

Abstract
Many studies have shown that the acidity of solid tumors contributes to local invasion and metastasis. Oral pH buffers can specifically neutralize the acidic pH of tumors and reduce the incidence of local invasion and metastatic formation in multiple murine models. However, this effect is not universal as we have previously observed that metastasis is not inhibited by buffers in some tumor models, regardless of buffer used. B16-F10 (murine melanoma), LL/2 (murine lung) and HCT116 (human colon) tumors are resistant to treatment with lysine buffer therapy, whereas metastasis is potently inhibited by lysine buffers in MDA-MB-231 (human breast) and PC3M (human prostate) tumors. In the current work, we confirmed that sensitive cells utilized a pH-dependent mechanism for successful metastasis supported by a highly glycolytic phenotype that acidifies the local tumor microenvironment resulting in morphological changes. In contrast, buffer-resistant cell lines exhibited a pH-independent metastatic mechanism involving constitutive secretion of matrix degrading proteases without elevated glycolysis. These results have identified two distinct mechanisms of experimental metastasis, one of which is pH-dependent (buffer therapy sensitive cells) and one which is pH-independent (buffer therapy resistant cells). Further characterization of these models has potential for therapeutic benefit.
Abbreviations: ECAR, extracellular acidification rate; MMPs, matrix metalloproteases; OCR, oxygen consumption rate; OXPHOS, oxidative Phosphorylation; pHe, extracellular pH
Introduction
Progression to metastatic disease remains the highest mortality risk for cancer patients, despite significant efforts to therapeutically target metastatic lesions [1,2]. Microenvironmental acidosis in a primary tumor increases cellular motility and invasiveness, leading to increased metastasis [3–5]. During primary tumor development, cell metabolism is often altered resulting in up-regulated glycolysis even in well-oxygenated environments [2,6]. Aberrant glycolytic flux, the “Warburg Effect”, results in intracellular lactate and proton pools, both of which are shuttled into the tumor microenvironment by monocarboxylate and proton transporters to maintain intracellular pH (pHi) at physiologic levels [7,8]. One such transporter, carbonic anhydrase-9 (CA-IX) is a negative prognostic indicator in many cancers [9,10]. In addition to excessive H+ secretion, the chaotic architecture of tumor vasculature results in a reduced capacity of tumors to remove cellular waste. As a result, the extracellular pH (pHe) of tumors is significantly more acidic (pH 6.5-6.9) than normal tissue (pH 7.2-7.5) [11,12]. As acidosis is a common phenotype in solid tumors, we have developed a strategy to neutralize acidity using orally ingested buffers [13]. We have shown previously that treatment of mice with orally available buffers is efficacious in reducing spontaneous and experimental metastasis by increasing pHe due to higher buffering capacity of blood [14]. This is a targeted effect, as it only brings into balance the pH of tissues that were previously out of the physiological range [13]. Increased tumor pHe after treatment with oral buffers has been confirmed through magnetic resonance imaging and microelectrodes [14,15]. Sodium bicarbonate (pKa = 6.4), 2-imidazole-1-yl-3-ethoxycarbonylpropionic acid (pKa = 6.9), and free-base lysine (pKa = 10) have all been shown to be effective in reducing metastases in vivo[14–16]. Reduction of metastasis is dependent upon buffering, as reducing the buffering capacity of free-base lysine by lowering the pH to pH 8.4 (below the second pKa) rendered the therapy significantly less effective [17]. Treatment with buffer therapy is non-toxic, as mice maintain their weight, blood pH, renal function and immune cell distribution throughout the course of treatment [14–16]. Notably, the efficacy of buffer therapy is not universally observed. Metastases in two cell lines; MDA-MB-231, human breast adenocarcinoma; and PC3M, prostate adenocarcinoma; are both inhibited by buffer therapy, while B16-F10 cells, murine melanoma, and LL/2 cells, murine lung carcinoma, were resistant to the same treatment [14–16]. Of the currently available buffers, lysine is the most efficacious as it has the highest pKa of the three [13].
The success of buffer therapy suggests that, at least in sensitive cells, there is a metastatic mechanism that has an acidic pH optimum. Furthermore, proteases contribute to tissue remodeling during the metastatic cascade, and overexpression of matrix metalloproteases (MMPs) has been observed in a number of tumors and is a predictor of progression to metastatic disease [18,19]. Another type of protease, cysteine cathepsins, are lysosomal in origin but can be excreted into the tumor microenvironment [20]. Acidity and proteases have been linked in the “acid-mediated invasion” hypothesis which proposes that acidification of the tumor microenvironment can be associated with cathepsin release to trigger matrix remodeling [21]. Cell migration through tissue can occur through different mechanisms, either as single cells or in a collective fashion [22,23]. Cell migration patterns have been further classified based on cellular morphology of rounded and elongated cell types in 3D culture systems as well as in-vivo tumor models [24]. Additionally, tumor acidity and expression of proton pumps have been associated with increased cellular migration and invasion in breast and melanoma cell lines [25–27].
This study investigates the mechanisms of buffer therapy resistance. The observation that buffering is not universally efficacious led us to hypothesize that resistant and sensitive lines utilize different metastatic mechanisms, one that is pH-independent and one that is pH-dependent. Metabolic profiling confirms that buffer-sensitive lines have a much more robust glycolytic phenotype, compared to resistant lines, and that this is coupled to increased tumor acidification. In contrast, resistant lines constitutively expressed proteases in a pH-independent fashion, compared to sensitive lines whose protease activities were low and pH-dependent. Acidic pHe results in morphological changes in sensitive cells, while resistant cells were unaffected. We propose that sensitive cells activate proteases and alter their morphology by acidifying their microenvironment, which can be inhibited by buffer therapy and that resistant cells have constitutively active protease release.
Materials and Methods
Animals
Animals were housed according to Institutional Animal Care and Use Committee protocol at the University of South Florida Vivarium within Moffitt Cancer Center. Four- to six-week-old SCID-beige (Charles River) or nu/nu mice (Harlan) were used in experimental metastasis models or for fluorescent imaging of subcutaneous tumors, respectively.
Cell Lines
PC-3M-Luc6 clone, B16-F10-G5 clone and LL/2-M38 clone luciferase expressing cells were obtained from Xenogen Caliper. MDA-MB-231 and HCT116 cells (American Type Culture Collection) stably expressing luciferase were generated using lentiviral transduction. Cells were cultured in typical incubation conditions (37°C and 5% CO2). Cell counts and diameters were measured with the Countess Automated Cell Counter (Invitrogen).
Experimental Metastasis Model and Bioluminescent Imaging
Experimental metastasis models and bioluminescent imaging was performed as described previously [16]. SCID-beige mice were pretreated with 200 mmol/l lysine or tap water for 1 week before injection, and continued throughout the study. 1 × 106 cells were injected intravenously in 100 μl PBS. Mice were imaged immediately after injection by bioluminescent imaging to confirm successful injections. Metastasis formation is inferred from bioluminescent signal, which is reported as mean log photons emitted/second ± SEM. MDA-MB-231 growth rate doubling times (DT) were determined by fitting three parameter Gompertz function [28,29]. Statistical significance using Log-DT as a descriptor for the groups was determined using one-sided ANOVA test.
