

Abstract
Homologous recombination (HR) is a high-fidelity DNA repair pathway that maintains genome integrity, by repairing double strand breaks (DSBs), single-stranded DNA (ssDNA) gaps, and supporting stalled/collapsed replication forks. The RecA/Rad51 family of proteins are the key enzymes in this homology-directed repair pathway, as they perform DNA strand invasion and exchange, in concert with a host of ancillary factors. In vitro, the RecA/Rad51 family of proteins share similar enzymatic activities including catalyzing ssDNA-stimulated ATP hydrolysis, formation of displacement loops (D-loops), and DNA strand exchange. After successful DNA strand invasion, DNA synthesis restores the lost genetic information using an undamaged DNA template. In this chapter, we describe two well-established biochemical assays to investigate the signature DNA strand transfer activity of RecA/Rad51 family of proteins: the D-loop assay and the DNA strand exchange reaction. Moreover, we describe a D-loop extension assay coupling D-loop formation with DNA synthesis, which can be used to define the roles of DNA polymerases in HR. Additionally, we present a protocol to investigate protein-mediated DNA annealing, a critical step in the synthesis-dependent strand annealing (SDSA) and double-Holiday junction (dHJ) pathways as well as the single-strand annealing (SSA) pathway. The quality of supercoiled plasmid DNA is critical in reconstituted HR reactions, and a protocol describing the preparation of this DNA substrate is included.
Keywords: D-loop, DNA polymerase, DNA strand exchange, DNA strand annealing, DNA synthesis, homologous recombination, Rad51, Rad52, RecA, supercoiled plasmid DNA
1. Introduction
The RecA/Rad51 family of proteins are the central enzymes in homologous recombination (HR), a high-fidelity DNA repair pathway that processes DNA double strand breaks (DSB) and single-stranded DNA (ssDNA) gaps and also supports replication forks in all domains of life (1) Krogh, 2004 #10747}. These proteins form filaments on ssDNA and catalyze homology search, DNA strand invasion, and DNA strand exchange using homologous double-stranded DNA (dsDNA) as a template. In vitro, two different biochemical assays have been developed to demonstrate this recombinational activity: DNA strand exchange and displacement loop (D-loop) formation. In this chapter, we describe protocols for DNA strand exchange and D-loop formation, developed for the budding yeast Rad51 protein (Figures 1 and 2). These assays have been critical to define the properties of the wild type and mutant proteins. Moreover, subtle modifications to the assays allow testing the functions of additional protein factors, such as mediator proteins or anti-recombination helicases, involved in Rad51-dependent recombination (2–4).
Figure 1. DNA strand exchange reaction.
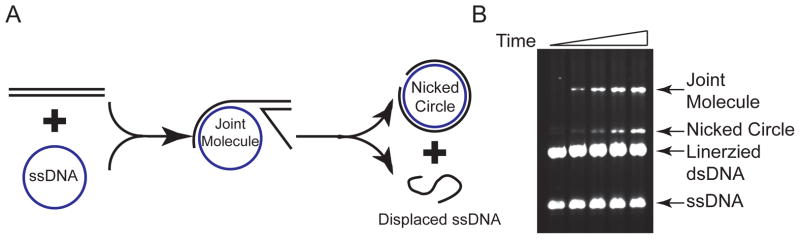
(A) Reaction scheme for the DNA strand exchange assay. Homologous circular ssDNA and linearized dsDNA are the substrates. Joint molecules are intermediates. Nicked circles and displaced ssDNAs are the final products. (B) Time course of a Rad51-catalyzed DNA strand exchange assay. 6.7 μM Rad51 was incubated with 20 μM (nt concentration) ϕX174 ssDNA for 15 min at 30 °C, then 1.11 μM RPA was added and incubated for another 30 min. Then 20 μM (bp concentration) PstI-linearized ϕX174 dsDNA was added to initiate the reaction. Samples were taken at 0, 30, 60, 90, 180 min, and immediately quenched by stop buffer. An ethidium bromide-stained agarose gel is shown.
Figure 2. D-loop formation and D-loop extension assays.

(A) Reaction scheme for the D-loop formation and D-loop extension assay. Rad51 forms nucleoprotein filaments on a short oligo nucleotide, which catalyzes D-loop formation with supercoiled plasmid dsDNA. The extension assay is initiated by the addition of both DNA polymerase and its accessory factors PCNA-RFC, as well as dNTPs. (B) D-loop formation and extension is monitored by analyzing DNA species on a 0.8% native agarose gel. (C) Two-dimensional native/denaturing agarose gel electrophoresis of a 1kb ladder (top panel) and D-loop extension products (bottom panel). Labels in B, C: a. free ssDNA, b. short extension products, dissociated from D-loops, c. unextended D-loops, d. partially extended D-loops, e. maximally extended D-loops.
After DNA strand invasion, the 3′-OH of the invading strand is positioned in the D-loop on an undamaged homologous template to initiate repair DNA synthesis. Our laboratory has recently developed a coupled reaction of D-loop formation and extension by DNA polymerase, using purified protein factors from Saccharomyces cerevisiae (Figure 2A) (5). This assay provides a tool to test the functional interplay between recombination proteins and replication factors, and to explore the roles of various DNA polymerases in the specific context of recombinational repair. In the D-loop formation and extension assays, supercoiled dsDNA serves as the homologous template, and its quality directly contributes to the minimization of experimental artifacts and interferences. Thus, we provide a protocol on how to prepare high-quality supercoiled plasmid dsDNA.
In vivo, DNA annealing is a late but critical step in two different subpathways of HR: the synthesis-dependent strand annealing (SDSA) and the double Holliday junction (dHJ) pathways (1). Strand annealing is also the central reaction of the single strand annealing (SSA) pathway (6). We describe a DNA strand annealing assay protocol based on the yeast Rad52 protein under physiologically relevant conditions that include free Mg2+ and sufficient amounts of the ssDNA binding protein RPA to saturate the ssDNA (Figure 3). This method can be used to test individual proteins, for example Rad52, and the modulation of their activity by other factors, such as Rad51 or Rad59 (7, 8).
Figure 3. DNA annealing assay.
