

Abstract
In ovarian cancer, loss of BRCA gene expression in tumors is associated with improved response to chemotherapy and increased survival. A means to pharmacologically downregulate BRCA gene expression could improve the outcomes of patients with BRCA wild-type tumors. We report that vascular endothelial growth factor receptor 3 (VEGFR3) inhibition in ovarian cancer cells is associated with decreased levels of both BRCA1 and BRCA2. Inhibition of VEGFR3 in ovarian tumor cells was associated with growth arrest. CD133+ ovarian cancer stemlike cells were preferentially susceptible to VEGFR3-mediated growth inhibition. VEGFR3 inhibition–mediated down-regulation of BRCA gene expression reversed chemotherapy resistance and restored chemosensitivity in resistant cell lines in which a BRCA2 mutation had reverted to wild type. Finally, we demonstrate that tumor-associated macrophages are a primary source of VEGF-C in the tumor microenvironment. Our studies suggest that VEGFR3 inhibition may be a pharmacologic means to downregulate BRCA genes and improve the outcomes of patients with BRCA wild-type tumors.
Introduction
Loss of BRCA gene expression is a double-edged sword. BRCA mutation carriers have a 40% to 80% lifetime risk of breast cancer and a 20% to 40% lifetime risk of ovarian cancer [1]. However, BRCA mutation carriers who develop breast or ovarian cancer have a better prognosis than non-BRCA mutation carriers; BRCA + patients with ovarian cancer will have a nearly 30% improvement in overall survival, whereas BRCA + patients with breast cancer will have a nearly 10% improvement in overall survival [2,3]. This improved outcome is presumed to be due to an increase in chemosensitivity to DNA-damaging chemotherapies such as cisplatin. When BRCA + patients develop chemotherapy-resistant disease, nearly 50% will have had a BRCA gene reversion [4]. Once a patient with ovarian cancer develops platinum-resistant disease, it is essentially universally fatal, with a 5-year survival of less than 10%.
In addition to genetic changes in tumor cells, host cells can contribute to chemotherapy resistance. Tumor-associated macrophages (TAMs) have been reported to have many roles in the tumor microenvironment. In addition to promoting angiogenesis and suppressing antitumor immunity, recent studies suggest that TAMs can promote chemotherapy resistance [5]. TAMs secrete numerous angiogenic factors including both vascular endothelial growth factor A (VEGF-A) and VEGF-C [6–10]. VEGF-A has a well-documented role in tumor angiogenesis, whereas VEGF-C has a primary role in lymphangiogenesis. Recently, VEGF proteins have been reported to directly impact cancer cells including cancer stemlike cells (CSCs). Vascular endothelial growth factor receptor 2 (VEGFR2), the primary receptor for VEGF-A, is preferentially expressed on glioma stem cells and promotes stem cell viability and growth, tumor cell migration, and vascular mimicry [11,12]. In breast cancer and glioma stem cells, treatment with anti–VEGF-A antibodies is associated with increased tumor hypoxia, resulting in the induction of hypoxia inducible factor proteins and increased stemness [13,14].
Less is known about the role of VEGF-C and VEGF-D in relation to their impact on cancer cells. VEGF-C levels are correlated with patient prognosis [15–21] and down-regulation of VEGF-C results in reduced lung and colon cancer metastases in mice [22]. Similarly, inhibition of VEGFR3 (primary receptor for VEGF-C/VEGF-D) is associated with reduced growth and metastasis in breast and pancreatic tumor models [23–25]. In specimens of patients with lung cancer, the level of expression of the CSC marker nestin correlated with lymphangiogenesis and nodal metastasis [26]. Most recently, soluble VEGFR3, used as a means to inhibit VEGF-C/VEGF-D, was found to reduce carcinogenesis in a murine model of skin carcinogenesis, suggesting a role for VEGF-C/VEGF-D in early tumor events [27].
One source of VEGF-C in the tumor microenvironment is a population of tumor-associated myeloid cells [28]. In ovarian cancer, we previously reported on an abundant population of tumor-associated myeloid cells termed vascular leukocytes (VLCs) [29,30]. Here, we report that VLCs produce high levels of VEGF-C, whereas tumor cells express VEGFR3 (little VEGF-D was detected in ovarian tumors). We demonstrate that VEGFR3 inhibition leads to preferential cell cycle arrest of CD133+ ovarian CSCs. Cell cycle arrest is associated with decreased p-extracellular signal-regulated kinase (p-ERK), E2F1, and both BRCA1 and BRCA2 expression. Furthermore, VEGFR3 inhibition and its resultant decreased expression of BRCA1 and BRCA2 were associated with significant increased chemosensitivity both in vitro and in vivo. This strongly supports VEGFR3 as a therapeutic target in ovarian cancer.
Materials and Methods
Tumor Cells and Cytotoxic Assays
Informed consent was obtained from patients for tissue procurement in accordance with the protocol approved by the University of Michigan’s Institutional Review Board. All tumors obtained were stage III or IV epithelial ovarian, fallopian tube, or primary peritoneal cancer of serous histology. Fresh tumor specimens were minced and processed into single-cell suspensions, and red cells were lysed as previously described [31]. For ovarian cancer ascites samples, cells were isolated through centrifugation, and red cells were lysed using ammonium-chloride-potassium (ACK) buffer (Lonza Walkersville Inc., Walkersville, MD). A2780 (wild-type p53, BRCA1, BRCA2), OVCAR8 (p53 null, wild-type BRCA1, BRCA2) [32], PEO1 (p53 mutant, BRCA1 wild type, BRCA2 null, p16 deleted), and PEO4 (p53 mutant, BRCA1 wild type, BRCA2 revertant to wild type, p16 deleted) [33,34] ovarian cancer cell lines were obtained from Susan Murphy (Duke University, Durham, NC). Isogenic murine cancer cell lines with and without BRCA1 deletion were a generous gift of Sandra Orsulic (Cedars-Sinai Cancer Center, Los Angeles, CA). Cell lines were cultured in RPMI-10 (10% fetal bovine and 1% streptomycin/penicillin; Invitrogen, Carlsbad, CA) for 24 hours and then treated with indicated doses of the VEGFR3 tyrosine kinase inhibitor Maz51 (Calbiochem, San Diego, CA,) daily for 3 days. Cell numbers and viability were then evaluated using the Cell Countess (Invitrogen). For chemosensitization assays, cells were treated with 5 μM Maz51 and 0.5 μg/ml cisplatin in the indicated sequence. For drug sequencing, A2780 or OVCAR8 cell replicates were treated with 1) DMSO (control), 2) Maz51 (5 μM) daily for 3 days, 3) 0.5 μg cisplatin for 3 days, 4) Maz51 for 3 days followed by cisplatin for 3 days, 5) cisplatin for 3 days followed by Maz51 for 3 days, or 6) cisplatin and Maz51 concurrent for 3 days. Each assay was repeated at least three times.
Fluorescence-Activated Cell Sorting
Cells from human ovarian cancer cell lines (A2780 and OVCAR8), human ascites, or primary ovarian tumors were processed and prepared as previously reported [35]. Cells were incubated with monoclonal anti-human VEGF R3-PE (R&D Systems, Minneapolis, MN) and CD133/2 (Allophycocyanin [APC]-tagged; Miltenyi Biotec, Gladbach, Germany) or for isotype control with Mouse IgG2A APC and Mouse IgG2A phycoerythrin (PE) (R&D Systems) and then ALDEFLUOR (Stemcell Technologies, Vancouver, Canada) as previously described [36]. Results were analyzed using Summit 6.0 (Beckman-Coulter, La Brea, CA).
