

Abstract
Introduction
We aimed to replicate a recent study which showed higher genetic risk load at 15 loci in men than in women with systemic lupus erythematosus (SLE). This difference was very significant, and it was interpreted as indicating that men require more genetic susceptibility than women to develop SLE.
Methods
Nineteen SLE-associated loci (thirteen of which are shared with the previous study) were analyzed in 1,457 SLE patients and 1,728 healthy controls of European ancestry. Genetic risk load was calculated as sex-specific sum genetic risk scores (GRSs).
Results
Our results did not replicate those of the previous study at either the level of individual loci or the global level of GRSs. GRSs were larger in women than in men (4.20 ± 1.07 in women vs. 3.27 ± 0.98 in men). This very significant difference (P < 10−16) was more dependent on the six new loci not included in the previous study (59% of the difference) than on the thirteen loci that are shared (the remaining 41%). However, the 13 shared loci also showed a higher genetic risk load in women than in men in our study (P = 6.6 × 10−7), suggesting that heterogeneity of participants, in addition to different loci, contributed to the opposite results.
Conclusion
Our results show the lack of a clear trend toward higher genetic risk in one of the sexes for the analyzed SLE loci. They also highlight several limitations of assessments of genetic risk load, including the possibility of ascertainment bias with loci discovered in studies that have included mainly women.
Introduction
The large sex bias in systemic lupus erythematosus (SLE) incidence has not been satisfactorily explained, although sex hormones, cell microchimerism and chromosome X dosage seem to be involved [1-4]. There are other aspects of SLE in addition to incidence in which patient sex is important [3-6]. They include an increased prevalence of renal disease, serositis and discoid lupus and a decrease in the mucocutaneous manifestations in men. Other differences between women and men have been reported with less consistency between studies as a higher prevalence of thrombocytopenia, anti–double-stranded DNA antibodies, decreased C3 and larger organ damage accrual in men than in women with SLE.
The difference in incidence is maximal during the fertile years. It is preceded by a more similar incidence in girls and boys before puberty and followed by a return toward equilibrium in incidence between the sexes in the eldery. This pattern is a powerful suggestion of the effect of sex hormones in SLE pathogenesis [1-4]. This hypothesis is supported by the results of estrogen suppression and estrogen administration experiments in SLE animal models, but the evidence is less clear in humans. There are not abnormal levels of sex hormones in most SLE patients of either sex. Only hyperprolactinemia is commonly found, but with uncertain involvement in disease pathogenesis. In addition, sex hormone therapy is associated with either no or very small increases in SLE or its severity. Other factors contributing to the increased SLE incidence in women could be lymphoid cell microchimerism as a consequence of pregnancy [1]. The incompatibility between the two lymphoid populations gives rise to autoimmunity in animal models, but the causal relationship in SLE patients is less clear, as microchimerism in damaged organs seems to increase as a consequence of tissue repair [7]. A third factor explaining the sexual dimorphism of SLE is suspected to reside in differences between the sex chromosomes [1-4]. The hypothesized mechanism involves a dose excess of genes promoting autoimmunity in women due to noninactivated X chromosomes. This hypothesis has received strong support based on the demonstration of an increased incidence of SLE in patients with Klinefelter’s syndrome who have an external male phenotype but carry the XXY sex chromosomes [8].
More recently, evidence supporting the possibility of a second genetic factor has been reported. Researchers who analyzed a very large collection of patients of European ancestry showed that the genetic risk load was very significantly larger in men than in women (P = 4.52 × 10−8), based on the sum genetic risk score (GRSs) of 15 SLE loci [9]. This result was interpreted as meaning that men require more genetic susceptibility than women to develop SLE. The difference in genetic risk load in that study was very dependent on two human leukocyte antigen (HLA) loci of the fifteen loci included. This was shown by the lack of significant difference between men and women when the two HLA single-nucleotide polymorphisms (SNPs) were excluded (P = 0.3). Our interest in the implications of a higher genetic risk load in men led us to replicate this previous study, but our results are contrary to those reported. We found that genetic risk load was very significantly higher in women than in men with SLE.
Material and methods
Clinical and genotype data
The participant samples used in this study have already been described [10-12]. Briefly, 17 recruiting centers in 10 European countries were asked for about 100 SLE patients who met the American College of Rheumatology criteria [13] and a similar number of healthy controls, all of whom have uniform ancestry from the country of recruitment. In our present study, samples overlapping with the Hughes et al. [9] report were considered separately. Recruiters asked each participant for his or her ancestry, and only those reporting uniform known ancestry from the respective countries were included. In addition, we used six top ancestry-informative markers for European population differentiation to determine whether there were differences between cases and controls from each recruitment center. Samples from two centers were excluded because cases and controls showed significant differences at any of the six top ancestry-informative markers analyzed, as described previously [11]. This selection process left us with a total of 1,457 SLE patients and 1,728 healthy controls from 15 collections and 8 countries. A large fraction of these samples did not overlap with the previous study of SLE genetic risk load. Specifically, the nonoverlapping samples were from 1,124 SLE patients and 1,422 healthy controls from 11 collections and 6 countries. All participants provided their written informed consent as approved by the respective local ethics committees: the Ethics Committee for Clinical Research of Galicia for samples in Santiago and Corunna, the Regional Ethics Committee for Clinical Research of Asturias for samples in Oviedo, the Garcia de Orta Hospital Ethics Committee for samples in Lisbon, the Ethics Committee of Evangelismos Athens General Hospital and Scientific Committee of the Euroclinic Hospital for samples in Athens, the Ethics Committee for Clinical Research of the Doce de Octubre University Hospital for samples in Madrid, the Ethical Committee of the Jessenius Medical Faculty for samples in Martin, the Ethics Committee for Clinical Research of the Vall d’Hebron Hospital for samples in Barcelona, the Ethics Committee of the Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico for samples in Milan, the Ethical Committee of the University Medical Center Groningen for samples in Groningen, the Ethics Committee Azienda Ospedaliera San Camillo–Forlanini for samples in Rome, the Ethical Committee of the Second University of Naples for samples in Naples, the Ethics Commission of the Medical School of Hannover for samples in Hannover, the Commission d’Ethique Biomédicale Hospitalo–Facultaire de l’Université Catholique de Louvain for samples in Brussels, the Institutional Ethics Committee of the Hospital of Hungarian State Railways for Hungarian samples and the Ethical Committee of the Institute of Rheumatology for samples in Prague. The overall study was approved by the Comite Etico de Investigacion Clinica de Galicia. The clinical characteristics of the patients were obtained at the same time. These data included the American College of Rheumatology classification criteria met, sex, age at time of disease onset and time to follow-up. The genotypes of 26 SNPs showing the greatest association at SLE loci identified in large studies [14-23] were available to us from previous studies [11,12,24]. All have been genotyped in the same laboratory and with the same technology: single-base extension using the SNaPshot Multiplex Kit (Applied Biosystems, Foster City, CA, USA), and have passed quality control filters. Nineteen of these SNPs were selected for analysis because they have reached association with SLE at the genome-wide association study significant level (P < 5 x 10-8) and because they were independently associated with SLE in our samples. Thirteen of these nineteen loci are shared with the fifteen SNPs included in the GRSs reported by Hughes et al. [9], and the other six loci have not been analyzed previously in the context of the sex differential genetic risk load. The thirteen loci shared between their studies and ours are listed in the top rows of Table 1, and the six new loci are listed in the bottom rows. Eleven of the thirteen shared loci were studied with the same SNPs used by Hughes et al.[9]; the remaining two loci, rs17266594 in BANK1 and rs10488631 in IRF5, were studied with highly correlated SNPs (r2 > 0.9 for rs10516487 and rs2070197, respectively, according to the HapMap data for the European population).