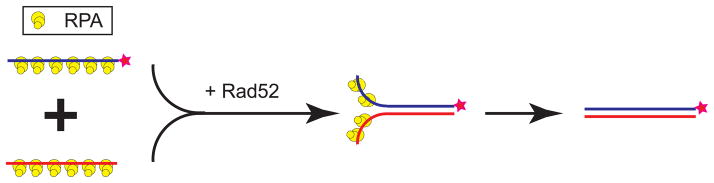
Reaction scheme for DNA strand annealing assay of RPA-covered ssDNA. Rad52 and its homolog in phage T4 (UvsY) and E. coli (RecO) can catalyze annealing between complementary ssDNA strands covered with RPA.
2. Materials
2.1. Preparation of Supercoiled Plasmid dsDNA by Detergent Lysis and CsCl Isopycnic Centrifugation
Liquid LB media (1 L): 10 g tryptone, 5 g yeast extract, and 5 g NaCl. Suspend all solids into ddH2O and autoclave it at 121 °C for 25 min. Fresh media is preferred at this scale.
STE buffer: 10 mM Tris-HCl (pH 8.0), 0.1 M NaCl, and 1 mM EDTA (pH 8.0). Sterilize the solution by passing through a 25 mm syringe filter with 0.22 μm pore size (Fisher Scientific), and store at 4 °C. All buffers described in this chapter can be sterilized in this manner. Use 200 mL for each 1 L cell culture.
Tris-Sucrose buffer: 25 % w/v sucrose and 50 mM Tris-HCl (pH 8.0). Sterilize by filtration and store at 4 °C. Use 100 mL for each 1 L cell culture.
0.5 M EDTA (pH 8.0), 10 % SDS, and 5 M NaCl.
Lysozyme solution: 10 mg/mL hen egg white lysozyme, 25 mM Tris-HCl (pH 8.0). Prepare fresh solution each time before use.
Sodium iodide solution (100 mL): 7.6 M NaI, 40 mM Tris-HCl (pH 8.0), and 20 mM EDTA. Sterilize by filtration and store at 4 °C.
3 M NaOAc (pH 5.2), isopropanol, 100 % and 70 % ethanol.
TE buffer (10 X): 100 mM Tris-HCl (pH 8.0) and 10 mM EDTA (pH 8.0). Sterilize by filtration and store at room temperature.
10 mg/mL ethidium bromide solution and solid cesium chloride (CsCl).
Beckman Coulter Allegra™ 6 Centrifuge: Swing-bucket bench-top centrifuge.
Beckman J2-MC Centrifuge and corresponding JA-20 and JA-14 rotors.
Beckman Optima™ LE-80K Ultracentrifuge: for CsCl gradient centrifugation.
Quick-Seal polyallomer 13.5 mL capacity centrifuge tubes (Beckman catalog number: 342413).
ISO-TIP quick charge soldering iron (Beckman, catalog number: 7740).
Agarose gel running apparatus and power supply.
10 X TBE buffer: 0.89 M Tris base, 0.89 M H3BO3, and 25 mM EDTA. Prepare 1 L solution by dissolving 109 g of Tris base, 55 g of M H3BO3, and 50 mL of 0.5 M EDTA (pH 8.0) into 1 L ddH2O.
Digital gel imaging system with UV illumination box.
Nanodrop ® spectrophotometer or alternative unit that can read the A260 of small volumes of DNA in solution.
2.2. DNA Strand Exchange Reaction
DNA Strand exchange buffer (5X SEB): 150 mM Tris-acetate (pH 7.5), 5 mM DTT, 250 μg/mL BSA, 12.5 mM ATP, 20 mM Mg(OAc)2, and 100 mM phosphocreatine (see Note 1). Sterilize by filtration and store at −20 °C.
Purified proteins from Saccharomyces cerevisiae: Rad51 and RPA (see Note 2) (5).
1 μg/μL creatine kinase stock solution, store at −20 °C.
100 mM spermidine (pH 7.4) stock solution, store at −20 °C.
Stop buffer: 0.714% SDS, 357 mM EDTA, and 4.3 mg/ml proteinase K. Prepare 84 μL solution freshly by mixing 6 μL 10% SDS, 60 μL 0.5 M EDTA, and 18 μL 20 mg/ml Proteinase K.
DNA gel loading buffer (10X): 0.25 % bromophenol blue (w/v), 50% glycerol (omit xylencyanol). Store at 4 °C.
ϕX174 ssDNA (virion) is purchased from NEB (catalog number: N3023S) (see Note 3).
RFI ϕX174 dsDNA (RF I) is purchased from NEB (catalog number: N3021S) or prepared by the protocol in Section 3.1.
TBE-Agarose gel running buffer, apparatus, and imaging system, as listed in Section 2.1.
ImageQuant software (version 5.1, GE Healthcare).
2.3. D-loop Assay
T4 polynucleotide kinase reaction buffer (10x): 70 mM Tris-HCl (pH 7.6), 10 mM MgCl2, and 5 mM DTT.
T4 polynucleotide kinase
D-loop buffer (5X): 150 mM Tris-acetate (pH 7.5), 5 mM DTT, 250 μg/mL BSA, 20 mM ATP, 25 mM Mg(OAc)2, and 100 mM phosphocreatine (see Note 1). Sterilize by filtration and store at −20 °C.
Purified proteins from Saccharomyces cerevisiae: Rad51, Rad54, and RPA (see Note 2) (5).
Creatine kinase and spermidine: as described in Section 2.2.
Stop buffer and DNA loading buffer (10 X): as described in Section 2.2.
95-mer DNA oligonucleotides: The sequence of PstI-95 mer is 5′-TgCAggCATgCAAgCTTggCgTAATCATggTCATAgCTgTTTCCTgTgTgAAATTgTTATCCgCTCACAATTCCACACAACATACgAgCCggAAg -3′. It shares homology with pUC19 plasmid DNA.
pUC19 supercoiled plasmid DNA: Prepare according to the protocol in Section 3.1. without alkaline cell lysis.
10 X TBE buffer: as described in Section 2.1.
Mini Spin Oligo Column (Roche Applied Science: catalog # 11814397001). These spin columns are used to remove unincorporated radioactive nucleotides from the labeled oligo substrates.
TBE-Agarose gel running buffer, apparatus, and imaging system, as listed in Section 2.1.
DE81 paper (Whatman): a thin DEAE cellulose paper about 1 mm in width. It has weakly basic anion exchangers coupled with diethylaminoethyl groups, resulting in low retention of DNA during the gel drying process.
