

Abstract
Protein characterization using top-down approaches emerged with advances in high-resolution mass spectrometers and increased diversity of available activation modes: collision induced dissociation (CID), infrared multiphoton dissociation (IRMPD) electron capture dissociation (ECD) and electron transfer dissociation (ETD). Nevertheless, top-down approaches are still rarely used for glycoproteins. Hence, this work summarized the capacity of top-down approaches to improve sequence coverage and glycosylation site assignment on the glycoprotein Ribonuclease B (RNase B). The glycan effect on the protein fragmentation pattern was also investigated by comparing the fragmentation patterns of RNase B and its non-glycosylated analog RNase A. The experiments were performed on a Bruker 12-T-Qh/FTICR SolariX mass spectrometer using vibrational (CID/IRMPD) and radical activation (ECD/ETD) with/without pre- or post-activation (IRMPD or CID, respectively).
The several activation modes yielded complementary sequence information. The radical activation modes yielded the most extensive sequence coverage that was slightly improved after a CID pre-dissociation-activation event. The combination of the data made it possible to obtain 90% final sequence coverage for RNase A and 86% for RNase B. Vibrational and radical activation modes showed high retention of the complete glycan moiety (>98% for ETD and ECD) facilitating unambiguous assignment of the high-mannose glycosylation site. Moreover, the presence of the high-mannose glycan enhanced fragmentation around the glycosylation site.
Keywords: glycoprotein, high mannose glycan, electron capture dissociation (ECD), electron transfer dissociation (ETD), infrared multiphoton dissociation (IRMPD)
1. Introduction
One of the most common post-translational modifications (PTMs) of proteins is glycosylation, which is reported to occur on over 50% of all proteins [1]. Glycosylation can significantly alter protein conformations and consequently their functional activity in many biological processes, such as molecular recognition, inter- and intracellular signaling, development, immune defense, cell adhesion and division [2,3]. Glycosylation is divided into two classes: (i) N-glycosylation linked to Asn in a consensus sequence Asn-X-Ser/Thr (occasionally Cys), in which X may be any amino acid except Pro [4,5] and (ii) O-glycosylation at Ser or Thr residues. Although there is no such well-defined consensus sequence for O-glycosylation, there is some recent evidence for preferential O-glycosylation effects from neighboring residues, e.g., a Pro residue in the n − 1, n + 1, and/or n + 3 position(s) in relation to the Ser/Thr attachment site (n) without consensus sequence [6]. Each glycosylation site can contain a distribution of glycoforms, resulting in a large heterogeneity. Hence, the full characterization glycoproteins is still a real challenge.
Tandem mass spectrometry (MS/MS) following the bottom-up approach is already a powerful method for glycoproteomics analysis [7,8] while the top-down approach is rarely used [9,10,11,12]. The top-down approach consists in directly analyzing intact glycoprotein ions by MS/MS without prior digestion. This method has numerous advantages: (i) limited need for sample preparation, short analysis time and avoidance of artifacts related to the digestion, (ii) direct information on the molecular mass of the intact protein, (iii) facility to preserve and assign the sites of all PTMs on a specific proteoform [10]. This approach became feasible with the advances of very high resolving power mass spectrometers (e.g., mostly FTICR or Orbitrap) which make it possible to assign MS/MS product ions without ambiguity. In an early demonstration of the effectiveness of delayed extraction for MALDI-TOF MS, Juhasz et al. showed that the glycoforms of intact RNAse B could be clearly resolved [13]. A few studies have also demonstrated the capacity for using newer, even higher resolving power ESI- or MALDI-TOF instruments for what is (sometimes erroneously) called a “top-down” approach for analysis of protein glycoforms, including ca. 150-kDa immunoglobulins, but these measurements have largely been limited to accurate molecular weight profiling of the intact proteoforms. [14,15]. An ESI-Orbitrap ETD study of an IgG provided substantial amino acid sequence information starting from the N- and C-termini but did not include glycan MS/MS site localization [16].
Numerous investigations on glycopeptide characterization already reported the utility of diversity and complementary activation modes such as collision-induced dissociation (CID) [17], infrared-multiphoton dissociation (IRMPD) [18], electron-capture dissociation (ECD) [19] and electron transfer dissociation (ETD) [20].
CID and IRMPD cause vibrational excitation of gas-phase molecular ions and thus yield similar types of product ions (b/y ions) and tend to remove most or all of the glycan from the peptide [8,21]. It should be noted that both resonant and non-resonant CID yield b/y ions although their activation processes differ. The former, which is mostly performed in a quadrupole ion trap, consists to the application, to the end-caps, of a high radio-frequency potential corresponding to the oscillation frequency of the precursor ion. The second mode, mostly performed in a hexapole linear ion trap, consists in the application, to the end-caps, of a low frequency; this results in a simultaneous excitation of all ions in the collision cell. Thus, a richer fragmentation pattern is usually obtained in the non-resonant CID mode. On the other hand, ECD and ETD are radical activation modes, and yield complementary information by causing different types of cleavages to form c/z• product ions and mostly preserve even labile PTMs [8,22,23,24]. Nonetheless, it should be noted that some reports have also shown the capacity of ETD to cleave a few glycan substituents [25]. Another advantage of the radical mode cleavage methods (ECD and ETD) is their capability to offer more extensive protein sequence coverage than the vibrational activation modes (CID/IRMPD) [19]. Nevertheless, improvements were still required to maximize the efficiency of fragmentation and sequence coverage. Hence, ion activation has been combined with ECD and ETD processes (AIECD and AIETD, respectively) for (glyco)peptides during the last decade [26, 27, 28, 29]. Although comparisons have been made and the complementarities of each activation mode have been widely described in the literature for glycopeptides, little information has been reported regarding the fragmentation of intact glycoproteins. Usually, investigations of intact glycoforms have been made solely to achieve information on the molecular mass distributions of the glycoforms of intact glycoproteins, without performing MS/MS experiments directly on the intact glycoprotein [30,31].