Tumor Sphere Assays
Five thousand cells in 3 ml of supplemented MEBM (Lonza) were plated onto each well of six-well ultralow adherence plates (Corning, Acton, MA). After 24 hours, cells were treated with media alone (control) or Maz51 in media, at 1, 2.5, or 5 μM concentrations. After 3 days, fresh media were added without Maz51. Media were changed every 3 to 4 days, and tumor spheres were counted after 2 weeks. Images of spheres were photographed and quantified using the Olympus MicroSuite Biological Suite software (Center Valley, PA), as previously described [37].
Western Blot Analysis
A2780 and OVCAR8 ovarian cancer cells were grown to 80% confluence. To detect VEGF-C, protein transport inhibitor GolgiPlug (BD Biosciences, San Jose, CA) was added to media (1 μl/ml) for 4 hours, and then cells were lysed with Radioimmunoprecipitation assay buffer (RIPA buffer) (Invitrogen) with complete proteinase inhibitor and phosphatase inhibitor (Roche, Basel, Switzerland). Insoluble material was removed by centrifugation at 16,000g at 4°C for 15 minutes. Protein concentrations were determined using the Bradford Protein Assay Kit (Bio-Rad Laboratories, Hercules, CA). To detect phosphoproteins after treatment with VEGF-C (PeproTech, Rocky Hill, NJ), A2780 and OVCAR8 cells were grown to 60% confluence, serum starved for 16 hours, and then treated with 50 ng/ml VEGF-C for 30 minutes. Cells were washed with ice-cold phosphate-buffered saline (PBS) and then lysed and processed as above. Standard Western blot analysis was performed with 10 μg of total protein per sample. Antibodies used for Western blot analysis include anti–VEGF-C (1:800 dilution; Santa Cruz Biotechnology, Santa Cruz, CA), anti-VEGFR3 (1:1000 dilution; Chemicon, Temecula, CA), anti–p-VEGFR3 (1:1000 dilution; Calbiochem, Madison, WI), anti–p-ERK 1/2 and anti-ERK (1:1000 dilution; Cell Signaling Technology, Danvers, MA), and anti–β-actin (1:10,000 dilution; Sigma-Aldrich, Saint Louis, MO). Bands were visualized using the ECL Kit (Pierce/Thermo Scientific, Rockford, IL), anti–p-p38 and anti-p38 (1:1000 dilution; Cell Signaling Technology), anti–p-jnk and anti-jnk (1:1000 dilution; Cell Signaling Technology), anti–p-AKT and anti-AKT (1:1000 dilution; Cell Signaling Technology), and anti–p-stat3 and anti-stat3 (1:1000 dilution; Cell Signaling Technology).
Apoptosis and Cell Cycle
A population of 5 × 104 A2780 or OVCAR8 cells was fixed in 70% cold ethanol. After washing with cold PBS, the cells were stained with PBS containing 50 μg/ml propidium iodide (Sigma-Aldrich) and treated with 100 μg/ml RNase A for 30 minutes at room temperature. Cells were analyzed using a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ). Apoptosis was characterized using the PI/annexin V FITC kit (BD Biosciences) as previously described [38]. The cell apoptosis distributions were determined on a FACSCalibur flow cytometer (BD Biosciences) and analyzed by CellQuest Pro software program (Becton, Dickinson and Company, Franklin Lakes, NJ).
Bromodeoxyuridine Proliferation Assays
For bromodeoxyuridine (BrdU) labeling, 0.1 × 105 A2780 cells and 0.5 × 105 OVCAR8 cells were plated onto an eight-well chamber slide (Lab-Tek, Scotts Valley, CA) in RPMI-10 (10% fetal bovine and 1% streptomycin/penicillin; Invitrogen) and incubated in 37°C 5% CO2/air mixture for 24 hours. Cells were treated with plain media (control) or Maz51 (5 μM) daily for 3 days, then incubated with 10 μM BrdU (Sigma-Aldrich) for 2 hours, and then 10% formalin fixed. BrdU incorporation was assessed through immunofluorescence. Sixteen high-power images (× 100) from each treatment group were compared versus control using the two-sided Student’s t test.
Quantitative Real-Time Reverse Transcription–Polymerase Chain Reaction
Cells were treated with plain media + DMSO (control) or 5 μM Maz51 or U0126 for 24 hours; then, total RNA was extracted from cells with the PureLink Total RNA Purification System (Invitrogen) with on-column DNAse treatment. RNA quality was confirmed on 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). First-strand cDNA was synthesized from 2 μg of total RNA with the SuperScript III First-Strand Synthesis System for reverse transcription–polymerase chain reaction (RT-PCR) (Invitrogen) according to manufacturer recommendations. SYBR Green–based array PCR was performed using the 7900 HT Sequence Detection System (Applied Biosystems, Grand Island, NY) and the indicated primer (Table W1).
Animal Studies
Six- to eight-week-old nude NOD-SCID mice were purchased from Charles River Laboratories (Wilmington, MA). All experiments were performed with approval of the University of Michigan Committee on Use and Care of Animals. Tumors were initiated with 5 × 105 A2780 or 1 × 107 OVCAR8 cells combined with 100 μl of phosphate buffered saline (PBS) and 100 μl of Matrigel (BD Biosciences), implanted subcutaneously into the axillae (n = 10 tumors per treatment group). Three days after tumor inoculation, mice were treated through intraperitoneal injection with 1) control DMSO (40 μl) or 2) Maz51 (8 mg/kg in 40 μl of DMSO). Tumor growth was measured using calipers, and tumor volume was calculated on the basis of the following modified ellipsoid formula: (L × W × W)/2.
Immunohistochemistry
A portion of each tumor was fixed in 10% formalin for 2 hours at room temperature and then transferred to 70% ethanol. Tumors were then paraffin embedded and stained at the histology core of the University of Michigan using EDTA-based antigen retrieval and either mouse anti-aldehyde dehydrogenase (anti-ALDH) antibody (clone 44/ALDH; 1:100; BD Biosciences) or anti-Ki67 antibody (Abcam, Cambridge, UK; No. 15580, 1:2000). For stain quantification, 8 to 10 sections from four to five tumors per treatment group were analyzed, and counts were compared using the two-sided Student’s t test.
Results
Expression of VEGF-C, VEGF-D, and VEGFR3 in Ovarian Cancer
We evaluated the expression of VEGF-C, VEGF-D, and their primary receptor, VEGFR3, in primary tumor endothelial cells (TECs), VLCs, and ovarian cancer cells. Quantitative real-time RT-PCR (qRT-PCR) evaluation of VEGF-C expression demonstrated 5 to 10 × higher expression in VLCs compared to TECs and tumor cells (Figure 1A). Western blot confirmed the highest levels of VEGF-C protein in VLCs (Figure 1C). In contrast, qRT-PCR demonstrated the greatest expression of VEGFR3 in tumor cells (Figure 1B). FACS and immunocytochemistry of both A2780 and OVCAR8 ovarian cancer cell lines confirmed VEGFR3 protein expression (Figure 1D and data not shown). Immunohistochemistry performed on primary human tumors confirmed expression of VEGFR3 in tumor cells and vascular cells (Figure 1E). In normal ovary, VEGFR3 expression was found primarily in vessels and in some stromal cells, with no expression appreciated on ovarian surface epithelial cells. Treatment of both A2780 and OVCAR8 cell lines with VEGF-C resulted in phosphorylation of VEGFR3, indicating that VEGF-C signaling is active in ovarian cancer cells (Figure 1F).
