

Abstract
Mammalian development requires cytosine methylation, a heritable epigenetic mark of cellular memory believed to maintain a cell’s unique gene expression pattern. However, it remains unclear how dynamic DNA methylation relates to cell-type specific gene expression and animal development. Here, by mapping base resolution methylomes in 17 adult mouse tissues at shallow coverage, we identify 302,864 tissue-specific differentially methylated regions (tsDMRs) and estimate that >6.7% of the mouse genome is variably methylated. Supporting a prominent role for DNA methylation in gene regulation, most tsDMRs occur at distal cis-regulatory elements. Surprisingly, some tsDMRs mark enhancers dormant in adult tissues but active in embryonic development. These “vestigial” enhancers are hypomethylated and lack active histone modifications in adult tissue, but nevertheless exhibit activity during embryonic development. Our results provide new insights into the role of DNA methylation at tissue-specific enhancers and suggest that epigenetic memory of embryonic development may be retained in adult tissues.
Introduction
DNA methylation is a heritable epigenetic mark that is critical for mammalian development1–5. The DNA methylation state at promoters of many developmental regulators such as Pou5f1/Oct4 is correlated with stable silencing of these genes during development, and overcoming this barrier is believed to be a main step during cellular reprogramming1,2,6,7. Recent genome-wide analysis of DNA methylation in human cells has revealed a widespread distribution of this epigenetic mark, and paradoxically hypermethylation at gene bodies of actively transcribed genes8,9. More recently, hypomethylation has also been found at active enhancers9,10. Together, these studies have underscored the diverse roles DNA methylation plays in gene regulation, and the need for systematic mapping and characterization of DNA methylomes in different tissues and cell types.
The DNA methylome is remodeled extensively during mammalian development and in different tissue lineages11,12. Previous studies, employing mainly technologies that examine a subset of the genome, have revealed many tissue-specific differentially methylated regions (tsDMRs)13. These regions are located in intergenic sequences and appear to be most variably methylated in cancer cells14. However, due to limited resolution and the absence of functional annotation data, the identity and biological role of these previously identified tsDMRs remains unclear. Recent progress in functional annotation of the mouse genome provides an opportunity to characterize tsDMRs and link dynamic methylation to gene expression patterns in different tissues. Further, advances in base resolution analysis of DNA methylomes permit a more comprehensive definition of tissue specific tsDMRs in the genome.
To explore the epigenetic variation of normal tissues, we performed shallow sampling of the methylomes of 17 mouse tissues spanning all three germ layers and extra-embryonic placenta. Given this limited set of tissues and sequencing depth, we estimate that tsDMRs span at least 6.7% of the genome. tsDMRs are short regions that are hypomethylated in a small number of tissues, and are predominantly localized to regulatory elements. Furthermore, we uncover a class of tsDMRs inactive in adult tissues but active during development, suggesting that some enhancers retain an epigenetic memory of their developmental history, and can be identified in adult cells by virtue of their DNA hypomethylation status.
Results
Whole genome bisulfite sequencing of 17 mouse tissues
To create unbiased genome-wide maps of DNA methylation, we performed whole genome bisulfite sequencing in 17 tissues spanning all three germ layers and extra-embryonic placenta derived from a single pregnant female mouse. We sequenced to an average depth of 8.2-fold genome coverage per tissue (Supplementary Fig. 1a), spanning on average 79.7% of the CpG dinucleotides in the mouse genome (Supplementary Fig. 1b). Sequencing of control unmethylated lambda DNA spiked into each sampled verified efficient bisulfite conversion, averaging 99.4%.
Visual examination reveals that somatic methylomes appear concordant on a >100kb scale (Fig. 1a): in general, the genome is highly methylated, with occasional drops in methylation corresponding to CpG islands. For example, the CpG-rich HOXA locus, as well as the CpG-island containing promoters for Skap2 and Evx1, are uniformly demethylated in most somatic tissues compared to surrounding regions. To examine the global distribution of DNA methylation, we partitioned the genome into 10-kb bins and quantified CpG methylation (mCG) (see Methods) (Fig. 1b). Confirming our visual observations, somatic methylomes are highly methylated (average median: 78.3% mCG), with blood and ectodermal cells containing the greatest abundance of mCG. In contrast, extra-embryonic placental tissue is globally hypomethylated (median: 45.4% mCG). These results are consistent with previous observations of the global abundance of DNA methylation in somatic tissues15 and hypomethylation in placenta16,17.
Figure 1. Distinct tissue-specific methylomes.

(a) UCSC Genome Browser snapshot of DNA methylomes, derived from tissues of a single mouse, near the HoxA locus. Each track spans %mCG values between 0% and 100%. Tracks are colored by tissue type, as determined by the clustering in (c). (b) Boxplots of the %mCG distributions for each tissue, calculated from non-overlapping 10-kb bins spanning the mouse genome. Boxplot edges indicate the 25th and 75th percentiles, and whiskers indicate non-outlier extremes. (c) Dendrogram constructed from 1-kb regions exhibiting significant tissue-specific methylation (p = 0.001, χ2 test). Distance is measured as 1 − Pearson correlation coefficient. (d) Global abundance of methylation in non-CG context, expressed as the difference between the fraction of non-CG cytosines that are methylated and the bisulfite non-conversion rate. (e) Abundance of large domains bearing low (L), medium (M), or high (H) methylation, as determined by hidden Markov models independently trained on each tissue.
Since DNA methylation patterns may store memory of lineage specification and differentiation, we expected methylomes of different tissues to cluster based on germ layer. Indeed, clustering analysis partitions the 17 tissues into four distinct lineage-related groups (Fig. 1c): blood-producing tissues (spleen, thymus, bone marrow), endoderm-derived tissues (colon, intestine, pancreas, stomach, liver), mostly mesodermal-derived tissues (heart, lung, kidney, uterus, skin), and ectoderm-derived tissues (cerebellum, cortex, olfactory bulb). Interestingly, in addition to being derived from the mesoderm, the kidney and uterus belong to the urogenital system, which develops together from the intermediate mesoderm18. In agreement with this developmental similarity, the methylomes from kidney and uterus cluster closest to each other. These results confirm that the DNA methylome contains cell type specific and lineage-specific information.