Metabolic Profile Analysis
Metabolic profiles were determined using the Seahorse Extracellular Flux (XF-96) analyzer (Seahorse Bioscience Chicopee, MA) as described previously [16]. Briefly, assay media was supplemented with 11 mmol/l glucose, 0.5 mmol/l sodium pyruvate and 2 mmol/l glutamine for mitochondrial stress tests and glucose-free media for glycolysis stress tests. Cells were treated with 1 μmol/l oligomycin, carbonyl cyanide 4-(Trifluoromethoxy) Phenylhydrazone, rotenone and antimycin during mitochondrial stress test. Glycolysis stress test treatments include 11 mmol/l glucose, 1 μmol/l oligomycin and 100 mmol/l 2-deoxyglucose. ECAR and oxygen consumption rate (OCR) values were standardized to mg/protein and reported as the mean ± SD.
Electrode Measurement of Tumor pH
pH measurements were performed as described previously [15]. Briefly, a reference electrode was placed in a non-tumor site. A needle microelectrode (optical density 0.8 mm with a beveled end) was inserted into the center of the tumor and was held in place until readings stabilized. pH was measured at three locations and reported as mean ± SEM.
In Vivo Protease Activity Measurements
nu/nu Mice were provided with either tap water or 200 mmol/l free base lysine 7 days before inoculation (Sigma Aldrich St. Louis, MO). 1 × 106 LL/2 or PC3M cells were injected as bilateral subcutaneous flank injections in PBS solution. Twenty-four hours before imaging, mice were injected with activatable fluorescent probes, MMPsense680 and Prosense750EX (Perkin Elmer Waltham, MA), intravenously. In vivo measurements were obtained using the FMT2500 (Perkin Elmer) tomographic imaging system. Fluorescent signal from each probe was quantified based on an internal standard. Data were reported as mean ± SD.
Quantitative PCR (qPCR)
RNA was isolated from cell pellets using RNeasy Mini Kit (Qiagen). qPCR reactions were carried out with iScript One-Step RT-PCR kit with SYBR Green (Bio-Rad) using Applied Biosystems StepOne PCR system (Applied Biosystems Grand Island, NY). MMP-2 and MMP-9 primers were obtained from S. Chellepan (Moffitt Cancer Center) (Supplemental Table 1). Data were analyzed using ΔΔCt, with the gene of interest normalized to β-actin.
Invasion Assay
Cells were pre-labeled with 10 μg/ml of DilC12(3) (BD Biosciences San Jose, CA) before seeding cells in serum free media into the apical chambers of the BD BioCoat™ Tumor Invasion System (BD Biosciences). Media containing serum was used as a chemo-attractant in the basal chambers. Fluorescence readings were obtained every 6 hours using a BioTek Synergy HT plate reader (BioTek Instruments Winooski, VT).
Microscopy Studies
Cell migration assays were conducted using a 96-well plate WoundMaker™ (Essen BioScience Ann Arbor, MI) to create homogenous 700- to 800-μm wounds. Images were recorded every 30 minutes and wound properties, including % Relative Wound Density (%RWD, shown below), were measured with IncuCyte Software (Essen BioScience).
Migration videos were recorded with a JuLi microscope using a 10 × objective lens (NanoEnTek) and generated with ImageJ software. Cell morphology studies were performed as previously described [24]. Confocal images were obtained with an Olympus FV1000 MPE multiphoton laser scanning microscope through a 60 × LUM Plan FI/IR 0.9N.A. water immersion lens (Olympus). 405 diode and Red HeNe lasers were used to excite the samples. Images were prepared using the FV10-ASW Version 03.00.01.15 software (Olympus).
Data Analysis
Data were analyzed using GraphPad Prism v6.02 (GraphPad Software, Inc. GraphPad Software, Inc. (La Jolla, CA) and Matlab (MathWorks, Inc. Natick, MA). Statistics were performed using an unpaired 2-tailed Student’s t test with Welch’s correction. Data are reported as mean ± SD or ± SEM.
Results
We have previously shown that experimental metastasis of PC3M cells was inhibited by 200 mmol/l lysine (pH = 10.1) (Supplemental Figure 1, A–C) [16,17]. Importantly, neutralized lysine (pH = 8.4) was much less effective in preventing metastases than lysine (pH = 10.1), showing the effect was due to buffering capacity [17]. In the current study, we sought to identify additional lysine-sensitive and -resistant cell lines using experimental metastasis models. Mice were pretreated with 200 mmol/l lysine or tap water for 1 week before intravenous injection of Firefly-luciferase expressing cells. Consistent with previous work, B16-F10 metastasis were unaffected by lysine treatment (Figure 1A)[14]. LL/2 cells were similarly unresponsive to lysine treatment, showing no difference in metastasis formation or survival benefit with treatment (Figure 1B and Supplementary Figure S1D). MDA-MB-231 cells experienced significantly lower metastatic burden after therapy (P < .05), which translated into a significant survival benefit (P < .05) (Figure 1D). HCT116 cells had not previously been tested using an experimental metastasis model, but had shown a reduction in local invasion in a window-chamber model when treated with 200 mmol/l bicarbonate [21]. In contrast to those results, treatment of mice with lysine had no effect on HCT116 metastatic formation or survival (Figure 1C and Supplementary Figure S1E). From this point on, cells will be identified as resistant or sensitive to lysine therapy with subscripts (R = resistant; S = sensitive).
Figure 1.

Effect of lysine on metastasis and survival. Efficacy of lysine was determined by pre-treating SCID-beige mice for a week before tail vein injection of cells stably expressing Firefly-luciferase in an experimental metastasis model. Treatment with 200 mmol/l lysine was administered continuously throughout the experiment. Metastasis formation was measured by bioluminescent imaging, reported as log photons per second ± SEM, and representative bioluminescent images of one mouse per cohort for each experiment are shown at the times indicated. Bioluminescent imaging of B16-F10 metastasis (Tap n = 10, Lysine n = 10)(A), LL/2 metastasis (Tap n = 10, Lysine n = 10) (B), and HCT116 metastasis (Tap n = 8, Lysine n = 10) (C) shows buffer therapy is ineffective. (D) Bioluminescent imaging of MDA-MB-231 metastasis shows buffer therapy is effective in reducing metastatic formation (upper panel) leading to a significant increase in survival (bottom panel) (Tap n = 5, Lysine n = 8). (E) Average cell growth curves measured over 72 hours indicates significant growth rate differences between resistant and sensitive cells and correlate with in vivo tumor growth rates. Data shown as mean cell number ± SD. (F) Cell diameter measurements of single cells in suspension show resistant cells are significantly smaller than sensitive cells. Data shown as mean cell diameter (μm) ± SD. Cells will be identified as resistant or sensitive to treatment with subscripts (R = Resistant; S = Sensitive). *P < .05; ***P < .001; ****P < .0001.