Figure 1.

Expression of VEGF-C, VEGF-D, and VEGFR3 in ovarian cancer. (A and B) qRT-PCR evaluation of VEGF-C mRNA and VEGFR3 mRNA in VLCs, TECs, and primary ovarian tumor cells (TCs) is presented. (C) Western blot confirms high VEGF-C protein expression in VLCs. (D) FACS analysis of VEGFR3 and CD133 in A2780 and OVCAR8 cells is presented. (E) Immunohistochemistry demonstrating VEGFR3 expression in human ovary is primarily in vessels and rare stromal cells, whereas VEGFR3 expression in primary ovarian tumors is in vascular structures and tumor cells (original magnification, × 100). (F) p-VEGFR3 Western blot of A2780 tumor cells treated with VEGF-C is shown.
VEGFR3 Inhibition Results in Tumor Cell Growth Arrest
We next tested the impact of the VEGFR3 inhibitor Maz51 on OVCAR8 and A2780 cell growth. OVCAR8 demonstrated a dose-dependent reduction in cell growth, with an IC50 of approximately 2.5 μM, and nearly complete growth arrest at 5 μM (Figure 2A). A2780 demonstrated a biphasic response, with a slight increase in growth at 1 μM Maz51, modest growth suppression at 2.5 μM, and nearly complete growth arrest at 5 μM (Figure 2A). Cell cycle analysis with Maz51 demonstrated a significant decrease in the number of cells in S/M phase, with a concurrent increase in the number of cells in G0/G1 (Figure 2B). Treatment was not associated with an increase in the number of sub-G0/apoptotic cells. In addition, annexin/PI analysis did not demonstrate apoptosis (Figure W1). Treatment with the VEGFR2 inhibitor Sutent was not associated with cell cycle arrest (data not shown), suggesting that the impact of Maz51 was not due to cross-reactivity with the VEGFR2. To further confirm that the impact of VEGFR3 inhibition on tumor cell growth was due to cell cycle arrest, we evaluated BrdU incorporation into both cell lines in the presence and absence of VEGFR3 inhibition. As anticipated, VEGFR3 inhibition was associated with an about two to three times reduction in BrdU incorporation (Figure 2C). Similarly, we evaluated CFSE dilution. Cells treated with Maz51 demonstrated slower CFSE dilution compared to untreated controls (data not shown). Taken together, these data indicate that Maz51 induces growth arrest, but not death, of ovarian cancer cells.
Figure 2.

VEGFR3 inhibition results in ovarian cancer cell growth arrest and preferentially targets CD133+ cells. (A) OVCAR8 and A2780 tumor cell number (relative to untreated controls) after treatment with the indicated doses of Maz51 is presented. (B) Propidium iodide FACS cell cycle analysis of control and Maz51-treated OVCAR8 and A2780 cells is presented. (C) Quantification of BrdU incorporation in control and Maz5-treated OVCAR8 and A2780 cells is presented. (D) Total cell numbers (normalized to untreated control) of FACS-isolated CD133+versus CD133− OVCAR8 and A2780 cells after treatment with the indicated doses of Maz51 are presented. (E) Quantification and representative images of tumor spheres formed from three independent primary ovarian cancer cell specimens in the absence and presence of increasing doses of Maz51 are shown.
We next tested the impact of the VEGFR3 inhibitor Maz51 on OVCAR8 and A2780 cell growth. OVCAR8 demonstrated a dose-dependent reduction in cell growth, with an IC50 of approximately 2.5 μM, and nearly complete growth arrest at 5 μM (Figure 2A). A2780 demonstrated a biphasic response, with a slight increase in growth at 1 μM Maz51, modest growth suppression at 2.5 μM, and nearly complete growth arrest at 5 μM (Figure 2A). Cell cycle analysis with Maz51 demonstrated a significant decrease in the number of cells in S/M phase, with a concurrent increase in the number of cells in G0/G1 (Figure 2B). Treatment was not associated with an increase in the number of sub-G0/apoptotic cells. In addition, annexin/PI analysis did not demonstrate apoptosis (Figure W1). Treatment with the VEGFR2 inhibitor Sutent (Pfizer, New York, NY) was not associated with cell cycle arrest (data not shown), suggesting that the impact of Maz51 was not due to cross-reactivity with the VEGFR2. To further confirm that the impact of VEGFR3 inhibition on tumor cell growth was due to cell cycle arrest, we evaluated BrdU incorporation into both cell lines in the presence and absence of VEGFR3 inhibition. As anticipated, VEGFR3 inhibition was associated with an about two to three times reduction in BrdU incorporation (Figure 2C). Similarly, we evaluated Carboxyfluorescein succinimidyl ester (CFSE) dilution. Cells treated with Maz51 demonstrated slower CFSE dilution compared to untreated controls (data not shown). Taken together, these data indicate that Maz51 induces growth arrest, but not death, of ovarian cancer cells.
VEGFR3 Inhibition Preferentially Impacts CD133+ Cells
VEGFR3 inhibition consistently had greater impact on OVCAR8 cells versus A2780 cells. More than 90% of OVCAR8 cells express the CSC marker CD133, whereas ~ 5% to 10% of A2780 cells express CD133+ [35,39]. We therefore evaluated the impact of VEGFR3 inhibition specifically on CD133+ ovarian tumor cells. Treatment of A2780 cells with increasing doses of Maz51 was associated with decreasing percentages of CD133+ cells (data not shown). To directly test the impact of Maz51 on CD133+ cells, we treated FACS-isolated CD133+ and CD133− cells from OVCAR8 and A2780. In both cell lines, CD133+ cells were sensitive to lower doses of Maz51 (Figure 2D).
To confirm a potential impact on primary human ovarian CSC growth, we performed tumor sphere assays on tumor cells directly isolated from patients with ovarian cancer in the presence and absence of Maz51. Similar to that seen with cell line data, low doses (1 μM) of Maz51 were associated with slight increases in tumor sphere number, whereas 5 μM doses of Maz51 were associated with nearly complete inhibition of tumor sphere formation (Figure 2E).
Impact of VEGFR3 Inhibition on Cell Signaling
We next evaluated downstream targets of VEGFR3 activation. Phosphoprotein Western blot analysis in both A2780 and OVCAR8 cells demonstrated that VEGF-C treatment of cells was associated with increased p-ERK levels that could be blocked by Maz51 treatment (Figure 3A, i). In the more sensitive OVCAR8 cells, VEGFR3 inhibition was also associated with decreased levels of p-JNK and p38 (Figure 3A, i).
Figure 3.

