

ABSTRACT
Zn2+ is an essential micronutrient and an important ionic signal whose excess, as well as scarcity, is detrimental to cells. Free cytoplasmic Zn2+ is controlled by a network of Zn2+ transporters and chelating proteins. Recently, lysosomes became the focus of studies in Zn2+ transport, as they were shown to play a role in Zn2+-induced toxicity by serving as Zn2+ sinks that absorb Zn2+ from the cytoplasm. Here, we investigated the impact of the lysosomal Zn2+ sink on the net cellular Zn2+ distribution and its role in cell death. We found that lysosomes played a cytoprotective role during exposure to extracellular Zn2+. Such a role required lysosomal acidification and exocytosis. Specifically, we found that the inhibition of lysosomal acidification using Bafilomycin A1 (Baf) led to a redistribution of Zn2+ pools and increased apoptosis. Additionally, the inhibition of lysosomal exocytosis through knockdown (KD) of the lysosomal SNARE proteins VAMP7 and synaptotagmin VII (SYT7) suppressed Zn2+ secretion and VAMP7 KD cells had increased apoptosis. These data show that lysosomes play a central role in Zn2+ handling, suggesting that there is a new Zn2+ detoxification pathway.
KEY WORDS: Zinc, Golgi, Lysosome, Metallothionein, Exocytosis, Zn2+ transport, ZnT, Slc30a
INTRODUCTION
Cellular Zn2+ dyshomeostasis has been linked to a number of human pathologies including growth defects (Prasad, 2013), impaired immune function (Rink and Gabriel, 2000), diabetes (Jansen et al., 2009) and neurodegenerative diseases (Forsleff et al., 1999; Rulon et al., 2000; Lee et al., 2002; Vinceti et al., 2002). Regulation of cellular Zn2+ levels involves controlling its influx, export and chelation. In general, Zn2+ transport is regulated by ZnT (also known as solute carrier family 30) and ZIP (also known as solute carrier family 39) transporters, and it is chelated by Zn2+-binding metallothioneins.
In addition to Zn2+ evacuation across the plasma membrane by the Zn2+ transporter ZnT1 (SLC30A1) (Palmiter and Findley, 1995), Zn2+ is exported from the cytoplasm into the organelles by the dedicated ZnT transporters such as ZnT6 (SLC30A6) for the Golgi (Huang et al., 2002), and ZnT2 (SLC30A2) and ZnT4 (SLC30A4) for the lysosome (Palmiter et al., 1996; Huang and Gitschier, 1997; Falcón-Pérez and Dell'Angelica, 2007; McCormick and Kelleher, 2012). This organellar Zn2+ export lowers potentially toxic cytoplasmic Zn2+ concentrations in pathophysiological conditions such as neurodegeneration (Kanninen et al., 2013) and breast cancer (Lopez et al., 2011). Moreover, it provides Zn2+ to organellar processes that require it, such as the maturation of enzymes like the lysosomal acid sphingomyelinase (Schissel et al., 1996), and for the secretion of Zn2+ under normal physiological conditions such as synaptic transmission (Frederickson and Bush, 2001) and lactation (Kelleher et al., 2009).
The upregulation of Zn2+ chelation and transport machinery following the activation of the transcription factor MTF-1 by Zn2+ binding (Andrews, 2001) requires time for transcription, translation and protein processing. It is tempting to speculate that Zn2+ export into organelles serves as a first line of defense to provide temporary Zn2+ storage, giving cells time to upregulate Zn2+ chelators and transporters. Our recent data on the role of lysosomes in Zn2+ handling, as well as some recently published results suggest that lysosomes play a role as such Zn2+ sinks, temporarily storing Zn2+ (Hwang et al., 2008; Kukic et al., 2013). In this paper, we sought to delineate the role of lysosomes in protection against Zn2+-induced toxicity.
