

Abstract
As a prerequisite to clinical application, we determined the long-term therapeutic effectiveness and safety of adeno-associated viral (AAV) S100A1 gene therapy in a preclinical, large animal model of heart failure. S100A1, a positive inotropic regulator of myocardial contractility, becomes depleted in failing cardiomyocytes in humans and various animal models, and myocardial-targeted S100A1 gene transfer rescues cardiac contractile function by restoring sarcoplasmic reticulum calcium Ca2+ handling in acutely and chronically failing hearts in small animal models. We induced heart failure in domestic pigs by balloon-occlusion of the left circumflex coronary artery, resulting in myocardial infarction. After 2 weeks, when the pigs displayed significant left ventricular contractile dysfunction, we administered through retrograde coronary venous delivery, AAV9-S100A1 to the left ventricular non-infarcted myocardium. AAV9-luciferase and saline treatment served as control. At 14 weeks, both control groups showed significantly decreased myocardial S100A1 protein expression along with progressive deterioration of cardiac performance and left ventricular remodeling. AAV9-S100A1 treatment prevented and reversed this phenotype by restoring cardiac S100A1 protein levels. S100A1 treatment normalized cardiomyocyte Ca2+ cycling, sarcoplasmic reticulum calcium handling and energy homeostasis. Transgene expression was restricted to cardiac tissue and extra-cardiac organ function was uncompromised indicating a favorable safety profile. This translational study shows the pre-clinical feasibility, long-term therapeutic effectiveness and a favorable safety profile of cardiac AAV9-S100A1 gene therapy in a preclinical model of heart failure. Our study presents a strong rational for a clinical trial of S100A1 gene therapy for human heart failure that could potentially complement current strategies to treat end-stage heart failure.
INTRODUCTION
Heart failure (HF) represents the common endpoint of a variety of cardiac diseases such as a myocardial infarction (MI) or genetic cardiomyopathies and is a major cause of morbidity and mortality worldwide (1). Essentially, loss of myocardium triggers a sequence of molecular, cellular and physiological responses leading to ventricular remodeling and the inability of the left ventricle to maintain an output of blood sufficient for the metabolic requirements of the tissues of the body. Patients with HF show a characteristic maladaptive neurohumeral response including an activation of the renin-angiotensin-aldosteron system (RAAS) and an increased catecholamine concentration in the blood, both causing a temporary increase in cardiac output but long-term detrimental effects such as water- and sodium-retention or adverse cardiac remodeling (2). Current standard of care for patients with HF is either a symptomatic therapy, e.g. by use of diuretics, or targets the above mentioned vicious cycle by use of β-adrenergic receptor (β-AR) blockers, angiotensin-converting-enzyme (ACE) inhibitors or aldosterone antagonists causing a moderate increase in survival, limited increase of cardiac ejection fraction (EF) and myocardial reverse remodeling (2–7). Despite of this clear benefit, conventional drug therapies have provided to patients suffering from HF, the disease progresses relentlessly and more than 50% of patients with end-stage HF still die within 5 years (2,8). This dilemma reflects a lack of innovative therapies targeting the underlying causes of the disease (1,2,8,9).
Coordinated regulation of Ca2+ cycling in the cardiomyocyte is required during each cycle of cardiac relaxation and contraction. Cytosolic Ca2+ is sequested into the SR lumen by cardiac sarcoplasmic reticulum (SR) Ca2+-ATPase (SERCA2a), permitting muscle relaxation, subsequently the stored Ca2+ is released through the ryanodine receptor (RyR2) to activate myofilament contraction. Abnormal cardiomyocyte calcium (Ca2+) handling is a key factor in HF pathogenesis (10). Among recently discovered molecules controlling this process, the Ca2+-sensor protein S100A1 has emerged as an attractive target for genetically-targeted HF therapy due to its molecular profile (11). S100A1 is a key regulator of a calcium-controlled integrative network in cardiomyocytes that regulates SR, sarcomeric and mitochondrial function through modulation of the RyR2, SERCA2, titin and mitochondrial F1-ATPase activity (11–18). As a result, cardiomyocytes and hearts exhibiting increased S100A1 expression show increased systolic and diastolic performance due to improved Ca2+ transient amplitudes emanating from augmented SR Ca2+ load and subsequent systolic Ca2+ release together with decreased diastolic SR Ca2+ leak and enhanced Ca2+-resequestration (11,13,15,17). Concurrently, S100A1 increases mitochondrial high energy phosphate production, and thus adapts energy supply to increased ATP-demand which is due to enhanced cardiomyocyte Ca2+-turnover (11,12,15).
Reduced S100A1 expression in the myocardium has been documented in humans and various animal models in HF, while numerous studies have established a clear association between depleted S100A1 protein levels in cardiomyocytes and reduced contractile function, corroborating its pathophysiological significance (15,19–25). Repairing this molecular defect in small animal HF models by cardiac-targeted S100A1 gene transfer reverses malfunction of the SR and mitochondria and restores cardiac function in the long-term (15,20–22). Despite this therapeutic profile in rodent models, clinical translation of this cardiac molecular therapeutic approach requires large animal HF models, which more closely approximate human physiology, function and anatomy (26). This is necessary since a mouse heart operates close to its theoretical maximum and beats about 10 times faster than the human heart indicating differences in the cardiac inotropic reserve. Mechanistical differences with regard to the regulation of cardiomyocyte Ca2+ cycling exists since Ca2+ removal from the cytosol in mice relies almost exclusively on SERCA2a (92% of total), whereas in humans Na+/Ca2+ exchanger (NCX) activity accounts for approximately one third of Ca2+ removal, with SERCA2a function largely responsible for the remainder (10, 27). The basic myocardial sarcomeric proteins are also different, with α-myosin heavy chain predominating in adult mice but β-myosin heavy chain predominating in humans (28). Moreover, technical barriers and safety standards for myocardial gene delivery differ significantly between rodent models and clinically relevant large animal models. The target volume of the myocardium necessary to achieve therapeutic effects is more than a hundred times larger in human compared to rats illustrating the need for highly efficient gene delivery. Moreover, cardiac gene transfer should be achieved through a percutaneous approach using certified and approved material. Thus, as a prerequisite to clinical application, we determined long-term therapeutic efficacy, safety and feasibility of AAV9-S100A1 gene therapy in a postischemic pig model of HF that recapitulates key clinical features of human HF (26,29) and explored mechanistic features of its action.
RESULTS
Myocardial infarction of pig hearts resulted in a preclinical heart failure model
Percutaneous, catheter-based intermittent balloon occlusion of the proximal left circumflex coronary artery (LCX) resulted in a reproducible perfusion defect of the lateral LV wall (30±4%, n=32) with a transmural infarction (Figure 1A–E). Infarcted pigs demonstrated systolic LV dysfunction after 2 weeks with transition to failure (ejection fraction (EF)=39±4%, n=18) and remodeling over a follow-up period of 14 weeks (Figure 1F–I).