3M Whatman filter paper.
Gel dryer with vacuum pump, dedicated for use with radioisotopes.
Phosphorimaging screen and Storm 860 (Molecular Dynamics) PhosphorImaging system.
ImageQuant software (version 5.1, GE healthcare).
2.4. D-Loop Extension Assay
All the materials listed above in Section 2.3.
Stock solution of dNTP mix containing: 1 mM each dATP, dCTP, dTTP, dGTP.
Purified proteins from Saccharomyces cerevisiae: RFC, PCNA, and polymerase δ (see Note 2) (5).
Denaturing agarose gel buffer: 50 mM NaOH, 1 mM EDTA.
DNA gel loading dye (6X): 1X TE, 18% ficoll (w/v), 0.15% bromocresol green (w/v), 50mM EDTA.
γ-32P-ATP-labeled 100 bp and 1 kb ladders.
50 °C water bath.
2.5. DNA Strand Annealing
Oligo A1 (5′-GCAATTAAGCTCTAAGCCATCCGCAAAAATGACCTCTTATCAAAAGGA) and Oligo A2 (5′-TCCTTTTGATAAGAGGTCATTTTTGCGGATGGCTTAGAGCTTAATTGC) either ordered PAGE-purified or purified by electrophoresis using 15 % polyacrylamide gels containing 8 M urea. The nucleotide concentrations of oligo A1 and A2 can be measured using extinction coefficients of 1.0 × 104 and 9.6 × 103 M−1 cm−1 at 260 nm, respectively.
DNA strand annealing buffer (5X): 150 mM Tris-acetate (pH 7.5), 25 mM magnesium acetate, 5 mM DTT. Sterilize by filtration and store at −20 °C.
Purified proteins from Saccharomyces cerevisiae: Rad52, RPA (see Note 2) (9).
Stop buffer: 1.54 μM unlabeled oligo A2, 0.77 % SDS, and 1.54 mg/ml proteinase K. Make fresh stop buffer each time to reach this final concentration. Prepare 30 μL of fresh stop buffer by mixing 10 μL of 20 μM unlabeled oligo A2, 10 μL of 10% SDS, and 10 μL of 20 mg/ml Proteinase K. Mix 3 μL fresh stop buffer with 10 μL sample aliquot rapidly. Store 10% SDS at room temperature and Proteinase K at −20 °C.
For radioactive samples: polyacrylamide gel running apparatus, power supply, and TBE buffer, as listed in section 2.1.
For fluorescent dye method: 4′,6-diamidino-2-phenylindole (DAPI, Invitrogen). The stock concentration of DAPI is measured using a molar extinction coefficient of 3.3 × 104 M−1·cm−1 at 345 nm.
For fluorescent dye method: a SLM8000 spectrofluorimeter or other similar spectrofluorimeter. Additionally, a 700 μL cuvette is needed, but other smaller cuvettes can be considered to minimize protein consumption.
3. Methods
Many biochemical assays including D-loop formation, DNA strand exchange, and DNA-binding use plasmid-based circular dsDNA as substrate. Commercial plasmid DNA is usually available as the product of an alkaline lysis method, the norm in industrial-scale plasmid preparation. However, alkaline treatment creates both nicked circles and locally melted regions in the dsDNA plasmid, which causes artifacts in these biochemical assays. For example, local melting of the dsDNA increases background for spontaneous D-loop formation, because of annealing between the ssDNA supplied to the assay as the invading strand and the ssDNA in the melted zones. The nicked circular form of dsDNA migrates very close to D-loops during agarose gel electrophoresis, interfering with data analysis. Equally relevant, an intact supercoiled dsDNA form will stabilize D-loops and therefore minimize D-loop dissociation during electrophoresis. Thus, a clean preparation of plasmid DNA in high-quality, supercoiled form provides a solid foundation for the success of D-loop formation and D-loop extension assays in Section 3.3 and 3.4. In Section 3.1, we describe a protocol for the preparation of intact supercoiled plasmid dsDNA from E. coli that uses detergent lysis as an alternative to alkaline lysis. The supercoiled form is recovered by isopycnic centrifugation in CsCl gradients.
As the major ssDNA-binding protein in eukaryotes, RPA binds ssDNA rapidly with high affinity and saturates ssDNA, protecting it from nuclease digestion and removing inhibitory secondary structure. In vitro, when Rad51 is added before the addition of RPA, Rad51 cannot form a saturated nucleoprotein filament because of inhibitory secondary structures in ssDNA. The subsequent addition of RPA to partially formed Rad51-ssDNA filaments promotes the fully assembled Rad51-ssDNA filament via the slow displacement of RPA by Rad51. The end result is a direct increase of product yield in the DNA strand exchange reaction. This sequence is used in the protocols described in Section 3.2. – 3.4. However, if the order-of-addition is reversed by adding RPA before Rad51, the strong and cooperative binding of RPA on ssDNA usually inhibits filament formation of Rad51 onto ssDNA, which results in an inhibition of Rad51-dependent DNA strand invasion. This order-of-addition is useful to demonstrate the function of mediator proteins, such as Rad52 protein, which facilitates Rad51-ssDNA filament formation under suboptimal conditions such as high salt, low Rad51 concentration, and RPA inhibition (4, 10). In general, mediator proteins may facilitate the nucleation or enhance stability of Rad51 filaments onto RPA-covered ssDNA. Consequently, Rad51 can propagate and form active nucleoprotein filaments to initiate homology search. For more details about this “order of addition” phenomenon the reader is referred to references (4, 10, 11).
In vivo, D-loop formation represents the successful invasion of the ssDNA into a homologous duplex DNA; the stability and processing of D-loops determines the outcome of competition between several HR subpathways. As the central intermediate of DNA strand exchange, the D-loop can be disrupted, migrated, expanded and/or it may lead to double Holliday junctions through second end capture. Thus, the functional characterizations of various protein factors in Rad51-dependent D-loop formation will define the mechanisms that differentiate the SDSA, dHJ, and break-induced replication (BIR) pathways of HR. We describe a D-loop assay in Section 3.3. that uses a short 95-mer as the ssDNA substrate, and the homologous pUC19 plasmid dsDNA as the target of D-loop formation (Figure 2A). Our laboratory has also developed a reconstituted D-loop extension assay, described in Section 3.4 (Figure 2A), to investigate the interplay between recombination proteins, DNA repair polymerases and their accessory factors (5).
