune system sees the tumor as harmless self and represses the initial antitumor response. In both cases, α radiation delivered through RIT would be of great therapeutic interest. Indeed, the high cytotoxicity of α particles could reduce tumor load, and at the same time, α-irradiated tumor cells could activate the immune response and tip the balance toward an effective antitumor response. Additional studies using less immunogenic tumors will however be required to determine whether α radiation would be as efficient in triggering an immune response in such settings.
To depict the mechanisms that could contribute to activation of T cells in vivo, we pursued in vitro studies on DCs. Their role in anticancer immunity is indeed crucial. When activated with the adequate stimuli, DCs can cross-present tumor-derived antigens to T cells and secrete stimulatory cytokines, which will lead to the establishment of an effective cell-mediated antitumor response. Conversely, if not properly activated, DCs may promote tolerance and T cell unresponsiveness. In fact, in a majority of cancer, the antitumor response is repressed by the host's immune regulatory cells [29]. Here, we exposed immature BMDCs to conditioned media from irradiated MC-38 tumor cells and observed significant changes in both DC morphology and expression profile of several DC activation markers, demonstrating the potency of α-irradiated tumor cells in triggering immune activation.
To further investigate the molecular processes that could contribute to the activation of DCs in vitro, we conducted ELISA tests on the conditioned media of α-irradiated tumor cells to detect the presence of well-characterized DAMPs [30]. In vivo, as irradiated tumor cells were washed in PBS before vaccination, the molecules involved in immune cells activation cannot be irradiated molecules from the culture medium and have to be secreted by the tumor cells after exposure to 213Bi. Besides, irradiated culture medium alone did not induce DC activation. We showed that 213Bi causes the release of Hsp70 and HMGB1 from MC-38 cells. Such molecules, which are normally endogenous, get released in the extracellular environment after a stress or nonphysiological cell death and can activate immune cells [26,31]. HSP are chaperone proteins capable of binding numerous peptides. The HSP-peptide complexes released from dying cells can be taken up by APC through the common receptor CD91 to allow for antigen processing and re-presentation to T cells [32]. HMGB1 is a ubiquitous nonhistone nuclear factor that mediates inflammation and immune responses when released from dying cells [33]. HMGB1 production in patients with cancer has conversely been reported in association with both good [34] and poor prognoses [35,36]. HMGB1 has also been involved in numerous processes facilitating tumor progression, such as proliferation [37], metastasis [38,39], angiogenesis [40], and chronic inflammation [41]. The differential activity of HMGB1 is related to the balance of its different redox states (i.e., all-thiol, disulfide, and sulfonated) that are produced in the extracellular environment [42]. In the case of radiotherapy, a rapid and local increase of HMGB1 might act differently on the immune system than a chronic systemic secretion. Notably, HMGB1 has been shown to be mandatory for cross-presentation of antigen derived from dying tumor cells to T cells after radiotherapy, leading to efficient antitumor immune response [43]. Further studies will be required to determine the importance of these DAMPs in the establishment of an immune response in our settings. Using larger screening techniques (i.e., mass spectrometry), it will also be important to identify other species produced after irradiation contributing to such bystander phenomena.
All these data underline the importance of the immune system in response to radiotherapy of cancer. Nevertheless, most studies on RIT efficacy are performed on xenograft models in immunodeficient mice. Therefore, the participation of the immune system in response to therapy is completely overseen. In the future, it will be of prime importance to assess the response to cancer therapy in both immunodeficient and immunocompetent models to take into account the effect of radiation on both tumor cells and the immune environment.
In conclusion, the data presented here show that irradiation of tumor cells with 213Bi induces the release of DAMPs, promotes DC activation, and leads to a systemic and long-lasting antitumor response. A few studies have investigated the impact of γ or β irradiation on the immune system. It has been shown that external beam [44–46] or β emitters like 90Y [21] or 153Sm [47] could elicit effective antitumor response when combined with vaccine or CTL transfer. Our study is in line with these investigations and is the first to demonstrate that irradiation of tumors using an α particle emitter can also lead to the establishment of an efficient antitumor immune response in vivo. Even though the impact of α irradiation on the immune response to cancer will have to be further characterized, this study brings new insights on the mechanism of action of α particles and further supports the interest in developing the use of such emitters for cancer therapy.
Acknowledgments
We thank Marie-Hélène Gaugler for critical review of the manuscript. We also thank Sandrine Minault (CRCNA [Centre de Recherche en Cancérologie Nantes-Angers]) for her technical help, as well as the staff of Unité Thérapeutique Expérimentale (UTE), Cytocell, and Radioactivity facilities (Structure Fédérative de Recherche [SFR] François Bonamy, Institut de Recherche en Santé de l'Université de Nantes [IRS-UN], University of Nantes).