Figure 1. Postischemic porcine HF model.
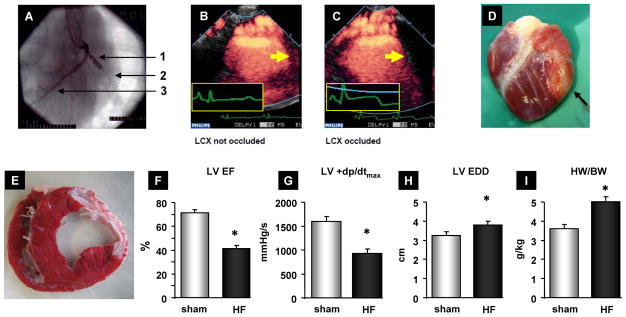
(A) Radioscopic image showing catheter-based left circumflex coronary artery (LCX) occlusion. 1, inflated balloon; 2, guided wire; 3, left anterior descending coronary artery (LAD). Perfusion-echocardiography showing normal (B) and defective (C) lateral LV wall perfusion corresponding to an open (B) or occluded LCX (C). (C, inset) Note the concomitant ST-segment elevation. (D) Representative infarcted pig heart 2 weeks after LCX occlusion (black arrow, infarcted area). (E) Triphenyltetrazolium chloride (TTC)-stained, mid-ventricular section demonstrating scar formation 14 weeks after MI. (F–I) Decreased LV function as assessed by echocardiography (EF, ejection fraction) and LV catheterization (+dp/dt) as well as LV dilation (EDD, enddiastolic diameter) and LV hypertrophy (HW/BW, heart weight/body weight) compared to sham operated pigs (n=13) 14 weeks after MI. *P<0.05 HF (n=23) vs. sham. Data are presented as mean±SEM. Sham pigs received a cardiac catheterization procedure without occlusion of the LCX.
Retrograde coronary venous AAV9-S100A1 gene delivery enhances S100A1 expression levels in failing hearts
Two weeks after MI, pigs (n=32) were randomized to saline (n=14), control virus (AAV9-luc, n=9) and S100A1 gene-based treatment (n=9). The AAV9-S100A1 vector carrying the human S100A1 cDNA under the control of a cardiomyocyte-specific promoter (CMV-MLC) was delivered at a dosage of 1.5×1013 total virus particles (tvp) per animal (~5×1011 tvp/kg), targeting non-infarcted anterior and septal LV myocardium (Figure 2A and B), while AAV9-CMV-MLC2-luc and saline served as control groups. 14 weeks after MI, myocardial S100A1 protein expression was decreased in control treatment groups compared to sham operated, non-infarcted pigs (Figure 2C). S100A1 gene therapy restored expression of the S100A1 protein in targeted non-infarcted LV myocardium (Figure 2D). Assessment of S100A1 protein abundance in extra-cardiac tissues including brain, lung, skeletal muscle and liver 3 months after gene delivery showed unchanged S100A1 protein expression both in control and AAV9-S100A1 groups (Figure S1). Importantly, the predominant presence of luciferase activity in the anterior LV wall verifies the feasibility of cardiac-restricted and targeted gene delivery (Figure 2E) by our minimally invasive, catheter-based delivery technique combined with a cardiomyocyte-specific gene expression vector system.
Figure 2. Cardiac-targeted AAV9-S100A1 gene therapy.

(A) Radioscopic image showing the retroperfusion catheter (1) to deliver AAV’s and the guide-wires in the ACV (2) and in the left LAD (3). (B) Location of the non-infarcted posterior LV wall, which is a non-targeted area (4), and the anterior wall, which is a targeted-area (5) for gene delivery. (C and D) Representative western blots showing decreased myocardial S100A1 expression 14 weeks after MI in HF-luc [48±19%, P<0.05 HF-luc (n=9) vs. sham (n=13)] and reconstituted S100A1 protein expression after AAV9-S100A1 gene delivery [3.2±0.3 times higher, P<0.05 in HF-S100A1 (n=9) vs. HF-luc (n=9)] (D). Equal loading for each tissue was confirmed by calsequestrin (CSQ) staining, a Ca2+ binding protein with unaltered expression in HF. (E) Analysis of luciferase activity of targeted (anterior) and non-targeted (posterior) LV wall in HF-luc myocardium. *P<0.05 vs. posterior LV wall and liver (n=9 for HF-luc myocardial and liver samples). Data are presented as mean±SEM.
S100A1 therapy rescues cardiac function and reverses remodeling in failing pig myocardium in vivo
12 weeks after gene therapy, AAV9-S100A1 treated HF animals showed significant improvement both in systolic and diastolic LV performance compared to HF control groups, demonstrating progressive deterioration of LV function (Figure 3A to D). In line with the long-term improvement of global cardiac function, the elevated heart rate seen in HF control groups was normalized by S100A1 therapy (Figure 3E). Analysis of LV remodeling revealed profound anti-hypertrophic effects after S100A1 gene therapy as reflected by significantly lower adjusted heart weight, smaller end-diastolic LV diameter as well as reversed LV expression of BNP (Figure 3F to H) compared to HF control groups. Increased cardiac contractile function of S100A1-treated HF pigs was maintained with a maximum dosage of β-AR stimulation indicating preserved β-AR signalling, and thus an increased inotropic cardiac reserve compared to HF control groups. Sham pigs showed enhanced inotropic reserve compared to all HF groups, including HF/S100A1 (Table 1).
Figure 3. AAV9-S100A1 gene therapy rescues cardiac function and reverses myocardial remodelling in failing myocardium.

(A–E) 12 weeks after AAV9-S100A1 treatment, failing hearts with reconstituted S100A1 expression (n=9) exhibit restored LV function, compared to HF-saline (n=14) or HF-Luc (n=9). (F–H) S100A1 gene therapy attenuated LV remodelling, which was corroborated by reversed fetal gene activation. †P<0.05 sham vs. HF-saline and HF-Luc, *P<0.05 HF-S100A1 vs. HF-saline and HF-Luc, **P<0.05 sham vs. all other groups. mRNA levels were assessed in the LV anterior wall of 5 animals in each group and normalized to sham. EF, ejection fraction; EDP, enddiastolic pressure; HW/BW, heart weight/body weight; EDD, enddiastolic diameter; BNP, brain natriuretic peptide. Data are presented as mean±SEM.
Table 1.