Zn2+ is transported from the cytoplasm into lysosomes by ZnT2 and ZnT4 (Palmiter et al., 1996; Huang and Gitschier, 1997; Falcón-Pérez and Dell'Angelica, 2007). Zn2+ can also be delivered to the lysosomes through endocytosis or autophagy (Lee and Koh, 2010; Cho et al., 2012). What happens to Zn2+ absorbed by the lysosomes? A recent series of work from several laboratories indicate that Zn2+ buildup in the lysosomes is toxic. It leads to lysosomal membrane permeabilization (LMP), to the release of the lysosomal enzymes such as cathepsins and to cell death (Hwang et al., 2008; Chung et al., 2009; Lee et al., 2009; Hwang et al., 2010). As such, the lysosomal Zn2+ accumulation might constitute a cell death mechanism during normal remodeling of Zn2+-rich tissues, such as the mammary gland (Kelleher et al., 2011), as well as in pathological conditions. With this in mind, we sought to answer whether or not accumulation of Zn2+ in the lysosome is the terminal depot for cellular Zn2+.
Alternatively, it is possible that lysosomal Zn2+ dissipates and lysosomes constitute only a temporary Zn2+ storage site. Our recently published data suggest that the lysosomal ion channel transient receptor potential mucolipin 1 (TRPML1, also known as mucolipin1, encoded by the MCOLN1 gene) is at least partly responsible for dissipating lysosomal Zn2+ into the cytoplasm (Kukic et al., 2013). It should be noted that lysosomes fuse with the plasma membrane through a process involving a specific SNARE complex, which includes the VAMP7 protein and synaptotagmin VII (SYT7) (Martinez-Arca et al., 2000; Braun et al., 2004; Rao et al., 2004; Logan et al., 2006; Mollinedo et al., 2006). It has recently been proposed that such secretion contributes to the excretion of undigested/indigestible products inside lysosomes (Medina et al., 2011). In the course of the present study, we used VAMP7 and SYT7 knockdown (KD) to suppress lysosomal secretion and assess its role in Zn2+ clearance from the cells.
Here, we aimed to establish the functional context of the lysosomal Zn2+ accumulation. Our findings indicate that lysosomes actively absorb Zn2+ and secrete it across the plasma membrane, given that suppressing the lysosomal Zn2+ absorption or secretion causes Zn2+ buildup in the cytoplasm, Golgi and mitochondria, leading to apoptosis.
RESULTS
In order to test the role of the lysosomal Zn2+ sink on cellular Zn2+ handling, we blocked the lysosomal H+ pump in HeLa cells using 1 µM Baf and exposed cells to 100 µM ZnCl2 for 3 hours. The resulting cytoplasmic Zn2+ spikes were measured using live-cell confocal microscopy and FluoZin-3,AM as described previously (Kukic et al., 2013) Fig. 1A shows that the exposure of Baf-treated cells to Zn2+ caused a significantly higher FluoZin-3,AM response than the exposure of untreated cells to Zn2+. Although Baf has been shown to decrease cytoplasmic pH, potentially affecting Zn2+ binding to cytoplasmic proteins, or FluoZin-3,AM fluorescence, the magnitude of the observed effects appear to be incompatible with the quantitative estimates of changes induced by Baf. Thus, the effect of Baf on cytoplasmic pH appears to be small, within only tenths of pH units (Heming et al., 1995). The degree of pH change necessary to cause an effect on Zn2+ handling, by contrast, significantly exceeds that reported to be caused by Baf. A pH drop below 6.7 is required to trigger an increase in intracellular Zn2+ according to one set of studies (Kiedrowski, 2012), whereas another set has shown that metallothioneins release Zn2+ only after cytoplasmic pH drops below 5.0 (Jiang et al., 2000). Thus, the increase in cytoplasmic Zn2+ caused by Baf likely correlates with the loss of lysosomal function, rather than cytosolic pH changes.
Fig. 1.

Inhibition of lysosomal Zn2+ sink function by Baf increases cytoplasmic Zn2+ levels. (A) Confocal images of HeLa cells treated with 100 µM ZnCl2 and/or 1 µM Baf for 3 hours then loaded with FluoZin-3,AM and LysoTracker. Note disappearance of LysoTracker staining, indicative of lysosomal deacidification, and an increase in FluoZin-3,AM staining intensity, indicative of increased Zn2+. (B) Confocal images of control or ZnT siRNA transfected HeLa cells (48 hours post-transfection) treated with 100 µM ZnCl2 for 3 hours then loaded with FluoZin-3,AM. Images represent at least three separate experiments and at least three images per condition in each experiment. Scale bars: 20 µm.