We subsequently performed in vitro studies to identify potential mechanisms of resistance. Profiling cell lines in vitro confirmed that cultured cells resemble cells grown in vivo, giving us confidence that mechanisms identified in vitro translate in vivo. For example, growth rates of resistant cells (B16-F10R, LL/2R and HCT116R) were significantly higher compared to sensitive cells (PC3MS and MDA-MB-231S) (P < .001) (Figure 1E). The in vitro growth curves closely followed in vivo growth rates, with B16-F10R and LL/2R expanding significantly faster than PC3MS and MDA-MB-231S (P < .0001) (compare Figure 1, A, B, D and S1C with Figure 1E), and HCT116R growing at an intermediate rate (Figure 1, C and E). Cell size was measured to estimate their size in circulation, showing resistant cells were significantly smaller in suspension than were the sensitive cells (P < .05) (Figure 1F), which may allow for more rapid extravasation during metastasis.
Buffer therapy selectively increases the pHe of tumors; hence, we sought to determine the effect of pH on invasion in vitro[14,15]. Using fluorescently labeled cells in a Boyden chamber system, cell invasion was quantitatively measured over 48 hours in either pH 6.8 or pH 7.4 media. Uptake of fluorescent dye had no adverse effect on cell proliferation (data not shown). Invasion rates were measured for each cell line and analyzed to determine the differential rate of invasion between each pH condition, in order to self-normalize for differences in uptake of the fluorescent dye across cell lines. Resistant cells, B16-F10R, LL/2R, and HCT116R showed no significant change in their rates of invasion between pH 6.8 and pH 7.4 (Figure 2). MDA-MB-231S cells, however, had a significantly increased rate of invasion at pH 6.8, relative to pH 7.4, when compared to B16-F10R (P < .05), LL/2R (P < .05), and HCT116R (P < .005) (Figure 2). While not statistically significant, PC3MS cells followed the same trend as MDA-MB-231S cells of having an increased rate of invasion at pH 6.8 relative to pH 7.4 (Figure 2). As growth rates are suppressed under these acute acidic conditions for each cell line (data not shown), we conclude that the increased invasion rate in sensitive cell lines is a real phenomenon and not the result of growth rate differences. Together, these data suggest that resistant and sensitive cells utilize different invasive mechanisms, pH-independent, and pH-dependent mechanisms, respectively.
Figure 2.
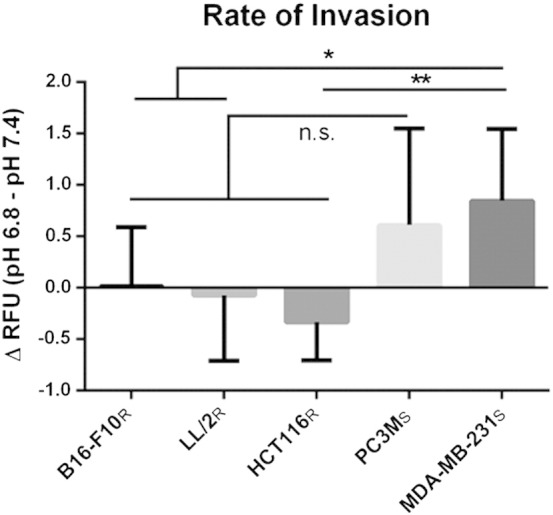
Effect of pHe on invasion rates of resistant and sensitive cells. In vitro invasion assay using a Boyden chamber coated with Matrigel. Fluorescently labeled cells were measured every 6 hours for 48 hours for invasion through Matrigel layer. Data shown is the result of two biologic experiments (n = 6/sample) normalized to wells lacking serum attractant (n = 2/sample). Data is presented as the mean difference in the rate of invasion of cells cultured in pH 6.8 and cells cultured in pH 7.4 ± SD. The rate of invasion of sensitive cells increases in pH 6.8 compared to resistant lines. *P < .05; **P < .005. R, resistant; S, sensitive.
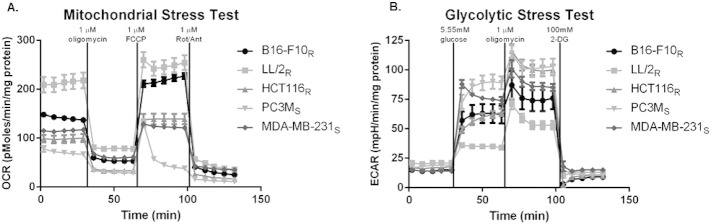
The Warburg Effect is a common phenomenon in solid tumors that contributes to acidification of the tumor microenvironment. Originally, we hypothesized resistant lines would produce acid at a higher rate, implying that increasing the buffer load could overcome buffer resistance. To test this, we examined the effect of 400 mmol/l bicarbonate on experimental B16-F10R metastasis formation and observed no effect (data not shown), suggesting resistant cells were not merely producing acid at a higher rate. This was verified by metabolic profiling of resistant and sensitive cells using a Seahorse XF® analyzer which measures real-time H+ production and oxygen consumption rate over a monolayer of cells in a transient microchamber. Metabolic profiling assays were performed in parallel and normalized to either cell number or protein concentration, to confirm that normalized results were not an artifact of cell size differences (data not shown). To determine glycolytic activity, a “glycolytic stress test” was performed, which includes measuring extracellular acidification rates (ECAR) after sequential addition of glucose to measure basal glycolysis, a mitochondrial poison (oligomycin) to estimate total glycolytic capacity, and 2-deoxyglucose to measure non-glycolytic ECAR. Interestingly, sensitive cells had significantly higher basal glycolytic rates, compared to resistant cells (P < .0001) (Figure 3A). Glycolytic reserve is calculated by measuring the difference in the maximal glycolytic capacity, after treatment with oligomycin, and basal glycolysis. Possibly as a consequence of their high basal rates, the sensitive cells showed significantly lower amounts of glycolytic reserve, compared to resistant cells (P < .0001), suggesting that they are near maximum glycolytic capacity in their basal metabolic state (Figure 3B).
Figure 3.