Protein-mediated annealing of complementary ssDNA molecules is a key step in the SDSA pathway and in second-end capture of the dHJ pathway. Moreover, annealing is the central reaction of the SSA pathway of DSB repair. Rad52 is a prototype protein for catalyzing annealing of complementary ssDNA under physiologically relevant conditions (ssDNA fully saturated with the cognate ssDNA-binding protein RPA, presence of free Mg2+; (9)). In Section 3.5., we describe two DNA annealing assays using purified Rad52 and RPA proteins.
For all assays described in this chapter, the reactions are terminated by a rapid inactivation of the proteins in the samples by metal ion chelation and SDS denaturation. This step is essential for reliable results that allow comparison of parallel conditions.
3.1. Preparation of Supercoiled pUC19 Plasmid DNA
Grow 1 L DH5α cells containing pUC19 plasmid in LB media with 100 μg/mL ampicillin, overnight at 37 °C. Be sure to inoculate the culture with a single colony from a fresh transformation.
Collect cells by centrifugation at 1,540 × g for 15 min using a JA-14 rotor. Discard supernatant and resuspend pellets in 200 mL ice-cold STE buffer. Collect cells at 1,540 × g for 15 min at 4 °C using a JA-14 rotor. Pellet can be stored at −80 °C.
Resuspend the cells in 100 mL ice-cold Tris-sucrose solution. Add 20 mL lysozyme solution (10 mg/mL) and 40 mL of 0.5 M EDTA (pH 8.0). Mix well by inverting gently, and incubate on ice for 10 min.
Add 40 mL of 10% SDS and mix immediately but gently into the solution with a glass rod. Add 60 mL of 5 M NaCl (to 1 M final concentration). Mix with glass rod, and incubate on ice for at least 1 hr. The incubation time on ice can be extended for enhanced yield and purity.
Centrifuge at 13,870 × g for 30 min at 4 °C using a JA-14 rotor. Then transfer the supernatant into four ultracentrifuge tubes, and centrifuge at 4 °C, 70,400 × g for 30 min using a Ti45 rotor.
Add 2 volumes of 100 % ethanol to the supernatant. Mix well and incubate at room temperature for 2 hrs.
Centrifuge at 13,870 × g for 20 min at 4 °C using a JA-14 rotor. Save the pellet, and wash with 1 volume of 70 % ethanol.
Centrifuge the resuspended pellet at 13,870 × g for 20 min at 4 °C using a JA-14 rotor. Air dry pellet, and resuspend in 10 mL TE (pH 8.0) buffer. At this step, the sample can be stored at −20 °C. Take OD260 reading to determine the DNA concentration using a molar extinction coefficient (bp) εM = 6500 M−1·cm−1 for dsDNA. Analyze the sample on a 0.8% native agarose-TBE gel to establish the quality of the sample.
Add 1.28 volumes of NaI buffer, and incubate for 5 min at room temperature.
Add 0.6 to 1.0 volume of isopropanol. Mix well and incubate for 10 to15 min at room temperature.
Centrifuge at 13,870 × g for 20 min at 4 °C using a JA-14 rotor. Save the pellet and wash out the isopropanol with 1 volume of 70 % ethanol.
Centrifuge at 13,870 × g for 20 min at 4 °C using a JA-14 rotor. Save the pellet and air dry.
Resuspend the dried pellet in 3 mL TE (pH 8.0) buffer. Add 10 mg/mL RNaseA to 3 mL sample in TE (pH 8.0) buffer to a final concentration of 50 ug/mL RNaseA, and incubate at 37 °C for 1 hr. Centrifuge again at 13,870 × g for 20 min at 4 °C using a JA-14 rotor. Keep the clear supernatant and discard small white precipitates observed at the bottom of the tube.
Add 0.1 volume of 3 M NaOAc (pH 5.2). Mix well.
Add 0.6 to 1 volume of isopropanol. Mix well, and incubate at room temperature for ~ 30 min. Centrifuge at 18,800 × g for 30 min at 4 °C using a JA-14 rotor.
Wash with 1 volume of 70 % ethanol. Centrifuge at 4 °C, 18,800 × g for 20 min using a JA-14 rotor.
Air dry the pellet, and resuspend in 1 mL TE (pH 8.0) buffer. Save 10 μL sample to be checked for by native agarose gel electrophoresis in later steps.
Add TE buffer to the sample from step 17 to reach a total volume of 19 mL. Add 20.8 g CsCl into the solution and dissolve well. Once CsCl is dissolved, add 1 mL 10 mg/mL ethidium bromide into the solution. Weigh 1 mL of this solution; the mass should be ~ 1.55 g. Adjust the density with CsCl or TE, as necessary (See Note 4).
Vortex the tube to mix well, and centrifuge at 2,280 × g for 15 min using the Beckman Coulter Allegra ™6 Bench-Top centrifuge. After centrifugation, take only the clear supernatant solution to fill in the Quick-Seal tubes in the next step. Discard the cloudy part at the top and the precipitants at the bottom of the tube.
Use an 18 gauge needle and a 5 mL syringe to transfer the supernatant into two Quick-Seal polyallomer 13.5 mL capacity tubes (Beckman cat. NO: 342413) up to the neck. Avoid any debris. Balance the tubes carefully, and then seal the tube by melting the neck of the tube using ISO-TIP quick charge soldering iron (Beckman, catalog number: 7740). It is important to check the integrity of the seal, to prevent leakage and ethidium bromide contamination of the centrifuge.
Centrifuge at 160,400 × g for 20–24 hrs (minimum for 10 hrs), at 20 °C using a Ti65 rotor. This centrifugation step is critical to separate the intact supercoiled plasmid form from other forms of DNA.
Use a syringe with an 18 gauge needle to pierce the Quick-Seal tube and to extract the bright orange band corresponding to supercoiled DNA in the middle of the tube, about 2–4 mL from each tube. Apply the sample into a new centrifuge tube. Remove only the center of the band containing supercoiled plasmid DNA to avoid contamination by nicked circular DNA. Only use an 18 gauge needle, since a smaller needle might shear DNA.