Table 1.
List of the 19 systemic lupus erythematosus loci included in this study and their association with systemic lupus erythematosus in our samples a
| SNP | Gene locus b | Chromosome | Position | SLE c | Control c | OR (95% CI) | P-value |
|---|---|---|---|---|---|---|---|
| rs2476601 |
PTPN22 |
1 |
114377568 |
0.099 |
0.074 |
1.37 (1.15 to 1.63) |
4.3 × 10−4 |
| rs1801274 |
FCGR2A |
1 |
161479745 |
0.509 |
0.475 |
1.15 (1.04 to 1.27) |
6.2 × 10−3 |
| rs2205960 |
TNFSF4 |
1 |
173191475 |
0.264 |
0.217 |
1.29 (1.15 to 1.45) |
1.0 × 10−5 |
| rs7574865 |
STAT4 |
2 |
191964633 |
0.323 |
0.233 |
1.57 (1.41 to 1.76) |
1.2 × 10−15 |
| rs6445975 |
PXK |
3 |
58370177 |
0.268 |
0.231 |
1.22 (1.09 to 1.37) |
6.2 × 10−4 |
| rs17266594 |
BANK1d |
4 |
98488450 |
0.749 |
0.711 |
1.21 (1.09 to 1.36) |
6.6 × 10−4 |
| rs3131379 |
MSH5 |
6 |
31721033 |
0.145 |
0.072 |
2.18 (1.85 to 2.58) |
4.2 × 10−21 |
| rs729302 |
IRF5 |
7 |
122930164 |
0.744 |
0.690 |
1.31 (1.17 to 1.46) |
2.5 × 10−6 |
| rs10488631 |
IRF5d |
7 |
128594183 |
0.183 |
0.101 |
1.99 (1.72 to 2.31) |
1.8 × 10−20 |
| rs10954213 |
IRF5 |
7 |
128589427 |
0.680 |
0.641 |
1.19 (1.07 to 1.32) |
1.1 × 10−3 |
| rs13277113 |
C8orf13-BLK |
8 |
11349186 |
0.308 |
0.247 |
1.35 (1.21 to 1.51) |
8.1 × 10−8 |
| rs4963128 |
KIAA1542 |
11 |
589564 |
0.698 |
0.656 |
1.21 (1.09 to 1.34) |
5.0 × 10−4 |
| rs1143679 |
ITGAM |
16 |
31276811 |
0.236 |
0.158 |
1.65 (1.45 to 1.87) |
7.0 × 10−15 |
| rs2304256 |
TYK2 |
19 |
10475652 |
0.766 |
0.725 |
1.24 (1.11 to 1.39) |
2.0 × 10−4 |
| rs5754217 |
UBE2L3 |
22 |
21939675 |
0.253 |
0.216 |
1.24 (1.10 to 1.39) |
3.6 × 10−4 |
| rs2230926 |
TNFAIP3 |
6 |
138196066 |
0.074 |
0.040 |
1.89 (1.52 to 2.35) |
8.5 × 10−9 |
| rs573775 |
ATG5 |
6 |
106764866 |
0.297 |
0.267 |
1.16 (1.04 to 1.29) |
8.8 × 10−3 |
| rs2187668 |
HLA-DQA1 |
6 |
32605884 |
0.200 |
0.106 |
2.11 (1.83 to 2.43) |
1.2 × 10−25 |
| rs10798269 | 1q25.1 | 1 | 173309713 | 0.729 | 0.685 | 1.24 (1.11 to 1.38) | 1.3 × 10−4 |
aSLE, Systemic lupus erythematosus; SNP, Single-nucleotide polymorphism. bThe 13 first loci are shared with those studied by Hughes et al. [1]. cRisk allele frequencies in patients with SLE (n = 1,457) and healthy controls (n = 1,728). dr2 = 0.94 with rs10516487 used for BANK1 by Hughes et al. [9] and r2 = 0.94 for rs2070197 used for IRF5 by Hughes et al. [9].
Analysis of sex-specific genetic risk load
We compared SNP allele frequencies between SLE patients and controls, women together with men and women and men separately. Also, allele frequencies were compared between women and men with SLE. These comparisons were done by performing χ2 tests. GRSs were obtained by applying the same approach used by Hughes et al.[9]. In brief, the number of risk alleles (none, one or two) carried by a patient at each locus was multiplied by the natural logarithm of the sex-specific odds ratio (OR) of that locus. The products corresponding to all loci were summed to obtain the GRSs for this patient. Only patients with valid genotypes at all loci were included in this analysis (1,247 women and 125 men with SLE, of whom 981 women and 96 men were nonoverlapping with those in the previous study). GRSs corresponding to women and men were compared using Student’s t-test. These analyses were done in a customized version of Statistica 7.0 software (StatSoft, Tulsa, OK, USA). Power analysis was done using the Power and Sample Size software [25].