Metabolic profile analysis of resistant and sensitive cells and tumor pH. In vitro oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of resistant and sensitive cells, measured using Seahorse XF-96 Instrument. Metabolic data are presented as mean ± SD. (A) ECAR measurements in response to 5.55 mmol/l glucose stimulation indicate basal glycolytic activity of cells. Sensitive cells are significantly more glycolytic than resistant cells. (B) The increase in ECAR of cells in response to treatment with 1 μmol/l oligomycin minus basal glycolytic activity indicates the glycolytic reserve of cells. Sensitive cells have significantly reduced glycolytic reserve compared resistant cells.(C) Basal OCR measurements show significantly higher oxidative phosphorylation flux in resistant cells compared to sensitive cells. (D) OCR contributing to production of ATP during oxidative phosphorylation is measured by the difference of basal OCR and the OCR of cells after treatment with 1 μmol/l oligomycin, a mitochondrial ATP synthase inhibitor. The amount of OCR contributing to the production of ATP by oxidative phosphorylation is significantly higher in resistant cells compared to sensitive cells. (E) The OCR/ECAR ratio of cells during basal metabolism indicates that sensitive cells are significantly more glycolytic than resistant cells. (F) Intratumoral pH measurements of subcutaneous tumors using pH electrodes shows that increased glycolytic activity of sensitive cells, PC3MS, contributes to a more acidic tumor microenvironment than resistant tumors, B16-F10R. pH data are presented as mean of independent measurements (B16-F10Rn = 10; PC3MSn = 5) ± SEM. *P < .05; ****P < .0001. R, resistant; S, sensitive.
The rate of decrease in O2 can be converted to an OCR. The “mitochondrial stress test” initially determines basal respiration. We observed that resistant cells had significantly higher basal OCR compared to sensitive cells (P < .05) (Figure 3C). We suspect that the higher basal OCR in resistant cells is most likely related to the energy demands of a higher proliferation rate (Figure 1E). Treatment with an inhibitor of the F0 subunit of mitochondrial adenosine triphosphate (ATP) synthase, oligomycin, provided OCR attributed directly to ATP production. These data showed that buffer therapy resistant cells had significantly higher OCR that attributed to ATP production (P < .0001) (Figure 3D). These mitochondrial stress test data suggest that resistant cells rely on mitochondrial oxidative metabolism (OXPHOS) for their energy needs.
Metabolic analysis of OXPHOS and glycolytic pathways in resistant and sensitive cells showed distinct metabolic profiles. This can be directly shown by expressing data as basal OCR/ECAR ratios, which are self-normalized and showed that sensitive cells were significantly (P < 0.0001) more glycolytic than resistant cells (Figure 3E). These differences in metabolic profiles were related to the ability of buffer therapy to inhibit metastasis (Figure 1, A–D, Supplemental Figure 1, A–C), and correlate with distinct phenotypic differences in their in vivo and in vitro proliferation rates (Figure 1E), and size (Figure 1F). pHe measurements of PC3MS and B16-F10R tumors support the metabolic evidence presented, with PC3MS tumors being more acidic than B16-F10R tumors (Figure 3F). Previous studies, using MRS imaging with the pH indicator 3-aminopropylphosphonate (3-APP) and fluorescent ratio imaging with SNARF-1, have shown that MDA-MB-231 tumors are similarly acidic and can be manipulated with buffer therapy to increase the tumor pH [14]. We have previously observed sensitivity of HCT116-GFP cells to bicarbonate in window-chamber studies [21], but observed clear resistance of HCT-116R-Luc cells in our current studies. Interestingly, HCT116-GFP cells had a different metabolic profile than the HCT116R-Luc cells used in the in vivo experimental metastasis model herein, suggesting the presence of two phenotypically distinct populations of these cells, supported by differences in their OCR/ECAR ratios (P < .0001) (Supplementary Figure 3). Importantly, DNA fingerprinting confirmed that both lines used were HCT116 colorectal cells (data not shown). Notably, the glycolytic HCT-116-GFP cells were inhibited by buffer therapy [21]; whereas the oxidative HCT-116-luc cells were not (Figure 1C).
Invasion kinetics and metabolic profiling suggest that resistant cells invade via a mechanism that is pH-independent. Proteases have been identified as key enzymes involved in the metastatic cascade. Prior data have shown that low pH significantly stimulated the release of active cathepsin-B from buffer-sensitive MDA-MB-231 cells in 2-D and 3-D culture [14,30]. Hence, we hypothesize that resistant cells may release active proteases in a constitutive, pH-independent fashion. To investigate this, we imaged mice bearing tumors using fluorescent indicators that are activated by protease activity. ProSense 750EX and MMPsense 680 fluoresce upon cleavage by Cathepsins B, L, S and Plasmin (ProSense 750EX) or MMPs-2, -3, -9 and -13 (MMPsense 680). Using a tomographic near-IR fluorescence imaging system, FMT2500, activated probes were imaged 24 hours post-probe injection followed by fluorescent signal integration over the tumor region of interest (ROI). B16-F10R tumors were not used due to high melanin levels that quench fluorescence. Thus, we measured the in vivo protease activity of the most resistant (LL/2R) and the most sensitive (PC3MS) cell lines (Figure 1B and Supplemental Figure S1, A–C). Quantification of activated ProSense 750EX in LL/2R tumors showed significantly higher cysteine cathepsin activity compared to PC3MS under control conditions (P < .0001) (Figure 4, A,C). Although treatment reduced ProSense 750EX activation in LL/2R tumors (P < .05), activity still remained significantly higher than PC3MS cells under either pH condition (Figure 4, A,C) [20]. Conversely, MMPsense 680 was visibly activated in both LL/2R and PC3MS tumors in control mice (Figure 4, B,D). Buffer treatment increased MMP activity in LL/2R tumors (P < 0.05), and reduced MMP activity 2-fold in PC3MS tumors, but was not statistically significant (Figure 4, B,D). Resistant LL/2R tumors had higher intrinsic cathepsin activity than did PC3MS tumors and elevated MMP activity after buffer therapy. Therefore, we can conclude that resistant lesions have more protease activity compared to sensitive lesions, which may be contributing to buffer therapy resistance.
Figure 4.

Resistant tumors have increased in-vivo protease activity. Mice bearing LL/2R (Tap n = 7, Lysine n = 8) and PC3MS (Tap n = 7, Lysine n = 5) tumors were injected with activatable probes, ProSense 750EX and MMPsense 680, which report Cathepsin and MMP activity, respectively. Representative images of fluorescent tomographic imaging showing cathepsin activity through ProSense 750EX signal (A) through MMPsense 680 (B) in LL/2R and PC3MS tumors in mice receiving either tap water or lysine buffer. (C) Quantitation of ProSense 750EX activated signal in tumors, normalized to tumor size. (D) Quantitation of MMPsense 680 activated signal in tumors, normalized to tumor size. Data are presented as mean nanomolar concentration ± SD. *P < .05; **P < .01; ****P < .0001. R, resistant; S, sensitive.
To confirm in vivo protease activity results, cell cultures exposed to media at pH 7.4 or pH 6.8 were analyzed for MMP-2, -3, -9, and -13 (MMPs that activate MMPsense 680) mRNA expression. We chose to focus on MMP expression due to the significant increase of MMP activity observed upon treatment with lysine in LL/2R tumors (Figure 4, B,D). Both LL/2R and PC3MS cells exhibited an increased expression of MMP-3 and -13 at pH 6.8 (Figure 5A). To further compare differences, transcript expression in LL/2R cells was normalized to PC3MS cultures. LL/2R cells have higher MMP expression compared to PC3MS cells for each of the transcripts analyzed, with the exception of MMP-9 (Figure 5B). Importantly, there were no differences in the fold change of each transcript relative to PC3MS when exposed to acidic or physiological conditions, showing that higher proteolytic expression of resistant cells was pH-independent. MMP transcript analysis confirms our hypothesis that buffer therapy resistant lines are constitutively proteolytic in vitro (Figure 5) as well as in vivo (Figure 4).