VEGFR3 inhibition decreases BRCA1 and BRCA2 gene expression through p-ERK and E2F1. (A) (i) Phosphoprotein Western blot analysis of control and VEGF-C–treated A2780 and OVCAR8 cells in the presence or absence of Maz51 showing VEGF-C treatment is associated with increased p-ERK. (ii) Western blot demonstrating Maz51 treatment is associated with decreased BRCA1 and E2F1 in both A2780 and OVCAR8 cells. (B) qRT-PCR demonstrating treatment with (i) Maz51 or (ii) MEK inhibition is associated with down-regulation of BRCA1 and BRCA2 mRNA. (C). Comparison of cell growth inhibition of Maz51 in (i) two sets of isogenic control and BRCA1 knockout murine ovarian cancer cell lines and in (i) BRCA2 mutant PEO1 and BRCA2 revertant PEO4 cells.
Given that VEGFR3 inhibition was associated with cell cycle arrest and that BRCA genes are known to regulate cell cycle, we next specifically evaluated the impact of VEGFR3 inhibition on BRCA1 and BRCA2 expression. In both cancer cell lines (A2780 and OVCAR8) and primary cells, we observed that VEGFR3 inhibition was associated with approximately three- to nine-fold down-regulation of BRCA1 and BRCA2 mRNA (Figure 3B). Western blot analysis confirmed that Maz51 treatment of A2780 and OVCAR8 cells decreased expression of BRCA1 protein (Figure 3A, ii). With Maz51 treatment, we also observed decreased levels of E2F1 in both cell lines, increased levels of p53 in A2780 (OVCAR8 cells are p53 null), and increased levels of cyclin-dependent kinase inhibitor 1B/p27) in OVCAR8 (Figure 3A, ii).
To determine if phosphorylation of ERK plays a central role in VEGFR3 inhibition–mediated inhibition of BRCA gene expression, we treated A2780, OVCAR8, and primary ovarian tumor cells with the mitogen-activated protein kinase kinase 1 (MEK) inhibitor U0126 and assessed BRCA1 and BRCA2 mRNA levels. MEK inhibition reduced both BRCA1 and BRCA2 mRNA levels albeit somewhat less effectively than Maz51 (Figure 3B, ii).
To confirm an important role of BRCA gene down-regulation important role in Maz51 mediated growth arrest, we treated murine ovarian tumor cell lines with and without BRCA1 mutation. Murine tumor cells with wild-type BRCA1 are more sensitive to Maz51 treatment than are syngeneic cell lines with mutant BRCA1 (Figure 3C, i). We also tested human PEO1 ovarian cancer cells (BRCA2 deficient due to a nonsense mutation) and PEO4 ovarian cancer cells (BRCA2 revertant cells derived from the same patient as PEO1 cells) [40]. BRCA2-deficient chemotherapy-sensitive PEO1 cells were less responsive to Maz51 therapy, whereas BRCA2 wild-type chemotherapy-resistant PEO4 cells were more responsive to Maz51 (Figure 3C, ii).
VEGFR3 Inhibition Induces Chemosensitization In Vitro and In Vivo
Loss of BRCA function in patients is associated with increased chemosensitivity and significantly improved outcome in ovarian cancer. We therefore tested the impact of Maz51 on chemotherapy response of ovarian cancer cell lines. We treated OVCAR8 and A2780 cell lines with Maz51 before, concurrent with, or after cisplatin chemotherapy. Pretreatment with Maz51 followed by chemotherapy was the most effective means to delay tumor regrowth (Figure 4A). We also assessed chemotherapy sensitivity in PEO1 and PEO4 cells. BRCA2-null PEO1 cells are sensitive to cisplatin, whereas BRCA2-revertant PEO4 cells are resistant to cisplatin. However, chemotherapy resistance in PEO4 was reversed in the presence of Maz51 (Figure 4B).
Figure 4.

VEGFR3 inhibition with Maz51 chemosensitizes in vitro in a BRCA-dependent manner. (A) Absolute tumor cell number of OVCAR8 and A2780 cells treated with Maz51 only, cisplatin chemotherapy only, cisplatin followed by Maz51, Maz51 and cisplatin concurrently, or Maz51 followed by cisplatin is presented. (B) Normalized cell counts of PEO1 (BRCA2 mutant) and PEO4 cells (BRCA2 revertant) treated with the indicated agents are presented.
We next tested the impact of Maz51 treatment on A2780 and OVCAR8 tumor growth in vivo. Single-agent Maz51 treatment demonstrated profound growth inhibitory effects on the OVCAR8 tumor cell line, which is > 90% CD133+ (Figure 5A). However, A2780 tumors, which have a small percentage of CD133+ cells (~ 5%-10%), demonstrated no response to single-agent Maz51 therapy (Figure 5A). We next tested the impact of Maz51 in combination with cisplatin chemotherapy in A2780 cells. Given our in vitro sequencing studies, we initiated Maz51 treatment before weekly chemotherapy treatment with cisplatin. Maz51 treatment was followed with low-dose chemotherapy that resulted in a significant reduction in A2780 tumor growth (Figure 5B, i) compared to cisplatin therapy alone. Evaluation of Maz51-treated tumors demonstrated significant reductions in Ki67 and reduced levels of LYVE1+ vessels (Figure 5B, ii, and data not shown). No differences were observed in CD31+ vessels, indicating no effects from VEGFR2 inhibition (data not shown). FACS analysis of tumors at the time of killing demonstrated a higher percentage (Figure 5C, i), but modest decreased absolute number of CD133+ cells in Maz51 + cisplatin versus cisplatin only–treated tumors (Figure 5C, ii). CD133− cells demonstrated ~ 10-fold decrease in absolute number with combined Maz51 + cisplatin treatment versus cisplatin treatment alone (Figure 5C).
Figure 5.

VEGFR3 inhibitor restricts tumor growth and increases chemosensitivity. (A and B) Tumor growth curves for OVCAR8 and A2780 cell–derived tumors treated with single agent Maz51 are presented. (B) (i) Tumor growth curves for A2780 tumors treated with cisplatin or Maz51 before and concurrent with cisplatin. Arrows indicate doses of cisplatin. Red bars indicate timing of daily Maz51 treatment. (ii) IHC analysis and quantification of Ki67 in cisplatin- and cisplatin + Maz51–treated A2780 tumors are presented. (C) (i) FACS evaluation of CD133 and ALDH expression in cisplatin- and Maz51 + cisplatin–treated tumors and (ii) absolute numbers of CD133+ and ALDH + cells in cisplatin- and Maz51 + cisplatin–treated tumors are presented.
Discussion
Our studies indicate a critical role for VEGF-C regulation of ovarian cancer cell gene expression. VEGF-C blockade results in down-regulation of both BRCA1 and BRCA2, leading to chemosensitization. The primary source of VEGF-C in the tumor microenvironment is tumor-associated myeloid cells.
These studies indicate another critical role for myeloid cells in the tumor microenvironment. Populations of tumor-associated myeloid cells, defined by the expression of various molecules (CD14, CD11b, GR, Tie2, and F4/80), have been reported to promote angiogenesis and suppress antitumor immunity [41]. In addition, myeloid cells have been linked with increased rates of tumor metastasis and establishment of a metastatic niche [42–44]. More recently, myeloid cells have been reported to directly impact cancer stemlike cells [45,46]. Our findings that ovarian cancer–associated myeloid cells produce high levels of VEGF-C are consistent with previous reports that CD14+CD11b+ cells produce high levels of VEGF-C to produce robust but aberrant lymphangiogenesis [28]. Our studies indicate that VEGF-C, in addition to its well-known effects on lymphangiogenesis, has direct effects on the cancer cell.