Recent genome-wide bisulfite sequencing experiments have unveiled two epigenetic phenomena of unknown biological significance: non-CG methylation and partially methylated domains9,19 (PMDs). The diversity of the tissues examined here offers an opportunity to explore the tissue-specificity of these events. Besides methylation in CpG context, the methylomes of ES cells9, neural progenitors cells10, and cortex tissue20 have recently been shown to exist in non-CG context. We observe that non-CG methylation is exclusively enriched in ectoderm-derived cells (Fig. 1d), is globally elevated in ectodermal tissues relative to other somatic tissues (Supplementary Fig. 1c–d), and overlaps significantly (pCHG < 1E-15, pCHH < 1E-15, normal distribution; random are normally distributed, Shapiro-Wilk test) (Supplementary Fig. 1e) with regulatory elements (Supplementary Fig. 1f). PMDs are large genomic regions (>10kb) of depleted DNA methylation (<70% mCG) typically found in somatic cell lines9,21 and tumor cells22. Using a hidden Markov model (HMM) approach to segment each tissue into methylation domains (see Methods), we indeed observe large domains of intermediate methylation (Supplementary Fig. 2a–b) that span a significantly (p = 0.0036, Wilcoxon) greater percentage of non-ectodermal somatic tissues (average 67.7%) than ectodermal tissues (average 36.4%) (Fig. 1e). These regions overlap in non-ectodermal tissues significantly more often than expected by chance (p < 1E-15, Wilcoxon) (Supplementary Fig. 2c–e). Consistent with previous observations22, these pre-PMDs contain more DNA methylation than classically defined PMDs9, but like PMDs they overlap extensively with lamina-associated23 and late-replicating24 domains (Supplementary Fig. 2f).
Identification of tissue-specific differentially methylated regions (tsDMRs)
The above analysis suggests that tissue-specific DNA methylation patterns reflect cell lineage identity. To comprehensively identify tsDMRs, we devised a χ2-based statistic to capture tissue-specific DNA methylation in CpG context, and employed an HMM on this statistic to segment the genome (Fig. 2a) (see Methods). This analysis excluded the globally hypomethylated placenta. Visual inspection indicates that tsDMRs are distributed throughout the genome as small, discrete segments. For example, near the ubiquitously expressed Wipf2 gene, we identify eight tsDMRs, each hypomethylated in a unique subset of tissues (Fig. 2b). Interestingly, Wipf2 is most highly expressed in brain tissue, and several tsDMRs (sites 2/3/7) are specifically hypomethylated in ectodermal tissue, raising the possibility that these tsDMRs might be involved in regulation of Wipf2.
Figure 2. Identification of tissue-specific methylated regions.

(a) UCSC Genome Browser snapshot of tsDMRs identified by HMM segmentation of the Chi-squared statistic for methylation variation (top two tracks). The tissue-specificity of highlighted tsDMRs is indicated above. (b) Quantification of DNA methylation abundance at tsDMRs from (a). (c) A hidden Markov model was used to segment the mouse genome into regions of low (L, grey), medium (M, white), and high (H, red) tissue-specificity of DNA methylation. Shown is the fraction of the genome spanned by these regions, with the H group denoting tsDMRs. (d) The size distribution of tsDMRs (H, red) and non-tsDMRs (L, grey). Boxplot edges indicate the 25th and 75th percentiles, and whiskers indicate non-outlier extremes. (e) The distribution of average (left) and standard deviation (right) of %mCG, for tsDMRs (H, red) and non-tsDMRs (L, grey). Dashed lines indicate the median, m.
Altogether, the initial HMM segmentation identified 341,975 potential tsDMRs (high tissue specificity) (Fig. 2c), at a false discovery rate of 3.96E-5. The median length of tsDMRs is 454 bp, significantly shorter than regions having low tissue specificity (non-tsDMRs) (median length = 1290 bp, p < 1E-15, Wilcoxon) (Fig. 2d, Supplementary Fig. 3a). In addition, the average methylation level of tsDMRs across tissue samples (60.4%) is significantly lower than non-tsDMRs (82.8%) (p < 1E-15, Wilcoxon), and the methylation variance is significantly greater in tsDMRs (p < 1E-15, Wilcoxon) (Fig. 2e, Supplementary 3b). These results suggest that tsDMRs are short, tissue-specifically hypomethylated genomic elements in a larger background of large non-tsDMRs that are uniformly highly methylated. To increase specificity, we filtered tsDMRs to those that exhibit high tissue methylation variance and significant tissue-specific entropy compared to non-tsDMRs. This results in 302,864 high-confidence tsDMRs (Supplementary Table 1), which we analyze below. Given our limited selection of tissues and sequencing depth, we estimate that at least 6.7% of the mouse epigenome is variably methylated in adult tissue.
Tissue-specific DMRs are predominantly regulatory elements
Previous analyses of tissue-specific DMRs showed that these regions are largely intergenic and their DNA methylation state is highly variable among different cell types. However, it was unclear what function these regions might carry out13. A recent study examining DNA methylation between mouse embryonic stem cells and neural precursors found that lowly methylated regions demonstrate features of distal regulatory elements and correspond to cell-type specific transcription factor binding sites10. We therefore investigated the possibility that the tsDMRs identified above may correspond to cis-regulatory elements by comparing them to publicly available genomic annotations. Consistent with an epigenetic signature at active enhancers25–27, in liver the histone modifications28 H3K4me1 and H3K27ac (but not promoter-specific H3K4me3) are abundant (log2(ChIP/input) > 1)) and more enriched for liver-specific tsDMRs than non-liver-specific tsDMRs (Fig. 3a, Supplementary Fig. 3b and 4). Both active as well as poised27,29,30 enhancers display hypomethylation (Supplementary Fig. 5a). In addition, tsDMRs are hypomethylated (Fig. 3a, c, Supplementary Fig. 4) and evolutionarily conserved31 (Fig. 3b, Supplementary Fig. 4). Given our current sequencing depth, we find that the vast majority of tsDMRs (average = 74.2%) are found near known distal regulatory elements (enhancers and CTCF) (Fig. 3c, right). Furthermore, as the remaining tsDMRs are highly conserved (Supplementary Fig. 5c) and enriched in sequence motifs of known transcription factors (Supplementary Table 2), they are likely regulatory elements that have escaped current detection methods. Consistent with the expectation that enhancers are highly cell-type specific, the vast majority of tsDMRs are specifically hypomethylated in either one or two tissues (Fig. 3c, left; Supplementary Fig. 5b), and instances of tsDMR hypomethylation in multiple tissues are generally confined to tissues of the same lineage.