Results
The 19 SNPs that we investigated were significantly different between SLE patients and controls in the unstratified analysis of our 1,457 SLE patients and 1,728 healthy controls (Table 1). The direction of change in all loci was the same as that previously reported [14-22,24]. In the sex-stratified analysis, the 19 loci were associated in women (1,321 SLE patients and 1,188 controls) and 9 in men (136 SLE patients and 540 controls), in part reflecting the decrease in sample size of the male subgroup (Table 2). Comparison of risk allele frequencies between women and men with SLE revealed a significant difference only in rs1143679, the ITGAM SNP, which was more frequent in men than in women with SLE. Our results at this locus and at other three loci are in contrast with those reported by Hughes et al. [9], who found that the ITGAM SNP was not different and the other three loci showed significant differences between women and men with SLE. These differences were an excess of the risk alleles of two of them in men, rs3131379 in MSH5 (HLA) and rs10488631 in IRF5, and an excess of the third, rs4963128 in KIAA1542, in women. None of these changes were observed in our samples (Table 2).
Table 2.
Comparisons of the risk allele frequencies of 19 systemic lupus erythematosus autosomal loci in patients and controls stratified by sex and between men and women with SLE a
| |
|
Men
b
|
Women
b
|
SLE men/SLE women |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP c | Gene locus | SLE | Control | OR (95% CI) | P-value | SLE | Control | OR (95% CI) | P-value | OR (95% CI) d | P-value |
| rs2476601 |
PTPN22 |
0.114 |
0.070 |
1.70 (1.09 to 2.64) |
0.017 |
0.097 |
0.076 |
1.31 (1.08 to 1.60) |
7.2 × 10−3 |
1.19 (0.80 to 1.77) |
0.4 |
| rs1801274 |
FCGR2A |
0.493 |
0.474 |
1.08 (0.82 to 1.41) |
0.6 |
0.511 |
0.474 |
1.15 (1.03 to 1.29) |
0.011 |
0.93 (0.72 to 1.20) |
0.6 |
| rs2205960 |
TNFSF4 |
0.257 |
0.209 |
1.31 (0.96 to 1.79) |
0.09 |
0.264 |
0.220 |
1.27 (1.12 to 1.45) |
3.1 × 10−4 |
0.97 (0.72 to 1.29) |
0.8 |
| rs7574865 |
STAT4 |
0.306 |
0.209 |
1.67 (1.24 to 2.25) |
7.1 × 10−4 |
0.325 |
0.243 |
1.49 (1.32 to 1.69) |
2.8 × 10−10 |
0.92 (0.70 to 1.20) |
0.5 |
| rs6445975 |
PXK |
0.311 |
0.241 |
1.42 (1.06 to 1.91) |
0.018 |
0.264 |
0.226 |
1.23 (1.08 to 1.40) |
2.2 × 10−3 |
1.26 (0.96 to 1.66) |
0.09 |
| rs17266594 |
BANK1e |
0.730 |
0.715 |
1.07 (0.80 to 1.45) |
0.6 |
0.751 |
0.708 |
1.24 (1.09 to 1.41) |
7.5 × 10−4 |
0.89 (0.67 to 1.19) |
0.4 |
| rs3131379 |
MSH5 (HLA) |
0.132 |
0.071 |
1.98 (1.30 to 3.02) |
1.2 × 10−3 |
0.146 |
0.072 |
2.20 (1.82 to 2.66) |
9.6 × 10−17 |
0.89 (0.62 to 1.29) |
0.5 |
| rs729302 |
IRF5 |
0.711 |
0.689 |
1.11 (0.83 to 1.49) |
0.5 |
0.748 |
0.691 |
1.33 (1.17 to 1.50) |
1.0 × 10−5 |
0.83 (0.63 to 1.10) |
0.2 |
| rs10488631 |
IRF5e |
0.184 |
0.111 |
1.81 (1.26 to 2.61) |
1.2 × 10−3 |
0.183 |
0.097 |
2.09 (1.76 to 2.48) |
1.0 × 10−17 |
1.01 (0.73 to 1.40) |
0.96 |
| rs10954213 |
IRF5 |
0.639 |
0.638 |
1.01 (0.76 to 1.33) |
0.97 |
0.685 |
0.643 |
1.21 (1.07 to 1.36) |
1.9 × 10−3 |
0.82 (0.63 to 1.06) |
0.13 |
| rs13277113 |
C8orf13-BLK |
0.274 |
0.250 |
1.13 (0.84 to 1.53) |
0.4 |
0.311 |
0.245 |
1.39 (1.23 to 1.58) |
2.8 × 10−7 |
0.84 (0.63 to 1.11) |
0.2 |
| rs4963128 |
KIAA1542 |
0.715 |
0.645 |
1.38 (1.03 to 1.85) |
0.03 |
0.696 |
0.662 |
1.17 (1.04 to 1.32) |
0.010 |
1.10 (0.83 to 1.45) |
0.5 |
| rs1143679 |
ITGAM |
0.299 |
0.169 |
2.10 (1.54 to 2.85) |
1.6 × 10−6 |
0.230 |
0.153 |
1.65 (1.42 to 1.90) |
1.2 × 10−11 |
1.43 (1.08 to 1.88) |
0.011 |
| rs2304256 |
TYK2 |
0.735 |
0.723 |
1.06 (0.79 to 1.44) |
0.7 |
0.770 |
0.727 |
1.26 (1.11 to 1.43) |
4.8 × 10−4 |
0.83 (0.62 to 1.11) |
0.2 |
| rs5754217 |
UBE2L3 |
0.244 |
0.209 |
1.22 (0.89 to 1.67) |
0.2 |
0.254 |
0.218 |
1.22 (1.07 to 1.39) |
2.8 × 10−3 |
0.95 (0.71 to 1.27) |
0.7 |
| rs2230926 |
TNFAIP3 |
0.070 |
0.039 |
1.87 (1.07 to 3.27) |
0.026 |
0.074 |
0.041 |
1.87 (1.45 to 2.40) |
7.0 × 10−7 |
0.95 (0.58 to 1.55) |
0.8 |
| rs573775 |
ATG5 |
0.256 |
0.263 |
0.96 (0.71 to 1.30) |
0.8 |
0.301 |
0.269 |
1.17 (1.04 to 1.33) |
0.012 |
0.80 (0.60 to 1.06) |
0.12 |
| rs2187668 |
HLA-DQA1 |
0.184 |
0.107 |
1.87 (1.30 to 2.69) |
6.0 × 10−4 |
0.201 |
0.105 |
2.15 (1.82 to 2.52) |
7.4 × 10−21 |
0.89 (0.65 to 1.23) |
0.5 |
| rs10798269 | 1q25.1 | 0.733 | 0.656 | 1.20 (0.89 to 1.62) | 0.2 | 0.729 | 0.680 | 1.26 (1.12 to 1.43) | 1.7 × 10−4 | 1.02 (0.77 to 1.36) | 0.9 |
aSLE, Systemic lupus erythematosus; SNP, Single-nucleotide polymorphism. bSLE men = 136; control men = 540; SLE women = 1,321; Control women = 1,188. cThe 13 first loci are the same as those studied by Hughes et al. [9]. dOR > 1.0 indicates higher frequency of the risk allele in men than in women. er2 = 0.94 with rs10516487 used for BANK1 and r2 = 0.94 for rs2070197 used for IRF5[9].