Figure 5.
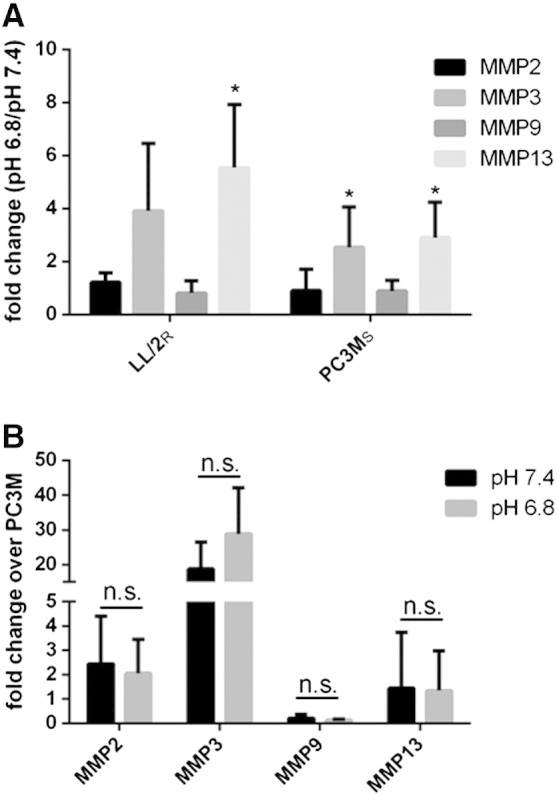
Elevated MMP expression in resistant cells. Quantitation of MMP expression in sensitive and resistant cells grown in physiological or acidic pH media for 24 hours. Transcripts were normalized to β-actin expression before analysis. (A) Ratio of expression of MMP-2, -3, -9 and -13 in LL/2R and PC3MS cultured in acidic media relative to cells cultured in physiological media. (B) Ratio of expression of MMP-2, -3, -9, and -13 in LL/2R cells relative to expression in PC3MS cells cultured in acidic and physiological media. Data are the average of three independent experiments and is reported as mean ± SD. *P < .05. R, resistant; S, sensitive.
Recent research has focused on identifying different modes of migration and invasion during metastasis [22]. To understand migratory differences between LL/2R and PC3MS, we monitored cell motility in a wound healing assay. Cell migration was observed by imaging cells for 18 hours after wound formation. Percent relative wound density was calculated by measuring the density of cells that migrated into the original wound. LL/2R cells were significantly more migratory than PC3MS cells cultured in physiologic pH (p < 0.0001) (Figure 6, A–B). Interestingly, exposure to low pH had differential effects on cell migration for each of the cell lines. LL/2R migration across the wound was significantly retarded under acidic conditions (P < .01), while PC3MS cell migration was significantly accelerated under acidic conditions (P < .0001) (Figure 6, A–B). Additionally, sensitive and resistant cells exhibited different modes of migration. PC3MS cells moved across the wound as a mass following leading cells, suggestive of a collective cell migration phenotype, while LL/2R cells moved as single cells, which is characteristic of single-cell or multicellular streaming invasive phenotypes (Supplemental Video). While studying the movement of cells in a 2D culture environment is useful, 3D cultures more closely resemble physiological obstacles encountered during metastasis. Using phalloidin staining, we studied the morphology of LL/2R and PC3MS cells in a thick (500–1000 μm) layer of Matrigel® under physiological or acidic conditions. In a 3D matrix LL/2R and PC3MS cells have distinctly different cellular morphologies. PC3MS cells have an elongated phenotype with multiple protrusions into the local matrix (Figure 7 and Supplemental Figure 4). In response to acidic conditions, PC3MS cells maintained an elongated phenotype although completely void of the protrusions that were observed under physiological conditions, which may allow for greater invasive potential. In contrast, LL/2R cells had a rounded morphology that, consistent with the phenotypes we characterized above, remain unchanged from physiologic to acidic pH (Figure 7 and Supplemental Figure 4).
Figure 6.

Migratory patterns of LL/2R and PC3MS cells; 700- to 800-μm wounds were created in confluent cell cultures exposed to physiologic or acidic media 24 hours before wound formation, and during the duration of the experiment. Samples were imaged in 30-minute intervals for 18 hours. (A) Representative microscopy images of LL/2R (upper panel) and PC3MS (lower panel) show movement across a wound at 0 and 18 hours in pH 7.4 or pH 6.8 media. Scale bars represent 300 μm. (B) Percent relative wound density was determined by measuring the density of cells within the original wound site at each of the time points imaged. Data are shown as the mean ± SEM and are representative of three independent experiments. **P < .01; ****P < .0001; R, resistant; S, sensitive.
Figure 7.

Morphologies in a 3D matrix in vitro. LL/2R and PC3MS cells were seeded onto a thick (500-1000 μm) layer of polymerized Matrigel and invaded into the matrix over a period of 72 hours in pH 6.8 or pH 7.4 media. Single cells were imaged using confocal microscopy for morphological studies. Representative images of PC3MS cells (upper panels) and LL/2R cells (lower panels) show a 3D reconstruction of at least 20 slices. Phalloidin (F-actin) is shown in red, Hoechst nuclear stain is shown in blue. Scale bars represent 10 μm. R, resistant; S, sensitive.
Discussion
In our previous studies, we demonstrated that an acidic microenvironment is critical for carcinogenesis and tumor invasion. Furthermore, we have found that systemic buffers reduce intra- and peri-tumoral acidity, inhibit carcinogenesis in transgenic mice, and inhibit metastatic growth in a wide range of cell lines in-vivo[14,21,31]. However, as with most therapeutic regimens, efficacy of buffer therapy was not universally observed. The current work focused on identifying molecular and metabolic phenotypes of resistant cells, with an expectation that such data could identify additional biomarkers to stratify tumors for their response to buffer therapy. Using a panel of cells representing different cancers, we have characterized two responsive cell lines, PC3MS and MDA-MB-231S, and three resistant cell lines, B16-F10R, and LL/2R, HCT116R. In addition to faster growth rates in vivo and in vitro, resistant cells were significantly smaller in diameter than sensitive cells, which may allow increased access to invade the extracellular space, either through more efficient extravasation or secondary site colonization. Faster growth and smaller size may be enough to render resistant cells too aggressive for buffer therapy to be effective.