Figure 3. Tissue-specific methylated regions are predominantly regulatory elements.

(a) Enrichment of H3K4me1 (top left), H3K4me3 (bottom left), H3K27ac (top right), and DNA methylation (bottom right) in liver cells for liver tsDMRs (red) and tsDMRs identified in other tissues (grey). Bone marrow is immediately below liver in the H3K4me3 plot. (b) Average PhastCons conservation scores relative to tsDMRs. Higher values indicate more conservation. (c) (left) Heatmap representing the abundance of DNA methylation for tsDMRs in all tissues. Each row indicates a tsDMR, which are grouped into horizontal blocks that represent the tsDMRs of a tissue, with the number of elements in the block displayed in the middle. A given tsDMR may appear more than one block. (right) The percentage of tsDMRs in each block within 500bp of promoters, distal regulatory elements (enhancers and CTCF binding sites), genic regions, and intergenic regions. The genome-wide average is indicated below.
A recent report documented that evolutionary conservation of gene expression levels in mammals depends on tissue type, with tissues of the nervous system displaying the most conservation32. In agreement with this observation, we find that evolutionary conservation of both promoter-proximal and promoter-distal tsDMRs (Fig. 4a–b) is also dependent on cell lineage, with ectodermal tissues being most conserved, followed by mesodermal tissues. Notably, distal-regulatory elements of the uterus are significantly more conserved than those of other mesodermal tissues (pheart < 1E-15, pkidney < 1E-15, pskin < 1E-15, Wilcoxon test on all PhastCons scores within 100bp to tsDMR CpG), which attests to the evolutionary importance of the uterus.
Figure 4. Tissue-specific conservation of regulatory elements.
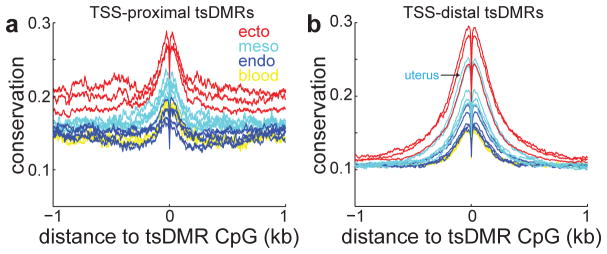
Average PhastCons conservation scores relative to tsDMRs that are (a) proximal (within 2.5-kb) or (b) distal (beyond 2.5-kb) to annotated transcription start sites (TSS). Higher values indicate more conservation.
To further show that tsDMRs are regulatory sequences, we examined them for enrichment of transcription factor binding motifs33. Indeed, consensus motifs for known lineage specific master regulators are significantly enriched at tsDMRs found in the specific tissues (Fig. 5, Supplementary Fig. 4). For example, motifs for the hematapoietic transcription factors SPI1/PU.1and RUNX1 are specifically enriched in tsDMRs found in blood-producing organs, the endodermal forkhead transcription factor FOXA1 is most enriched in tsDMRs found in the endodermal tissues, and the neuronal differentiation factors NEUROD1 and MEF2A are significantly enriched in tsDMRs found in the ectodermal tissues. The above results indicate that the vast majority of tissue-specific DMRs correspond to cis-regulatory elements in each tissue. Similar analysis reveals that regions of intermediate tissue-specific DNA methylation exhibit weaker enrichment of active chromatin, evolutionary conservation, distal regulatory elements, and tissue-specific motifs (Supplementary Fig. 3c–f).
Figure 5. Transcription factor binding motif enrichment near tsDMRs.

(a) Heatmap representing the enrichment of transcription factor binding motifs for the tsDMRs identified in each tissue. Each row represents a motif, and the corresponding transcription factors for selected motifs are labeled on the right. (b) Predicted regulatory elements in heart (p300 binding sites (grey), chromatin-predicted enhancers (blue), tsDMRs (red)) were overlapped, and the density of known tissue-specific motifs (EWS, FOXO1, GATA4, JUN, MEF2A, NF1, SOX6, STAT3, TEAD4) relative to these aligned intersected sites is displayed. (c) The cumulative density of graphs in (b) with respect to the absolute distance to the predicted regulatory element. The horizontal dashed line indicates 50% cumulative density, and vertical dotted lines indicate the resolution in base pairs of each set of predicted regulatory elements to achieve 50% cumulative density.
To assess the resolution of tsDMRs in predicting enhancers, we compared to approaches involving ChIP-Seq of histone modifications25,26,28 or the transcriptional co-activator p30025,26,34. In heart, we identified a common set of regulatory elements overlapped by all methods, and searched for motifs on overlapped loci with factors specific to the tissue. We find that while all methods exhibit a clustering of tissue-specific motifs near the predicted site of a regulatory element, p300 binding sites and tsDMRs exhibited the highest degree of clustering while enhancers predicted by chromatin modifications demonstrated the least (Fig. 5b). To more precisely quantify resolution, we calculated the cumulative distribution of distance between tissue-specific motif occurrences and predicted regulatory sites for distances less than 500 bp. We then define d50 as the distance such that 50% of motif occurrences are within the set of predicted regulatory sites. Thus, small values of d50 indicate higher resolution, with 250 bp indicative of a null model. In heart, d50 values for p300 binding sites (189 bp) and tsDMRs (191 bp) are clearly separated from those for chromatin-predicted enhancers (243 bp) (Fig. 5c). These results suggest that identifying tsDMRs is an alternative method to define putative regulatory elements at high resolution in vivo. However, identifying enhancers using tsDMRs requires the presence of CpG sequences, thus precluding the identification of enhancers with no CpGs (Supplementary Fig. 6c) (see Supplemental Text). Future work using higher depth will be required to identify tsDMRs at CpG-poor regions and to assess their genomic function.