We have previously shown that Zn2+ transporters ZnT2 and ZnT4 colocalize with the lysosomal ion channel TRPML1 in HeLa cells (Kukic et al., 2013). We suggested that these transporters play a role in loading of the lysosomes with Zn2+. In order to test this assumption, we performed ZnT2 and ZnT4 KD using siRNA as described previously, and tested the resulting changes in Zn2+ handling using FluoZin-3,AM. Fig. 1B shows that ZnT2 and ZnT4 KD increased cytoplasmic Zn2+ levels observed in these cells after a 3-hour long treatment with 100 µM ZnCl2. These results are in agreement with the previously published data on the dependence of ZnTs activity on the acidic environment of the lysosomes (Chao and Fu, 2004; Ohana et al., 2009) for Zn2+ binding and transporting activity.
The upregulation of MTF-1-dependent, Zn2+-responsive genes such as metallothionein 2A (MT2a) and ZnT1 (Saydam et al., 2002) indicates elevated cytoplasmic Zn2+. MT2a mRNA was used previously in our studies of the role of TRPML1 in Zn2+ handling. We measured the expression of the mRNA of these genes using qRT-PCR (Fig. 2). An increase in MT2a and ZnT1 mRNA responses to Zn2+ in cells treated with Baf is evident. With MT2a mRNA levels in DMSO-treated (no Zn2+) cells set as 100%, MT2a mRNA levels were 816.3%9±73.61 (in cells treated with DMSO and Zn2+ (n = 4; P<0.001), and 1174.61%±94.20 (mean±s.e.m., n = 4; P<0.05 relative to DMSO-only, and DMSO plus Zn2+ controls) in cells treated with Baf and Zn2+ (Fig. 2A). ZnT1 mRNA increased to 323.43%±23.32 (n = 4; P<0.001) in cells treated with DMSO plus Zn2+, and to 552.09%±40.46 (n = 4; P<0.05 relative to DMSO-only, and DMSO plus Zn2+ controls) of control values in cells treated with Baf plus Zn2+ (Fig. 2B). In addition to confirming the elevated cytoplasmic Zn2+ levels due to Baf, as assessed by FluoZin-3,AM in Fig. 1A, these data also corroborate MTF-1 activation due to cytoplasmic Zn2+ buildup.
Fig. 2.
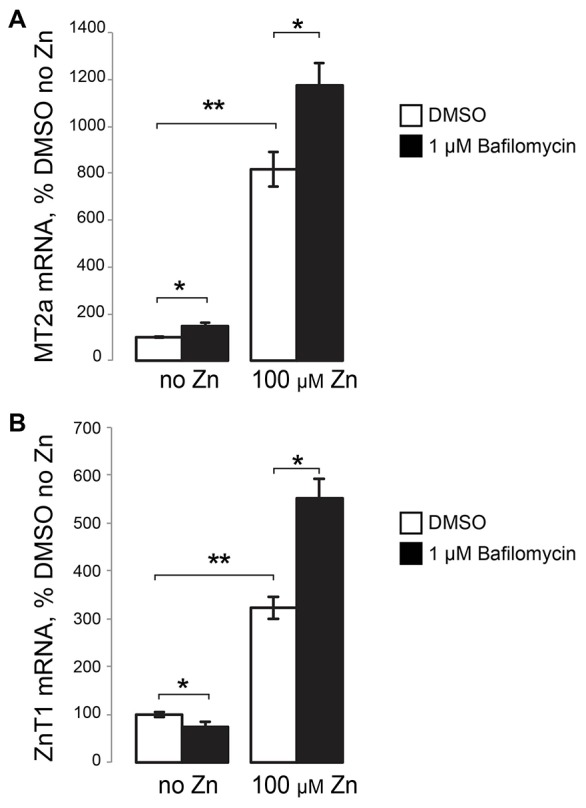
Inhibition of lysosomal Zn2+ sink function by Baf increases the transcriptional response of Zn2+-responsive genes. qRT-PCR results of MT2a (A) and ZnT1 (B) mRNA, shown as percentage of DMSO-only-treated (no Zn2+) cells. RNA was isolated from HeLa cells treated for 3 hours with either DMSO or 1 µM Baf alone or with 100 µM ZnCl2. Results are mean±s.e.m.; *P<0.05; **P<0.001.