We found that inhibition of VEGFR3 in both ovarian cancer cell lines (OVCAR8 and A2780) and primary tumor cells dramatically restricted tumor cell growth. CD133+ ovarian cancer stemlike cells were preferentially sensitive to VEGFR3 inhibition. At a dose of 5 μM, we found essentially 100% growth inhibition. In all cells tested, Maz51 treatment–associated growth restriction was linked to reductions in p-ERK, E2F, and BRCA gene expression. Treatment with a MEK inhibitor partially reduced BRCA gene expression, suggesting that signaling from VEGFR3 through p-ERK regulates transcription factors, such as E2F, which at least partly contribute to the regulation of BRCA gene transcription [47]. Supporting a critical role for BRCA genes, cells with mutant BRCA1 or BRCA2 genes were two- to three-fold more resistant to VEGFR3 inhibitor therapy. In A2780 cells, growth reduction could also potentially be associated with stabilization of p53. In OVCAR8 cells, which do not express a functional p53 null, increased p27 could also account for some growth arrest.
With VEGFR3 inhibitor therapy, we observed increased chemosensitivity both in vitro and in vivo. This is initially somewhat surprising given the observation that VEGFR3 inhibition is inducing cell cycle arrest and nondividing cells are felt to be resistant to DNA-damaging chemotherapies such as cisplatin. However, chemosensitization was greatest when cells were treated in sequence (VEGFR3 inhibition, followed by cisplatin therapy), rather than concurrently. Thus, VEGFR3 inhibition could induce cellular quiescence, and then when cells resumed proliferation after withdrawal of VEGFR3 inhibitor, chemotherapy then killed the cells as they entered the cell cycle. Killing is presumably increased initially in the presence of BRCA proteins. Consistent with this, FACS evaluation of tumors treated with Maz51 and cisplatin demonstrated an increased percentage of CD133+ cells but a reduced absolute number of CD133+ cells. CD133− cells had both a decreased percentage, and 10-fold decrease and absolute number of cells. This suggests that the CD133+ cells remained quiescent, whereas the CD133− progenitor cells, which are normally highly proliferative, are restricted by chemotherapy.
Our findings have important clinical implications. In both breast and ovarian cancers, patients with BRCA mutations are known to have a better clinical outcome [2,3]. This has been presumed to be due to increased sensitivity to chemotherapy. Indeed, when BRCA mutation carriers' tumors become resistant to cisplatin therapy, this can be associated with reversion of one of the BRCA genes back to wild type [4,40,48]. Our studies indicate that VEGFR3 inhibition may represent a therapeutic means to downregulate BRCA expression in BRCA wild-type tumors, thus making them mimic BRCA-deficient tumors. Given a nearly 30% improvement in the overall survival rate of patients with germline BRCA mutations over BRCA wild-type patients with stage III/IV ovarian cancer, this simple intervention could dramatically improve outcome. Furthermore, for those patients who develop chemotherapy-resistant disease, it is possible that VEGFR3 inhibition could reverse this phenomenon to improve progression-free and overall survival.
Preclinical evidence supports VEGFR3 as a clinical target. VEGFR3 inhibitors have been found to both inhibit angiogenesis/lymphangiogenic activities and reduce metastases [8,22,49]. Interestingly, a soluble version of VEGFR3 has been identified [50]. Several studies have now used sVEGFR3 as a “VEGF-C Trap” and reported significant reductions in lymphangiogenesis and tumor metastases [27,51,52].
Clinical evidence also supports a potentially meaningful role for VEGFR3 inhibitors. Recent studies suggested an ~ 5-month progression-free survival for the VEGFR2/VEGFR3 inhibitor pazopanib [53]. This is significantly better than the 8- to 10-week progression-free survival advantage for the anti–VEGF-A antibody bevacizumab [54,55]. We speculate that this could be due to the significant anti–VEGFR3 inhibitory actions of pazopanib. In this study, pazopanib was used only as a consolidation agent after chemotherapy and not concurrent with chemotherapy. Our data would suggest that use of pazopanib before or concurrent with chemotherapy would dramatically enhance response rates. There is currently an ongoing trial of another VEGFR3 inhibitor, BIBF-1120 (nintedanib), for the treatment of ovarian cancer. In this trial, patients receive BIBF-1120 or placebo concurrent with standard chemotherapy and as a consolidation agent. Our results would suggest that this trial would show a significant benefit for the experimental arm. If this or other trials with VEGFR3 inhibitors show a significant clinical benefit compared to standard therapy targeting the VEGF/VEGFR2 pathway, VEGFR3 expression on cancer cells could be an important biomarker of patient response.
Our study also provides rationale for combining VEGFR3 inhibition with poly(ADP-ribose) polymerase (PARP) inhibitor therapy. PARP inhibitor therapy has shown promise in BRCA mutation carriers [56,57]. However, with the development of platinum-resistant disease, nearly 50% of patient tumors demonstrate BRCA gene reversions [4]. Use of VEGFR3 inhibitors in combination with PARP inhibitors could restore the activity of PARP inhibitor therapy. This hypothesis will be indirectly tested in an ongoing phase II clinical trial comparing response rates for patients with ovarian cancer treated with the PARP inhibitor olaparib and the pan VEGF receptor inhibitor cediranib.
Our study is limited in that it was not performed with a clinically available compound. Unlike the compounds discussed above that target VEGFR1 to VEGFR3, Maz51 targets primarily VEGFR3 in the concentrations used in this study. It remains to be determined if VEGFR3 inhibition in the presence of VEGFR1/2 inhibition will have similar activity. Similarly, our study was limited to ovarian cancer. Further work will be necessary to see if a BRCA expression is also regulated by VEGFR3 in breast cancer or other solid tumors.
In conclusion, we have found that tumor-associated myeloid cells signal to cancer cells through VEGF-C interactions with VEGFR3. VEGFR3 blockade results in down-regulation of BRCA genes, the induction of cell cycle arrest, and chemosensitization. Given the observation that BRCA mutant ovarian tumors have a much better prognosis than BRCA wild-type tumors, our results imply that VEGFR3, through down-regulation of BRCA, is a critical clinical target for ovarian cancer. VEGFR3 inhibition could allow BRCA wild-type patients to benefit from the improved clinical outcomes observed for BRCA mutation carriers. In addition, VEGFR3 inhibition could allow chemosensitization for BRCA patients in whom BRCA mutations have reverted to wild type.
Footnotes
This work was supported by grant 1-R01-CA163345-01 from the National Institutes of Health. Core facilities used to perform this work were supported by the National Institutes of Health through the University of Michigan Cancer Center Support grant (P30 CA046592). The authors have no conflicts of interest to report.
This article refers to supplementary materials, which are designated by Table W1, Table W2 and Figure W1 and are available online at www.neoplasia.com.
Appendix.
Figure W1.