Preserved cardiac inotropic reserve and blood biomarkers 14 weeks after AAV9-S100A1 and control AAV9 vector treatment (n=13 for sham, n=9 each for HF/luc and HF/S100A1 and n=14 for HF/saline). Leukocyte numbers of 9–15.7/nl reflect normal values in pigs (48).
| Sham | HF-Saline | HF-Luc. | HF-S100A1 | |
|---|---|---|---|---|
| Cardiac inotropic reserve (Dobutamin 100 μg/kg BW): | ||||
| HR (min−1) | 120±7 | 123±3 | 121±3 | 118±4 |
| LV +dP/dtmax (mmHg/s) | 6662±237*, †, ‡ | 5101±137 | 5146±120 | 5897±134*, † |
| LV −dP/dtmin (mmHg/s) | 3326±344* | 2402±140 | 2248±54 | 2846±61*, † |
| LVEDP (mmHg) | 4.7±0.8*, †, ‡ | 9.2±0.8 | 8.8±0.5 | 6.5±0.8*, † |
| LVESP (mmHg) | 125±3.7*, † | 111±2.6 | 115±2.5 | 126±2.6*, † |
| Blood parameters: | ||||
| Sodium (140–160 mmol/l) | 141±0.8 | 142±0.6 | 141±0.8 | 141±0.5 |
| Potassium (3.5–4.5 mmol/l) | 3.9±0.1 | 4.1±0.11 | 3.9±0.12 | 4.0±0.1 |
| Leucocytes (4.1–15.7 nl) | 15.7±2.1 | 15.4±2.2 | 14.0±1.5 | 12.8±1.8 |
| Erythrocytes (6.0–12.5 pl) | 6.9±0.3 | 6.2±0.1 | 6.4±0.2 | 6.1±0.3 |
| Platelets (280–650 nl) | 345±39 | 394±36 | 392±37 | 371±47 |
| Hemoglobin (8.0–13.8 g/dl) | 12.1±0.8 | 10.8±0.6 | 11.3±0.6 | 10.8±0.5 |
| Glucose (80–120 mg/dl) | 128±3.5 | 117±6.8 | 114±3.9 | 116±1.6 |
| Creatinine (0,6–1,7 mg/dl) | 2.03±0.16 | 1.73±0.12 | 1.68±0.12 | 1.58±0.13 |
| Urea (20–53 mg/dl) | 31±1.8 | 29±5.1 | 22±1.6 | 31±2.2 |
| Lipase (15–60 u/l) | 22±3 | 20±2.5 | 16±0.8 | 17±1.2 |
| γ-Glutamyl-Transferase (58–106 U/l) | 82±10 | 71±8 | 73±13 | 71±9 |
| Glutamate-Oxalacetate- Transaminase (GOT) (20–66 U/l) | 46±4 | 51±4 | 43±3 | 58±10 |
P<0.05 vs. HF-Luc,
P<0.05 vs. HF-saline,
P<0.05 vs. HF-S100A1
S100A1 gene therapy restores cardiomyocyte and SR calcium handling in failing myocardium
Isolated cardiomyocytes and SR vesicles from HF control groups, 14 weeks after MI, exhibited decreased systolic Ca2+ transient amplitudes, diastolic Ca2+ overload as well as diminished SR Ca2+ uptake and enhanced Ca2+ leak (Figure 4A–F). In contrast, cardiomyocytes from S100A1-treated myocardium showed significantly higher Ca2+-transient amplitudes and lower diastolic Ca2+ levels than did control HF cardiomyocytes (Figure 4A to D). Assessment of Ca2+ fluxes in isolated LV SR vesicles from AAV9-S100A1-treated, failing myocardium yielded significantly reduced Ca2+-leak and enhanced Ca2+-uptake compared to HF controls (Figure 4E and F). Immunoprecipitation of both RyR2 and SERCA2 from S100A1-treated and control failing myocardium showed significantly enhanced S100A1/RyR2 and S100A1/SERCA2 binding ratios in the treated group (Figure 5A–F). Consistently, SR vesicles from AAV9-S100A1 treated myocardium exhibited significantly lower 3H-ryanodine binding at 150 nM free Ca2+ concentrations ([Ca2+]) than did myocardium from failing control groups, indicating improved diastolic RyR2 closure in the presence of S100A1. Addition of human recombinant S100A1 protein (500 nM) partially reversed abnormal 3H-ryanodine binding in SR vesicles from HF control groups (Figure 5G and H)(20).
Figure 4. AAV9-S100A1 gene therapy rescues SR Ca2+ handling in failing myocardium.

(A) Representative Ca2+-tracings from cardiomyocytes isolated from the anterior LV wall of sham, HF-saline, AAV9-luc (HF-luc) and AAV9-S100A1 (HF-S100A1) treated pig hearts. (B) Light microscopy of isolated pig cardiomyocytes (bar, 100 μm). (C and D) S100A1-treated cardiomyocytes exhibit greater Ca2+-transient amplitude and decreased diastolic Ca2+ levels compared to the HF control group (n=30 cells from 3 different animals in each group). (E and F) Fluo-3 based assessment of Ca2+ leakage and uptake in SR vesicles isolated from LV anterior wall segments reveals improved SR function in AAV9-S100A1 treated failing myocardium compared with HF control groups (n=5 SR preparations from different animals in each group). †P<0.05 sham vs. HF-saline and HF-Luc, *P<0.05 HF-S100A1 vs. HF-saline and HF-Luc, **P<0.05 HF-S100A1 vs. all other groups. Data are presented as mean±SEM.
Figure 5. AAV9-S100A1 gene therapy reverses abnormal S100A1/SR target protein ratios and improves SR Ca2+ handling in failing myocardium.

(A and B) Representative immunoprecipitation (IP) for SERCA2 and RyR2 from sham, saline, AAV9-luc and AAV9-S100A1 treated myocardium and corresponding western blots for co-precipitation of S100A1 protein. (C and D) HF control groups contain significantly lower SERCA2 but equal RyR2 protein levels than sham and S100A1-treated myocardium. SR from 5 different hearts (LV anterior wall) was prepared for each group and IP’s were conducted in duplicate from each SR preparation with equal amounts of protein (300 μg). (E and F) Abnormally low S100A1/SERCA2 and S100A1/RyR2 binding ratios in HF control groups are restored after AAV9-S100A1 treatment. (G) Significantly increased 3[H]-ryanodine binding at 150 nm free Ca2+ in HF control groups indicates greater SR Ca2+ leak compared to sham and HF/S100A1. (H) Addition of human recombinant S100A1 protein to SR vesicles from HF control groups significantly reverses abnormally high 3[H]-ryanodine binding. †P<0.05 sham vs. HF/saline and HF/Luc, *P<0.05 HF/S100A1 vs. HF/saline and HF/Luc. ‡P<0.05 HF/saline and HF/luc + S100A1 protein vs. corresponding HF/saline or HF/luc. n=5 in each group. Internal control of immunoprecipitation is shown in Fig. S3.
Cardiomyocyte-targeted AAV9-S100A1 gene therapy expression reconstitutes energy homeostasis in failing myocardium
Myocardium from HF control groups showed reduced energy homeostasis (Figure 6A and B), while assessment of bioenergetic surrogate markers including high energy phosphate and dinucleotide content revealed significantly higher phosphocreatine/adenosine triphosphate (PC/ATP) and nicotinamide adenine dinucleotide phosphate/nicotinamide adenine dinucleotide (NADH/NAD) ratios in S100A1-treated failing myocardium compared to HF control groups (Figure 6A and B).