In this study, we explored the effects of activation on an intact high-mannose N-linked glycoprotein often used as model for such PTM studies (RNase B) [9,32,33]. Common or new activation modes (non-resonant CID, IRMPD, ECD and ETD with/without pre- or post-activation events) that have so far been used only for glycopeptides were explored to determine their capacities and their complementarities for the top-down approach.
Moreover, this glycoprotein is also a suitable model to study the effects from the presence of a high mannose N-linked glycan on the MS/MS fragmentation pattern because of the ready availability of the corresponding non-glycosylated protein (RNase A). The glycan’s effect on the fragmentation pathway was, therefore, evaluated by comparing the sequence coverages obtained around the glycosylation site of RNase B and around the equivalent position on RNase A.
2. Materials and methods
2.1. Chemical and Sample Preparation
Bovine pancreas ribonuclease A and B (RNase A and B), dithiothreitol (DTT) and ammonium bicarbonate were purchased from Sigma-Aldrich (St. Louis, MO, USA). The disulfide bonds of RNase B and RNase A were reduced by using DTT prior to alkylation of thiols with iodoacetamide. The reduced and alkylated samples were purified using a C-18 reversed-phase micro-column (Zip-Tip™ C-18, Millipore, Billerica, MA, USA) Finally, the protein solutions were analyzed by MS after being diluted to 2 μM with electrospray solvent consisting of 50/50/0.5 H2O/ACN/FA.
2.2. Mass Spectrometry
All experiments were performed with a top-down approach by using a 12-T Qh/FTICR hybrid MS (SolariX, Bruker, Billerica, MA, USA). The ions were generated by Bruker’s nano-spray source operated in the positive mode with a flow rate of 150 nL/min. The ions were transferred through the quadrupole which was operated in the rf-only mode for recording the MS1 spectra and was operated as a mass selective quadrupole, with an isolation window of 2 u, for recording spectra in the MS/MS mode. The ions were accumulated for 0.5 s in the hexapole that served as the collision cell in MS/MS mode. The ions were then transferred to the ICR cell, where they were captured by dynamic trapping. The spectra were acquired over the range m/z 172 – 3000 during a transient for which 1M points provided a mass resolving power around 67,000 (at m/z 800), after FFT processing (total time per scan was 2 s). The external calibration was carried out using NaTFA clusters.
2.2.1. CID and IRMPD
RNase A and B were activated using two different kinds of vibrational activation modes: CID and IRMPD. All CID and IRMPD spectra were acquired for 40 scans, in triplicate. The CID experiments were carried out in the hexapole linear ion trap under non-resonant conditions. The pressure in the hexapole was maintained at 7 × 10−6 mbar using argon as the inert target gas. The excitation voltage was applied from 0 V to 10 V for both RNase A and B.
The IRMPD experiments were performed, in ICR cell, using a CO2 laser (10.6 μm) set at 95% power with irradiation times at 300 ms, 600 ms and 800 ms.
2.2.2. ETD and ECD
Radical activation was performed using ETD in the collision cell or ECD in the ICR cell. ETD and ECD spectra were recorded during 200 scans, in triplicate.
ETD activation was performed using radical negative ions of fluoranthene as reagent. The reagent was produced in the negative chemical ionization (nCI) source operated at 60 °C to sublime the fluoranthene, which was then directed to the nCI chamber that contained methane ions generated using 70 eV ionization energy and emission filament current 3 μA. After an optimization of ETD conditions (Supplemental data S1), the reagent accumulation time in the collision cell was fixed at 30 ms.
For ECD experiments, the typical event sequence was: precursor ions were accumulated in the collision cell, and then transferred through the hexapole ion guide and captured in the ICR cell with trapping conditions described above. Afterwards, the precursor ions were irradiated with electrons for 7 ms with a filament bias voltage 1.5 V and cathode heater current 1.6 A.
2.2.3. Activated Ions on ETD or ECD (AIEXD with X=T or C)
Ion activation in conjunction with the EXD processes (ETD or ECD) was carried out by pre- or post-activation using CID and IRMPD, respectively. These modes are noted as pre-AIEXD(CID) and post-AIEXD(IRMPD) with X = T or C. The resulting spectra were recorded during 200 scans, in triplicate.
Pre-AIETD(CID)
The pre-activation of ions by CID was performed during the introduction of the positive ions of the analyte into the hexapole. Then, the ions were activated by following the classical ETD event described above.
Post-AIETD(IRMPD)
The post-activation by IRMPD consisted of applying a classical ETD event before activating the ions with the CO2 laser in the ICR cell, with the irradiation times set at 300 ms or 600 ms.
Pre-AIECD(CID)
During the pre-activation by CID event, the precursor ions were activated in the hexapole before their transfer into the ICR cell and exposure to the electron beam.
Post-AIECD(IRMPD)
This supplemental excitation can be applied before, simultaneous with, or after the ECD activation [26]. For experiments reported herein, the post-AIECD approach was preferred, in order to preserve the ECD efficiency because the ions could be expected to remain in a position on-axis with respect to the electron beam for the ECD process, without having to consider the impact of ion magnetron motion on ECD [9]. Hence, the event of AIECD using IRMPD was typically initiated by ECD activation of precursor ions in the ICR cell, under conditions similar to those described for activation by ECD only. Then, the precursor ions and resulting ions were irradiated with the CO2 laser for 300 ms or 600 ms.
2.3. Data Analysis and Sequencing Analysis
Data were analyzed and deconvoluted using Data Analysis software (v 4.0, SP2, Bruker) and the SNAP algorithm which allowed the assignment of the monoisotopic molecular mass of single protonated species. Spectra were recorded in triplicate and treated by MS-Stat (free software built by M. Bourgoin, 2010). Only ions obtained in ≥ two of three experiments were used for analysis. Sequence analysis of the deconvoluted and filtered data was performed using BUPID top-down (Boston University Protein Identifier), an in-house developed MS data interpretation software [34]. The product ions were assigned with errors ≤ 5 ppm (A shift of +1.00336 Da (Δ[12C-13C]) could be considered for species with higher molecular mass than 6,000 Da) [35, 36]. Finally, sequence coverage (expressed in %) for RNase A and RNase B was determined as the percent of the residues assigned in each sequence (See supplemental data 2a and 2b, respectively).