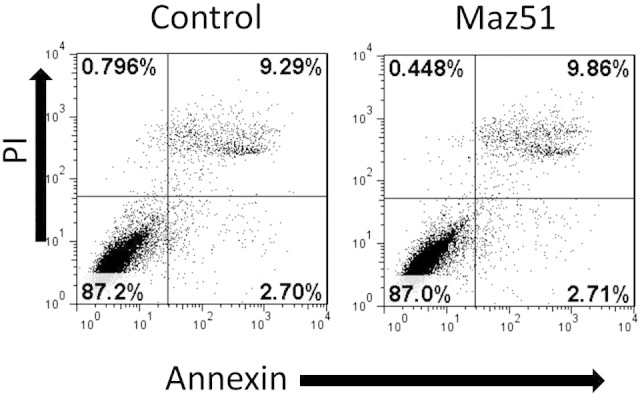
Maz51 does not induce apopotosis. PI/Annexin FACS analysis of control and Max51 treated OVCAR8 cells demonstrating no increase in Annexin stain with Maz51 treatment.
Supplemental Table 1.
Information for Antibodies Used
| Antibody | Conjugation | Source | Clone |
|---|---|---|---|
| FACS | |||
| Anti-human CD133 | APC | Miltenyi Biotec | 293C3 |
| Anti-human VEGF R3 | PE | R&D system | LGC03 |
| Mouse IgG | APC | R&D Systems | 20102 |
| Mouse IgG | PE | R&D Systems | 20102 |
| IHC | |||
| Rat anti-mouse CD31 | E-Bioscience | 390 | |
| Rabbit polyclonal to LYVE1 | Abcam | polyclonal | |
| Mouse anti-human Ki67 | Abcam | B126.1 | |
| Mouse anti-Human VEGF R3 | Chemicon | 9D9F9 | |
| Goat polyclonal antibody to VEGFC | Santa Cruz | 8 B1.3 | |
| Rat monoclonal to BRDU | Abcam | BU1/75 (ICR1) | |
| Rabbit anti-rat | biotinylated | Vector | Secondary Ab |
| Goat anti-rabbit | biotinylated | Vector | Secondary Ab |
| Horse anti-mouse | biotinylated | Vector | Secondary Ab |
| Rabbit anti-goat | biotinylated | Vector | Secondary Ab |
| Goat anti-Rat IgG | Alexa 488 | Invitrogen | Secondary Ab |
| Antibody | Source | Clone |
|---|---|---|
| Western blot | ||
| Mouse anti-human VEGF R3 | Chemicon | 9D9F9 |
| Mouse anti-human p-VEGF R3 | Calbiochem | pc460 |
| Goat polyclonal antibody to VEGFC | Santa Cruz | 8 B1.3 |
| Rabbit mAb Erk1/2 | Cell Signaling Technology | D13.14.4E |
| Rabbit mAb P38 | Cell Signaling Technology | D3F9 |
| Rabbit mAb JNK | Cell Signaling Technology | 81E11 |
| Rabbit polyclonal AKT | Cell Signaling Technology | polyclonal |
| Rabbit mAb stat3 | Cell Signaling Technology | D3Z2G |
| Rabbit mAb p-Erk1/2 | Cell Signaling Technology | 137F5 |
| Rabbit mAb p-P38 | Cell Signaling Technology | D13E1 |
| Rabbit mAb p-JNK | Cell Signaling Technology | 56G8 |
| Rabbit polyclonal p- | Cell Signaling Technology | polyclonal |
| Rabbit polyclonal p-stat3 | Cell Signaling Technology | polyclonal |
| Mouse monoclonal β-Actin | Santa Cruz | AC-15 |
| Horse anti-mouse IgG (H&L) antibody | Cell Signaling Technology | Secondary Ab |
| Purified goat anti-rabbit IgG (H&L) antibody horseradish peroxidase-tagged | Cell Signaling Technology | Secondary Ab |
Supplemental Table 2.
Sequences of oligonucleotides used for transcript quantitation
| VEGF-C |
Mouse F:CAAGGCTTTTGAAGGCAAAG R:TCCCCTGTCCTGGTATTGAG |
|
Human F: CACGAGCTACCTCAGCAAGA R:GCTGCCTGACACTGTGGTA | |
| VEGF-D |
Human F:TGTAAGTGCTTGCCAACAGC R:GTGGATTTTCCTCCTGCAAA |
| VEGFR3 |
Human F:GTACATGCCAACGACACAGG R:TGATGAATGGCTGCTCAAAG |
| BRCA1 |
Human F:CTCAAGGAACCAGGGATGAA R:GCTGTAATGAGCTGGCATGA |
| BRCA2 |
Human F:AGGCTTCAAAAAGCACTCCA R:GTGCGAAAGGGTACACAGGT |
| HPRT1 |
Human F: ATGCTGAGGATTTGGAAAGG R: CAGAGGGCTACAATGTGATGG |
| Transferrin receptor |
Mouse F:TGCAGAAAAGGTTGCAAATG R:AGTGCAAGGTCTGCCTCAAC |
References
- 1.Lindor NM, McMaster ML, Lindor CJ, Greene MH, National Cancer Institute, Division of Cancer Prevention, Community Oncology and Prevention Trials Research Group Concise handbook of familial cancer susceptibility syndromes—second edition. J Natl Cancer Inst Monogr. 2008;(38):1–93. doi: 10.1093/jncimonographs/lgn001. [DOI] [PubMed] [Google Scholar]
- 2.Rubin SC, Benjamin I, Behbakht K, Takahashi H, Morgan MA, LiVolsi VA, Berchuck A, Muto MG, Garber JE, Weber BL. Clinical and pathological features of ovarian cancer in women with germ-line mutations of BRCA1. N Engl J Med. 1996;335:1413–1416. doi: 10.1056/NEJM199611073351901. [DOI] [PubMed] [Google Scholar]
- 3.Verhoog LC, Brekelmans CT, Seynaeve C, van den Bosch LM, Dahmen G, van Geel AN, Tilanus-Linthorst MM, Bartels CC, Wagner A, van den Ouweland A. Survival and tumour characteristics of breast-cancer patients with germline mutations of BRCA1. Lancet. 1998;351:316–321. doi: 10.1016/s0140-6736(97)07065-7. [DOI] [PubMed] [Google Scholar]
- 4.Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, Karlan BY, Taniguchi T, Swisher EM. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29:3008–3015. doi: 10.1200/JCO.2010.34.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73:1128–1141. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzalez FJ, Vicioso L, Alvarez M, Sevilla I, Marques E, Gallego E, Alonso L, Matilla A, Alba E. Association between VEGF expression in tumour-associated macrophages and elevated serum VEGF levels in primary colorectal cancer patients. Cancer Biomark. 2007;3:325–333. doi: 10.3233/cbm-2007-3606. [DOI] [PubMed] [Google Scholar]
- 7.Bolat F, Kayaselcuk F, Nursal TZ, Yagmurdur MC, Bal N, Demirhan B. Microvessel density, VEGF expression, and tumor-associated macrophages in breast tumors: correlations with prognostic parameters. J Exp Clin Cancer Res. 2006;25:365–372. [PubMed] [Google Scholar]
- 8.Schoppmann SF, Fenzl A, Nagy K, Unger S, Bayer G, Geleff S, Gnant M, Horvat R, Jakesz R, Birner P. VEGF-C expressing tumor-associated macrophages in lymph node positive breast cancer: impact on lymphangiogenesis and survival. Surgery. 2006;139:839–846. doi: 10.1016/j.surg.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 9.Schoppmann SF, Birner P, Stöckl J, Kalt R, Ullrich R, Caucig C, Kriehuber E, Nagy K, Alitalo K, Kerjaschki D. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am J Pathol. 2002;161:947–956. doi: 10.1016/S0002-9440(10)64255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salvesen HB, Akslen LA. Significance of tumour-associated macrophages, vascular endothelial growth factor and thrombospondin-1 expression for tumour angiogenesis and prognosis in endometrial carcinomas. Int J Cancer. 