Add 1 volume of n-Butanol saturated with TE to extract the ethidium bromide from the purified plasmid DNA. Vortex and centrifuge at 2,280 × g for 15 min using the Beckman Coulter Allegra ™6 Bench-Top centrifuge.
Discard the upper layer (n-Butanol + ethidium bromide) into a designated ethidium bromide hazardous waste container.
Repeat steps 23–24 five times until the top organic phase is transparent.
Transfer the sample into a 35 mL centrifuge tube. Rinse the tube from step 24–25 twice with 1 volume of sterile H2O each time to wash any residual plasmid off the tube walls. Transfer the H2O into the same 35 mL centrifuge tube with the sample.
Add 6 volumes of cold 100 % ethanol (stored at −20 °C). Vortex to mix and keep it at 4 °C over night, since CsCl precipitates at −20 °C.
Centrifuge the sample at 28,300 × g for 1 hr at 4 °C using a JA-20 rotor.
Rinse the pellet with 10 mL of cold 70 % ethanol. Centrifuge at 28,300 × g for 1 hr at 4 °C using a JA-20 rotor.
Repeat step 29. Air dry the pellet, and resuspend it into 2 mL of TE buffer. A smaller amount of TE buffer can be used to dissolve the pellet to achieve a more concentrated DNA stock solution.
Take OD260 reading to determine the DNA concentration (bp) using a molar extinction coefficient (bp) εM= 6500 M−1·cm−1 for dsDNA. Run the saved samples in a 0.8 % native agarose-TBE gel to establish the quality of the samples and to monitor the purification results.
3.2. DNA Strand Exchange Reaction
This assay describes a DNA strand exchange reaction catalyzed by the yeast Rad51 protein (Figure 1A).
-
Prepare PstI-linearized ϕX174 dsDNA:
Set up a 100 μL restriction digestion reaction by incubating 26 μg ϕX174 RFI dsDNA with 50 units of PstI, in NEB buffer #3 supplemented with 100 μg/mL BSA, at 37 °C for 2 hrs.
After incubation, remove PstI enzyme using the Qiagen PCR Clean Up Kit as described in the manufacturer’s instructions.
Measure the nucleic acid absorbance at A260 and determine the sample concentration using the molar extinction coefficient (bp) εM = 6500 M−1·cm−1 at 260 nm.
Set up a 12.5 μL reaction by incubating 10 μM Rad51 protein (see Note 5) with 30 μM (in nucleotides) ϕX174 ssDNA (see Note 6) in 1X SEB buffer supplemented with 0.1 μg/μL creatine kinase (see Note 7) and 2.4 mM spermidine, at 30 °C for 15 min. Notice that the total reaction volume will be 12.5 μL, and the total volume at this step should be 10μL.
Dilute the RPA stock to add 1.8 μM RPA in 0.5 μL into the reaction. Incubate at 30 °C for 30 min.
Initiate the reaction by adding 15 μM (in base pairs) PstI-linearized ϕX174 dsDNA and incubate at 30 °C for 4 hrs. If Rad54 is added, 0.2 μM Rad54 is added with the dsDNA. Dilute the stock solution of both ϕX174 dsDNA and Rad54 such that the total added volume is 2 μL. At this point, the reaction is fully assembled with all the substrates and proteins, and the total volume should be 12.5 μL.
Stop the reaction by adding 2 μL stop buffer and incubate at 30 °C for 30 min.
Add 2 μL loading buffer, and separate the samples by running a 0.8 % agarose gel at a low voltage (25–30 volts) for 12–20 hrs.
Stain gel with 0.5 μg/mL ethidium bromide solution for 20 min, and destain with H2O for 10 min (see Note 8).
Place gel on the UV light box in the digital gel imaging system, and take image under 300 nm (UV) illumination. Avoid adding ethidium bromide into the agarose gel before pouring in step 6, which creates uneven background and decreases the sensitivity needed.
After gel visualization, quantitate the intensity of substrate and product bands using densitometry with ImageQuant software (Version 5.1). As shown in Figure 1A, there are joint molecule (JM) intermediates and nicked circle (NC) products. The yield of DNA strand exchange can be calculated through the equation: .
3.3. D-loop Formation Assay
This assay describes the formation of D-loops catalyzed by the S. cerevisiae Rad51 protein using a short oligo and a homologous supercoiled dsDNA substrate. D-loop formation by yeast Rad51 is dependent on Rad54. The D-loop yield is very time-dependent and declines typically after 10 min, presumably caused by the motor activity of Rad54 protein (12).
To 5′ end-label the 95-mer with 32P, set up a labeling reaction as follows: 0.5 μg of gel-purified 95-mer, 2 μL of 10X T4 polynucleotide kinase reaction buffer, 25 units of T4 polynucleotide kinase, 5 μL of [γ-32P]ATP (3000 Ci/mmol), and add H2O to a final volume of 20 μL. Incubate the reaction at 37 °C for 1 hr and inactivate the kinase by incubating at 65 °C for 30 min (see Note 9).
Separate end-labeled oligonucleotides from unincorporated [γ-32P]ATP through a Mini Spin Oligo Column (Roche Applied Science). These spin columns are ready-to-use, disposable, and microcentrifuge-compatible. Prepare the column according to the manufacturer instructions, and load the sample carefully into the center of the column bed. After centrifugation at 1,000 × g for 4 min in a microcentrifuge, recover the eluate containing the end-labeled oligonucleotides. Quantitate 32P labeled 95-mer by counting 1 μL of a 1:10 diluted sample in a scintillation counter, and expect between 20–100 × 106 cpm (Cerenkov).
Set up a 10 μL reaction by incubating 0.67 μM Rad51 (1:3 Rad51/nucleotide) (see Note 6) with 2 μM 95-mer ssDNA at 30 °C for 10 min, in 1X D-loop buffer with 100 ng/μL creatine kinase.
Add 0.1 μM RPA into the reaction. Mix and incubate for 10 min at 30°C. The addition of RPA stabilizes and stimulates D-loop formation.
Add 100 nM Rad54 into the reaction with 56.6 μM (base pair) supercoiled pUC19 dsDNA to initiate D-loop formation. In the presence of Rad54, time points at 0, 2, 5, 10, and 20 min are usually taken. D-loop yield is very time-sensitive (12). Thus, we recommend performing full time courses for each experiment.