3. Results and Discussion
As RNase B is the N-linked glycoprotein containing the same protein backbone as the protein RNase A, the details of the fragmentation observed in the spectra obtained for these biomolecules were closely compared to determine the effects of the PTM, N-linked glycosylation. We investigated fragmentations of RNase A and B by top-down approaches using both vibrational (CID/IRMPD) and radical activation (ECD, ETD) with/without a pre-or post-activation event to explore the following aspects:
sequence coverage and complementary sequence information
glycan structure and glycosylation site
glycan effect on the fragmentation pattern.
As RNase A and B both have a high pI value (9.7) [32] and their common protein amino acid sequence contains numerous basic residues (14), the analysis of each reduced species in the positive ionization mode favored achieving charge states high enough to undergo efficient fragmentations, an aspect known to be especially important for ETD and ECD activations. The ESI-MS of reduced and alkylated RNase B (Figure 1) showed a major distribution at high charge states (z = 11 – 19), facilitating the ETD and ECD processes and yielding the determined mono-isotopic molecular mass 15,354 Da for the most abundant glycoform, GlcNAc2Man5. The MS/MS experiments were performed for this GlcNAc2Man5 glycoform.
Figure 1.

NanoESI-MS spectrum of reduced RNase B. (Sum of 20 scans)
One of the main goals of this investigation was to compare the fragmentation pattern of a protein bearing a glycan chain (RNase B) with the pattern from its non-glycosylated analog (RNase A); to minimize the variables, we deliberately focused on a single charge state and selected a value (z = 16+) which represented what we felt was the best compromise between a high charge state and precursor ion abundance sufficient to produce excellent sensitivity in the MS/MS spectra obtained. Thus, activation was performed on the [M + 16H]16+ species (m/z 885 for RNase A and m/z 961 for RNase B), using similar conditions for CID, IRMPD, ETD and ECD, with or without pre- or post-activation events. It may be noted that an earlier CAD, IRMPD and ECD study had used exclusively the 13+ charge state of RNase A [37]. The S/N achieved with our higher field instrument (12 T vs. 6 T) and our choice of a higher charge state (16+ vs. 13+) allowed us to make more fragment ion assignments, but the overall patterns were consistent. Even so, we must point out that the spectrum of a single charge state has now been recognized to display the sum of products generated from multiple structures that vary in the charge site localizations [38,39].
High mannose structures such as those found on RNase B have been established to be more stable than other kinds of glycan chains such as complex or hybrid chains. Nevertheless, depending on the activation mode used, the results in terms of cleavages of the glycosidic bonds between sugar residues and even the amide bond between Asn and the high mannose glycan could compete with cleavage of peptide bonds. This is especially true in the case of the glycopeptides of RNase B that would be analyzed within a bottom-up approach, even if some steps were taken to minimize such cleavages. However this is not the case in the top-down approach as shown in our data or in the results published by Reid et al. [9]. For example, Adamson et al. [21] who showed that IRMPD, shown previously to preferentially cleave glycosidic linkages rather than peptide linkages [22], when applied to glycopeptides bearing the substantially stable high mannose moiety such as that found in RNase B, induced peptide backbone fragmentation that competed with glycosidic cleavages. Similar conclusions regarding the behaviour of the glycopeptides of RNase B were also drawn for the CID activation mode by Alley et al. [40]. Moreover, RNase B presented a suitable model to compare the fragmentation pattern of a glycoprotein (RNase B) with its corresponding non glycosylated analog (RNase A), and therefore, evaluate the effect of a simple sugar on the fragmentation pattern in such activation modes.
3.1. Sequence Coverage and Complementarities
3.1.1. Classical Activation Modes (CID/IRMPD/ETD/ECD)
The vibrational modes (CID, IRMPD) are known to operate as ergodic processes yielding b/y ions that compete with both glycosidic cleavages that remove complete glycans from glycopeptides and stepwise elimination of monosaccharide residues or other small neutrals [7,21]. As expected, our non-resonant CID experiments performed on [M + 16H]16+ showed exclusively b/y ions (Figure 2) and common neutral losses, e.g., water (− 18 u) and ammonia (− 17 u). Sequence results obtained from spectra recorded under similar CID excitation conditions (excitation voltage set at 8 V) of RNase A and B yielded analogous results in terms of protein sequence coverage (23% and 21%, respectively), ion types and specific fragmentation orientation. The b ions were mostly produced and localized in the middle and toward the C-terminus of the sequence while y ions were less favored and originated primarily from cleavages near the C-terminus. The maximum coverage information was obtained with the excitation voltage set at 6 V for RNase A (25%) and 10 V for RNase B (23%) (data not shown). Up to 6 V for RNase A and up to 10 V for RNase B, use of the higher CID excitation voltage values did not improve the sequence coverage, presumably because the CID occurred in non-resonant mode and facilitated formation of secondary ions, leading to formation of internal ions (ions not included in the sequence coverage maps). IRMPD experiments showed similar results, with poor information in terms of fragment ions and sequence coverage (Figure 2).
Figure 2.

Sequence coverage obtained from CID (collision energy 8 eV, 40 scans), IRMPD (IR time 800 ms, 40 scans), ETD and ECD experiments for reduced (200 scans) (a) RNase A, (b) RNase B. Reproducible sequence coverage was achieved from triplicate analyses.