Some tsDMRs correspond to dormant developmental enhancers
While many tsDMRs identified in adult tissues are marked by H3K4me1 and H3K27ac in the same tissue (Fig. 3a), a closer examination reveals a gradient of active chromatin with some tsDMRs exhibiting an inactive chromatin state (Fig. 6a). For the 10 tissues with available histone modification ChIP-Seq data28, we therefore used chromatin to partition tsDMRs into two distinct groups: those that contain active chromatin (ADult-Active tsDMR, denoted AD-A tsDMRs) and those that do not (ADult-Inactive tsDMR, denoted AD-I tsDMRs) (Fig. 6b) (see Methods) (Supplementary Table 3). Both groups of enhancers are depleted of DNA methylation (Fig. 6b).
Figure 6. Adult tissue-inactive (AD-I) tsDMRs demonstrate features of dormant enhancers.

(a) Heatmap representing the density of H3K4me1 and H3K27ac relative at all tsDMRs identified in heart (left), kidney (middle), and olfactory bulb (right). Also indicated are adult tissue-active (AD-A, grey) tsDMRs and adult tissue-inactive (AD-I, red) tsDMRs. (b) AD-A and AD-I tsDMRs were identified for 10 tissues where histone modification data are publicly available28 (spleen, thymus, bone marrow, intestine, liver, heart, kidney, cerebellum, cortex, olfactory bulb). Shown are the average profiles of H3K4me1 (left), H3K27ac (middle), and DNA methylation (right). Each curve represents the average over a tissue/replicate pair. (c) Heatmap representing the enrichment of transcription factor binding motifs for the AD-I and AD-A tsDMRs of each tissue. Each row represents a motif, and the several rows are labeled on the right. The number of AD-A and AD-I tsDMRs is indicated in parentheses. (d) Heatmap representing the enrichment of GO biological process terms obtained by running the GREAT tool35 on AD-I tsDMRs. (e) Average PhastCons conservation scores for AD-A (grey) and AD-I (red) tsDMRs identified in cerebellum (left), kidney (middle), and heart (right). Higher values indicate more conservation.
Given the lack of active chromatin features at AD-I tsDMRs, we wondered if these regions are potentially false predictions. To test this possibility, we searched for known motifs specific to either AD-A or AD-I tsDMRs in each tissue, reasoning that false predictions would not be enriched for motifs. AD-A-specific motifs are enriched for transcription factors found in differentiated cells, including the liver regulator HNF1 and signal transducer STAT3 (Fig. 6c). Interestingly, AD-I-specific motifs belong to many developmental transcription factors such as the trophoblast differentiation factor EOMES, members of the HOX family, and the neuronal differentiation factor TCF4 (Fig. 6c). These results hold for AD-A and AD-I tsDMRs defined by different cutoffs (Supplementary Fig. 7a–b). Furthermore, not only do AD-I tsDMRs harbor motifs for developmental factors, but their putative targets are also enriched for developmental genes. Using the GREAT tool35 on AD-I tsDMRs (Fig. 6d), we find that more than half (54.2%) of all significantly enriched GO biological process terms were related to development, compared to 15.7% for AD-A tsDMRs. Notably, terms for kidney development including mesoderm morphogenesis and kidney smooth muscle cell differentiation are specific to AD-I tsDMRs found in kidney. These results suggest that AD-I tsDMRs may be enhancers of developmental importance. In other words, these regions may correspond to enhancers that are dormant in adult tissues but active during embryonic development.
To test this hypothesis, we first turned to evolutionary conservation. Consistent with the notion that the regulation of developmental gene expression is highly conserved through evolution36, we find that AD-I tsDMRs in cerebellum and kidney are significantly more conserved than AD-A tsDMRs (Fig. 6e) (pcerebellum < 1E-15, pkidney = 1.6E-127, Wilcoxon test on all PhastCons scores within 100bp to tsDMR CpG). In addition, consistent with a previous study indicating that developmental heart enhancers are less conserved37, we find that AD-I tsDMRs are less conserved than AD-A tsDMRs (Fig. 6e). Next, we focused on AD-I tsDMRs identified in adult cerebellum. Comparing chromatin modifications in adult cerebellum with those in whole brains of E14.5 fetuses38, we find a significant enrichment of active enhancer modifications H3K4me1 (p < 1e-15, Wilcoxon) and H3K27ac (p < 1e-15, Wilcoxon) in fetal tissue compared to adult tissue (Fig. 7a). Similar observations also hold for other tissues of the adult brain (cortex, olfactory bulb) compared to developing brain (Fig. 7a, Supplementary Fig. 8a), and for adult heart compared to developing heart (Fig. 7a): in all cases examined, AD-I tsDMRs in developing tissue were significantly enriched in active enhancer chromatin modifications compared to the same loci in respective adult tissue. In line with this increase in active chromatin modifications, genes near AD-I tsDMRs are transcribed at significantly higher levels in early developmental time-points than in adult tissues (Fig. 7b) (p = 3.3E-18, Wilcoxon). These results are reminiscent of previous observations of counteracting epigenetic states at enhancer/promoter pairs39.
Figure 7. AD-I tsDMRs are active during development.