Stop the reaction at desired time points through deproteinization by adding 2 μL of stop buffer into a 10 μL sample reaction, and incubate at 37 °C for 20 min.
Add 1.5 μL 10X loading dye into each sample, and analyze by electrophoresis in a 1 % agarose-TBE gel for 2.5 h at 100V. Further details can be found in Zhang & Heyer in this book.
Dehydrate the gel by placing it onto a DE81 membrane, on top of a piece of 3M Whatman filter paper and a stack of paper towels. Cover the gel with a layer of plastic wrap, and put a plastic plate on top with a 1 L bottle on top as extra weight to facilitate dehydration. Allow to dehydrate for no more than 1 hr, otherwise bands will diffuse.
Place the wrapped gel with DE81 and filter paper underneath into the gel dryer to dry for 60 min at 80 °C.
Put the dried gel with plastic wrap into the phosphor image screen cassette for exposure. The exposure time is usually about 1–10 hrs, based on the specific activity of the isotope.
After exposure, place the screen face down into the PhosphorImager system, such as a Storm 860 (Molecular Dynamics), and scan the selected area to obtain the image.
Analyze and quantitate the joint molecule (D-loop) yield using densitometry with a program such as ImageQuant (version 5.1). Calculate the yield as a percentage of the input ssDNA.
3.4. D-Loop Extension Assay
The D-loop extension assay tests the ability of DNA polymerases to prime DNA synthesis from the invading strand in a D-loop formed by Rad51, capturing the essence of recombinational DSB in vitro. The size of the newly synthesized DNA in the extended D-loop can be determined by two-dimensional gel electrophoresis (native/denaturing), as shown in Figure 2B (native first dimension) and 2C (denaturing second dimension).
The initial steps of the D-loop extension assay are identical to the D-loop assay. Set up a 10 μL reaction by incubating 0.67 μM Rad51 (1:3 Rad51/nucleotide) with 2 μM 95-mer ssDNA at 30°C for 10 min, in 1X D-loop buffer supplemented with 100 ng/μL creatine kinase and 100 μM each of dATP, dGTP, dTTP, and dCTP (see Note 10).
Add 0.1 μM RPA into the reaction. Mix and incubate for 10 min at 30 °C. Then add 72 nM Rad54 into the reaction with 56.6 μM (base pair) supercoiled pUC19 dsDNA, and incubate for 2 min at 30 °C.
Add 20 nM RFC and 20 nM PCNA into the reaction, and incubate an additional 2 min.
To initiate DNA synthesis, add DNA polymerase to 20 nM final assay concentration. Typically, aliquots are sampled at 0, 2, 5, and 10 min.
Stop the reaction at desired time points through deproteinization by adding 2 μL of stop buffer into a 10 μL sample reaction, and incubate at 30 °C for 20 min.
As described in Section 3.3., analyze samples through electrophoresis using a 1% agarose-TBE gel.
For a two-dimensional gel involving denaturing electrophoresis in the second dimension (Figure 2C), run the first dimension native agarose-TBE gel as described. After finishing, slice individual gel lanes carefully.
Prepare a 1.5 % denaturing agarose gel by adding 360 mL H2O to 6 grams of agarose and heating to 95 °C until the agarose is completely melted. Add 40 mL 10 X denaturing agarose gel buffer and mix well. Equilibrate in a 50 °C water bath.
In a 4 °C cold room, place the cut gel slices at the top of a gel mold. Pour the agarose solution around the slices, taking care to ensure that the slices remain in the desired positions. Allow the gel to solidify.
Run the gel in a 4 °C cold room at low voltage (about 2 V/cm), for several hrs until the bromocresol green dye marker migrates about halfway through gel.
Repeat step 9–13 in section 3.3. for gel handling, image collection, and data analysis.
3.5. DNA Strand Annealing Assay
For DNA strand annealing, we provide two protocols: one is based on radioisotope-labeled substrates, the second on fluorescent dye intercalation (such as DAPI). Both methods will be described separately, and the scheme in Figure 3 depicts a radioactive substrate. Additionally, several different ssDNA oligos with various lengths and composition can be used as substrates, such as short oligos, poly dT and poly dA, or full-length heat-denatured PstI-linearized pUC19 DNA. In this chapter, we will only discuss the annealing activity of Rad52 protein on short oligo substrates, since they are the most common substrates in use. Details on using longer DNA substrates can be found in the reference (9).
For Radioactive Substrates
End label gel-purified oligo A2 using [γ-32P]ATP as described in Section 3.3. Leave complementary oligo A1 unlabeled, as shown in Figure 3.
Set up a 60 μL reaction by mixing 200 nM (nt) of radioactive oligo A2 and 200 nM (nt) of unlabeled oligo A1 in the 1X DNA strand annealing buffer containing 30 mM Tris-acetate (pH 7.5), 5 mM magnesium acetate, and 1 mM DTT, in a total current volume of 54 μL (see Note 11). Assemble the reaction on ice to minimize spontaneous annealing between the complementary oligos.
Add 30 nM RPA and incubate at 30 °C for 15 min (see Note 11). The addition of saturating or over-saturating amounts of RPA decreases the spontaneous annealing between complementary short oligos efficiently. 1 RPA heterotrimer per 25 nts is considered to be a saturating amount.
Initiate the reaction by adding 20 nM Rad52 protein and incubate at 30 °C. Keep the total volume added for RPA and Rad52 to 6 μL, thereby reaching a 60 μL final reaction volume.
At 2, 4, 6, 8, and 10 min, remove a 10 μL sample aliquot and quench it rapidly by mixing it with 3 μL stop buffer. Incubate the mixture for 15 min at 30°C. For the zero-time point sample, proteins and DNA are mixed in the stop buffer.
Mix the sample with 1 μL of 10X loading dye. Pre-run a 1 mm-thick 10% polyacrylamide gel in TBE buffer at 100 V for 20 min to eliminate ammonium persulfate (APS) in the gel, which might interfere with the electrophoresis of DNA oligonucleotides.
After the pre-run, load the samples into the wells and start electrophoresis at 100 V for 1–2 hrs to separate the radioactive annealed product from the substrates.
For the 1 mm-thick polyacrylamide gel, there is no need to dehydrate. Place the gel onto a DE81 membrane on top of a 3M Whatman filter paper, and cover the gel with a layer of plastic wrap. Place the wrapped gel with filter paper underneath into the gel dryer to dry for 60 min at 80 °C.