Contrary to vibrational activation modes, radical activation modes (ETD, ECD) have been shown to cleave protein backbones at N-αC, generating c/z• fragments that provide more extensive sequence coverage and preserve labile PTMs, including glycosylation [19]. As expected, our ETD and ECD spectra (Figure 2) showed extensive amino acid sequence coverage for both RNase A (ETD: 62%, ECD: 64%) and RNase B (ETD: 58%, ECD: 65%) by producing mostly c/z• ions and by preserving the RNase B glycan chain. It should be noted that this extensive sequence coverage observed by ETD could be achieved after optimization of reagent accumulation times (+8%) (See supplemental data S1). In addition to evidence that the radical (ETD, ECD) and vibrational (CID, IRMPD) activation modes led to different ion types (c/z• and b/y, respectively), the results showed that the sequence coverages from these modes was totally different. Vibrational activation modes produced mainly fragmentations oriented in the sequence region that resisted activation by radical modes, i.e., in middle (Y76STMSIT82) and near the C-terminus (G112NPYVP116). Contrary to the important neutral losses of water and ammonia in CID spectra, ETD and ECD spectra showed preferential loss of the carboamidomethyl side chain on the alkylated cysteine (−57.0214 u). Even though ETD and ECD produced similar ions types (c/z•), some differences between them, in terms of sequence coverage and efficiency, were observed (See supplemental data S3). ECD processes produced more extensive ions leading to slightly higher sequence coverage (2 or 7%) from ETD experiments (Figure 2). The resulting fragmentation patterns also showed slightly different distributions of the sequence coverage. The ECD process produced z• ions in the middle (Y76STMSIT82) and in the C-terminal region (Q101ANK104) of the sequence that were not observed in the ETD experiments. ECD also favored the formation of c ions in the N-terminal region of the sequence (A19ASS22), while the ETD process led to fewer complementary c ions in the middle of the sequence (C65KNGQ69). Drastic differences between the ETD and ECD processes were also shown directly in the profiles of the ETD and ECD mass spectra. The charge-reduced species were favored in ETD experiments (Figure S3a) while the number of product ions was larger in the ECD spectra (Figure S3b). The disparities observed between ETD and ECD spectra were presumably related to the following parameters: energy, reaction times and/or pressure (See supplemental data S3).
3.1.2. Activated Ion on EXD (X = T or C)
The activated ion (AI) effect on the ECD and ETD fragmentation patterns of RNase A and B was investigated by performing pre-activation by CID (AIEXD(CID)) and or post-activation by IRMPD (AIEXD(IRMPD)).
Pre-AIEXD(CID)
Figure 3 illustrates the results in terms of sequence coverage and observed product ion types for CID ion activation in ETD/ECD for RNase A and RNase B. An improvement for sequence coverage (+2 – 4%) was found at low excitation voltage (2 V or 4 V), whereas, at higher CID excitation voltage, a reduction of sequence coverage appeared to be accompanied by an abrupt decrease in the formation of c/z• ions. We propose two reasons for this outcome: (1) formation of both fragment ion types (b/y and c/z•) and (2) production of complementary c/z• ions especially at the sequence extremities (Figure 4). The pre-AIETD(CID) results (+2–4%) suggested that at least a part of the ion population had no time to dissipate internal energy before the ETD process. Nevertheless, since the cooling time needed to achieve internal energy equilibrium through cooling collisions (100–250 ms) [41] that is shorter than the ion accumulation time (500 ms), we assume that only the last ions introduced in the collision cell remained activated until the ETD process. The similar results obtained for pre-activation before ECD (+4%) suggested that at least one population of the ions were still activated until their entrance in the ICR cell process, perhaps because the cooling effect during the transfer is limited by the high vacuum and kinetic energy. Although this pre-activation event presented the advantage of slight improvements in the sequence coverage efficiency at low excitation voltage (+2 – 4%), this method should be used with precaution because higher excitation voltage decreased it. Two possible reasons may explain this observation: (1) secondary fragments are favored or (2) precursor ion abundance is decreased. As there was no evidence for internal fragments and the resulting spectra were poorer in terms of product ions, the effect of the precursor ion abundance would appear to be the important factor.
Figure 3.
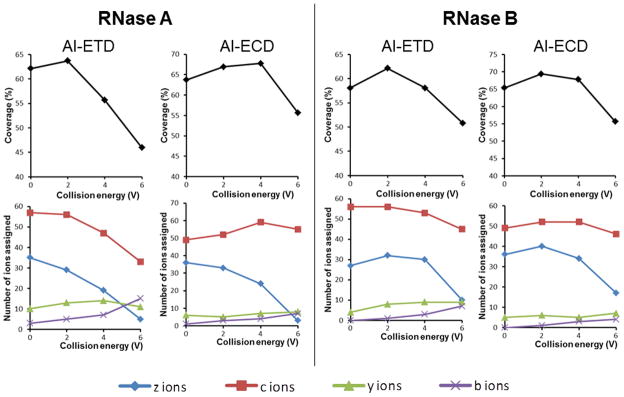
Effect of CID pre-activation event on sequence coverage and observed product ion types for reduced and alkylated RNase A and RNase B. The coverage and number of different ion types obtained from pre-AIETD(CID) and pre-AIECD(CID) experiments at different CID excitation voltages are indicated (Number of common product ions obtained from triplicate analyses).
Figure 4.
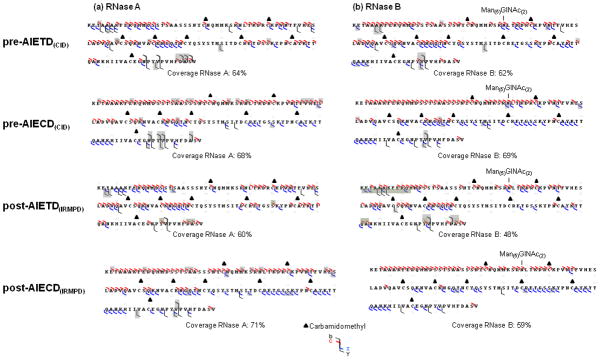
Sequence obtained from pre- or post-activation event after ETD or ECD (CID/IRMPD) AIETD(IRMPD-300ms) for reduced and alkylated (a) RNase A and (b) RNase B. Grey highlights indicated complementary ions observed in these experiments, as compared to those obtained from classical ECD and ETD.