In addition to increasing cytoplasmic Zn2+, suppression of the lysosomal function caused redistribution of Zn2+ storage pools. Concentration of FluoZin-3 fluorescence in intracellular inclusions was noted in Baf-treated cells exposed to Zn2+. At least some of these inclusions were positive for the Golgi marker GalT–mCherry (Fig. 3A). Under these conditions, Zn2+ also accumulated in the mitochondria, which was shown using the mitochondrial Zn2+ dye RhodZin-3,AM, whose signal was brighter in Baf-treated than in control Zn2+-treated cells (Fig. 3B). Therefore, suppression of lysosomal function leads to the loss of Zn2+-buffering capacity and to a spike in cytoplasmic Zn2+ when cells are exposed to Zn2+. In the absence of Zn2+ buffering by the lysosomes, Zn2+ is redirected to other organelles.
Fig. 3.

Inhibition of lysosomal Zn2+ sink function by Baf redistributes cellular Zn2+ pools to the Golgi and the mitochondria. (A) Confocal images of GalT–mCherry-transfected HeLa cells treated with 100 µM ZnCl2 and/or 1 µM Baf for 3 hours, then loaded with FluoZin-3,AM. The black-and-white panes show overlap between the green and the red channels, obtained using the RG2B function of ImageJ. (B) Confocal images of HeLa cells treated with 100 µM ZnCl2 and/or 1 µM Baf for 3 hours, then loaded with RhodZin-3,AM. Plot profiles on the right show intensity profiles of RhodZin-3,AM fluorescence recorded along the lines indicated in the corresponding image. Note increased RhodZin-3,AM florescence with Baf-treated cells. Images represent at least three separate experiments and at least three images per condition in each experiment. Scale bars: 20 µm.
As previously shown, high Zn2+ is toxic to the cells owing to its buildup in the cytoplasm and in the organelles (Medvedeva et al., 2009). If the lysosomes are a Zn2+-buffering sink, then inhibiting that function should result in cell death. Our findings in Fig. 4A support this: although HeLa cells were fairly resistant to the effects of 100 µM ZnCl2 or 1 µM Baf alone, their combination caused pronounced caspase-3 (Cas3) activation, indicative of apoptosis. An exposure of cells to 100 µM ZnCl2 for 48 hours increased Cas3 activity by 43.90%±14.25 (mean±s.e.m., n = 3; P<0.05) and exposure to 1 µM Baf increased Cas3 activity by 179.5%±68.04 (n = 3, P<0.05). A combination of Zn2+- and Baf-exposure increased Cas3 activity by 387.87%±36.70 (n = 3, P<0.001 relative to untreated control), suggesting that Baf enhances the pro-apoptotic effects of Zn2+. Given that Baf is conventionally used to block the lysosomal H+ pump, we think that the simplest interpretation of these data are as diminished cytoprotective capacity of the lysosomal Zn2+ sink in Baf-treated cells.
Fig. 4.
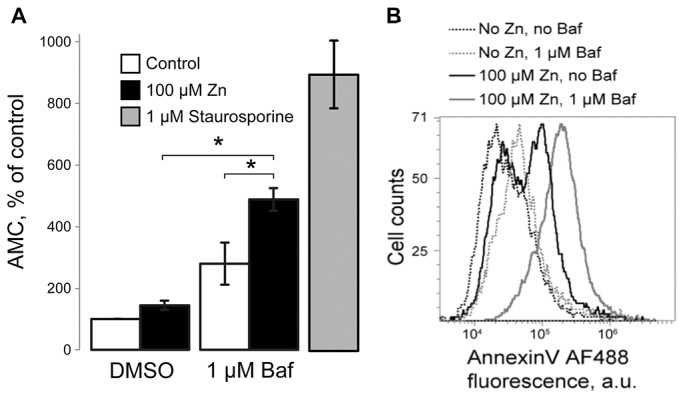
Inhibition of lysosomal function leads to increased cell death upon high Zn2+ exposure. (A) Caspase-3 (Cas3) activity assay showing increased Cas3 activation upon a 48-hour 100 µM ZnCl2 and 1 µM Baf treatment in HeLa cells. A 3-hour long exposure to 1 µM staurosporine was used as a positive control, which increased Cas3 activity by 796.72%±109.06 (n = 3, P<0.001). Cas3 activity is shown as AMC fluorescence and a percentage of untreated controls. Results are mean±s.e.m.; *P<0.05. (B) Flow cytometry data showing increased AnnV staining of cells treated with 100 µM ZnCl2 and/or 1 µM Baf for 48 hours. Data represent three experiments.