Place the dried gel with plastic wrap into the phosphorimage screen cassette for exposure. The exposure time is usually about 1–10 hrs, based on the specific activity of the isotope.
After exposure, put the screen face down into the PhosphorImager system, such as a Storm 860 (Molecular Dynamics), and scan the selected area to obtain the image.
Analyze and quantitate the annealed duplex and ssDNA oligonucleotide substrates using densitometry with a program such as ImageQuant (version 5.1). Calculate the annealing efficiency as a percentage of the annealed duplex to the input ssDNA amount.
For Unlabeled Substrates Using DAPI Dye
This protocol describes a real-time and quantitative assay of DNA annealing by Rad52 protein, based on the unique fluorescent property of DAPI. DAPI binds to the minor groove of dsDNA specifically and exhibits an enhanced fluorescent signal at 467 nm, when excited at 345 nm (13, 14). The limitation of this method is the requirement of a spectrofluorimeter and the demand for a larger quantity of protein, compared to the assay as performed with radioactively labeled substrate.
Assemble a 400 μL reaction by adding 200 nM oligo A1 and 200 nM oligo A2 in the 1X DNA strand annealing buffer containing 30 mM Tris-acetate (pH 7.5), 5 mM magnesium acetate, and 1 mM DTT with 0.2 μM DAPI into the cuvette (see Note 11).
Set the excitation and emission wavelengths of the SLM8000 spectrofluorimeter to 345 and 467 nm, respectively. Set the slit widths for excitation and emission light to 1 and 4 mm, respectively. The DAPI fluorescence signal is proportional to the dsDNA concentration up to 10 μM (bp) at a 0.2 μM DAPI concentration (9).
Place the cuvette into the holder of the SLM8000 spectrofluorimeter, and adjust the temperature control to 30 °C.
Add 30 nM of RPA (see Note 11) and incubate at 30 °C for 15 min. The addition of saturating amounts of RPA decreases the spontaneous annealing between complementary short oligonucleotides efficiently.
Initiate the reaction by adding 20 nM Rad52 protein to the reaction at 30 °C, and begin to record the signal at 10 or 20 sec intervals continuously with the above setting for 10 min. Usually, there is a 3–5 sec delay between mixing Rad52 into reaction solution and starting data collection. The total volume added for RPA and Rad52 should be equal to 10 μL to reach a 400 μL final reaction volume.
The increase in DAPI fluorescence signal reflects the annealing of complementary ssDNA oligonucleotides and the formation of dsDNA products. To calculate the percentage of annealing, divide the dsDNA formed by the total input DNA. Define background fluorescence signal on ssDNA substrates as 0 %, and maximum fluorescence increase over ssDNA background on fully annealed dsDNA product as 100 %.
Plot the percentage of annealing over time to demonstrate the annealing role of Rad52, compared to spontaneous annealing in the no protein control.
Acknowledgments
We thank Kirk Ehmsen, William Wright, Clare Fasching, Ryan Janke, Erin Schwartz, Shannon Ceballos, Damon Meyer, Xiao-Ping Zhang, and Margarita Alexeeva for helpful comments on the manuscript. Our work is supported by the NIH (GM58015, CA92276), the DoD (BC083684), an NIH training grant fellowship (5T32CA108459) to J.S., and a TRDRP Postdoctoral fellowship (17FT-0046) to J.L.
Footnotes
The presence of free Mg2+ is critical, since Mg2+-ATP is the substrate of the RecA/Rad51-like proteins. For each enzyme, the optimum concentration of free Mg2+ is different. Ususally, a 1–5 mM free Mg2+ is used for the yeast and human Rad51 proteins. For the human RAD51 protein, the presence of Ca2+ stimulates the DNA strand exchange activity dramatically, compared to Mg2+ alone, by locking hRAD51 into the active, ATP-bound configuration (15). When using Ca2+, its possible effect on other reaction components must be considered.
Store purified proteins at −80 °C. Thaw and dilute immediately before use, to avoid loss of activity. Frequent freeze/thaw cycles can decrease protein activity. To minimize this loss, aliquot purified protein stocks into small volumes prior to storage. For protocols to ensure the absence of relevant contamination in preparations of HR proteins see chapter by Zhang & Heyer (this volume).
Yeast Rad51 prefers linearized ϕX174 dsDNA with 3′ overhang (e.g. PstI-linearized) as the substrate in the DNA strand exchange assay, whereas human RAD51 prefers linearized ϕX174 dsDNA with 5′ overhangs (e.g. ApaL1-linearized). Furthermore, while E. coli RecA and bacteriophage T4 UvsX catalyze efficient DNA strand exchange using phage M13mp18 DNA substrates, the efficiency of DNA strand exchange with M13mp18 substrates is poor when using eukaryotic Rad51 proteins. The reasons for this substrate preference are unknown. Furthermore, the end products in DNA strand exchange for these proteins are quite different as well. RecA catalyzes the DNA strand exchange reaction highly efficiently, accumulating nicked circles as the final product. UvsX catalyzes the same reaction very fast and through multiple invasions, leading to the formation of large DNA networks (shown as aggregates in the well) as the major product. Yeast Rad51 catalyzes DNA strand exchange much less efficiently than RecA, accumulating joint molecule intermediates and fewer nicked circle products. Finally, human Rad51 protein is even less efficient, since nicked circle formation is very low and joint molecules are the major species.
After the addition of ethidium bromide, the sample tube should be wrapped with foil and work should be performed in low light until after the ethidium bromide is removed, to avoid nicking the DNA. Be sure to wear gloves for the steps involving ethidium bromide and CsCl. Ethidium bromide is a mutagen and carcinogen and direct contact with it should be avoided. Cesium is a heavy metal, and exposure to it should be avoided. If there is a spill, rinse the contaminated area with a large amount of H2O.
The addition of corresponding protein storage buffer to match the ionic strength in the no protein control to the ionic strength of the experimental samples is critical. The D-loop and DNA strand exchange assays catalyzed by RecA/Rad51 family proteins are very sensitive to the ionic strength. Artifacts can be avoided by ensuring that the actual buffer components in the reaction are identical.