Post-AIEXD(IRMPD)
We also applied post-AIETD(IRMPD) using an IR irradiation time of 300 ms which minimized secondary fragmentations. A decrease of global sequence coverage upon increase in IR irradiation time was evident for both RNase A and B, presumably due to generation of secondary ions (Figure 4). Post-AIETD(IRMPD) sequencing coverage indicated complementary ions, especially a few c ions generated by cleavages around the C-terminus for RNaseA and B and z• ions from cleavages around the N-terminus for RNase B, while c ions were disfavored in this region (grey highlights, Figure 4). Hence, post-AIETD(IRMPD) is still a useful approach to complete the protein sequence information obtained from ETD by producing complementary ions. Nevertheless, as the activation decreased the global sequence coverage, a combination of results obtained +/− post-activation event seems to be advisable. Exploration of post-AIECD(IRMPD) on RNase A and B showed effects slightly different from those obtained after ETD. The sequence coverage obtained after 300 ms irradiation (Figure 4) presented some complementary information described below and yielded improved sequence coverage for RNase A, but lower for RNase B. The complementary ions observed by activating the ECD process by 300 ms of IR irradiation were principally c ions resulting from cleavages near the N-terminus, and z ions formed near the C-terminus for RNase A, resulting in an extensive coverage. RNase B gave different results, in that only z• ions generated near to the C-terminus seemed favored whereas a non-negligible decrease in the coverage was obtained (65% to 59%). In both cases, neither b/y ions nor glycan loss were favored. The peptide backbone cleavage corresponding to formation of c ions near the C-terminus (e.g., c34) of the RNase B glycosylation site was produced with or without activation for RNase B, but appeared only with post-activation for RNase A.
As expected [19], the radical modes (ETD, ECD) yielded different dissociation pathways (c/z• ions) and more extensive sequence coverage than those obtained from vibrational modes (CID, IRMPD) (b/y ions) [19]. Although this substantial difference in terms of sequence coverage could be in part due to the higher number of scans recorded for ETD/ECD/AIEXD spectra, this result can be mostly attributed to the fact that the vibrational mode dissociation is an ergodic process leading to more specific cleavages [42], primarily on the weakest bonds because of randomization of the energy along the peptide backbone as described in the mobile proton model [43,44]. In fact, we are confident that any artifactual contribution due to the number of scans is quite negligible, because we tested in a few conditions the acquisition of CID/IRMPD spectra with scan numbers higher than 40 (40–100) without noting any significant improvements in terms of sequence coverage. The coverage of ETD and ECD was significantly lower for 40 scans than for 200 scans. As ETD and ECD are non-ergodic processes, their fragmentation patterns are more random than those of the CID and IRMPD ergodic processes that more specifically cleave the weakest bonds, and thus we usually needed more scans to achieve sufficient product ion sensitivity in ECD/ETD/AIEXD spectra.
In contrast, the radical activation mode is a non-ergodic process because dissociation happens prior the vibrational energy distribution [19,24,45,46]. These properties yielded extensive cleavage of the protein backbone at N-αC generating c/z• ions and preservation of the glycosylation. Nevertheless, the use of both vibrational and radical modes is advised in such studies because they provided bound to ions useful to improve overall sequence coverage [19,24,42,45,46]. In our study, we confirmed this view for fragmentations in the middle (Y76STMSIT82) and near the C-terminus (G112NPYVP116) that resulted only from vibrational activation. Few complementary product ions were observed for the radical activation modes with and without ion activation event (Figure 4). As complementarities became apparent from the several types of experiment and the maximum coverage from any one experiment did not exceed 71% or 69% for RNase A and B, respectively, we combined the results obtained from all the activation modes presented in this work. This combination improved the total sequence coverage (+19 % and +17 % for RNase A and B, respectively) so that the observed sequence coverage reached 90% and 86% for RNase A and RNase B, respectively.
3.2. Glycan Cleavage and Glycan Binding Site on RNase B
No glycan cleavage
Although vibrational activation modes have been shown to fully or partially cleave glycans from glycopeptides [21,40], both vibrational and radical activation modes performed directly on the intact glycoprotein RNase B mostly preserve the glycan binding. Indeed, only a few, low abundance glycan losses accompanied by protein backbone cleavage were observed (Table 1), except in the CID spectrum where the loss of Man4, accompanying formation of the y122 (Man1GlcNAc2) ion, was significant (relative abundance 19%). IRMPD activation performed on the same species preserved the glycan chain: only low abundance glycan losses were observed (<3%). This low competition between protein backbone and glycan cleavages was already observed on intact RNase B during ion/ion proton-transfer reactions followed by resonant CID [9]. The retention of the N-linked sugar was presumably due to two factors: First, the competition from the facile amide bond cleavage channels effectively protects the N-linked glycosidic bond from cleavage [10], a protection significantly increased on the whole glycoprotein as compared to glycopeptides. Secondly, the lack of glycan cleavage could also be attributed, in part, to the relative stability of the high mannose glycan [21].
Table 1.
Fragment ions resulting from glycan losses observed from a non-resonant CID experiment on RNase B with excitation at 8 eV.
| Residual glycan motif attached to the protein | Fragment ions observed with residual glycan motif | Relative Abundance (%) |
|---|---|---|
| Man(4)GlcNAc(2) | b116 | <5 |
| Man(3)GlcNAc(2) | b118 | <5 |
| Man(2)GlcNAc(2) | y116 | <5 |
| Man(1)GlcNAc(2) | y121 y122 |
<5 19 |
| GlcNAc(2) | y121 | <5 |
| GlcNAc(1) | b90 | <5 |
| GlcNAc(0) | b86 | <5 |
Glycan binding site
The capacity to preserve the whole glycan chain on the protein facilitated the assignment of the glycan position. However, retention alone is not sufficient for unambiguous assignment of the glycan site; complete amino acid sequence coverage around it is also required. For this, the radical vibrational modes (ETD, ECD), +/− pre- or post-activation, proved more appropriate than vibrational activation modes (CID/IRMPD). Coverage obtained from vibrational activation modes yielded specific fragmentations focused preferentially in the middle and near the C-terminus, far away from the glycan binding site, whereas radical activation modes yielded extensive fragment ions randomized over the whole sequence, providing crucial information on the glycan binding site. Indeed, sequence coverage around this site was total, made very explicit by the diagnostic ions: c33/z90 ions without the Man5GlNAc2 motif and c34/z91 ions presenting the Man5GlcNAc2 chain, allowing us to confirm assignment of the glycan binding site at N-34.