1999;84:538–543. doi: 10.1002/(sici)1097-0215(19991022)84:5<538::aid-ijc17>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 11.Hamerlik P, Lathia JD, Rasmussen R, Wu Q, Bartkova J, Lee M, Moudry P, Bartek J, Jr., Fischer W, Lukas J. Autocrine VEGF–VEGFR2–Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J Exp Med. 2012;209:507–520. doi: 10.1084/jem.20111424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yao X, Ping Y, Liu Y, Chen K, Yoshimura T, Liu M, Gong W, Chen C, Niu Q, Guo D. Vascular endothelial growth factor receptor 2 (VEGFR-2) plays a key role in vasculogenic mimicry formation, neovascularization and tumor initiation by Glioma stem-like cells. PLoS One. 2013;8(3):e57188. doi: 10.1371/journal.pone.0057188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, Clouthier SG, Wicha MS. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci U S A. 2012;109:2784–2789. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishida N, Yano H, Komai K, Nishida T, Kamura T, Kojiro M. Vascular endothelial growth factor C and vascular endothelial growth factor receptor 2 are related closely to the prognosis of patients with ovarian carcinoma. Cancer. 2004;101:1364–1374. doi: 10.1002/cncr.20449. [DOI] [PubMed] [Google Scholar]
- 16.Ueda M, Terai Y, Kumagai K, Ueki K, Yamaguchi H, Akise D, Ueki M. Vascular endothelial growth factor C gene expression is closely related to invasion phenotype in gynecological tumor cells. Gynecol Oncol. 2001;82:162–166. doi: 10.1006/gyno.2001.6229. [DOI] [PubMed] [Google Scholar]
- 17.Sapoznik S, Cohen B, Tzuman Y, Meir G, Ben-Dor S, Harmelin A, Neeman M. Gonadotropin-regulated lymphangiogenesis in ovarian cancer is mediated by LEDGF-induced expression of VEGF-C. Cancer Res. 2009;69:9306–9314. doi: 10.1158/0008-5472.CAN-09-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sinn BV, Darb-Esfahani S, Wirtz RM, Faggad A, Weichert W, Buckendahl AC, Noske A, Müller BM, Budczies J, Sehouli J. Vascular endothelial growth factor C mRNA expression is a prognostic factor in epithelial ovarian cancer as detected by kinetic RT-PCR in formalin-fixed paraffin-embedded tissue. Virchows Arch. 2009;455:461–467. doi: 10.1007/s00428-009-0851-6. [DOI] [PubMed] [Google Scholar]
- 19.Yokoyama Y, Charnock-Jones DS, Licence D, Yanaihara A, Hastings JM, Holland CM, Emoto M, Umemoto M, Sakamoto T, Sato S. Vascular endothelial growth factor-D is an independent prognostic factor in epithelial ovarian carcinoma. Br J Cancer. 2003;88:237–244. doi: 10.1038/sj.bjc.6600701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van der Bilt AR, van der Zee AG, de Vries EG, de Jong S, Timmer-Bosscha H, ten Hoor KA, den Dunnen WF, Hollema H, Reyners AK. Multiple VEGF family members are simultaneously expressed in ovarian cancer: a proposed model for bevacizumab resistance. Curr Pharm Des. 2012;18:3784–3792. doi: 10.2174/138161212802002661. [DOI] [PubMed] [Google Scholar]
- 21.Schultheis AM, Lurje G, Rhodes KE, Zhang W, Yang D, Garcia AA, Morgan R, Gandara D, Scudder S, Oza A. Polymorphisms and clinical outcome in recurrent ovarian cancer treated with cyclophosphamide and bevacizumab. Clin Cancer Res. 2008;14:7554–7563. doi: 10.1158/1078-0432.CCR-08-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khromova N, Kopnin P, Rybko V, Kopnin BP. Downregulation of VEGF-C expression in lung and colon cancer cells decelerates tumor growth and inhibits metastasis via multiple mechanisms. Oncogene. 2012;31:1389–1397. doi: 10.1038/onc.2011.330. [DOI] [PubMed] [Google Scholar]
- 23.Lee E, Koskimaki JE, Pandey NB, Popel AS. Inhibition of lymphangiogenesis and angiogenesis in breast tumor xenografts and lymph nodes by a peptide derived from transmembrane protein 45A. Neoplasia. 2013;15:112–124. doi: 10.1593/neo.121638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roberts N, Kloos B, Cassella M, Podgrabinska S, Persaud K, Wu Y, Pytowski B, Skobe M. Inhibition of VEGFR-3 activation with the antagonistic antibody more potently suppresses lymph node and distant metastases than inactivation of VEGFR-2. Cancer Res. 2006;66:2650–2657. doi: 10.1158/0008-5472.CAN-05-1843. [DOI] [PubMed] [Google Scholar]
- 25.Alam A, Blanc I, Gueguen-Dorbes G, Duclos O, Bonnin J, Barron P, Laplace MC, Morin G, Gaujarengues F, Dol F. SAR131675, a potent and selective VEGFR-3-TK inhibitor with antilymphangiogenic, antitumoral, and antimetastatic activities. Mol Cancer Ther. 2012;11:1637–1649. doi: 10.1158/1535-7163.MCT-11-0866-T. [DOI] [PubMed] [Google Scholar]
- 26.Chen Z, Wang T, Luo H, Lai Y, Yang X, Li F, Lei Y, Su C, Zhang X, Lahn BT. Expression of nestin in lymph node metastasis and lymphangiogenesis in non-small cell lung cancer patients. Hum Pathol. 2010;41:737–744. doi: 10.1016/j.humpath.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 27.Alitalo AK, Proulx ST, Karaman S, Aebischer D, Martino S, Jost M, Schneider N, Bry M, Detmar M. VEGF-C and VEGF-D blockade inhibits inflammatory skin carcinogenesis. Cancer Res. 2013;73:4212–4221. doi: 10.1158/0008-5472.CAN-12-4539. [DOI] [PubMed] [Google Scholar]
- 28.Jeon BH, Jang C, Han J, Kataru RP, Piao L, Jung K, Cha HJ, Schwendener RA, Jang KY, Kim KS. Profound but dysfunctional lymphangiogenesis via vascular endothelial growth factor ligands from CD11b+ macrophages in advanced ovarian cancer. Cancer Res. 2008;68:1100–1109. doi: 10.1158/0008-5472.CAN-07-2572. [DOI] [PubMed] [Google Scholar]
- 29.Conejo-Garcia JR, Buckanovich RJ, Benencia F, Courreges MC, Rubin SC, Carroll RG, Coukos G. Vascular leukocytes contribute to tumor vascularization. Blood. 2005;105:679–681. doi: 10.1182/blood-2004-05-1906. [DOI] [PubMed] [Google Scholar]
- 30.Conejo-Garcia JR, Benencia F, Courreges MC, Kang E, Mohamed-Hadley A, Buckanovich RJ, Holtz DO, Jenkins A, Na H, Zhang L. Tumor-infiltrating dendritic cell precursors recruited by a β-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat Med. 2004;10:950–958. doi: 10.1038/nm1097. [DOI] [PubMed] [Google Scholar]