Figure 6. AAV9-S100A1 gene therapy restores high energy phosphate and dinucleotide content in failing myocardium.
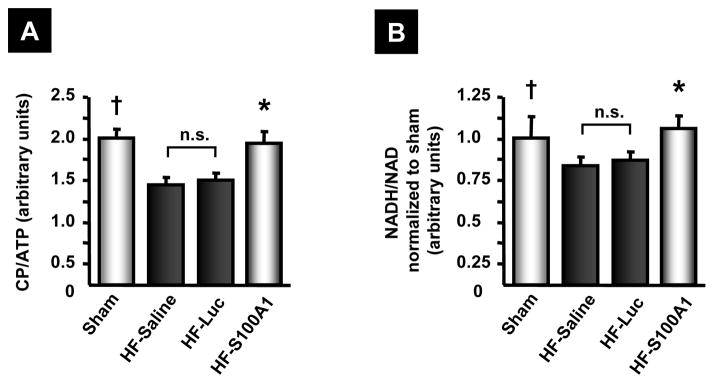
(A and B) Assessment of the bioenergetic marker CP/ATP (A) and NADH/NAD (B) revealed significantly decreased values in HF control groups compared to sham. AAV9-S100A1 gene therapy restores CP/ATP and NADH/NAD ratios to normal, indicative of reconstituted energy homeostasis and redox-state in failing hearts (n=6 different animals in each group, each measurement was carried out in triplicate). †P<0.05 sham vs. HF-saline and HF-Luc, *P<0.05 HF-S100A1 vs. HF-saline and HF-Luc. Data are presented as mean±SEM.
Cardiac-targeted S100A1 gene therapy yields a favorable safety profile
12 weeks after AAV9-S100A1 gene therapy, leukocyte, erythrocyte and platelet counts as well as hemoglobin concentrations were similar in sham and HF animals whether treated with saline, AAV9-luc or AAV9-S100A1 (Table 1). Accordingly, in all groups, sodium, potassium and glucose blood concentrations were similar and pancreas enzymes, kidney retention parameters, and serum liver enzymes were within the physiological normal range for pigs (Table 1).
DISCUSSION
S100A1 controls intracellular Ca2+ cycling by increasing SERCA2a activity and regulating the open probability of the RyR2 causing a reduced diastolic SR Ca2+ leak and increased systolic contractility of the cardiomyocyte in various species including human (11,30). Concurrently, S100A1 enhances ATP-generation via mitochondrial F1-ATPase. This understanding together with the evolution of safe and efficient gene transfer technologies that overcome previous barriers such as the lack of gene delivery efficiency and use of invasive techniques provide a rational for the use of S100A1 gene therapy to treat HF (11,19,31,32). Our data show long-term therapeutic effectiveness and a favorable safety profile of AAV9-S100A1 gene therapy in a preclinical large animal HF model using a clinically relevant catheter-based gene delivery approach.
Significant insight into the molecular and cellular basis of S100A1 cardiovascular biology and therapeutic potential has come from rodent models (11). However, significant differences exist between small animals and humans with regard to safety and technical aspects. Anatomically, the target area for gene transfer is much larger in humans requiring a highly efficient gene delivery technique. Clinical application of the S100A1 transgene should be achieved in a non-surgical, minimal-invasive percutaneous approach using certified catheters. In addition cardiac characteristics differ such as energy homeostasis, the β1/β2-AR ratio and key aspects of excitation-contraction coupling (26,33). The significantly lower heart rate in humans accentuates pulsatile ejection of the blood, and thus affects cardiac diastolic and systolic contractile properties. At the molecular level, this is reflected by a difference of the expression of the predominant sarcomeric protein. β-myosin heavy-chain is predominant in humans showing lower ATPase activity, a reduced maximum velocity but a higher tension-time integral compared to the α-isoform (28). The NCX/SERCA2a ratio is significantly increased in humans and large animals compared to rodents being highly relevant for therapeutic strategies targeting cardiomyocytes Ca2+ cycling (10,27). Ablation of phospholamban, a protein also involved in cardiomyocyte Ca2+ cycling by regulating SERCA2a activity, did show opposite effects in humans compared to mouse models (34). Therefore, large animal models of HF such as pig, sheep and dog, which more closely approximate human physiology, function and anatomy, are a critical link in translating basic concepts into clinical therapies (26).
As shown in human HF, myocardial S100A1 protein expression is diminished in our postischemic pig HF model concomitant with progressive deterioration of cardiac function, an enhanced heart rate and adverse remodeling of the myocardium as well as impaired PC/ATP and NADH/NAD ratios. To correct reduced S100A1 expression in non-infarcted failing myocardium, we employed a minimally invasive, percutaneous gene delivery technique developed by Boekstegers et al. (32). We employed an AAV9 vector driving expression of the human S100A1 cDNA under control of a cardiomyocyte-specific promoter to achieve long-term restoration of S100A1 expression in targeted LV regions (21,35). Control experiments excluded extra-cardiac expression of the therapeutic transgene demonstrating both selectivity and feasibility of the expression system and delivery technique. Of note, biodistribution of AAV vectors shown by human S100A1 cDNA amplified from genomic DNA further corroborates biosafety since only in the liver we detected modest human S100A1 cDNA while in all other non-target organs human S100A1 cDNA was not detectable (Fig. S2).
Genetically targeted correction of reduced myocardial S100A1 expression, a hallmark of advanced HF, not only prevented progressive deterioration but resulted in long-term rescue of global cardiac function. In line with other previous studies, the therapeutic effects of S100A1 are a result of its molecular profile to improve both systolic and diastolic SR Ca2+ handling (9,11,19). We have previously shown that S100A1 binds to and enhances opening of the RyR2 during systole thereby, augmenting SR Ca2+ release independent of Ca2+ load (9,11,14,20). Prevention of diastolic RyR2 leakage by S100A1 together with an increased SERCA2 activity to remove Ca2+ from the cytosol into the SR, causes decreased diastolic cytosolic Ca2+ levels and an increased SR Ca2+ load (9,11,15,17,20,36). Accordingly, isolated LV cardiomyocytes from AAV9-S100A1 treated failing pig hearts exhibited restoration of intracellular Ca2+ cycling reflecting improved function of RyR2 and SERCA2. In particular, our molecular results show improved diastolic RyR2 closure in AAV9-S100A1 treated myocardium and correction of defective systolic RyR2 function by recombinant S100A1 protein in failing control groups.