Therefore, the top-down approach performed on the high mannose glycoprotein proved adequate to efficiently preserve the high mannose glycan chain and to easily define the glycan binding site by providing complete sequence coverage around the glycosylation site. While the top-down approach facilitated the assignment of glycan attachment site, the characterization of the detailed glycan structure was hindered by the absence of fragmentation within the glycan. To achieve this determination, other strategies including bottom–up or glycan digestion experiments and glycopeptide enrichment should be carried out in parallel with the top-down analyses to obtain more information on the glycan structure. Extension of this study to include further classes of glycoproteins that contain multiple glycosylation sites and/or complex or hybrid glycans, is underway to determine the impact of the glycan nature on retention of the glycan chain in the intact glycoprotein by top-down approach.
3.3. Glycan Effect on Fragmentation Pattern
Comparison of the fragmentation patterns of RNase A and B suggested that the presence of the glycan chain affected the gas phase stability of the analyte (Supplemental data S4) and, more surprisingly, favored fragmentation around the glycosylation site by radical activation (ETD, ECD) pathways. The RNase B glycan seemed to orient the fragmentation pathway around the glycosylation site, N-34 (Figure 2, ETD and ECD). In the presence of the glycan chain, fragments on the N-terminal side (c33/z91 ions) and the C-terminal side (c34/z90) of N-34 were observed in both the ETD and ECD experiments. Strikingly, fragmentations on the C-terminal side (c34/z90) were never produced from classical ETD and ECD experiments when the amino acid N-34 was devoid of glycan (RNase A). Following the hydrogen capture model of electron activation mechanism [46] the capacity of the glycan chain to favor c/z• ion generation at C-terminal side of N-34 from classical ETD and ECD experiments suggested that protonation of the carbonyl group on the Asn residue was favored. We hypothesize that the glycan attachment may favor this protonation from different pathways:
the glycan motif generates steric hindrance that modifies the three-dimensional structure of reduced RNase B, making it more suitable for Asn protonation. (Although 3-D structures of native RNase A and B glycoforms have been reported to be identical [47], no information is available in the literature on the tertiary structures of the reduced species.)
the glycan motif generates a higher polarizability resulting in higher proton affinity of the Asn.
In order to obtain a better understanding of the phenomenon, supplemental investigations of the glycan effect on high mannose glycoproteins with multiple glycosylation sites and an extension of this study to other kinds of glycoproteins will also be very useful.
4. Conclusions
Our study performed on the glycoprotein (RNase B) and the unmodified analogous protein (RNase A) combined the top-down approach with high resolution detection and generated different fragmentation pathways induced by the multiple activation modes. MS/MS experiments performed using both vibrational (CID/IRMPD) and radical activation (ECD, ETD) with/without a pre- or post-activation event, showed different amino acid sequence coverage in terms of efficiencies and complementarities. Sequence coverage obtained was more extensive with radical activation modes (58 – 65%) to vibrational activation modes (12 – 23%). We showed also that the CID pre-activation event prior ETD or ECD could slightly increase the sequence coverage (62% – 69%). The activation mode showed complementarities that were more significant between radical and vibrational activation modes. Thus, combination of the set of results is very useful to complete the sequence information and thereby obtain high sequence coverage (90% and 86% for RNase A and RNase B, respectively), whereas the maximum coverage obtained from any one experiment did not exceed 71% or 69% for RNase A and B, respectively.
This report demonstrates also that top-down MS experiments mostly protect the glycan chain from vibrational activation modes that favor glycan cleavage in glycopeptides. Preservation of the glycan chain was presumably due to the high mass of the species and perhaps aided by the stability of the high mannose glycan. Moreover, radical activation modes yielded extensive fragment ions randomized over the whole protein backbone and provided complete sequence coverage around the glycan binding site, including the diagnostic ions that confirm without ambiguity the glycan attachment at N-34.
Comparison of the fragmentation patterns of the glycoprotein and its corresponding protein suggested that the presence of the glycan chain increased the gas phase stability of the analyte and favored fragmentation occurring around the glycosylation site, thus facilitating the unambiguous assignment of the glycan site, presumably because glycosylation facilitated protonation on the Asn. Nevertheless, the mechanism is not yet elucidated and further investigations of the glycan effect on glycopeptides/proteins will be necessary to explain this phenomenon. This study focused on the comparison of the behaviors of a high mannose glycoprotein and its non-glycosylated analog. An extension of this research to include glycoproteins with multiple glycosylation sites and more labile components, e.g., hybrid and/or complex glycans, is underway.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge financial support from the NIH grants P41 RR010888/GM104603 and S10 RR025082 and NHLBI Contract HHSN268201000031C.
Abbreviations
- ACN
Acetonitrile
- AI
Activated Ions
- CID
collision-induced dissociation
- ECD
electron capture dissociation
- ESI
Electrospray ionization
- ETD
electron transfer dissociation
- FA
Formic acid
- FTICR
Fourier Transform Ion Cyclotron Resonance
- IRMPD
infrared multiphoton dissociation
- MALDI
Matrix-assisted laser desorption/ionization
- NaTFA
Sodium trifluoroacetate
- PTM
post-translational modifications
- Qh
Quadrupole - hexapole
- RNase
Ribonuclease
- TOF
Time-of-flight
Footnotes
The authors have no conflict of interest.
Supporting Information. This material is available free of charge via the Internet at xxx.