- 31.Pulaski HL, Spahlinger G, Silva IA, McLean K, Kueck AS, Reynolds RK, Coukos G, Conejo-Garcia JR, Buckanovich RJ. Identifying alemtuzumab as an anti-myeloid cell antiangiogenic therapy for the treatment of ovarian cancer. J Transl Med. 2009;7:49. doi: 10.1186/1479-5876-7-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun. 2013;4:2126. doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP, Wurz K, Higgins J, Villegas E, Taniguchi T. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009;69:6381–6386. doi: 10.1158/0008-5472.CAN-09-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cooke SL, Ng CKY, Melnyk N, Garcia MJ, Hardcastle T, Temple J, Langdon S, Huntsman D, Brenton JD. Genomic analysis of genetic heterogeneity and evolution in high-grade serous ovarian carcinoma. Oncogene. 2010;29:4905–4913. doi: 10.1038/onc.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silva IA, Bai S, McLean K, Yang K, Griffith K, Thomas D, Ginestier C, Johnston C, Kueck A, Reynolds RK. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 2011;71:3991–4001. doi: 10.1158/0008-5472.CAN-10-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burgos-Ojeda D, McLean K, Bai S, Pulaski H, Gong YS, Silva I, Skorecki K, Tzukerman M, Buckanovich RJ. A novel model for evaluating therapies targeting human tumor vasculature and human cancer stem-like cells. Cancer Res. 2013;73:3555–3565. doi: 10.1158/0008-5472.CAN-12-2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shank JJ, Yang K, Ghannam J, Cabrera L, Johnston CJ, Reynolds RK, Buckanovich RJ. Metformin targets ovarian cancer stem cells in vitro and in vivo. Gynecol Oncol. 2012;127:390–397. doi: 10.1016/j.ygyno.2012.07.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coffman L, Mooney C, Lim J, Bai S, Silva I, Gong Y, Yang K, Buckanovich RJ. Endothelin receptor-A is required for the recruitment of antitumor T cells and modulates chemotherapy induction of cancer stem cells. Cancer Biol Ther. 2013;14:184–192. doi: 10.4161/cbt.22959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baba T, Convery PA, Matsumura N, Whitaker RS, Kondoh E, Perry T, Huang Z, Bentley RC, Mori S, Fujii S. Epigenetic regulation of CD133 and tumorigenicity of CD133 + ovarian cancer cells. Oncogene. 2009;28:209–218. doi: 10.1038/onc.2008.374. [DOI] [PubMed] [Google Scholar]
- 40.Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McLean K, Buckanovich RJ. Myeloid cells functioning in tumor vascularization as a novel therapeutic target. Transl Res. 2008;151:59–67. doi: 10.1016/j.trsl.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 42.Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006;25:315–322. doi: 10.1007/s10555-006-9001-7. [DOI] [PubMed] [Google Scholar]
- 43.Schmid MC, Varner JA. Myeloid cell trafficking and tumor angiogenesis. Cancer Lett. 2007;250:1–8. doi: 10.1016/j.canlet.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peinado H, Rafii S, Lyden D. Inflammation joins the “niche”. Cancer Cell. 2008;14:347–349. doi: 10.1016/j.ccr.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jinushi M, Baghdadi M, Chiba S, Yoshiyama H. Regulation of cancer stem cell activities by tumor-associated macrophages. Am J Cancer Res. 2012;2:529–539. [PMC free article] [PubMed] [Google Scholar]
- 46.Yang J, Liao D, Chen C, Liu Y, Chuang TH, Xiang R, Markowitz D, Reisfeld RA, Luo Y. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells. 2013;31:248–258. doi: 10.1002/stem.1281. [DOI] [PubMed] [Google Scholar]
- 47.Polager S, Kalma Y, Berkovich E, Ginsberg D. E2Fs up-regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene. 2002;21:437–446. doi: 10.1038/sj.onc.1205102. [DOI] [PubMed] [Google Scholar]
- 48.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 49.Chen H, Ding X, Gao Y, Jiang X, Liu X, Chen Y, Gao J, Zhou X, Cai Z, Sun Q. Inhibition of angiogenesis by a novel neutralizing antibody targeting human VEGFR-3. MAbs. 2013;5:956–961. doi: 10.4161/mabs.26239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Albuquerque RJ. The newest member of the VEGF family. Blood. 2013;121:4015–4016. doi: 10.1182/blood-2013-03-490367. [DOI] [PubMed] [Google Scholar]
- 51.Yang H, Kim C, Kim MJ, Schwendener RA, Alitalo K, Heston W, Kim I, Kim WJ, Koh GY. Soluble vascular endothelial growth factor receptor-3 suppresses lymphangiogenesis and lymphatic metastasis in bladder cancer. Mol Cancer. 2011;10:36. doi: 10.1186/1476-4598-10-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sallinen H, Anttila M, Narvainen J, Koponen J, Hamalainen K, Kholova I, Heikura T, Toivanen P, Kosma VM, Heinonen S. Antiangiogenic gene therapy with soluble VEGFR-1, -2, and -3 reduces the growth of solid human ovarian carcinoma in mice. Mol Ther. 2009;17:278–284. doi: 10.1038/mt.2008.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Du Bois A, Floquet A, Kim JW, Rau J, Del Campo JM, Friedlander M, Pignata S, Fujiwara K, Vergote I, Colombo N. Vol. 31. 2013. Randomized, double-blind, phase III trial of pazopanib versus placebo in women who have not progressed after first-line chemotherapy for advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer (AEOC): results of an international Intergroup trial (AGO-OVAR16) p. LBA5503. (ASCO Meeting Abstracts). [Google Scholar]
- 54.Burger RA, Brady MF, Bookman MA, Fleming GF, Monk BJ, Huang H, Mannel RS, Homesley HD, Fowler J, Greer BE. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365:2473–2483. doi: 10.1056/NEJMoa1104390. [DOI] [PubMed] [Google Scholar]
- 55.Perren TJ, Swart AM, Pfisterer J, Ledermann JA, Pujade-Lauraine E, Kristensen G, Carey MS, Beale P, Cervantes A, Kurzeder C. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365:2484–2496. doi: 10.1056/NEJMoa1103799. [DOI] [PubMed] [Google Scholar]
- 56.Itamochi H, Kigawa J. Clinical trials and future potential of targeted therapy for ovarian cancer. Int J Clin Oncol. 2012;17:430–440. doi: 10.1007/s10147-012-0459-8. [DOI] [PubMed] [Google Scholar]
- 57.Marchetti C, Imperiale L, Gasparri ML, Palaia I, Pignata S, Boni T, Bellati F, Benedetti Panici P. Olaparib, PARP1 inhibitor in ovarian cancer. Expert Opin Investig Drugs. 2012;21:1575–1584. doi: 10.1517/13543784.2012.707189. [DOI] [PubMed] [Google Scholar]