GRSs were obtained separately for women and men with SLE using the risk alleles and sex-specific ORs of the 19 loci (Figure 1). They showed very significantly higher values in women than in men (mean GRSs ± SD = 4.15 ± 1.07 vs 3.22 ± 1.0, respectively; P < 10−16 by Student’s t-test). This result was opposite to that observed by Hughes et al. [9], that is, higher GRSs in men than in women. The difference was very significant regardless of consideration of the 77.1% nonoverlapping samples (mean GRSs ± SD in women = 4.20 ± 1.07 vs 3.27 ± 0.98 in men; P = 8.9 × 10−16) or of the 22.9% samples overlapping with those studied by Hughes et al. (3.98 ± 1.07 in women vs 3.05 ± 1.02 in men; P = 1.3 × 10−5). The mean difference in GRSs between the sexes, 0.93, was the same in the two subgroups of patients. This result of the overlapping samples was not reflected in the Hughes et al. study, because they were only a minor fraction (7.9%) of the samples included in that study and they were considered together with all others.
Figure 1.
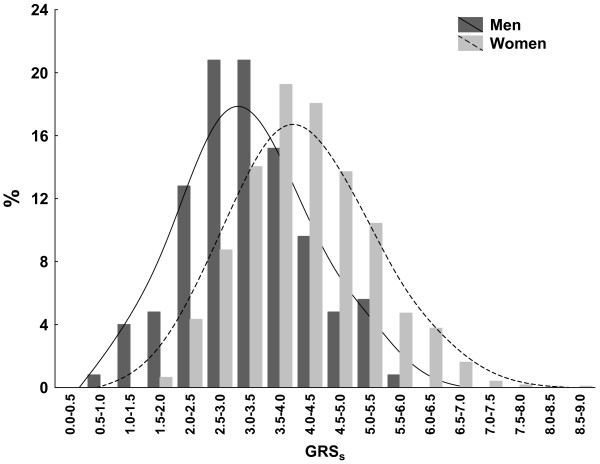
Sum of genetic risk scores in women and men with systemic lupus erythematosus. The y-axis represents percentages of participants in each of the two groups with the sum of genetic risk scores (GRSs) at the indicated intervals along the x-axis. This analysis was done with GRSs obtained for each of the 1,247 women and 125 men with systemic lupus erythematosus (SLE) and 100% genotype success at the 19 SLE loci. The GRSs of each patient with SLE is the sum of the products of the natural logarithm of the sex-specific OR by the number of risk alleles at each locus carried by the patient, as described by Hughes et al. [9]. Histograms and distance-weighted least-squares fitting lines for women (light gray bars and discontinuous line) and for men (dark gray bars and continuous line) are shown.
Differences in the loci investigated between the two studies include three components: six loci included in our study that were not in the Hughes et al. study, thirteen loci shared by the two studies and two loci in the Hughes et al. that are absent from our study. The six loci included only in our study showed either no difference or a trend toward higher risks in women (Table 2). Comparison of the GRSs between women and men restricted to these six loci gave a very significant excess of risk load in women (mean GRSs for these six loci = 1.29 ± 0.55 in women and 0.74 ± 0.46 in men; P < 10−16), which accounted for most of the difference between the two sexes in our study (0.55, or 59% of the 0.93 difference with the 19 loci). The 13 loci that are shared by the two studies also contributed to the contrasting results, but less markedly so (mean GRSs of the 13 shared loci = 2.86 ± 0.82 in women and 2.48 ± 0.77 in men; P = 6.6 × 10−7). Their effect is reflected in the remaining 41% of the mean difference with the 19 loci (0.38 of the 0.93 difference). Therefore, whereas each of the six loci included only in our study contributed 0.09 to the mean difference in GRSs, each of the loci included in both studies contributed only 0.03 to the average score. We cannot exclude the possibility that the two loci included in the previous report that we did not study might have contributed to the very contrasting results.
Two HLA loci accounted for a large fraction of the previously reported increased GRSs in men with SLE [9]. We also included two strongly SLE-associated HLA loci in our analyses (rs3131379 and rs2187668), but they did not account for a particularly large fraction of the difference between sexes, as demonstrated by the similar result obtained after excluding these two SNPs (mean GRSs except HLA = 3.61 ± 0.81 for women and 2.82 ± 0.76 for men with SLE (P < 10−16), corresponding to 85% of the difference in GRSs with the 19 loci).
Discussion
Our analysis shows a very significantly higher genetic risk load in women than in men with SLE. This result is in striking contrast to the previous analysis by Hughes et al. [9] and highlights important limitations of this type of study that prevent firm conclusions about the relative genetic risk load of the two sexes until these limitations can be addressed.