S1. Optimization of ETD conditions
S2a. Table of assigned product ions for RNase A
S2b. Table of assigned product ions for RNase B
S3. Comparison ETD and ECD processes
S4. Effect of high mannose glycan on the gas phase stability of proteins
References
- 1.Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 2.Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dwek RA. Glycobiology: toward understanding the function of sugars. Chem Rev. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 4.Kornfeld R, Kornfeld S. Assembly of Asparagine-Linked Oligosaccharides. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 5.Vance BA, Wu W, Ribaudo RK, Segal DM, Kearse KP. Multiple dimeric forms of human CD69 result from differential addition of N-glycans to typical (Asn-X-Ser/Thr) and atypical (Asn-X-Cys) glycosylation motifs. J Biol Chem. 1997;272:23117–23122. doi: 10.1074/jbc.272.37.23117. [DOI] [PubMed] [Google Scholar]
- 6.Halim A, Rüetschi U, Larson G, Nilsson J. LC MS/MS Characterization of O glycosylation sites and glycan structures of human cerebrospinal fluid glycoproteins. J Proteome Res. 2013;12(2):573–584. doi: 10.1021/pr300963h. [DOI] [PubMed] [Google Scholar]
- 7.Geyer H, Geyer R. Strategies for analysis of glycoprotein glycosylation. Biochimica et Biophysica Acta (BBA) - Proteins & Proteomics. 2006;1764(12):1853–1869. doi: 10.1016/j.bbapap.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Wuhrer M, Catalina MI, Deelder AM, Hokke CH. Glycoproteomics based on tandem mass spectrometry of glycopeptides. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;849(1–2):115–128. doi: 10.1016/j.jchromb.2006.09.041. [DOI] [PubMed] [Google Scholar]
- 9.Reid GE, Stephenson JL, Jr, McLuckey SA. Tandem mass spectrometry of Ribonuclease A and B:N-linked glycosylation site analysis of whole protein ions. Anal Chem. 2002;74:577–583. doi: 10.1021/ac015618l. [DOI] [PubMed] [Google Scholar]
- 10.Siuti N, Kelleher NL. Decoding protein modifications using top-down mass spectrometry. Nature Meth. 2007;4(10):817–821. doi: 10.1038/nmeth1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui W, Rohrs HW, Gross ML. Top-down mass spectrometry: recent developments, applications and perspectives. Analyst. 2011;136:3854–3864. doi: 10.1039/c1an15286f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gault J, Malosse C, Duménil G, Chamot-Rooke J. A combined mass spectrometry strategy for complete posttranslational modification mapping of Neisseria meningitidis major pilin. J Mass Spectrom. 2013;48:1199–1206. doi: 10.1002/jms.3262. [DOI] [PubMed] [Google Scholar]
- 13.Vestal ML, Juhasz P, Martin SA. Delayed extraction matrix-assisted laser desorption time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 1995;9:1044–1050. [Google Scholar]
- 14.Taichriba A, Pelzingb M, Pellegrinoc C, Rossic M, Neusüß C. High resolution TOF MS coupled to CE for the analysis of isotopically resolved intact proteins. J Proteomics. 2011;74:958–966. doi: 10.1016/j.jprot.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y, Sinaiko AR, Nelsestuen GL. Glycoproteins and glycosylation_ Apolipoprotein C3 glycoforms by top-down MALDI-TOF mass spectrometry. Methods Mol Biol. 2012;909:141–150. doi: 10.1007/978-1-61779-959-4_10. [DOI] [PubMed] [Google Scholar]
- 16.Fornelli L, Damoc E, Thomas PM, Kelleher NL, Aizikov K, Denisov E, Makarov A, Tsybin YO. Analysis of intact monoclonal antibody IgG1 by electron transfer dissociation Orbitrap FTMS. Mol Cell Proteomics. 2012;11(12):1758–67. doi: 10.1074/mcp.M112.019620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Senko MW, Speir JP, McLafferty FW. Collisional activation of large multiply charged ions using Fourier transform mass spectrometry. Anal Chem. 1994;66:2801–2808. doi: 10.1021/ac00090a003. [DOI] [PubMed] [Google Scholar]
- 18.Little DP, Spier JP, Senko MW, O’Connor PB, McLafferty FW. Infrared multiphoton dissociation of large multiply charged ions for biomolecule sequencing. Anal Chem. 1994;66:2809–2815. doi: 10.1021/ac00090a004. [DOI] [PubMed] [Google Scholar]
- 19.Zubarev RA. Electron-capture dissociation tandem mass spectrometry. Curr Opin Biotechnol. 2004;15:12–16. doi: 10.1016/j.copbio.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci USA. 2004;101(26):9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adamson JT, Håkansson K. Infrared multiphoton dissociation and electron capture dissociation of high-mannose type glycopeptides. J Proteome Res. 2006;5:493–501. doi: 10.1021/pr0504081. [DOI] [PubMed] [Google Scholar]
- 22.Håkansson K, Cooper HJ, Emmet MR, Costello CE, Marshall AG, Nilson CL. Electron capture dissociation and infrared multiphoton dissociation MS/MS of an N-glycosylated tryptic peptide to yield complementary sequence information. Anal Chem. 2001;73:4530–4536. doi: 10.1021/ac0103470. [DOI] [PubMed] [Google Scholar]
- 23.Wiesner J, Premsler T, Sickmann A. Application of electron transfer dissociation (ETD) for the analysis of posttranslational modifications. Proteomics. 2008;8:4466–4483. doi: 10.1002/pmic.200800329. [DOI] [PubMed] [Google Scholar]
- 24.Mikesh LM, Ueberheide B, Chi A, Coon JJ, Syka JEP, et al. The utility of ETD mass spectrometry in proteomic analysis. Biochim Biophys Acta. 2006;1764:1811–1822. doi: 10.1016/j.bbapap.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Catalina MI, Koeleman CAM, Deelder AM, Wuher M. Electron transfer dissociation of N-glycopeptides: loss of the entire N-glycosylated asparagine side chain. Rapid Commun Mass Spectrom. 2007;21:1053–1061. doi: 10.1002/rcm.2929. [DOI] [PubMed] [Google Scholar]