Our in vivo data are corroborated by the S100A1-induced reversion of Ca2+ cycling defects in isolated LV cardiomyocytes and SR vesicles. These changes recapitulate characteristic abnormalities of human failing cardiomyocytes concerning Ca2+ handling such as a reduced Ca2+ transient amplitude, increased cytosolic diastolic Ca2+ and an increased diastolic Ca2+ leak as well as a reduced PC/ATP ratio consistent with a previous report by Zhang et al using a similar HF model (29,37).
The S100A1-mediated long-term improvement of myocardial function was accompanied by improvements in cardiac remodelling, as reflected by a decreased heart-to-body weight ratio. The significant decrease both in end-diastolic LV pressure and diameter in S100A1-treated HF pigs indicates that diminished diastolic wall stress eventually attenuated hypertrophic paracrine signalling and maladaptive growth. The normalized heart rate of S100A1 treated HF pigs suggests abrogated maladaptive β-AR sympathetic overdrive due to improved cardiac output (38). This important effect could not be observed in small animal HF models underscoring the clinical relevance of the pig HF model used here (15,21,33). Consistent with previous reports revealing improved β-AR-dependent contractile reserve in S100A1-treated rodent HF models (20–22), the S100A1-mediated increase in global contractile function was preserved under stimulation with the β-adrenergic agonist dobutamine in failing pig hearts.
From landmark large clinical trials of current standard HF therapy such as β-blockers, ACE-inhibitors and aldosterone antagonists aiming to limit the progress of the disease, we learned that ejection fraction of patients with severe HF might slightly increase showing that cardiac function cannot be rescued by current conventional drug therapy and rendering a large percentage of highly symptomatic patients with dyspnoe (1–8). Combining current standard of care with S100A1 gene therapy may translate into clinical benefit of patients with HF since S100A1 significantly increases cardiac contractile function. Importantly, therapeutic effects of S100A1 gene therapy were preserved under β-blocker treatment using metoprolol offering the possibility of ameliorated cardiac reverse remodelling, inotropic action and antiarrhythmic effects (21).
Inotropic therapeutic interventions in HF based on manipulation of cAMP-dependent signaling acutely increase cardiac function but show long-term detrimental consequences on heart rate and energy homeostasis and are therefore controversially discussed (39,40). It is important to point out that S100A1 does not depend on cAMP-dependent protein kinase A (PKA) and calmodulin-dependent kinase (CaMK) signalling (13,14,20). In contrast, long-term S100A1 gene therapy enhanced the PC/ATP ratio, indicative of restored energy flux in failing cardiomyocytes, and improved the NADH/NAD ratio, suggesting restored mitochondrial function (41,42).
This study further demonstrates a clinically relevant, favorable therapeutic window for postischemic S100A1 gene therapy. Patients suffering from cardiac ischemia may not necessarily require immediate molecular-guided intervention but can be treated after the clinical event even after already experiencing cardiac dysfunction. Of note, the body-weight adjusted AAV-vector dosage used in our study matches the range used in preclinical AAV-vector toxicology studies in pigs revealing no signs of toxicity by clinical or histopathological assessment (43). In addition, our study highlights the safety of S100A1 gene therapy showing cardiac-restriced biodistribution of the S100A1 transgene without adverse effects on organs such as blood, kidney, pancreas, liver and others up to 3 months after treatment.
As a final step before clinical application, our translational study shows long-term therapeutic efficacy together with a favorable safety profile of AAV9-S100A1 gene therapy with a clinically-relevant delivery approach in a preclinical HF model that approximates human cardiovascular physiology and anatomy. Clinical application of S100A1 genetically targeted therapy will have to be tested in a human HF trial and may potentially complement future treatment of end-stage HF.
MATERIALS AND METHODS
All animal procedures and experiments were performed in accordance with the “Guide for the Care and Use of Laboratory Animals” (NIH) and approved by the local “Animal Care and Use Committee” of Baden-Württemberg, Germany.
Animal procedures, echocardiography and catheter-based cardiac gene therapy
Postischemic HF was induced in german farm pigs (Bräunling; mean body weight: 30±4 kg) by temporary (2 hours) occlusion of the proximal LCX with a percutaneous transluminal coronary angioplasty balloon (Boston Scientific). On the day of cardiac gene transfer and at the end of the study (2 and 14 weeks after MI) we performed echocardiography and LV catheter-based hemodynamic measurements with a Philips Sonos 5500 imaging system (Philips) and a 5-F pressure catheter (SPC-350; Millar instruments). To examine LV inotropic reserve dobutamine (100 μg/kg body weight) was administrated via an ear vein. Myocardial perfusion echocardiography was used to verify comparability of area at risk during occlusion of the LCX and was performed similar to described protocols (44–45). Myocardial contrast imaging performed was in a mid short axis view in a harmonic power Doppler mode (Sonos 5500; Philips) using a broad-band transducer (S3) with a mean transmission frequency of 1.2 MHz, a receiving frequency of 3.0 MHz, a mechanical index of 1.2 and a 1:3 systolic trigger rate. 1 cc per minute of a commercially available second generation contrast agent (SonoVue, Bracco) was infused intravenously during image acquisition. Complete myocardial area and the perfusion defect area were traced to allow calculation of their ratio in the mid short axis view.
Myocardial gene transfer of HF pigs (2 weeks post MI) was achieved by selective retroinfusion as described with some modifications (32). Briefly, a 7-F balloon wedge pressure catheter (Arrow) was placed in the anterior cardiac vein (ACV). While the left anterior descending coronary artery (LAD) was occluded distal of the first diagonal branch, with a 3.5×8mm balloon, 3 times each for 3 minutes to stop antegrade blood flow, 1.5×1013 tvp of the AAV9 constructs in 50 cc saline were retrogradely injected in the ACV. Afterwards, the integrity of the LAD was checked by angiography.
AAV9 vector production
High titer vectors were produced with a triple transfection approach of 293T cells in cell stacks (Corning, Munich, Germany), harvested after 48h, purified by filtration and Iodixanol as described before (35).
Western blot analysis and immunoprecipitation
Immunoprecipitation for RyR2 and SERCA2 was carried out with A/G-PLUS-Agarose beads (sc-2003, Santa Cruz Biotechnology) as previously described (20). Co-precipitated S100A1 was stained (Acris #SP5355P, 1:1000) and reprobed for RyR2 (ABR, MA3-916, 1:1000) and SERCA2 (Santa Cruz Biotechnology sc-73022, 1:500) and appropriate secondary antibodies coupled to fluorescent 680 or 800 dyes (1:2000). Western blot was carried out as described using anti-S100A1-Ab (SA 5632, 1:1000, custom-made; detecting the human and the pig isoform of S100A1) (23).
Isolation of Adult Porcine Failung Cardiomyocytes
Adult porcine cardiomyocytes were isolated from myocardial tissue (2×2 cm) of the anterior basal LV wall 12 weeks after S100A1 gene therapy (14 weeks after MI) with a standard enzymatic digestion procedure based on described methods (46). Cardiomyocytes used for Ca2+-measurements were plated at a density of 20,000 cells/cm2 on laminin-coated glass dishes and cultivated as described (20).