Footnotes
Conflict of interest statement: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Authors' contributions: Conception and design—J.-B.G., F.D., and J.G. Development of methodology—J.-B.G. and J.G. Acquisition of data—J.-B.G., J.M., S.G., C.M., A.F.-C., A.M., and F.B. Analysis and interpretation of data—J.-B.G., J.M., S.G., M.C., F.D., and J.G. Writing, review, and/or revision of the manuscript—J.-B.G., J.M., S.G., Y.G., M.C., F.D., and J.G. Study supervision—F.D. and J.G.
Grant support: This work was supported by grants from La Ligue Contre le Cancer and from the Pays de la Loire Council “Nucléaire pour la Santé” (NucSan). J.-B.G. and J.M. are supported by grants from the French Ministry of Research and Higher Education. A.M. and F.B. are supported by the European Commission.
References
- 1.Goldberg Z., Lehnert B.E. Radiation-induced effects in unirradiated cells: a review and implications in cancer. Int J Oncol. 2002;21:337–349. [PubMed] [Google Scholar]
- 2.Morgan W.F. Is there a common mechanism underlying genomic instability, bystander effects and other nontargeted effects of exposure to ionizing radiation? Oncogene. 2003;22:7094–7099. doi: 10.1038/sj.onc.1206992. [DOI] [PubMed] [Google Scholar]
- 3.Mothersill C., Seymour C.B. Radiation-induced bystander effects—implications for cancer. Nat Rev Cancer. 2004;4:158–164. doi: 10.1038/nrc1277. [DOI] [PubMed] [Google Scholar]
- 4.Barbet J., Bardiès M., Bourgeois M., Chatal J.F., Chérel M., Davodeau F., Faivre-Chauvet A., Gestin J.F., Kraeber-Bodéré F. Radiolabeled antibodies for cancer imaging and therapy. Methods Mol Biol. 2012;907:681–697. doi: 10.1007/978-1-61779-974-7_38. [DOI] [PubMed] [Google Scholar]
- 5.Sharkey R.M., Goldenberg D.M. Cancer radioimmunotherapy. Immunotherapy. 2011;3:349–370. doi: 10.2217/imt.10.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lücke-Huhle C. α-irradiation–induced G2 delay: a period of cell recovery. Radiat Res. 1982;89:298–308. [PubMed] [Google Scholar]
- 7.Yong K.J., Milenic D.E., Baidoo K.E., Brechbiel M.W. 212Pb-radioimmunotherapy induces G2 cell-cycle arrest and delays DNA damage repair in tumor xenografts in a model for disseminated intraperitoneal disease. Mol Cancer Ther. 2012;11:639–648. doi: 10.1158/1535-7163.MCT-11-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Søyland C., Hassfjell S.P. Survival of human lung epithelial cells following in vitro α-particle irradiation with absolute determination of the number of α-particle traversals of individual cells. Int J Radiat Biol. 2000;76:1315–1322. doi: 10.1080/09553000050151583. [DOI] [PubMed] [Google Scholar]
- 9.Humm J.L., Cobb L.M. Nonuniformity of tumor dose in radioimmunotherapy. J Nucl Med. 1990;31:75–83. [PubMed] [Google Scholar]
- 10.Sgouros G., Roeske J.C., McDevitt M.R., Palm S., Allen B.J., Fisher D.R., Brill A.B., Song H., Howell R.W., Akabani G. MIRD Pamphlet No. 22 (abridged): radiobiology and dosimetry of α-particle emitters for targeted radionuclide therapy. J Nucl Med. 2010;51:311–328. doi: 10.2967/jnumed.108.058651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seideman J.H., Stancevic B., Rotolo J.A., McDevitt M.R., Howell R.W., Kolesnick R.N., Scheinberg D.A. Alpha particles induce apoptosis through the sphingomyelin pathway. Radiat Res. 2011;176:434–446. doi: 10.1667/rr2472.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friesen C., Roscher M., Hormann I., Leib O., Marx S., Moreno J., Miltner E. Anti-CD33-antibodies labelled with the alpha-emitter Bismuth-213 kill CD33-positive acute myeloid leukaemia cells specifically by activation of caspases and break radio- and chemoresistance by inhibition of the anti-apoptotic proteins X-linked inhibitor of apoptosis protein and B-cell lymphoma-extra large. Eur J Cancer. 2013;49:2542–2554. doi: 10.1016/j.ejca.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Supiot S., Gouard S., Charrier J., Apostolidis C., Chatal J.F., Barbet J., Davodeau F., Cherel M. Mechanisms of cell sensitization to α radioimmunotherapy by doxorubicin or paclitaxel in multiple myeloma cell lines. Clin Cancer Res. 2005;11:7047s–7052s. doi: 10.1158/1078-0432.CCR-1004-0021. [DOI] [PubMed] [Google Scholar]