The most prominent outcome of our study is the lack of reproducibility of the results, both at the level of each locus and at the level of global genetic risk load measured by GRSs. At the locus level, lack of reproducibility is well known to have plagued genetic association studies in the past [26], and it has been notably common in studies claiming sex-specific associations [27]. Two of the limitations of these studies are especially relevant here: tolerant significance thresholds that do not account for the multiple tests involved and small sample sizes that lead to imprecision in the estimated effects. Regarding the significance thresholds, none of the loci showed a difference between sexes with P < 10−4 in the two studies. Therefore, all nonreproducible locus associations will be eliminated if a strict threshold is applied. This limitation does not apply, however, to the comparison of GRSs, because only a test was done in each study and the results obtained are very significantly different. In contrast, the second limitation—the decrease in sample size inherent to stratification by sex—has deleterious effects in both the analysis of each locus and the calculation of GRSs. The effect at the locus level can be shown by comparing, within each study, the power to detect differences between SLE patients and controls with the power to detect differences between women and men with SLE. Our study has enough power (1 − β > 0.8 for α = 0.05 and risk allele frequency = 0.2) to detect differences with OR = 1.19 in the first comparison, but only for OR > 1.52 in the second. Similarly, the Hughes et al. study [9] has enough power for detection with an OR > 1.12 in the patient–control analysis but an OR > 1.31 in the female–male comparison. The decrease in power affects the precision of the OR, which is used to calculate GRSs and, therefore, also has a negative effect in the reproducibility of the GRSs results. Thus, GRSs, as is true of other summary parameters, is less variable than the individual OR included in its calculation, but GRSs performance depends on the OR quality, which is determined by sample size and the minor allele frequency of the SNPs [28].
A second notable message derived from the data is that most known SLE loci are not clearly biased to more risk in one of the sexes. In our study, only one of the nineteen SNPs showed a significant difference between women and men with SLE. The risk allele of this SNP (rs1143679) in ITGAM was more associated with SLE in men than in women. Similar results were reported by Hughes et al. [9], who showed that only four loci (rs1270942 in the HLA, rs3131379 in MSH5 (HLA), rs10488631 in IRF5, and rs4963128 in KIAA1542) of the fifteen analyzed had significant differences between women and men with SLE. They found that the risk alleles of three of these loci were more common in men than in women with SLE, but the fourth (in KIAA1542) was more common in women than in men with SLE. In addition, when the Bonferroni correction is applied for the number of loci analyzed, only the two HLA loci included in the Hughes et al. study remain significantly different between the sexes in either of the two studies. Given this lack of clear bias for most SLE loci to excess risk in one of the sexes, differences in estimated global genetic risk load will be sensitive to the inclusion of loci showing small and inconsistent differences, with some showing a small bias toward one sex and others to the opposite sex. The balance between the two components will determine the global outcome. In addition, solving the question of the different genetic risk load in women and men will be difficult until we know a larger fraction of the heritability of SLE than the current 9% to 15% [16,29]. This fraction of heritability has be explained with up to 23 loci, but the most current estimates put the number of loci for complex diseases such as SLE at several hundred [30]. These estimates indicate that many of the yet undiscovered loci will show lower effects than those already known. However, it is clear that there is plenty of room for improvement. Larger coverage of SLE heritability will permit more reproducible assessment of differences in the total genetic burden between the sexes.
The very significant differences (P < 10−16 and P < 5 × 10−8) in genetic risk load in the two studies indicate that there are factors other than the imprecision of GRSs leading to their opposite directions. Two factors are likely to have been major contributors: the different sets of loci and the differences between the participants included. Our analysis shows that the different sets of loci in each study were an important factor. The six loci we explored that were not included in the previous study accounted for more than one-half of the excess risk in women, despite representing less than one-third of the analyzed loci. In addition, it is very likely that lack of the HLA SNP rs1270942 in our study also contributed to the contrasting results, because it accounted for the highest risk in men of all the loci included in the Hughes et al. study [9]. Regarding the differences between the study participants, they are shown by the significantly higher risk load in women than in men in our study (P < 6.6 × 10−7), based on the GRSs calculated for the 13 loci shared by the two studies. The differences between the two studies could include genetic heterogeneity affecting both patients with SLE and controls and clinical differences between the patients with SLE. Genetic heterogeneity is especially likely at the HLA SNPs, where large differences, even within subpopulations of the same ethnic group, are common. The two types of heterogeneity, genetic and clinical, have previously been shown to be reflected in the degree of association of SLE loci [11,31-34]. In this regard, we could compare the prevalence of only three SLE classification criteria, but they showed significant heterogeneity between the SLE patients included in the two studies. Men in the Hughes et al. study had a higher prevalence of renal disease (OR = 1.7, P = 1.2 × 10−5) than that in women, but not of serositis or neurologic involvement [9]. In contrast, the men in our study had a higher incidence of serositis than the women did (OR = 1.58, P = 0.017), but not renal (OR = 1.23, P = 0.3) or neurologic involvement. This type of inconsistency between studies regarding clinical differences between women and men with SLE are common [3-6].
There is an additional point to consider in the interpretation of this type of study: the possibility of ascertainment bias. This artefact is the systematic deviation from the true population value that is attributable to the sampling processes used to find SNPs and estimate their population-specific allele frequencies [35]. As all the SLE loci have been discovered in studies involving either only women or a very dominant fraction of women, they reflect more faithfully genetic susceptibility to SLE in women than in men. Therefore, they are more likely to show stronger associations with women than with men, in cases where the genetic susceptibility of the two sexes is different. The possibility of ascertainment bias means that the increased genetic risk load we found in women cannot be interpreted as a more marked genetic component in SLE susceptibility in women than in men.
Conclusions
Our results highlight that there is not a uniform trend in the known SLE loci toward higher genetic risk in one or the other of the sexes among Europeans. In addition, our results show the multiple limitations of studies in which investigators aim to establish sex-specific genetic risk loads in SLE. These limitations include the critical role of the assortment of loci that are considered in a particular study; the imprecision in GRSs estimates for men; heterogeneity between sets of participants, including genetic and clinical heterogeneity; and the possibility of ascertainment bias when analyzing loci identified predominantly in women. The limitations could someday be addressed in studies in which researchers include SNPs representing a larger fraction of the SLE genetic component of what is currently known and that have been identified in large samples of both women and men; however, such studies are not currently possible. Therefore, at present, these limitations prevent assessment with confidence of differences in genetic load between women and men with SLE.
Abbreviations
GRSs: Sum genetic risk score; HLA: Human leukocyte antigen; SLE: Systemic lupus erythematosus; SNP: Single-nucleotide polymorphism.
Competing interest
The authors declare that they have no competing interests.