Ca2+-transient Analyses of Isolated Adult Failing Pig Cardiomyocytes
Intracellular Ca2+-transients of Fura 2-AM loaded (1.0 μmol/L for 20 minutes at 37°C) adult pig cardiomyocytes were measured two hours following myocyte isolation as described (20,21). Measurements were carried out with an inverse Olympus microscope (IX 70) with a UV filter connected to a monochromator (Polychrome II, T.I.L.L. Photonics) with a biphasic electric pulse at 37°C. Twenty steady-state twitches for each cardiomyocyte at 1 Hz and 2.5 mM [Ca2+]e were averaged and analyzed by T.I.L.L. Vision software (version 4.01) (20). Myocytes were isolated from 3 pigs of each group.
Assessment SR Ca2+ handling and 3H-ryanodine binding
Preparation of SR vesicles from LV anterior wall samples and SR Ca2+ uptake and SR Ca2+ leak was assessed by the use of the fluorescent Ca2+ indicator Fluo-3 salt, as described, with the use of a heated spectrophotometer (15). For Ca2+ uptake measurements, SR vesicles (0.3 mg/ml) were incubated in 0.2 ml of reaction solution (RS) containing 0.15 mM potassium gluconate, 1 mM MgCl2, 0.2 mM EGTA-Ca2+ buffer (free [Ca2+] 0.3 μM), 10 mM NaN3, 20 mM MOPS, pH 7.0 and 10 μM ruthenium red was added to inhibit RyR2 opening. Free [Ca2+] was calculated by the use of MAX Chelator (47). 3[H]-ryanodine binding experiments were performed from SR vesicle preparations prepared from LV anterior wall samples from each group as described (20). Membranes were incubated at 37°C with 6 nM [3H]-ryanodine (Perkin Elmer) in 300 μl of 20 mM PIPES, pH 7.1, 150 mM KCL, 0.5 mM MgCl2, 15 mM NaCl, EGTA 3 mM, 10 mM caffeine, 1% phosphatase inhibitors (Sigma; inhibitor mix I/II), protease inhibitors (1tablet/5 ml) (Roche Applied Science; Mini Complete EDTA free protease inhibitors) and 150 nM free Ca2+. 500 nM human recombinant S100A1 protein was generated and purified as described (20) and added to HF control groups 30 min prior to 3[H]-ryanodine incubation. Nonspecific binding was determined using 1000-fold excess of unlabeled ryanodine.
Measurement of Luciferase Activity
Luciferase reporter activities were determined with the luciferase assay kit from Promega in a luminometer (Lumat LB9501; Berthold) as described (31).
Real-time RT-PCR
Genomic DNA and total RNA from tissue was isolated as described (20,22). Real-time RT-PCR was performed in duplicates with a 1:100 dilution of the cDNA on a MyIQ real time PCR detection system (BioRad) with the SYBR Green PCR master mix (Applied Biosystems). The following GenBank cDNA sequences were used to design oligonucleotide primers to examine expression of porcine BNP (M23596, forward primer 5′-cccgcagtagcatcttcca-3′, reverse primer 5′-ttgctttgaaggggagcag-3′) and human S100A1 (NM006271.1, forward primer 5′-cgatggagaccctcatcaa-3′, reverse primer 5′-tggaagtccacctccccgtc-3′). For normalization, 18S rRNA was used (forward primer 5′-tcaagaacgaaagtcggagg-3′, reverse primer 5′-ggacatctaagggcatcac-3′). PCR conditions were 95°C, 3 min, and 40 cycles of 95°C, 10 sec; 60.5°C, 45 sec. Specificity of PCR products were confirmed by gel electrophoresis.
Measurement of bioenergetic markers
Phosphocreatine (PC) and ATP in the LV anterior wall samples were assessed as described (15,20). NADH and NAD levels were enzymatically quantified with a kit from BioVision (#K337-100) following the manufacturer’s protocol.
Hematology, electrolytes and clinical chemisty
Analyses were carried out according to the International Federation of Clinical Chemistry primary reference procedures for the measurement of catalytic activity of enzymes at 37 degrees Celsius as well as standard methods using indirect potentiometry for electrolytes.
Statistical Analysis
Data are expressed as means ± SEM. Unpaired student’s t test was used when appropriate. To compare means among 3 or more independent groups, one way ANOVA measures were performed for statistical comparisons. A Bonferroni test was applied whenever multiple comparisons were conducted. For statistical analysis, Graph PadPrism software was used and the P-values shown are adjusted in function for the number of comparisons. For all tests, a value of P<0.05 was accepted as statistically significant.
Supplementary Material
Acknowledgments
We thank Renate Eudenbach and the DKFZ vector core production unit for generating high titer AAV vector stocks.
Funding: This work was supported by NIH grants: R01HL92130 and R01HL92130-02S1 (P.M.); P01HL075443, R01HL56205 and R01HL061690 (W.J.K.); Deutsche Forschungsgemeinschaft: 1654/3-2 (O.J.M. and H.A.K.), 562/1-1 (P.M. and S.T.P.); and the Bundesministerium für Bildung und Forschung: 01GU0527 (P.M., O.J.M., P.B., H.A.K.), BioFuture grant (RB).
Footnotes
Author contributions: S.T.P. and P.M. wrote the manuscript. S.T.P., C.S., J.K. J.A.K, P.R, W.J.K., H.A.K., O.J.M. and P.M. designed the experiments. S.T.P, C.S., J.K., R.B., P.B., R.H., S.S., B.L., J.L., C.W. and O.J.M performed the experiments and analyzed the data.
Competing interests: P.M. and H.A.K. have filed a U.S. and an EU patent application on the therapeutic use of the S100A1 protein to treat heart failure.