- 14.Seidl C., Port M., Gilbertz K.P., Morgenstern A., Bruchertseifer F., Schwaiger M., Röper B., Senekowitsch-Schmidtke R., Abend M. 213Bi-induced death of HSC45-M2 gastric cancer cells is characterized by G2 arrest and up-regulation of genes known to prevent apoptosis but induce necrosis and mitotic catastrophe. Mol Cancer Ther. 2007;6:2346–2359. doi: 10.1158/1535-7163.MCT-07-0132. [DOI] [PubMed] [Google Scholar]
- 15.Boyd M., Ross S.C., Dorrens J., Fullerton N.E., Tan K.W., Zalutsky M.R., Mairs R.J. Radiation-induced biologic bystander effect elicited in vitro by targeted radiopharmaceuticals labeled with α-, β-, and auger electron–emitting radionuclides. J Nucl Med. 2006;47:1007–1015. [PubMed] [Google Scholar]
- 16.Chakravarty P.K., Alfieri A., Thomas E.K., Beri V., Tanaka K.E., Vikram B., Guha C. Flt3-ligand administration after radiation therapy prolongs survival in a murine model of metastatic lung cancer. Cancer Res. 1999;59:6028–6032. [PubMed] [Google Scholar]
- 17.Demaria S., Ng B., Devitt M.L., Babb J.S., Kawashima N., Liebes L., Formenti S.C. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. 2004;58:862–870. doi: 10.1016/j.ijrobp.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 18.Golden E.B., Pellicciotta I., Demaria S., Barcellos-Hoff M.H., Formenti S.C. The convergence of radiation and immunogenic cell death signaling pathways. Front Oncol. 2012;2:88. doi: 10.3389/fonc.2012.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Formenti S.C., Demaria S. Combining radiotherapy and Cancer Immunotherapy: a Paradigm Shift. J Natl Cancer Inst. 2012;1–10 doi: 10.1093/jnci/djs629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cameron R.B., Spiess P.J., Rosenberg S.A. Synergistic antitumor activity of Tumor-Infiltrating Lymphocytes, Interleukin-2 and local tumor irradiation. J Exp Med. 1990;171:249–263. doi: 10.1084/jem.171.1.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chakraborty M., Gelbard A., Carrasquillo J.A., Yu S., Mamede M., Paik C.H., Camphausen K., Schlom J., Hodge J.W. Use of radiolabeled monoclonal antibody to enhance vaccine-mediated antitumor effects. Cancer Immunol Immunother. 2008;57:1173–1183. doi: 10.1007/s00262-008-0449-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nikula T.K., Curcio M.J., Brechbiel M.W., Gansow O.A., Finn R.D., Scheinberg D.A. A rapid, single vessel method for preparation of clinical grade ligand conjugated monoclonal antibodies. Nucl Med Biol. 1995;22:387–390. doi: 10.1016/0969-8051(94)00126-5. [DOI] [PubMed] [Google Scholar]
- 23.Dranoff G., Jaffee E., Lazenby A., Golumbek P., Levitsky H., Brose K., Jackson V., Hamada H., Pardoll D., Mulligan R.C. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte–macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delemarre F.G., Hoogeveen P.G., De Haan-Meulman M., Simons P.J., Drexhage H.A. Homotypic cluster formation of dendritic cells, a close correlate of their state of maturation. Defects in the biobreeding diabetes-prone rat. J Leukoc Biol. 2001;69:373–380. [PubMed] [Google Scholar]
- 25.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 26.Tang D., Kang R., Coyne C.B., Zeh H.J., Lotze M. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249:158–175. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nace G., Evankovich J., Eid R., Tsung A. Dendritic cells and damage-associated molecular patterns: endogenous danger signals linking innate and adaptive immunity. J Innate Immun. 2012;4:6–15. doi: 10.1159/000334245. [DOI] [PubMed] [Google Scholar]
- 28.Kroemer G., Galluzzi L., Kepp O., Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 29.Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4:941–952. doi: 10.1038/nri1498. [DOI] [PubMed] [Google Scholar]
- 30.Bianchi M.E. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 31.Garg A.D., Nowis D., Golab J., Vandenabeele P., Krysko D.V., Agostinis P. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta. 2010;1805:53–71. doi: 10.1016/j.bbcan.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 32.Basu S., Binder R.J., Ramalingam T., Srivastava P.K. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303–313. doi: 10.1016/s1074-7613(01)00111-x. [DOI] [PubMed] [Google Scholar]