(a) For AD-I tsDMRs identified in adult cerebellum (left), adult cortex (middle), and adult heart (right), shown is the average enrichment of H3K4me1 (top) and H3K27ac (bottom) in adult tissue (grey) and embryonic day 14.5 tissue (red). Two biological replicates for each sample are shown. (b) AD-A and AD-I tsDMRs were associated with genes within 50-kb. Shown is the distribution of expression for genes associated with AD-A tsDMRs (green) and AD-I tsDMRs (grey) in respective adult tissue. For AD-I tsDMRs for which RNA-Seq data exist at earlier developmental timepoints (cerebellum/cortex/olfactory bulb: brain E14.5; heart: heart E14.5; liver: liver E14.5), also shown is the distribution of expression of AD-I tsDMR-associated genes in developmental tissue (red). P-values, Wilcoxon. (c) Heatmap of the overlap enrichment of AD-I tsDMRs with enhancers predicted in developing embryos (left) and adult cells (right). Enrichment is determined relative to sets of random AD-I tsDMRs. (d) Boxplots of overlap enrichment from (C) of AD-I tsDMRs with enhancers predicted in cells in developing or adult cells. (e) Overlap of p300 binding sites34 identified in embyronic forebrain, midbrain, and limb with AD-I tsDMRs (red) identified in ectodermal tissue (cerebellum, cortex, olfactory bulb) and all other tissue. As a comparison, overlap was also performed against random sets of AD-I tsDMRs (grey). Error bars indicate standard deviation. (f) In vivo reporter assays of enhancer activity for VISTA enhancers mm447 and mm414, as obtained from the VISTA enhancer browser40. (g) UCSC Genome Browser snapshot of AD-I tsDMRs identified in cerebellum (highlighted), which overlaps with an in vivo validated enhancer active in developing mouse midbrain. Also shown are active histone modifications (H3K4me1, H3K4me3, H3K27ac) and DNase I hypersensitivity in developing mouse brains (E14.5, E18.5, adult) and cerebellum. For all boxplots, edges indicate the 25th and 75th percentiles, and whiskers indicate non-outlier extremes.
To further test the hypothesis that AD-I tsDMRs are active in development but dormant in adult tissues, we examined the enrichment of overlap between tsDMRs and enhancers defined by chromatin modifications28 in a panel of samples spanning embryonic stem cells (ES), developing fetuses, and adult tissues (Fig. 7c, Supplementary Fig. 8b). AD-I tsDMRs are generally depleted of enhancers in adult tissues but enriched for enhancers in developing cells. Examining this enrichment statistically, we find that AD-I tsDMRs are significantly more enriched for chromatin-defined enhancers in developmental rather than adult tissue (p = 3.1E-14, Wilcoxon) (Fig. 7d). Notably, AD-I tsDMRs are most enriched for developmental enhancers in the same lineage. For example, enhancers in E14.5 mouse heart are most enriched for AD-I tsDMRs found in adult mouse heart (Supplementary Fig. 8b). Similarly, mouse brain E14.5 enhancers are most enriched at AD-I tsDMRs in cerebellum, cortex, and olfactory bulb. Supporting these observations, comparison of p300 binding sites mapped in E11.5 forebrain and midbrain34 reveals that AD-I tsDMRs in brain are significantly more enriched than other adult tissue (Fig. 7e) (pforebrain p300, ecto AD-I < 1E-15, pmidbrain p300, ecto AD-I < 1E-15, normal distribution; random are normally distributed, Shapiro-Wilk test). These results can also be extended across evolution: mouse cerebellum AD-I tsDMRs that are conserved in human are more likely to show activity in human fetal brain tissue than non-brain tissue (Supplementary Fig. 8d).
To further demonstrate that dormant tsDMRs are active enhancers during embryonic development, we examined developmental mouse enhancers validated by transgenic mouse assays (VISTA database)40. Of the 172 enhancers showing enhancer activity at E11.5 stage of mouse development, 25 (14.5%) are classified as AD-I tsDMRs. For example, enhancer mm447, showing positive in vivo enhancer activity in E11.5 midbrain (Fig. 7f), overlaps with an AD-I tsDMR in adult cerebellum that is only 18.2% methylated (Fig. 7g). This enhancer is within 200-kb to Vax141 and Emx242, two genes encoding critical regulators of brain development that are highly expressed28 in developing E14.5 brain (RPKMVax1 = 2.05, RPKMEmx2 = 13.03) but not adult cerebellum (RPKMVax1 < 0.05, RPKMEmx2 = 0.088). In E14.5 whole brains, mm447 exhibits DNase I hypersensitivity43, but this is lost by E18.5 and remains lost in both whole brains and cerebellum of adults. Consistent with this loss of open chromatin, the enhancer is enriched for the active enhancer modifications H3K4me1 and H3K27ac in E14.5 whole brains but not in adult cerebellum28. These data support a model in which enhancer mm447 is active in E11.5-E14.5 midbrain, but inactive by E18.5 and into adult brain. However, in both embryonic and adult stages, the enhancer remains hypomethylated. A similar trend also holds for another VISTA enhancer mm414. Thus, the observation that some AD-I tsDMRs demonstrate enhancer activities in early embryos in transgenic assays supports the hypothesis that these sequences are embryonic enhancers.
Taken together, the above results support a model in which some enhancers active during development retain an epigenetic memory in adult tissues in the form of DNA hypomethylation. We term these “vestigial” enhancers. Previously, it has been shown that loss of transcription factor binding results in passive “filling in” of methylation, which would presumably restore vacated enhancers to a fully methylated state10,44. Since vestigial enhancers remain hypomethylated while existing in closed chromatin, the nucleosomes occupying these sequences may be incompatible with DNA methylation. Consistent with this hypothesis, H3K27me3 is present at significantly higher levels at vestigial enhancers (AD-I tsDMRs) than AD-A tsDMRs (Supplementary Fig. 8c). Previously, it has been shown that DNA methylation is antagonistic to Polycomb complex mediated histone modifications21,45,46. Although the exact mechanisms for this antagonism are not yet fully understood, it is conceivable that Polycomb complexes may be responsible for hypomethylation at the vestigial enhancers in adult tissues.
A subset of active developmental enhancers become vestigial in adult tissue
An active developmental enhancer can exist in several epigenetic configurations in adult tissue: it can remain active (H3K4me1+/H3K27ac+/mCG−), become inactive (H3K4me1−/H3K27ac−/mCG+), or become vestigial (H3K4me1−/H3K27ac−/mCG−). To further explore vestigial enhancers in the context of development, we examined 57,298 enhancers marked with active chromatin in whole brains of E14.5 mouse embryos28. In adult brain tissue (cerebellum, cortex, olfactory bulb), we find that the majority of these developmentally active enhancers become inactive (losing active chromatin and gaining DNA methylation, average 50.1%) or remain active (average 41.2%), while the remaining subset (average 8.6%) loses active chromatin and retains hypomethylation (Fig. 8a–b, Supplementary Table 4).