Author’s contributions
EAP and AG designed the study and wrote the first draft of the manuscript. EAP and MSG genotyped the samples. EAP, MSG, MC and AG analyzed the data. FJB, AS, MJS, CP, PC, RP, JOR, MM, FNS, MB, NB, GDS, SM, TW, BRL, AK, SR and JJGR provided samples from and clinical and demographic information of the participants. EAP, MSG, MC, FJB, AS, MJS, CP, PC, RP, JOR, MM, FNS, MB, NB, GDS, SM, TW, BRL, AK, SR, JJGR and AG contributed to the interpretation of the data and revised the final version of the manuscript. AG obtained funding, supervised the study and had complete access to all of the data. All authors read and approved the final version of the manuscript.
Contributor Information
Elisa Alonso-Perez, Email: mixyoelisa@gmail.com.
Marian Suarez-Gestal, Email: mariansg82@hotmail.com.
Manuel Calaza, Email: mcalaza@gmail.com.
Francisco J Blanco, Email: Francisco_Blanco@canalejo.org.
Ana Suarez, Email: anasua@uniovi.es.
Maria Jose Santos, Email: MJPS@netvisao.pt.
Chryssa Papasteriades, Email: cpapaste@gmail.com.
Patricia Carreira, Email: carreira@h12o.es.
Rudolf Pullmann, Email: pullmann@mfn.sk.
Josep Ordi-Ros, Email: jordi@vhebron.net.
Maurizio Marchini, Email: maurizio.marchini@unimi.it.
Fotini N Skopouli, Email: fskopouli@euroclinic.gr.
Marc Bijl, Email: M.Bijl@mzh.nl.
Nadia Barrizone, Email: barizzo@med.unipmn.it.
Gian Domenico Sebastiani, Email: gsebastiani@scamilloforlanini.rm.it.
Sergio Migliaresi, Email: sergio.migliaresi@unina2.it.
Torsten Witte, Email: witte.torsten@mh-hannover.de.
Bernard R Lauwerys, Email: Bernard.Lauwerys@uclouvain.be.
Attila Kovacs, Email: dr.kovacs.attila.phd@gmail.com.
Sarka Ruzickova, Email: Sarka.Ruzickova@ibt.cas.cz.
Juan J Gomez-Reino, Email: juan.jesus.gomez-reino.carnota@sergas.es.
Antonio Gonzalez, Email: antonio.gonzalez.martinez-pedrayo@sergas.es.
Acknowledgements
The authors thank Carmen Pena for taking care of the samples and helping with the genotyping assays. This work was supported by the Health Research Fund of the Instituto de Salud Carlos III (Spain) (grants 08/0744 and 11/01048) and by the RETICS Program (RD08/0075 and RD12/0009/0008), which that are partially financed by the European Regional Development Fund of the European Union. The following are other contributors to the European Consortium of SLE DNA Collections who are participating in this study: Myriam Liz, Laboratorio de Investigacion 10 and Rheumatology Unit, Instituto de Investigacion Sanitaria–Hospital Clinico Universitario de Santiago, Santiago de Compostela, Spain; Iris Kappou-Rigatou, Department of Histocompatibility and Immunology, Evangelismos Hospital, Athens, Greece; Lorenzo Beretta, Clinical Immunology, University of Milan, and Fondazione IRCCS Ospedale Maggiore Policlinico, Mangiagalli e Regina Elena, Milan, Italy; Eva Balada, Internal Medicine, Research Laboratory in Autoimmune Diseases Hospital Vall d’Hebron, Barcelona, Spain; Cees G Kallenberg, Department of Rheumatology and Clinical Immunology, University Medical Center Groningen, Groningen, the Netherlands; Filipe Vinagre, Rheumatology Department, Hospital Garcia de Orta, Almada (Portugal), and Rheumatology Research Unit, Molecular Medicine Institute, Faculty of Medicine, University of Lisbon, Lisbon, Portugal; Rudolf Pullmann Jr, Gerontology Research Center, National Institute on Aging, Baltimore, MD, USA; Maria Mavromati, Pathophysiology Department, Athens University Medical School, Athens, Greece; Carmen Gutierrez, Department of Functional Biology, Hospital Universitario Central de Asturias, Universidad de Oviedo, Oviedo, Spain; Ignacio Rego, Servicio de Reumatología, Complejo Hospitalario Universitario A Coruña, A Coruña, Spain; Sandra D’Alfonso, Department of Medical Sciences and Interdisciplinary Research Center of Autoimmune Diseases (IRCAD), University of Eastern Piedmont Amedeo Avogadro, Novara, Italy; Reinhold E Schmidt, Division of Clinical Immunology, Department of Internal Medicine, Hannover Medical School, Hannover, Germany; Emöke Endreffy, Pediatrics Department, Albert Szent-Györgyi Medical and Pharmaceutical Centre, University of Szeged, Szeged, Hungary; and Ctibor Dostal, Molecular Biology and Immunogenetics Department, Institute of Rheumatology, Prague, Czech Republic.