The ratio between Rad51 protein and DNA is key for high yield in D-loop and DNA strand exchange reactions. The nucleotide binding site size of Rad51 is n = 3, which means one Rad51 binds 3 nucleotides in a saturated nucleoprotein filament. Higher or lower Rad51 to ssDNA ratios decrease the yields in D-loop and DNA strand exchange reactions. However, suboptimal conditions (non-optimal Rad51:ssDNA ratio, elevated salt concentration, presence of RPA inhibition, order-of-addition changes) can reveal the functions and mechanisms of ancillary proteins. For example, Rad54 can significantly boost the DNA strand exchange activity of sub-saturating amounts of Rad51 (Rad51/nucleotide = 1:14) (2).
The ATP regenerating system is based on creatine kinase and phosphocreatine. Other regenerating systems can be used, such as pyruvate kinase and phosphoenolpyruvate (PEP). Commercial kinases are sometimes supplied as an ammonium sulfate suspension in buffer. This buffer might contain salts or chemicals that inhibit D-loop formation and DNA strand exchange activity. To avoid this, centrifuge a small aliquot of the kinase before each use, and resuspend the kinase pellet in the corresponding reaction buffer.
The ethidium bromide staining solution is stable for several months at 4 °C. If the dye concentration is low in this staining solution, add 10 μL of 10 mg/mL ethidium bromide solution into the staining solution before each new staining. In this way, ethidium bromide waste is minimized. ssDNA does not stain well by ethidium bromide, and a brief destaining for only 10 min will enhance the ssDNA signal.
We recommend to order PAGE-purified oligonucleotides or to purify the oligonucleotide using 10–16% polyacrylamide gels containing 8 M urea. For all steps below involving radioactive materials, wear double gloves throughout and work on designated radioactive benches. All waste must be monitored. Discard all radioactive waste into the designated solid or liquid waste containers for 32P.
It is important to heat inactivate Polynucleotide Kinase, to avoid subsequent inhibition of the D-loop formation by the binding of kinase onto labeled 95-mer oligonucleotides and labeling of nicked or linearized plasmid during D-loop formation and synthesis.
In the D-loop extension assay, it is important to consider the KM for dNTPs of the individual DNA polymerase. For example, translesion synthesis DNA polymerase η requires at least 100 μM each dNTP for efficient synthesis, while DNA polymerase δ can extend efficiently at a 10-fold lower concentration. If [α-32P]dCTP (0.22 μM, 6000 uCi/mmol, Perkin Elmer) is used instead of labeled 95-mer to increase sensitivity, be aware that some DNA polymerases are highly sensitive to unbalanced dNTPs pools.
The presence of Mg2+ is key to condense the ssDNA structure and suppress DNA breathing at the dsDNA ends. Thus, fewer artifacts from DNA annealing will be introduced through the initial protein-DNA binding and the following deproteinization treatment. The presence of saturating amount of RPA (1 RPA/25 nts) better mimics the physiologically relevant situation and avoids artifacts caused by DNA condensation (protein-mediated aggregation) and spontaneous annealing.
References
- 1.Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Solinger JA, Kiianitsa K, Heyer WD. Rad54, a Swi2/Snf2-like recombinational repair protein, disassembles Rad51:dsDNA filaments. Mol Cell. 2002;10:1175–1188. doi: 10.1016/s1097-2765(02)00743-8. [DOI] [PubMed] [Google Scholar]
- 3.Prakash R, Satory D, Dray E, Papusha A, Scheller J, Kramer W, Krejci L, Klein H, Haber JE, Sung P, Ira G. Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev. 2009;23:67–79. doi: 10.1101/gad.1737809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.New JH, Sugiyama T, Zaitseva E, Kowalczykowski SC. Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature. 1998;391:407–410. doi: 10.1038/34950. [DOI] [PubMed] [Google Scholar]
- 5.Li X, Stith CM, Burgers PM, Heyer WD. PCNA is required for initiating recombination-associated DNA synthesis by DNA polymerase δ. Mol Cell. 2009;36:704–713. doi: 10.1016/j.molcel.2009.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krogh BO, Symington LS. Recombination proteins in yeast. Annu Rev Genet. 2004;38:233–271. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- 7.Sugiyama T, Kantake N, Wu Y, Kowalczykowski SC. Rad52-mediated DNA annealing after Rad51-mediated DNA strand exchange promotes second ssDNA capture. EMBO J. 2006;25:5539–5548. doi: 10.1038/sj.emboj.7601412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu Y, Kantake N, Sugiyama T, Kowalczykowski SC. Rad51 protein controls Rad52-mediated DNA annealing. J Biol Chem. 2008;283:14883–14892. doi: 10.1074/jbc.M801097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sugiyama T, New JH, Kowalczykowski SC. DNA annealing by Rad52 Protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc Natl Acad Sci USA. 1998;95:6049–6054. doi: 10.1073/pnas.95.11.6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shinohara A, Ogawa T. Stimulation by Rad52 of yeast Rad51-mediated recombination. Nature. 1998;391:404–407. doi: 10.1038/34943. [DOI] [PubMed] [Google Scholar]
- 11.Sung P. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J Biol Chem. 1997;272:28194–28197. doi: 10.1074/jbc.272.45.28194. [DOI] [PubMed] [Google Scholar]
- 12.Bugreev DV, Hanaoka F, Mazin AV. Rad54 dissociates homologous recombination intermediates by branch migration. Nature Struct Mol Biol. 2007;14:746–753. doi: 10.1038/nsmb1268. [DOI] [PubMed] [Google Scholar]
- 13.Kapuscinski J, Szer W. Interactions of 4′, 6-diamidine-2-phenylindole with synthetic polynucleotides. Nucleic Acids Res. 1979;6:3519–3534. doi: 10.1093/nar/6.11.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kubista M, Akerman B, Norden B. Characterization of interaction between DNA and 4′,6-diamidino-2-phenylindole by optical spectroscopy. Biochemistry. 1987;26:4545–4553. doi: 10.1021/bi00388a057. [DOI] [PubMed] [Google Scholar]
- 15.Bugreev DV, Mazin AV. Ca2+ activates human homologous recombination protein Rad51 by modulating its ATPase activity. Proc Natl Acad Sci USA. 2004;101:9988–9993. doi: 10.1073/pnas.0402105101. [DOI] [PMC free article] [PubMed] [Google Scholar]