REFERENCES AND NOTES
- 1.AHA. Heart disease and stroke statistics - 2010 update. Circulation. 2010;21:e1–e170. [Google Scholar]
- 2.McMurray JJ. Clinical practice. Systolic heart failure. N Engl J Med. 2010;362:228–238. doi: 10.1056/NEJMcp0909392. [DOI] [PubMed] [Google Scholar]
- 3.Groenning BA, Nilsson JC, Sondergaard L, Fritz-Hansen T, Larsson HB, Hildebrandt PR. Antiremodeling effects on the left ventricle during beta-blockade with metoprolol in the treatment of chronic heart failure. J Am Coll Cardiol. 2000;36:2072–80. doi: 10.1016/s0735-1097(00)01006-8. [DOI] [PubMed] [Google Scholar]
- 4.The RESOLVED investigators. Effects of metoprolol CR in patients with ischemic and dilated cardiomyopathy: the randomized evaluation of strategies for left ventricular dysfunction pilot study. Circulation. 2000;101:378–84. doi: 10.1161/01.cir.101.4.378. [DOI] [PubMed] [Google Scholar]
- 5.Vizzardi E, D’Aloia A, Giubbini R, Bordonali T, Bugatti S, Pezzali N, Romeo A, Dei Cas A, Metra M, Dei Cas L. Effect of spironolactone on left ventricular ejection fraction and volumes in patients with class I or II heart failure. Am J Cardiol. 2010;106:1292–6. doi: 10.1016/j.amjcard.2010.06.052. [DOI] [PubMed] [Google Scholar]
- 6.Flather MD, Yusuf S, Køber L, Pfeffer M, Hall A, Murray G, Torp-Pedersen C, Ball S, Pogue J, Moyé L, Braunwald E. Long-term ACE-inhibitor therapy in patients with heart failure or left-ventricular dysfunction: a systematic overview of data from individual patients. Lancet. 2000;6:1575–81. doi: 10.1016/s0140-6736(00)02212-1. [DOI] [PubMed] [Google Scholar]
- 7.Greenberg B, Quinones MA, Koilpillai C, Limacher M, Shindler D, Benedict C, Shelton B. Effects of long-term enalapril therapy on cardiac structure and function in patients with left ventricular dysfunction. Results of the SOLVD echocardiography substudy. Circulation. 1995;15:2573–81. doi: 10.1161/01.cir.91.10.2573. [DOI] [PubMed] [Google Scholar]
- 8.Ho KK, Pinsky JL, Kannel WB, Levy D. The epidemiology of heart failure: the Framingham Study. J Am Coll Cardiol. 1993;22:6A–13A. doi: 10.1016/0735-1097(93)90455-a. [DOI] [PubMed] [Google Scholar]
- 9.Pleger ST, Boucher M, Most P, Koch WJ. Targeting myocardial beta-adrenergic receptor signaling and calcium cycling for heart failure gene therapy. J Card Fail. 2007;13:401–414. doi: 10.1016/j.cardfail.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 10.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 11.Kraus C, Rohde D, Weidenhammer C, Qiu G, Pleger ST, Voelkers M, Boerries M, Remppis A, Katus HA, Most P. S100A1 in cardiovascular health and disease: closing the gap between basic science and clinical therapy. J Mol Cell Cardiol. 2009;47:445–455. doi: 10.1016/j.yjmcc.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boerries M, Most P, Gledhill JR, Walker JE, Katus HA, Koch WJ, Aebi U, Schoenenberger CA. Ca2+-dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Mol Cell Biol. 2007;27:4365–4373. doi: 10.1128/MCB.02045-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Most P, Bernotat J, Ehlermann P, Pleger ST, Reppel M, Börries M, Niroomand F, Pieske B, Janssen PM, Eschenhagen T, Karczewski P, Smith GL, Koch WJ, Katus HA, Remppis A. S100A1: a regulator of myocardial contractility. Proc Natl Acad Sci USA. 2001;98:13889–13894. doi: 10.1073/pnas.241393598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Most P, Remppis A, Pleger ST, Löffler E, Ehlermann P, Bernotat J, Kleuss C, Heierhorst J, Ruiz P, Witt H, Karczewski P, Mao L, Rockman HA, Duncan SJ, Katus HA, Koch WJ. Transgenic overexpression of the Ca2+ binding protein S100A1 in the heart leads to increased in vivo myocardial contractile performance. J Biol Chem. 2003;278:33809–33817. doi: 10.1074/jbc.M301788200. [DOI] [PubMed] [Google Scholar]
- 15.Most P, Seifert H, Gao E, Funakoshi H, Völkers M, Heierhorst J, Remppis A, Pleger ST, DeGeorge BR, Jr, Eckhart AD, Feldman AM, Koch WJ. Cardiac S100A1 protein levels determine contractile performance and propensity toward heart failure after myocardial infarction. Circulation. 2006;114:1258–1268. doi: 10.1161/CIRCULATIONAHA.106.622415. [DOI] [PubMed] [Google Scholar]
- 16.Tsoporis JN, Marks A, Zimmer DB, McMahon C, Parker TG. The myocardial protein S100A1 plays a role in the maintenance of normal gene expression in the adult heart. Mol Cell Biochem. 2003;242:27–33. [PubMed] [Google Scholar]
- 17.Völkers M, Loughrey CM, Macquaide N, Remppis A, DeGeorge BR, Jr, Wegner FV, Friedrich O, Fink RH, Koch WJ, Smith GL, Most P. S100A1 decreases calcium spark frequency and alters their spatial characteristics in permeabilized adult ventricular cardiomyocytes. Cell Calcium. 2007;41:135–143. doi: 10.1016/j.ceca.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Gusev K, Ackermann GE, Heizmann CW, Niggli E. Ca(2+) signaling in mouse cardiomyocytes with ablated S100A1 protein. Gen Physiol Biophys. 2009;28:371–383. doi: 10.4149/gpb_2009_04_371. [DOI] [PubMed] [Google Scholar]
- 19.Wright NT, Cannon BR, Zimmer DB, Weber DJ. S100A1: Structure, Function, and Therapeutic Potential. Curr Chem Biol. 2009;3:138–145. doi: 10.2174/187231309788166460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Most P, Pleger ST, Völkers M, Heidt B, Boerries M, Weichenhan D, Löffler E, Janssen PM, Eckhart AD, Martini J, Williams ML, Katus HA, Remppis A, Koch WJ. Cardiac adenoviral S100A1 gene transfer rescues failing myocardium. J Clin Invest. 2004;114:1550–1563. doi: 10.1172/JCI21454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pleger ST, Most P, Boucher M, Soltys S, Chuprun JK, Pleger W, Gao E, Dasgupta A, Rengo G, Remppis A, Katus HA, Eckhart AD, Rabinowitz JE, Koch WJ. Stable myocardial-specific AAV6-S100A1 gene therapy results in chronic functional heart failure rescue. Circulation. 2007;115:2506–2515. doi: 10.1161/CIRCULATIONAHA.106.671701. [DOI] [PubMed] [Google Scholar]
- 22.Pleger ST, Remppis A, Heidt B, Völkers M, Chuprun JK, Kuhn M, Zhou RH, Gao E, Szabo G, Weichenhan D, Müller OJ, Eckhart AD, Katus HA, Koch WJ, Most P. S100A1 gene therapy preserves in vivo cardiac function after myocardial infarction. Mol Ther. 2005;12:1120–1129. doi: 10.1016/j.ymthe.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 23.Remppis A, Greten T, Schafer BW, Hunziker P, Erne P, Katus HA, Heizmann CW. Altered expression of the Ca(2+)-binding protein S100A1 in human cardiomyopathy. Biochim Biophys Acta. 1996;1313:253–257. doi: 10.1016/0167-4889(96)00097-3. [DOI] [PubMed] [Google Scholar]