Figure 8. Vestigial enhancers across development and strain.

(a) Heatmaps representing the enrichment of chromatin (me1 = H3K4me1, ac = H3K27ac) at predicted enhancers with active chromatin in E14.5 mouse brain that consistently (top) retain or (bottom) lose active chromatin in all adult brain tissue (cerebellum, cortex, olfactory bulb). The DNA methylation status in adult tissues is indicated on the right. (b) The percentage of E14.5 mouse brain enhancers belonging to various epigenetic states in adult brain tissue (cerebellum, cortex, olfactory bulb). E14.5 enhancers were overlapped with adult enhancers (A, light grey) and the remainder were overlapped with adult tsDMRs (D, red). The remaining fraction is labeled as inactive (I, dark grey). (c) Heatmap comparing the enrichment of transcription factor binding motifs for vestigial enhancers in brain tissue t1 with those in brain tissue t2. Each row represents a motif, and the several rows are labeled on the right. (d) (top) Heatmap of the DNA methylation status for various tissues and mouse strains, centered at vestigial enhancers identified in B6 mouse cortex. (bottom) Boxplots comparing B6 cortex vestigial enhancer methylation with that of each tissue/strain. Distance is measured as the absolute difference in methylation of each vestigial enhancer between two tissues. Boxplot edges indicate the 25th and 75th percentiles, and whiskers indicate non-outlier extremes. * indicates p < 1E-3 by Wilcoxon. (e) Average profiles of DNA methylation for cortical tissue from B6, C/129, and 129/C mice, centered at B6 cortex vestigial enhancers.
The establishment of vestigial enhancers during development can either be a stochastic or a regulated event. That vestigial and inactive enhancers both lose active chromatin but one set remains hypomethylated suggests this is a regulated event. Supporting this possibility, different sets of developmental enhancers become vestigial in distinct regions of the adult brain, even though all of these regions are derived from the developmental timepoint tissue (Fig. 8b). In addition, vestigial enhancers from distinct brain regions are enriched for different sets of developmental motifs (Fig. 8c), with cerebellum more enriched for members of the GATA and FOX transcription factor families compared with SOX family enrichment in olfactory bulb.
If the creation of vestigial enhancers is a regulated rather than a stochastic event, then one expectation is that a consistent set of enhancers becomes vestigial in different mice. Indeed, we find that the cortex vestigial enhancers identified in our study (C57BL/6 strain, denoted B6) are also hypomethylated in the cortices of two additional mice20 (Cast/129 strains) (Fig. 8d–e). The consistency of the epigenetic status of vestigial enhancers in diverse strains of mice further underscores the regulated nature of their establishment. The methylomes of these Cast/129 cortices at vestigial enhancers are significantly more similar to B6 cortex than any other B6 tissue, including other B6 non-cortical brain tissue (pCast/129 cortex, B6 olf = 3.29E-4, p129/Cast cortex, B6 olf = 8.06E-6, pCast/129 cortex, B6 cerebellum = 3.31E-57, p129/Cast cortex, B6 cerebellum = 1.29E-62, Wilcoxon) (Fig. 8d). Together, these results support the consistency of vestigial enhancers across different mice of diverse strains, and suggest that the unique epigenetic state of vestigial enhancers is created by a regulated event.
Discussion
DNA methylation dynamics have been characterized in mammalian development and in cancer cells3,6,13,14,19,21,22,47–49. During mammalian development, the genome undergoes two waves of genome-wide erasure50 and re-establishment11. In cancer cells, global hypomethylation and local hypermethylation have been well-documented49,51,52. Despite these general observations of developmental profiles of DNA methylation, it was still unclear how DNA methylomes vary across the wide spectrum of normal tissue types. In profiling the methylomes of a diverse panel of 17 normal adult tissues, we estimate that at least 6.7% of the genome undergoes dynamic methylation in a tissue-specific manner. These tsDMRs are generally tissue-specifically hypomethylated. Most strikingly, these sequences predominantly correspond to distal regulatory elements in the genome.
While DNA hypomethylation has previously been observed at enhancers9,10, the preponderance of tsDMRs corresponding to distal regulatory elements is surprising. These results raise further questions about the relationship between DNA methylation and enhancer activity. Our observation that vestigial enhancers lack DNA methylation in adult tissue but remain inactive indicates that DNA hypomethylation is not sufficient for enhancer activity. However, it is still unclear if DNA hypomethylation is required for enhancer activity. Recent evidence that CTCF binding of methylated DNA precedes demethylation suggests that methylation does not impede transcription factor binding10. Future study will be required to precisely define this relationship within the context of DNA hydroxymethylation and demethylation.
It is now clear that, in addition to reflecting the current transcriptional configuration of a cell25,53, the epigenome can also reflect potential future transcriptional states of a cell through the action of poised enhancers27,29,30. Our observations that a set of enhancers transition from being active during development to being dormant in adult tissue while retaining hypomethylation indicate that, besides reflecting the current cellular state, the epigenome can also reflect a cell’s past history of activities. As each methylome is derived from another during development, and as DNA methylation is faithfully copied during cell division by replication-coupled maintenance methylation54, incomplete erasure of a previous developmental state’s epigenetic information can potentially be passed on to subsequent generations. In this way, the methylome could potentially be used to unravel the developmental decisions made during differentiation.