Further exploration, however, has revealed a number of other important molecular and metabolic parameters that could contribute to resistance. Kinetic invasion assays suggest that there are distinct mechanisms used for invasion by these two groups. Sensitive line invasion is pH-dependent, allowing buffer therapy to intercept metastasis by neutralizing acidity in vivo. Resistant cell line invasion, on the other hand, is pH-independent, bypassing the need for acidosis to metastasize.
Metabolic alterations contribute to acidification of tumor microenvironments. Metabolic profiling showed that sensitive cells were unequivocally more glycolytic than the resistant cells. Cells with elevated glycolysis produce more acidic tumors. Previously, we have confirmed that buffer therapy is an effective method of increasing the pHe of tumors, which diminishes the ability of sensitive, but not resistant cells, to invade locally and metastasize [14–16]. While resistant tumors were less acidic than highly glycolytic sensitive tumors, they were nonetheless more acidic relative to normal tissues, likely due to poor perfusion. Neutralizing the tumor acidity in these tumors had less of an effect on their metastatic potential because they have upregulated mechanisms to bypass the need for acid-stimulated invasion.
Expression, release, and enzymatic activity of proteases are regulated by acidosis. While sensitive cells, PC3MS, have measureable expression of MMPs, resistant cells, LL/2R, have consistently higher expression of MMPs regardless of pH. Expression of MMPs correlated with protease activity in vivo. Interestingly, LL/2R tumors had a significant increase in MMP activity in response to treatment, suggesting buffer therapy could exacerbate the metastatic burden, although increases in metastatic formation in mice receiving buffer therapy was not observed. Protease activation in resistant tumors is not adversely affected by buffer therapy, and allows resistant lines to circumvent inhibition of metastasis by buffer therapy. Similar results were observed in a parallel study [30], in which acidic pHe increased pericellular active cysteine cathepsins in vitro, which was reduced after buffer therapy treatment in vivo. Furthermore, resistant and sensitive cells exhibited distinct morphological differences in 3D culture systems. Interestingly, acidic conditions resulted in the loss of protrusions in 3D culture of PC3MS, which may contribute to their increased invasiveness in acidosis. Resistant and sensitive cells consistently had differential responses to changes in extracelluar pH, regardless of the inclusion or absence of Matrigel matrix (migration, invasion and morphology studies), suggesting that the changes observed were due to pH alterations, rather than cell signaling pathways such as integrin signaling.
Although buffer therapy is not universally effective in reducing metastases, it does have potential advantages over targeted cytotoxic chemotherapies that are in current clinical practice. Tumors are heterogeneous, containing genetically distinct regional sub-populations that originate over a lifetime of tumor growth [32–34]. Intrinsic or acquired resistance to chemotherapy is a major obstacle of targeted therapies clinically, and will continue to remain so for the foreseeable future [35]. In contrast, treatment of solid tumors with buffer therapy targets tumor acidity, a common phenotypic consequence of tumor somatic evolution [36,37]. In addition to enhancing local invasion and metastatic potential, tumor acidosis contributes to drug resistance through ion-trapping of weakly basic chemotherapeutics, preventing active drug from reaching therapeutic doses within cells [38–40]. Therefore, buffer therapy may also be useful as an adjuvant to traditional chemotherapies. Notably, a clinical trial of buffer therapy in cancer patients has recently been initiated (NCT01846429).
In the current studies, buffer therapy was initiated before inoculation to prevent progression to metastatic disease. Previous studies show buffer therapy has little effect on reducing primary tumor growth, but significantly reduces spontaneous metastasis formation [14]. Similarly, in a transgenic prostate cancer model, buffer therapy prevented development of prostate adenocarcinoma when therapy was initiated immediately after weaning (4 weeks of age) [31]. Interestingly, if therapy was initiated later (10 weeks of age) development of prostate cancer was delayed, but was not inhibited, while progression to metastatic disease was still prevented with treatment. Our data shows that buffer therapy is an effective method of halting tumor progression and metastasis formation, but also indicates that the timing of therapy initiation is instrumental for maximal efficacy. Future studies on optimization of buffer therapy delivery and treatment schedules are necessary to harness the full potential of buffer therapy clinically.
Identifying distinct metastatic mechanisms of sensitive and resistant tumors allows for the identification of predictive biomarkers of buffer therapy response. FDG-PET screening may be an ideal method of screening patients to assess their glycolytic phenotype to predict response to buffer therapy. Clinically, FDG-PET imaging is used to diagnose up to 90% of primary tumors, indicating that the vast majority of patients have glycolytic tumors that may benefit from treatment with buffer therapy [41]. Further screening of additional cell lines for responsiveness to buffer therapy will help solidify potential biomarkers or alternative treatments for resistant tumors. A current limitation of our research was the use of five cell lines originating from different cancers. Deeper analysis into panels of cell lines originating from the same primary organ site will surely provide more insight and will need to be studied in the future. Such observations are commencing with the identification of two populations of HCT116 cells that have distinctly different metabolic and invasive behaviors.
Acknowledgments
Grant Support: This study was supported by grants U54 CA143970 (to R.A. Gatenby and R.J. Gillies); R01 CA 077575 (to R.J. Gillies and R.A. Gatenby); and 1S10RR031726-01 (to D.L. Morse).
Conflicts of Interest: We have a Sponsored Research Agreement with Seahorse Biosciences to develop methods for metabolic profiles of biopsy tissues. This is relevant to the current publication, as a related Seahorse instrument has been used to metabolically profile cells. The SRA is limited to support for an assistant lab member, and related supplies.
Appendix A.
Supplemental Figure 1.

Sensitivity of PC3MS to treatment with lysine buffer therapy. (A–C) Figure reprinted from [16]. (A) Representative bioluminescent images of PC3MS metastasis in SCID mice. (B) Ex vivo bioluminescent imaging of PC3MS metastasis. (C) Graphic representation of bioluminescent imaging of PC3MS metastasis. Data is reported as log photons/sec ± SEM. Kaplan Meier curves showing survival of experimental metastasis models of LL/2R (D) and HCT116R (E). (F) Box plot of log doubling time (DT) estimate of MDA-MB-231 experimental metastasis data. *P < 0.5. R, resistant.
Supplemental Figure 2.
Metabolic analysis of buffer therapy resistant and sensitive cells. A. Line graph showing sensitive and resistant cells oxygen consumption rates (OCR) through a mitochondrial stress test. Five basal measurements were obtained while cells were in media containing 5.55 mmol/l glucose, 2 mmol/l glutamine and 1 mmol/l pyruvate. Treatment of cells with oligomycin (1 μmol/l) led to a decrease in OCR that can be related to ATP production, followed by an increase in OCR to maximal respiration rates after treatment with FCCP (1 μmol/l). Finally, complete OXPHOS inhibition was achieved after treatment with Rotentone (1 μmol/l) and Antimycin A (1 μmol/l) B. Line graph showing sensitive and resistant cells extracellular acidification rates (ECAR) through a glycolytic stress test. Five basal measurements of cells that had been glucose starved for 2 hours shows non-glycolytic acidification. Stimulation of cells with glucose (5.55 mmol/l) results in basal glycolysis levels, which is increased to maximal glycolysis flux upon treatment with oligomycin (1 μmol/l). Glycolysis inhibition is achieved with treatment of 2DG (100 mmol/l). Data was normalized to mg protein and is reported as mean ± SD. R= Resistant, S= Sensitive.