- 33.Andersson U., Tracey K.J. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki Y., Mimura K., Yoshimoto Y., Watanabe M., Ohkubo Y., Izawa S., Murata K., Fujii H., Nakano T., Kono K. Immunogenic tumor cell death induced by chemoradiotherapy in patients with esophageal squamous cell carcinoma. Cancer Res. 2012;72:3967–3976. doi: 10.1158/0008-5472.CAN-12-0851. [DOI] [PubMed] [Google Scholar]
- 35.Chung H., Lee S.G., Kim H., Hong D., Chung J., Stroncek D., Lim J.B. Serum high mobility group box-1 (HMGB1) is closely associated with the clinical and pathologic features of gastric cancer. J Transl Med. 2009;7:38. doi: 10.1186/1479-5876-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang G.L., Zhang L.H., Bo J.J., Huo X.J., Chen H.G., Cao M., Liu D.M., Huang Y.R. Increased expression of HMGB1 is associated with poor prognosis in human bladder cancer. J Surg Oncol. 2012;106:57–61. doi: 10.1002/jso.23040. [DOI] [PubMed] [Google Scholar]
- 37.Riuzzi F., Sorci G., Donato R. The amphoterin (HMGB1)/receptor for advanced glycation end products (RAGE) pair modulates myoblast proliferation, apoptosis, adhesiveness, migration, and invasiveness. Functional inactivation of RAGE in L6 myoblasts results in tumor formation in vivo. J Biol Chem. 2006;281:8242–8253. doi: 10.1074/jbc.M509436200. [DOI] [PubMed] [Google Scholar]
- 38.Kuniyasu H., Oue N., Wakikawa A., Shigeishi H., Matsutani N., Kuraoka K., Ito R., Yokozaki H., Yasui W. Expression of receptors for advanced glycation end-products (RAGE) is closely associated with the invasive and metastatic activity of gastric cancer. J Pathol. 2002;196:163–170. doi: 10.1002/path.1031. [DOI] [PubMed] [Google Scholar]
- 39.Sasahira T., Akama Y., Fujii K., Kuniyasu H. Expression of receptor for advanced glycation end products and HMGB1/amphoterin in colorectal adenomas. Virchows Arch. 2005;446:411–415. doi: 10.1007/s00428-005-1210-x. [DOI] [PubMed] [Google Scholar]
- 40.van Beijnum J.R., Buurman W.A., Griffioen A.W. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1) Angiogenesis. 2008;11:91–99. doi: 10.1007/s10456-008-9093-5. [DOI] [PubMed] [Google Scholar]
- 41.Campana L., Bosurgi L., Rovere-Querini P. HMGB1: a two-headed signal regulating tumor progression and immunity. Curr Opin Immunol. 2008;20:518–523. doi: 10.1016/j.coi.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 42.Venereau E., Casalgrandi M., Schiraldi M., Antoine D.J., Cattaneo A., De Marchis F., Liu J., Antonelli A., Preti A., Raeli L. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209:1519–1528. doi: 10.1084/jem.20120189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Apetoh L., Ghiringhelli F., Tesniere A., Obeid M., Ortiz C., Criollo A., Mignot G., Maiuri M.C., Ullrich E., Saulnier P. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 44.Garnett C.T1., Palena C., Chakraborty M., Tsang K.Y., Schlom J., Hodge J.W. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004;64:7985–7994. doi: 10.1158/0008-5472.CAN-04-1525. [DOI] [PubMed] [Google Scholar]
- 45.Chakraborty M., Abrams S.I., Coleman C.N., Camphausen K., Schlom J., Hodge J.W. External beam radiation of tumors alters phenotype of tumor cells to render them susceptible to vaccine-mediated T-cell killing. Cancer Res. 2004;64:4328–4337. doi: 10.1158/0008-5472.CAN-04-0073. [DOI] [PubMed] [Google Scholar]
- 46.Reits E.A., Hodge J.W., Herberts C.A., Groothuis T.A., Chakraborty M., Wansley E.K., Camphausen K., Luiten R.M., de Ru A.H., Neijssen J. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203:1259–1271. doi: 10.1084/jem.20052494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chakraborty M., Wansley E.K., Carrasquillo J.A., Yu S., Paik C.H., Camphausen K., Becker M.D., Goeckeler W.F., Schlom J., Hodge J.W. The use of chelated radionuclide (samarium-153-ethylenediaminetetramethylenephosphonate) to modulate phenotype of tumor cells and enhance T cell–mediated killing. Clin Cancer Res. 2008;14:4241–4249. doi: 10.1158/1078-0432.CCR-08-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]