Methods
Whole genome bisulfite sequencing
Mouse tissues were harvested from a female C57Bl/6 mouse (Charles River) at 14.5 days of pregnancy. Laboratory animal care and use comply with federal regulations approved by the Institutional Animal Care and Use Committee of the University of California, San Diego. The number of sampled tissues was determined by the number collectable tissues, and not on statistical considerations. MethylC-Seq was performed as previously described9,21. Briefly, extracted DNA (DNeasy Kit, Qiagen) was spiked with unmethylated lambda DNA (Promega) at 0.5%, sonicated (Bioruptor, Diagenode), end-repaired, adenylated, and ligated to Illumina TruSeq sequencing adapters. After 2% agarose gel purification to select fragments of size 200–650 bp, samples were subjected to bisulfite conversion (MethylCode, Invitrogen) and PCR amplification with PfuTurbo Cx Hotstart DNA Polymerase (Agilent). After gel purification, libraries were sequenced on an Illumina Hi-Seq 2000. Reads were mapped to the computationally bisulfite-converted mouse genome (mm9) using bowtie55 and PCR duplicates were removed with the Picard tool. Only basecalls with Phred score ≥ 20 were considered for analysis.
External datasets
Previously published mouse methylomes (ES, NPC)10 were downloaded and remapped to allow more direct comparison to the mouse tissues analyzed here. Previously published RNA-Seq and ChIP-Seq data for histone modifications, CTCF, and p30038 were acquired from the mouse ENCODE Project43. CTCF binding sites were defined as in mouse ENCODE Project. Enhancers were predicted using a random forest strategy56 applied to H3K4me1, H3K4me3, and H3K27ac (Supplementary Table 5).
To assess if mouse AD-I tsDMRs are active in fetal human tissue, we used previously published human DNase I hypersensitivity data57 from the Roadmap Epigenomics Project.
PhastCons31 conservation scores from alignments of 29 vertebrate genomes with mouse were acquired from the UCSC Genome Browser58.
Methylomes for Cast/129 and 129/Cast mice were previously published20.
Identifying tissue-specific DNA methylation
For a given genomic interval, let mt be the number of methylated cytosines sequenced and dt be the depth of sequencing for tissue t. To capture the deviation of the methylomes from that expected if all tissues were uniformly methylated, we used a Chi-Squared test statistic. Denote the uniform methylation level in this interval as f = Σmt/Σdt. Then the expected number of methylated cytosines sequenced for each tissue is et = fdt. Thus, Chi-Squared test statistic is χ2 = Σ(mt − et)2/et, with the degrees of freedom equal to one less than the total number of tissues.
To identify tsDMRs, we calculated χ2 values for each CpG unit, defined as three consecutive CpGs, and employed a hidden Markov model (HMM) as implemented by pmtk3 to segment the genome, using methods as previously described59 with several alterations. Briefly, we trained a 4-state HMM, each consisting of a mixture of 2 Gaussians, with the Baun-Welch algorithm. States were estimated using the forward-backward algorithm, with the highest-valued χ2 state denoting a pre-filtered set of tsDMRs. To select for tsDMRs with a large magnitude of tissue methylation variance, we determined the intersection of tissue-specific and non-tissue-specific standard deviation distributions (Fig. 2e, right) and removed those sites with smaller values of this intersection point. The final set of tsDMRs are represented by 3 genomic loci: the left and right CpG boundaries as called by the HMM, along with the central CpG site having the highest χ2 value which represents an estimate of the most tissue-specifically methylated base. To estimate a false discovery rate, we repeated this analysis on ten random permutations of the dataset, each of which consists of random assignment of methylated base-calls within each tissue while maintaining base-level sequencing depth.
Each tsDMR is a genomic region with associated methylation abundances for each tissue. To identify the tissues for which a given tsDMR exhibits differential methylation, we utilized Shannon entropy60. Briefly, for a given tsDMR, we defined the relative methylation of a tissue as pt = Mt/ΣMt, where Mt represents the percent methylation of tissue t. Then, using standard formulations, the methylation entropy of the tsDMR is H = Σ − pt log2(pt), and the categorical tissue specificity is Qt = H − log2(pt). We then identified a global cutoff C such that all tissues with Qt ≥ C are labeled as tissue-specifically methylated. We empirically defined C by comparing the distributions of Qt for tsDMRs and non-tsDMRs: setting C = 8.7 bits, we estimate the false discovery rate to be 0.0193.
Partitioning the genome into methylation domains
To partition each tissue into domains of low (L), medium (M), and high (H) methylation, we split each methylome into non-overlapping 10-kb bins, each represented by the fraction of methylated basecalls in CpG context. For each methylome, we employed the R package RHmm to train a 3-state HMM of univariate Gaussians with 50 random initializations on chromosome 19. This trained model was then applied to all other chromosomes of the tissue. The resulting Viterbi states were then sorted by methylation abundance to yield L/M/H domains.
Assessing abundance of epigenetic modifications
The abundance of DNA methylation was measured as %mCG, the percentage of methylated cytosines in CpG context. The abundance of ChIP-Seq reads in a given genomic region was measured as a log-ratio of ChIP RPKM to input RPKM, each with a pseudocount of 0.05.
Motif analysis
We searched for enrichment of known motifs using the Homer tool33. To search for motifs within a single tissue, we used default parameters with a fragment size for motif searching of 200bp. To search for motifs enriched in one list of loci L1 over another L2, we used the “-bg” parameter, setting the background to L2. The known motifs used in our analysis comes from the Homer tool, and can be found in Supplementary Table 6.
Enrichment of AD-I sites at genomic loci
We randomly sampled the genome to create background sets of AD-I tsDMRs, maintaining the chromosomal distribution and the number of CpGs spanned by the random sites. For a given set of enhancers, enrichment was determined as the number of enhancers overlapping AD-I tsDMRs divided by the average overlap with random AD-I tsDMRs sites.
Identifying AD-A and AD-I tsDMRs
For a given tissue, we defined AD-A tsDMRs as TSS-distal tsDMRs having log2(H3K27ac RPKM/input RPKM) ≥ 1 for either of two biological replicates, corresponding to a 2-fold ChIP enrichment over input. AD-I tsDMRs are defined as TSS-distal tsDMRs having log2(H3K4me1 RPKM/input RPKM) ≤ 0.32 and log2(H3K27ac RPKM/input RPKM) ≤ 0.32 for both biological replicates, corresponding to a maximum of 25% ChIP enrichment over input. In both cases, to reduce false positives, RPKM enrichment is calculated with a pseudocount of 0.5.