Supplemental Figure 3.

Metabolic profile analysis of HCT116-Luc and HCT116-GFP cells. Glycolytic and mitochondrial stress tests show different metabolic profiles of HCT116-Luc and HCT116-GFP cells. (A) Extracellular acidification rates (ECAR) of cells after stimulation of glycolysis with glucose (5.55 mmol/l), oligomycin (1 μmol/l) and glycolysis inhibitor, 2DG (100 mmol/l). (B) Oxygen consumption rates (OCR) of cells before treatment with oligomycin (1 μmol/l), FCCP (1 μmol/l) and Rotentone (1 μmol/l) and Antimycin A (1 μmol/l). (C) OCR/ECAR ratio of cells during basal metabolism. (D) Representative brightfield microscopy images of HCT116-Luc and HCT116-GFP cells in pH 7.4 culture conditions. Data shown as mean ± SD. Scale bars represent 100 μm.
Supplemental Figure 4.

Morphologies in a 3D matrix in vitro. LL/2R and PC3MS cells were seeded onto a thick (500-1000 μm) layer of polymerized matrigel and invaded into the matrix over a period of 72 hours in pH 6.8 or pH 7.4 media. Single cells were imaged using confocal microscopy for morphological studies. Additional representative images of PC3MS cells (upper panels) and LL/2R cells (lower panels) show a 3D reconstruction of at least 20 slices. Phalloidin (F-actin) is shown in red; Hoechst nuclear stain is shown in blue. Scale bars represent 10 μm. R, resistant; S, sensitive.
Supplemental Table 1.
qPCR primer sequences.
| h/m MMP2 FW | 5’-AGATCTTCTTCTTCAAGGACCGCTT-3’ |
| h/m MMP2 RV | 5’-GGCTGGTCAGTGGCTTGGGGTA-3’ |
| hMMP3 FW | 5’-TCCAATCCTACTGTTGCTGTGCGT-3’ |
| hMMP3 RV | 5’-ACAAGGTTCATGCTGGTGTCCTCA-3’ |
| mMMP3 FW | 5’-AAGTTCCTCGGGTTGGAGATGACA-3’ |
| mMMP3 RV | 5’-ACCAACATCAGGAACACCACACCT-3’ |
| h/m MMP9 FW | 5’-CGGAGCACGGAGACGGGTATC-3’ |
| h/m MMP9 RV | 5’-GGCAGAGTAGGAGCGGCCCT-3’ |
| hMMP13 FW | 5’-TCCCAGTGGTGGTGATGAAGATGA-3’ |
| hMMP13 RV | 5’-TTCCCGCGAGATTTGTAGGATGGT-3’ |
| mMMP13 FW | 5’-TGGCTTAGAGGTGACTGGCAAACT-3’ |
| mMMP13 RV | 5’-TATTCACCCACATCAGGCACTCCA-3’ |
| hACTB FW | 5’-CCACACCTTCTACAATGAGC-3’ |
| hACTB RV | 5’-CATGATCTGGGTCATCTTCTCG-3’ |
| mACTB FW | 5’-ACCGCTCGTTGCCAATAGTGATGA-3’ |
| mACTB RV | 5’-TGAGAGGGAAATCGTGCGTGACAT-3’ |
Appendix B.
Time-lapse microscopy of wound healing assays. Cells were exposed to pH 6.8 or pH 7.4 media for 24 hours before wound generation, and continued for the duration of the experiment. (A) Recording of time-lapse microscopy of PC3MS cells movement across a wound. (B) Recording of time-lapse microscopy of LL/2R cells movement across a wound. Images were recorded in 5 minute intervals for a total of 18 hours post wound generation and shown at 12 fps. Scale bars represent 200 μm.
References
- 1.Chambers A.F., Groom A.C., MacDonald I.C. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Moellering R.E., Black K.C., Krishnamurty C., Baggett B.K., Stafford P., Rain M., Gatenby R.A., Gillies R.J. Acid treatment of melanoma cells selects for invasive phenotypes. Clin Exp Metastasis. 2008;25:411–425. doi: 10.1007/s10585-008-9145-7. [DOI] [PubMed] [Google Scholar]
- 4.Rofstad E.K. Microenvironment-induced cancer metastasis. Int J Radiat Biol. 2000;76:589–605. doi: 10.1080/095530000138259. [DOI] [PubMed] [Google Scholar]
- 5.Rofstad E.K., Mathiesen B., Kindem K., Galappathi K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res. 2006;66:6699–6707. doi: 10.1158/0008-5472.CAN-06-0983. [DOI] [PubMed] [Google Scholar]
- 6.Warburg O., Wind F., Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pinheiro C., Longatto-Filho A., Ferreira L., Pereira S.M., Etlinger D., Moreira M.A., Jube L.F., Queiroz G.S., Schmitt F., Baltazar F. Increasing expression of monocarboxylate transporters 1 and 4 along progression to invasive cervical carcinoma. Int J Gynecol Pathol. 2008;27:568–574. doi: 10.1097/PGP.0b013e31817b5b40. [DOI] [PubMed] [Google Scholar]
- 8.Pinheiro C., Longatto-Filho A., Scapulatempo C., Ferreira L., Martins S., Pellerin L., Rodrigues M., Alves V.A., Schmitt F., Baltazar F. Increased expression of monocarboxylate transporters 1, 2, and 4 in colorectal carcinomas. Virchows Arch. 2008;452:139–146. doi: 10.1007/s00428-007-0558-5. [DOI] [PubMed] [Google Scholar]
- 9.Ord J.J., Agrawal S., Thamboo T.P., Roberts I., Campo L., Turley H., Han C., Fawcett D.W., Kulkarni R.P., Cranston D. An investigation into the prognostic significance of necrosis and hypoxia in high grade and invasive bladder cancer. J Urol. 2007;178:677–682. doi: 10.1016/j.juro.2007.03.112. [DOI] [PubMed] [Google Scholar]
- 10.Hussain S.A., Ganesan R., Reynolds G., Gross L., Stevens A., Pastorek J., Murray P.G., Perunovic B., Anwar M.S., Billingham L. Hypoxia-regulated carbonic anhydrase IX expression is associated with poor survival in patients with invasive breast cancer. Br J Cancer. 2007;96:104–109. doi: 10.1038/sj.bjc.6603530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffiths J.R. Are cancer cells acidic? Br J Cancer. 1991;64:425–427. doi: 10.1038/bjc.1991.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wike-Hooley J.L., Haveman J., Reinhold H.S. The relevance of tumour pH to the treatment of malignant disease. Radiother Oncol. 1984;2:343–366. doi: 10.1016/s0167-8140(84)80077-8. [DOI] [PubMed] [Google Scholar]
- 13.Silva A.S., Yunes J.A., Gillies R.J., Gatenby R.A. The potential role of systemic buffers in reducing intratumoral extracellular pH and acid-mediated invasion. Cancer Res. 2009;69:2677–2684. doi: 10.1158/0008-5472.CAN-08-2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robey I.F., Baggett B.K., Kirkpatrick N.D., Roe D.J., Dosescu J., Sloane B.F., Hashim A.I., Morse D.L., Raghunand N., Gatenby R.A. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009;69:2260–2268. doi: 10.1158/0008-5472.CAN-07-5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ibrahim Hashim A., Cornnell H.H., Coelho Ribeiro Mde L., Abrahams D., Cunningham J., Lloyd M., Martinez G.V., Gatenby R.A., Gillies R.J. Reduction of metastasis using a non-volatile buffer. Clin Exp Metastasis. 2011;28:841–849. doi: 10.1007/s10585-011-9415-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ibrahim Hashim A., Wojtkowiak J.W., de Lourdes Coelho Ribeiro M., Estrella V., Bailey K.M., Cornnell H.H., Gatenbyl R.A., Gillies R.J. Free base lysine increases survival and reduces metastasis in prostate cancer model. J Cancer Sci Ther. 2011;S1 [PMC free article] [PubMed] [Google Scholar]