References
- McCombe PA, Greer JM, Mackay IR. Sexual dimorphism in autoimmune disease. Curr Mol Med. 2009;9:1058–1079. doi: 10.2174/156652409789839116. [DOI] [PubMed] [Google Scholar]
- Zandman-Goddard G, Peeva E, Shoenfeld Y. Gender and autoimmunity. Autoimmun Rev. 2007;6:366–372. doi: 10.1016/j.autrev.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Schwartzman-Morris J, Putterman C. Gender differences in the pathogenesis and outcome of lupus and of lupus nephritis. Clin Dev Immunol. 2012;2012:604892. doi: 10.1155/2012/604892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu LJ, Wallace DJ, Ishimori ML, Scofield RH, Weisman MH. Review: Male systemic lupus erythematosus: a review of sex disparities in this disease. Lupus. 2010;19:119–129. doi: 10.1177/0961203309350755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan TC, Fang H, Magder LS, Petri MA. Differences between male and female systemic lupus erythematosus in a multiethnic population. J Rheumatol. 2012;39:759–769. doi: 10.3899/jrheum.111061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy G, Isenberg D. Effect of gender on clinical presentation in systemic lupus erythematosus. Rheumatology (Oxford) 2013;52:2108–2115. doi: 10.1093/rheumatology/ket160. [DOI] [PubMed] [Google Scholar]
- Kremer Hovinga IC, Koopmans M, Baelde HJ, de Heer E, Bruijn JA, Bajema IM. Tissue chimerism in systemic lupus erythematosus is related to injury. Ann Rheum Dis. 2007;66:1568–1573. doi: 10.1136/ard.2007.070516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield RH, Bruner GR, Namjou B, Kimberly RP, Ramsey-Goldman R, Petri M, Reveille JD, Alarcón GS, Vilá LM, Reid J, Harris B, Li S, Kelly JA, Harley JB. Klinefelter’s syndrome (47, XXY) in male systemic lupus erythematosus patients: support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheum. 2008;58:2511–2517. doi: 10.1002/art.23701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes T, Adler A, Merrill JT, Kelly JA, Kaufman KM, Williams A, Langefeld CD, Gilkeson GS, Sanchez E, Martin J, Boackle SA, Stevens AM, Alarcón GS, Niewold TB, Brown EE, Kimberly RP, Edberg JC, Ramsey-Goldman R, Petri M, Reveille JD, Criswell LA, Vilá LM, Jacob CO, Gaffney PM, Moser KL, Vyse TJ, Alarcón-Riquelme ME, James JA, Tsao BP. BIOLUPUS Network et al. Analysis of autosomal genes reveals gene–sex interactions and higher total genetic risk in men with systemic lupus erythematosus. Ann Rheum Dis. 2012;71:694–699. doi: 10.1136/annrheumdis-2011-200385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Perez E, Suarez-Gestal M, Calaza M, Witte T, Papasteriades C, Marchini M, Migliaresi S, Kovacs A, Ordi-Ros J, Bijl M, Santos MJ, Ruzickova S, Pullmann R, Carreira P, Skopouli FN, D’Alfonso S, Sebastiani GD, Suarez A, Blanco FJ, Gomez-Reino JJ, Gonzalez A. for the European Consortium of SLE DNA Collections. Association of systemic lupus erythematosus clinical features with European population genetic substructure. PLoS One. 2011;6:e29033. doi: 10.1371/journal.pone.0029033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Perez E, Suarez-Gestal M, Calaza M, Sebastiani GD, Pullmann R, Papasteriades C, Kovacs A, Skopouli FN, Bijl M, Suarez A, Marchini M, Migliaresi S, Carreira P, Ordi-Ros J, Witte T, Ruzickova S, Santos MJ, Barizzone N, Blanco FJ, Lauwerys BR, Gomez-Reino JJ, Gonzalez A. European Consortium of SLE DNA Collections. Bias in effect size of systemic lupus erythematosus susceptibility loci across Europe: a case–control study. Arthritis Res Ther. 2012;14:R94. doi: 10.1186/ar3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Gestal M, Calaza M, Endreffy E, Pullmann R, Ordi-Ros J, Sebastiani GD, Ruzickova S, Santos MJ, Papasteriades C, Marchini M, Skopouli FN, Suarez A, Blanco FJ, D’Alfonso S, Bijl M, Carreira P, Witte T, Migliaresi S, Gomez-Reino JJ, Gonzalez A. European Consortium of SLE DNA Collections. Replication of recently identified systemic lupus erythematosus genetic associations: a case–control study. Arthritis Res Ther. 2009;11:R69. doi: 10.1186/ar2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- Sigurdsson S, Nordmark G, Göring HH, Lindroos K, Wiman AC, Sturfelt G, Jönsen A, Rantapää-Dahlqvist S, Möller B, Kere J, Koskenmies S, Widén E, Eloranta ML, Julkunen H, Kristjansdottir H, Steinsson K, Alm G, Rönnblom L, Syvänen AC. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet. 2005;76:528–537. doi: 10.1086/428480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunninghame Graham DS, Graham RR, Manku H, Wong AK, Whittaker JC, Gaffney PM, Moser KL, Rioux JD, Altshuler D, Behrens TW, Vyse TJ. Polymorphism at the TNF superfamily gene TNFSF4 confers susceptibility to systemic lupus erythematosus. Nat Genet. 2008;40:83–89. doi: 10.1038/ng.2007.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley JB, Alarcón-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ, Langefeld CD, Nath SK, Guthridge JM, Cobb BL, Mirel DB, Marion MC, Williams AH, Divers J, Wang W, Frank SG, Namjou B, Gabriel SB, Lee AT, Gregersen PK, Behrens TW, Taylor KE, Fernando M, Zidovetzki R, Gaffney PM, Edberg JC, Rioux JD, Ojwang JO. The International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN) et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S, Lee AT, Chung SA, Ferreira RC, Pant PV, Ballinger DG, Kosoy R, Demirci FY, Kamboh MI, Kao AH, Tian C, Gunnarsson I, Bengtsson AA, Rantapää-Dahlqvist S, Petri M, Manzi S, Seldin MF, Rönnblom L, Syvänen AC, Criswell LA, Gregersen PK, Behrens TW. Association of systemic lupus erythematosus with C8orf13–BLK and ITGAM–ITGAX. N Engl J Med. 2008;358:900–909. doi: 10.1056/NEJMoa0707865. [DOI] [PubMed] [Google Scholar]
- Kozyrev SV, Abelson AK, Wojcik J, Zaghlool A, Prasad Linga Reddy MV, Sanchez E, Gunnarsson I, Svenungsson E, Sturfelt G, Jönsen A, Truedsson L, Pons-Estel BA, Witte T, D’Alfonso S, Barizzone N, Danieli MG, Gutierrez C, Suarez A, Junker P, Laustrup H, Gonzalez-Escribano MF, Martin J, Abderrahim H, Alarcón-Riquelme ME. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:211–216. doi: 10.1038/ng.79. [DOI] [PubMed] [Google Scholar]