Supplementary Material
Acknowledgments
We thank Samantha Kuan, Zhen Ye and Lee Edsall for their assistance in sequencing and initial sequencing reads processing. This work is funded by the Ludwig Institute for Cancer Research and NIH (R01 HG003991, ES017166).
Footnotes
Accession codes
Bisulfite sequencing datasets generated in this study have been deposited to the Gene Expression Omnibus (GEO), accession GSE42836.
Author Contributions
G.C.H., N.R., and F.Y. performed bioinformatic analysis. G.C.H, Y.S., D.F.M., and M.D.D. performed experiments. G.C.H. and B.R. prepared the manuscript.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science. 2010;330:622–7. doi: 10.1126/science.1190614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 3.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 4.Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330:612–6. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okano M, Li E. Genetic analyses of DNA methyltransferase genes in mouse model system. J Nutr. 2002;132:2462S–2465S. doi: 10.1093/jn/132.8.2462S. [DOI] [PubMed] [Google Scholar]
- 6.Meissner A. Epigenetic modifications in pluripotent and differentiated cells. Nat Biotechnol. 2010;28:1079–88. doi: 10.1038/nbt.1684. [DOI] [PubMed] [Google Scholar]
- 7.Polo JM, et al. A Molecular Roadmap of Reprogramming Somatic Cells into iPS Cells. Cell. 2012;151:1617–32. doi: 10.1016/j.cell.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ball MP, et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27:361–8. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stadler MB, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–5. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- 11.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 12.Hemberger M, Dean W, Reik W. Epigenetic dynamics of stem cells and cell lineage commitment: digging Waddington’s canal. Nat Rev Mol Cell Biol. 2009;10:526–37. doi: 10.1038/nrm2727. [DOI] [PubMed] [Google Scholar]
- 13.Irizarry RA, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen KD, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768–75. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ehrlich M, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10:2709–21. doi: 10.1093/nar/10.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novakovic B, et al. DNA methylation-mediated down-regulation of DNA methyltransferase-1 (DNMT1) is coincident with, but not essential for, global hypomethylation in human placenta. J Biol Chem. 2010;285:9583–93. doi: 10.1074/jbc.M109.064956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schroeder DI, et al. The human placenta methylome. Proc Natl Acad Sci U S A. 2013;110:6037–42. doi: 10.1073/pnas.1215145110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilbert S. Developmental Biology. Sinauer Associates; Sunderland, MA: 2000. [Google Scholar]
- 19.Lister R, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie W, et al. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012;148:816–31. doi: 10.1016/j.cell.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hon GC, et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2011;22:246–58. doi: 10.1101/gr.125872.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berman BP, et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet. 2011;44:40–6. doi: 10.1038/ng.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peric-Hupkes D, et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell. 2010;38:603–13. doi: 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hiratani I, et al. Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res. 2010;20:155–69. doi: 10.1101/gr.099796.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heintzman ND, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–12. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heintzman ND, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–8. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 27.Rada-Iglesias A, et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2010;470:279–83. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen Y, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–20. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hawkins RD, et al. Dynamic chromatin states in human ES cells reveal potential regulatory sequences and genes involved in pluripotency. Cell Res. 2011;21:1393–409. doi: 10.1038/cr.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Creyghton MP, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–6. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siepel A, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–50. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brawand D, et al. The evolution of gene expression levels in mammalian organs. Nature. 2011;478:343–8. doi: 10.1038/nature10532. [DOI] [PubMed] [Google Scholar]
- 33.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Visel A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–8. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McLean CY, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reichert H. Evolutionary conservation of mechanisms for neural regionalization, proliferation and interconnection in brain development. Biol Lett. 2009;5:112–6. doi: 10.1098/rsbl.2008.0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blow MJ, et al. ChIP-Seq identification of weakly conserved heart enhancers. Nat Genet. 2010;42:806–10. doi: 10.1038/ng.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen Y, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 488:116–20. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taberlay PC, et al. Polycomb-repressed genes have permissive enhancers that initiate reprogramming. Cell. 2011;147:1283–94. doi: 10.1016/j.cell.2011.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35:D88–92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hallonet M, Hollemann T, Pieler T, Gruss P. Vax1, a novel homeobox-containing gene, directs development of the basal forebrain and visual system. Genes Dev. 1999;13:3106–14. doi: 10.1101/gad.13.23.3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muzio L, et al. Emx2 and Pax6 control regionalization of the pre-neuronogenic cortical primordium. Cereb Cortex. 2002;12:129–39. doi: 10.1093/cercor/12.2.129. [DOI] [PubMed] [Google Scholar]
- 43.A user’s guide to the encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011;9:e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thurman RE, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu H, et al. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science. 2010;329:444–8. doi: 10.1126/science.1190485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brinkman AB, et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res. 2012;22:1128–38. doi: 10.1101/gr.133728.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bocker MT, et al. Genome-wide promoter DNA methylation dynamics of human hematopoietic progenitor cells during differentiation and aging. Blood. 2011;117:e182–9. doi: 10.1182/blood-2011-01-331926. [DOI] [PubMed] [Google Scholar]
- 48.Doi A, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–3. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- 50.Monk M, Boubelik M, Lehnert S. Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Development. 1987;99:371–82. doi: 10.1242/dev.99.3.371. [DOI] [PubMed] [Google Scholar]
- 51.Gama-Sosa MA, et al. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983;11:6883–94. doi: 10.1093/nar/11.19.6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herman JG, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci U S A. 1994;91:9700–4. doi: 10.1073/pnas.91.21.9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dunham I, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bestor T, Laudano A, Mattaliano R, Ingram V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J Mol Biol. 1988;203:971–83. doi: 10.1016/0022-2836(88)90122-2. [DOI] [PubMed] [Google Scholar]
- 55.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rajagopal N, et al. RFECS: A Random-Forest Based Algorithm for Enhancer Identification from Chromatin State. PLoS Comput Biol. 2013;9:e1002968. doi: 10.1371/journal.pcbi.1002968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maurano MT, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–5. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meyer LR, et al. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dixon JR, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 485:376–80. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schug J, et al. Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol. 2005;6:R33. doi: 10.1186/gb-2005-6-4-r33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.