- 17.Ribeiro MdLC S.A., Bailey K.M., Kumar N.B., Sellers T.A., Gatenby R.A., Ibrahim-Hashim A., Gillies R.J. Buffer therapy for cancer. J Nutr Food Sci. 2012;S2 [PMC free article] [PubMed] [Google Scholar]
- 18.Kessenbrock K., Plaks V., Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deryugina E.I., Quigley J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9–34. doi: 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- 20.Turk V., Stoka V., Vasiljeva O., Renko M., Sun T., Turk B., Turk D. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta. 2012;1824:68–88. doi: 10.1016/j.bbapap.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Estrella V., Chen T., Lloyd M., Wojtkowiak J., Cornnell H.H., Ibrahim-Hashim A., Bailey K., Balagurunathan Y., Rothberg J.M., Sloane B.F. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013;73:1524–1535. doi: 10.1158/0008-5472.CAN-12-2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedl P., Locker J., Sahai E., Segall J.E. Classifying collective cancer cell invasion. Nat Cell Biol. 2012;14:777–783. doi: 10.1038/ncb2548. [DOI] [PubMed] [Google Scholar]
- 23.Friedl P., Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147:992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 24.Sahai E., Marshall C.J. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol. 2003;5:711–719. doi: 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- 25.Reshkin S.J., Bellizzi A., Albarani V., Guerra L., Tommasino M., Paradiso A., Casavola V. Phosphoinositide 3-kinase is involved in the tumor-specific activation of human breast cancer cell Na(+)/H(+) exchange, motility, and invasion induced by serum deprivation. J Biol Chem. 2000;275:5361–5369. doi: 10.1074/jbc.275.8.5361. [DOI] [PubMed] [Google Scholar]
- 26.Stock C., Gassner B., Hauck C.R., Arnold H., Mally S., Eble J.A., Dieterich P., Schwab A. Migration of human melanoma cells depends on extracellular pH and Na +/H + exchange. J Physiol. 2005;567:225–238. doi: 10.1113/jphysiol.2005.088344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Denker S.P., Barber D.L. Cell migration requires both ion translocation and cytoskeletal anchoring by the Na-H exchanger NHE1. J Cell Biol. 2002;159:1087–1096. doi: 10.1083/jcb.200208050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laird A.K. Dynamics of tumor growth. Br J Cancer. 1964;13:490–502. doi: 10.1038/bjc.1964.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehrara E., Forssell-Aronsson E., Johanson V., Kolby L., Hultborn R., Bernhardt P. A new method to estimate parameters of the growth model for metastatic tumours. Theor Biol Med Model. 2013;10:31. doi: 10.1186/1742-4682-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rothberg J.M., Bailey K.M., Wojtkowiak J.W., Bennun Y., Boygo M.S., Weber E., Moin K., Blum G., Mattingly R.R., Gillies R.J. Acidic extracellular ph increases contribution of cysteine cathepsins, including cathepsin B, to breast carcinoma-associated proteolysis. Neoplasia. 2013;15:1125–1137. doi: 10.1593/neo.13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ibrahim-Hashim A., Cornnell H.H., Abrahams D., Lloyd M., Bui M., Gillies R.J., Gatenby R.A. Systemic buffers inhibit carcinogenesis in TRAMP mice. J Urol. 2012;188:624–631. doi: 10.1016/j.juro.2012.03.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerlinger M., Rowan A.J., Horswell S., Larkin J., Endesfelder D., Gronroos E., Martinez P., Matthews N., Stewart A., Tarpey P. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wood L.D., Parsons D.W., Jones S., Lin J., Sjoblom T., Leary R.J., Shen D., Boca S.M., Barber T., Ptak J. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 34.Yachida S., Jones S., Bozic I., Antal T., Leary R., Fu B., Kamiyama M., Hruban R.H., Eshleman J.R., Nowak M.A. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaw A.T., Engelman J.A. ALK in lung cancer: past, present, and future. J Clin Oncol. 2013;31:1105–1111. doi: 10.1200/JCO.2012.44.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gatenby R.A., Gillies R.J. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 37.Gatenby R.A., Gillies R.J. A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008;8:56–61. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- 38.Jahde E., Glusenkamp K.H., Rajewsky M.F. Protection of cultured malignant cells from mitoxantrone cytotoxicity by low extracellular pH: a possible mechanism for chemoresistance in vivo. Eur J Cancer. 1990;26:101–106. doi: 10.1016/0277-5379(90)90290-a. [DOI] [PubMed] [Google Scholar]
- 39.Raghunand N., Mahoney B., van Sluis R., Baggett B., Gillies R.J. Acute metabolic alkalosis enhances response of C3H mouse mammary tumors to the weak base mitoxantrone. Neoplasia. 2001;3:227–235. doi: 10.1038/sj.neo.7900151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wojtkowiak J.W., Verduzco D., Schramm K.J., Gillies R.J. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol Pharm. 2011;8:2032–2038. doi: 10.1021/mp200292c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gambhir S.S. Molecular imaging of cancer with positron emission tomography. Nat Rev Cancer. 2002;2:683–693. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time-lapse microscopy of wound healing assays. Cells were exposed to pH 6.8 or pH 7.4 media for 24 hours before wound generation, and continued for the duration of the experiment. (A) Recording of time-lapse microscopy of PC3MS cells movement across a wound. (B) Recording of time-lapse microscopy of LL/2R cells movement across a wound. Images were recorded in 5 minute intervals for a total of 18 hours post wound generation and shown at 12 fps. Scale bars represent 200 μm.