- 26.Tsybin YO, Witt M, Baykut G, Kjeldsen F, Hakansson P. Combined infrared multiphoton dissociation and electron capture dissociation with a hollow electron beam in Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun Mass Spectrom. 2003;17:1759–1768. doi: 10.1002/rcm.1118. [DOI] [PubMed] [Google Scholar]
- 27.Gorshkov MV, Masselon CD, Nikolaev EN, Udseth HR, Paša-Tolic L, Smith RD. Considerations for electron capture dissociation efficiency in FTICR mass spectrometry. Int J Mass Spectrom. 2004;234:131–136. [Google Scholar]
- 28.Han H, Xia Y, Yang M, McLuckey SA. Rapidly alternating transmission mode Electron-transfer dissociation and collisional activation for the characterization of polypeptide Ions. Anal Chem. 2008;80:3492–3497. doi: 10.1021/ac7022734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campbell JL, Hager JW, Le Blanc JCY. On performing simultaneous electron transfer dissociation and collision-induced dissociation on multiply protonated peptides in a linear ion trap. J Am Soc Mass Spectrom. 2009;20:1672–1683. doi: 10.1016/j.jasms.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 30.Nagy K, Vékey K, Imre T, Ludányi K, Barrow MP, Derrick PJ. Electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry of human alpha-1-acid glycoprotein. Anal Chem. 2004;76:4998–5005. doi: 10.1021/ac040019a. [DOI] [PubMed] [Google Scholar]
- 31.Balaguer E, Neusüss C. Glycoprotein characterization combining intact protein and glycan analysis by capillary electrophoresis-electrospray ionization-mass spectrometry. Anal Chem. 2006;78:5384–5393. doi: 10.1021/ac060376g. [DOI] [PubMed] [Google Scholar]
- 32.Baek WO, Vijayalakshmi MA. Effect of chemical glycosylation of RNase A on the protein stability and surface histidines accessibility in immobilized metal ion affinity electrophoresis (IMAGE) system. Biochimica et Biophysica Acta. 1997;1336:394–402. doi: 10.1016/s0304-4165(97)00050-0. [DOI] [PubMed] [Google Scholar]
- 33.Tsybin YO, Hendrickson CL, Beu SC, Marshall AG. Impact of ion magnetron motion on electron capture dissociation Fourier transform ion cyclotron resonance mass spectrometry. Int J Mass Spectrom. 2006;256:144–149. [Google Scholar]
- 34.Tong RTW, Infusini G, Cui W, Perlman DH, Lin C, Mccomb ME, Costello CE. BUPID-Top-Down: database search and assignment of top-down MS/MS Data. Proceedings of the 57th American Society Conference on Mass Spectrometry and Allied Topics; 2009; Philadelphia, PA. [Google Scholar]
- 35.Horn DM, Zubarev RA, McLafferty FW. Automated reduction and interpretation of high resolution electrospray mass spectra of large molecules. J Am Soc Mass Spectrom. 2000;11:320–332. doi: 10.1016/s1044-0305(99)00157-9. [DOI] [PubMed] [Google Scholar]
- 36.Horn DM, Ge Y, McLafferty FW. Activated ion electron capture dissociation for mass spectral sequencing of larger (42 kDa) proteins. Anal Chem. 2000;72:4778–4784. doi: 10.1021/ac000494i. [DOI] [PubMed] [Google Scholar]
- 37.Zabrouskov V, Han X, Welker E, Zhai H, Lin C, van Wijk KJ, Scheraga HA, McLafferty FW. Stepwise deamidation of ribonuclease A at five sites determined by top down mass spectrometry. Biochemistry. 2006;45:987–992. doi: 10.1021/bi0517584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hall Z, Robinson CV. Do charge state signatures guarantee protein conformations? J Am Soc Mass Spectrom. 2012;23:1161–1168. doi: 10.1007/s13361-012-0393-z. [DOI] [PubMed] [Google Scholar]
- 39.Skinner OS, Breuker K, McLafferty FW. Charge site mass spectra: conformation-sensitive components of the electron capture dissociation spectrum of a protein. J Am Soc Mass Spectrom. 2013;24:807–810. doi: 10.1007/s13361-013-0603-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alley WR, Jr, Mechref Y, Novotny MV. Characterization of glycopeptides by combining collision-induced dissociation and electron-transfer dissociation mass spectrometry data. Rapid Commun Mass Spectrom. 2009;23:161–170. doi: 10.1002/rcm.3850. [DOI] [PubMed] [Google Scholar]
- 41.Konn DO, Murrell J, Despeyroux D. Comparison of the effects of ionization mechanism, analyte concentration, and ion “cool-times” on the internal energies of peptide ions produced by electrospray and atmospheric pressure matrix-assisted laser desorption ionization. J Am Soc Mass Spectrom. 2005;16:743–751. doi: 10.1016/j.jasms.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 42.Sleno L, Volmer DA. Ion activation methods for tandem mass spectrometry. J Mass Spectrom. 2004;39:1091–1112. doi: 10.1002/jms.703. [DOI] [PubMed] [Google Scholar]
- 43.Dongre AR, Jones JL, Somogyi A, Wysocki VH. Influence of peptide composition, gas-phase basicity, and chemical modification on fragmentation efficiency: evidence for the mobile proton model. J Am Chem Soc. 1996;118:8365–8374. [Google Scholar]
- 44.Paizs B, Suhai S. Fragmentation pathways of protonated peptides. Mass Spectrometry Reviews. 2005;24:508–548. doi: 10.1002/mas.20024. [DOI] [PubMed] [Google Scholar]
- 45.Zubarev RA, Kelleher NL, McLafferty FW. Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- 46.Zubarev RA, Haselmann KF, Budnik B, Kjeldsen F, Jensen F. Towards an understanding of the mechanism of electron capture dissociation: a historical perspective. Eur J Mass Spectrom. 2002;8(5):337–349. [Google Scholar]
- 47.Williams RL, Greene SM, McPherson A. The crystal structure of ribonuclease B at 2.5-A resolution. J Biol Chem. 1987;262(33):16020–16031. doi: 10.2210/pdb1rbb/pdb. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.