- Musone SL, Taylor KE, Lu TT, Nititham J, Ferreira RC, Ortmann W, Shifrin N, Petri MA, Kamboh MI, Manzi S, Seldin MF, Gregersen PK, Behrens TW, Ma A, Kwok PY, Criswell LA. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet. 2008;40:1062–1064. doi: 10.1038/ng.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath SK, Han S, Kim-Howard X, Kelly JA, Viswanathan P, Gilkeson GS, Chen W, Zhu C, McEver RP, Kimberly RP, Alarcón-Riquelme ME, Vyse TJ, Li QZ, Wakeland EK, Merrill JT, James JA, Kaufman KM, Guthridge JM, Harley JB. A nonsynonymous functional variant in integrin-αM (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet. 2008;40:152–154. doi: 10.1038/ng.71. [DOI] [PubMed] [Google Scholar]
- Sawalha AH, Webb R, Han S, Kelly JA, Kaufman KM, Kimberly RP, Alarcón-Riquelme ME, James JA, Vyse TJ, Gilkeson GS, Choi CB, Scofield RH, Bae SC, Nath SK, Harley JB. Common variants within MECP2 confer risk of systemic lupus erythematosus. PLoS One. 2008;3:e1727. doi: 10.1371/journal.pone.0001727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, Ortmann W, Kosoy R, Ferreira RC, Nordmark G, Gunnarsson I, Svenungsson E, Padyukov L, Sturfelt G, Jönsen A, Bengtsson AA, Rantapää-Dahlqvist S, Baechler EC, Brown EE, Alarcón GS, Edberg JC, Ramsey-Goldman R, McGwin G Jr, Reveille JD, Vilá LM, Kimberly RP, Manzi S, Petri MA, Lee A, Gregersen PK. et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. 2009;41:1228–1233. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunninghame Graham DS, Vyse TJ, Fortin PR, Montpetit A, Cai Y, Lim S, McKenzie T, Farwell L, Rhodes B, Chad L, Hudson TJ, Sharpe A, Terhorst C, Greenwood CM, Wither J, Rioux JD. CaNIOS GenES Investigators. Association of LY9 in UK and Canadian SLE families. Genes Immun. 2008;9:93–102. doi: 10.1038/sj.gene.6364453. [DOI] [PubMed] [Google Scholar]
- Ferreiro-Neira I, Calaza M, Alonso-Perez E, Marchini M, Scorza R, Sebastiani GD, Blanco FJ, Rego I, Pullmann R Jr, Pullmann R, Kallenberg CG, Bijl M, Skopouli FN, Mavromati M, Migliaresi S, Barizzone N, Ruzickova S, Dostal C, Schmidt RE, Witte T, Papasteriades C, Kappou-Rigatou I, Endreffy E, Kovacs A, Ordi-Ros J, Balada E, Carreira P, Gomez-Reino JJ, Gonzalez A. Opposed independent effects and epistasis in the complex association of IRF5 to SLE. Genes Immun. 2007;8:429–438. doi: 10.1038/sj.gene.6364407. [DOI] [PubMed] [Google Scholar]
- Dupont WD, Plummer WD Jr. Power and sample size calculations: a review and computer program. Control Clin Trials. 1990;11:116–128. doi: 10.1016/0197-2456(90)90005-m. [DOI] [PubMed] [Google Scholar]
- Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsopoulos NA, Tatsioni A, Ioannidis JP. Claims of sex differences: an empirical assessment in genetic associations. JAMA. 2007;298:880–893. doi: 10.1001/jama.298.8.880. [DOI] [PubMed] [Google Scholar]
- Che R, Motsinger-Reif AA. Evaluation of genetic risk score models in the presence of interaction and linkage disequilibrium. Front Genet. 2013;4:138. doi: 10.3389/fgene.2013.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So HC, Gui AH, Cherny SS, Sham PC. Evaluating the heritability explained by known susceptibility variants: a survey of ten complex diseases. Genet Epidemiol. 2011;35:310–317. doi: 10.1002/gepi.20579. [DOI] [PubMed] [Google Scholar]
- Stahl EA, Wegmann D, Trynka G, Gutierrez-Achury J, Do R, Voight BF, Kraft P, Chen R, Kallberg HJ, Kurreeman FAS, Diabetes Genetics R, Meta-analysis C, Meta-analysis C, Myocardial Infarction Genetics C, Kathiresan S, Wijmenga C, Gregersen PK, Alfredsson L, Siminovitch KA, Worthington J, de Bakker PIW, Raychaudhuri S, Plenge RM. Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat Genet. 2012;44:483–489. doi: 10.1038/ng.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Perez E, Suarez-Gestal M, Calaza M, Ordi-Ros J, Balada E, Bijl M, Papasteriades C, Carreira P, Skopouli FN, Witte T, Endreffy E, Marchini M, Migliaresi S, Sebastiani GD, Santos MJ, Suarez A, Blanco FJ, Barizzone N, Pullmann R, Ruzickova S, Lauwerys BR, Gomez-Reino JJ, Gonzalez A. European Consortium of SLE DNA Collections. Further evidence of subphenotype association with systemic lupus erythematosus susceptibility loci: a European cases only study. PLoS One. 2012;7:e45356. doi: 10.1371/journal.pone.0045356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez E, Nadig A, Richardson BC, Freedman BI, Kaufman KM, Kelly JA, Niewold TB, Kamen DL, Gilkeson GS, Ziegler JT, Langefeld CD, Alarcón GS, Edberg JC, Ramsey-Goldman R, Petri M, Brown EE, Kimberly RP, Reveille JD, Vilá LM, Merrill JT, Anaya JM, James JA, Pons-Estel BA, Martin J, Park SY, Bang SY, Bae SC, Moser KL, Vyse TJ, Criswell LA. et al. Phenotypic associations of genetic susceptibility loci in systemic lupus erythematosus. Ann Rheum Dis. 2011;70:1752–1757. doi: 10.1136/ard.2011.154104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson S, Nordmark G, Garnier S, Grundberg E, Kwan T, Nilsson O, Eloranta ML, Gunnarsson I, Svenungsson E, Sturfelt G, Bengtsson AA, Jönsen A, Truedsson L, Rantapää-Dahlqvist S, Eriksson C, Alm G, Göring HH, Pastinen T, Syvänen AC, Röonnblom L. A risk haplotype of STAT4 for systemic lupus erythematosus is over-expressed, correlates with anti-dsDNA and shows additive effects with two risk alleles of IRF5. Hum Mol Genet. 2008;17:2868–2876. doi: 10.1093/hmg/ddn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung SA, Tian C, Taylor KE, Lee AT, Ortmann WA, Hom G, Graham RR, Nititham J, Kelly JA, Morrisey J, Wu H, Yin H, Alarcón-Riquelme ME, Tsao BP, Harley JB, Gaffney PM, Moser KL, Manzi S, Petri M, Gregersen PK, Langefeld CD, Behrens TW, Seldin MF, Criswell LA. European population substructure is associated with mucocutaneous manifestations and autoantibody production in systemic lupus erythematosus. Arthritis Rheum. 2009;60:2448–2456. doi: 10.1002/art.24707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SNP FAQ Archive. NCBI Help Manual. Bethesda, MD: National Center for Biotechnology Information; 2005–2014. [Google Scholar]