- 24.Desjardins JF, Pourdjabbar A, Quan A, Leong-Poi H, Teichert-Kuliszewska K, Verma S, Parker TG. Lack of S100A1 in mice confers a gender-dependent hypertensive phenotype and increased mortality after myocardial infarction. Am J Physiol. 2009;296:H1457–1465. doi: 10.1152/ajpheart.00088.2008. [DOI] [PubMed] [Google Scholar]
- 25.Du XJ, Cole TJ, Tenis N, Gao XM, Köntgen F, Kemp BE, Heierhorst J. Impaired cardiac contractility response to hemodynamic stress in S100A1-deficient mice. Mol Cell Biol. 2002;22:2821–2829. doi: 10.1128/MCB.22.8.2821-2829.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dixon JA, Spinale FG. Large animal models of heart failure: a critical link in the translation of basic science to clinical practice. Circ Heart Fail. 2009;2:262–271. doi: 10.1161/CIRCHEARTFAILURE.108.814459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hove-Madsen L, Bers DM. Sarcoplasmic reticulum Ca2+ uptake and thapsigargin sensitivity in permeabilized rabbit and rat ventricular myocytes. Circ Res. 1993;73:820–8. doi: 10.1161/01.res.73.5.820. [DOI] [PubMed] [Google Scholar]
- 28.James J, Zhang Y, Wright K, Witt S, Glascock E, Osinska H, Klevitsky R, Martin L, Yager K, Sanbe A, Robbins J. Transgenic rabbits expressing mutant essential light chain do not develop hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2002;34:873–82. doi: 10.1006/jmcc.2002.2025. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Wilke N, Wang Y, Zhang Y, Wang C, Eijgelshoven MH, Cho YK, Murakami Y, Ugurbil K, Bache RJ, From AH. Functional and bioenergetic consequences of postinfarction left ventricular remodelling in a new porcine model. MRI and 31 P-MRS study. Circulation. 1996;94:1089–1100. doi: 10.1161/01.cir.94.5.1089. [DOI] [PubMed] [Google Scholar]
- 30.Brinks et al, accepted JACC
- 31.Müller OJ, Kaul F, Weitzman MD, Pasqualini R, Arap W, Kleinschmidt JA, Trepel M. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat Biotechnol. 2003;21:1040–1046. doi: 10.1038/nbt856. [DOI] [PubMed] [Google Scholar]
- 32.von Degenfeld G, Raake P, Kupatt C, Lebherz C, Hinkel R, Gildehaus FJ, Münzing W, Kranz A, Waltenberger J, Simoes M, Schwaiger M, Thein E, Boekstegers P. Selective pressure-regulated retroinfusion of fibroblast growth factor-2 into the coronary vein enhances regional myocardial blood flow and function in pigs with chronic myocardial ischemia. J Am Coll Cardiol. 2003;42:1120–1128. doi: 10.1016/s0735-1097(03)00915-x. [DOI] [PubMed] [Google Scholar]
- 33.Yutzey KE, Robbins J. Principles of genetic murine models for cardiac disease. Circulation. 2007;115:792–799. doi: 10.1161/CIRCULATIONAHA.106.682534. [DOI] [PubMed] [Google Scholar]
- 34.Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW, 2nd, MacLennan DH, Kremastinos DT, Kranias EG. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111:869–76. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Müller OJ, Leuchs B, Pleger ST, Grimm D, Franz WM, Katus HA, Kleinschmidt JA. Improved cardiac gene transfer by transcriptional and transductional targeting of adeno-associated viral vectors. Cardiovasc Res. 2006;70:70–78. doi: 10.1016/j.cardiores.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 36.Most P, Koch WJ. Sealing the leak, healing the heart. Nat Med. 2003;9:993–994. doi: 10.1038/nm0803-993. [DOI] [PubMed] [Google Scholar]
- 37.Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, Morgan JP. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70–76. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- 38.Koch WJ, Lefkowitz RJ, Rockman HA. Functional consequences of altering myocardial adrenergic receptor signaling. Annu Rev Physiol. 2000;62:237–260. doi: 10.1146/annurev.physiol.62.1.237. [DOI] [PubMed] [Google Scholar]
- 39.Felker GM, O’Connor CM. Inotropic therapy for heart failure: an evidence-based approach. Am Heart J. 2001;142:393–401. doi: 10.1067/mhj.2001.117606. [DOI] [PubMed] [Google Scholar]
- 40.Shin DD, Brandimarte F, De Luca L, Sabbah HN, Fonarow GC, Filippatos G, Komajda M, Gheorghiade M. Review of current and investigational pharmacologic agents for acute heart failure syndromes. Am J Cardiol. 2007;99:4A–23A. doi: 10.1016/j.amjcard.2006.11.025. [DOI] [PubMed] [Google Scholar]
- 41.Balaban RS. Cardiac Energy Metabolism Homeostasis: Role of Cytosolic Calcium. J Mol Cell Cardiol. 2002;34:1259–1271. doi: 10.1006/jmcc.2002.2082. [DOI] [PubMed] [Google Scholar]
- 42.Ventura-Clapier R, Garnier A, Veksler V. Energy metabolism in heart failure. J Physiol. 2004;555:1–13. doi: 10.1113/jphysiol.2003.055095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hajjar RJ, Zsebo K, Deckelbaum L, Thompson C, Rudy J, Yaroshinsky A, Ly H, Kawase Y, Wagner K, Borow K, Jaski B, London B, Greenberg B, Pauly DF, Patten R, Starling R, Mancini D, Jessup M. Design of a phase 1/2 trial of intracoronary administration of AAV1/SERCA2a in patients with heart failure. J Card Fail. 2008;14:355–367. doi: 10.1016/j.cardfail.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 44.Villanueva FS, Gertz EW, Csikari M, Pulido G, Fisher D, Sklenar J. Detection of coronary artery stenosis with power Doppler imaging. Circulation. 2001;103:2624–2630. doi: 10.1161/01.cir.103.21.2624. [DOI] [PubMed] [Google Scholar]
- 45.Bekeredjian R, Chen S, Frenkel PA, Grayburn PA, Shohet RV. Ultrasound-targeted microbubble destruction can repeatedly direct highly specific plasmid expression to the heart. Circulation. 2003;108:1022–1026. doi: 10.1161/01.CIR.0000084535.35435.AE. [DOI] [PubMed] [Google Scholar]
- 46.Peeters GA, Sanguinetti MC, Eki Y, Konarzewska H, Renlund DG, Karwande SV, Barry WH. Method for isolation of human ventricular myocytes from single endocardial and epicardial biopsies. Am J Physiol. 1995;268:H1757–1764. doi: 10.1152/ajpheart.1995.268.4.H1757. [DOI] [PubMed] [Google Scholar]
- 47.http://www.stanford.edu/~cpatton/maxc.html
- 48.http://www.nal.usda.gov/awic/pubs/swine/swine.htm
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.