As a complimentary cell death assay, Annexin V (AnnV) and Propidium Iodide (PI) staining were analyzed using flow cytometry. Fig. 4B and supplementary material Fig. S1 show an increase in the number of AnnV-positive, apoptotic cells when cells are treated for 48 hours with Zn2+ plus Baf. Taken together, these data show that suppression of the lysosomal Zn2+ sink facilitates the apoptotic cell death caused by Zn2+ exposure. They suggest that lysosomes play a crucial role in buffering cytoplasmic Zn2+ and in its detoxification. A loss of such a role exposes cells to the pro-apoptotic effects of Zn2+.
Zn2+ toxicity has been linked to LMP, resulting in cell death under some conditions (Hwang et al., 2008; Chung et al., 2009; Lee et al., 2009; Hwang et al., 2010). Why is the lysosomal Zn2+ sink cytoprotective under some, but not other conditions of Zn2+ exposure? We propose that the switch between pro- and anti-apoptotic effects of the Zn2+ sink is dictated by the rate of Zn2+ absorption by the lysosomes and/or the rate of its clearance from the lysosomes. If the rate of Zn2+ clearance exceeds the rate of its sequestration into the lysosomes, then the Zn2+ sink is cytoprotective. A rate of sequestration exceeding the rate of clearance leads to LMP and cell death (see model in Fig. 5). Zn2+ clearance might occur through a Zn2+ leak into the cytoplasm [as suggested by our recent publication (Kukic et al., 2013)], or through its secretion mediated by lysosomal fusion with the plasma membrane. The latter has recently gained a lot of attention as detoxification mechanism in the lysosomal storage diseases (Fraldi et al., 2010; Medina et al., 2011; Palmieri et al., 2011; Decressac et al., 2013; Pastore et al., 2013). The next set of experiments tested the role of lysosomes in Zn2+ secretion.
Fig. 5.
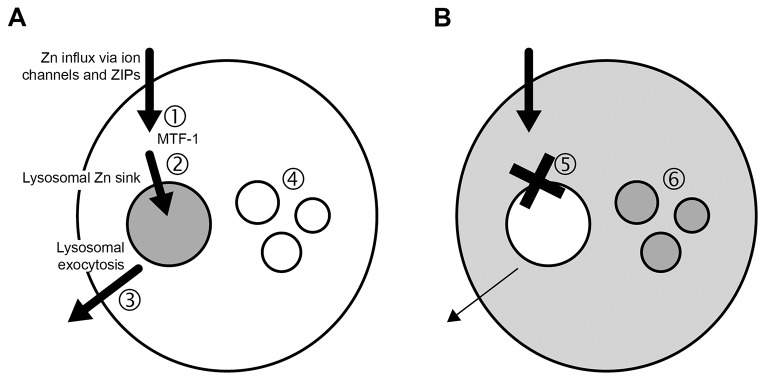
A model of lysosomal Zn2+ sink and its role in Zn2+ detoxification. (A) Under normal conditions, Zn2+ enters the cells through ion channels and ZIPs and is (1) recognized by the Zn2+-sensitive transcription factor MTF-1 that, once Zn2+-bound, translocates to the nucleus to upregulate ZnT1 and MT2a gene expression. (2) Zn2+ is transported out of the cytoplasm and into the lysosome through ZnT2 and ZnT4. Zn2+ that builds up in the lysosomal Zn2+ sink is then secreted across the plasma membrane (3) through a VAMP7-dependent mechanism. This prevents toxic Zn2+ buildup in the cytoplasm and other organelles (4). (B) Suppression of Zn2+ absorption by the lysosomal Zn2+ sink (5), leads to toxic levels of Zn2+ buildup in the cytoplasm as well as other organelles (6), such as the Golgi and mitochondria.
We suppressed lysosomal secretion by using siRNAs against two lysosomal SNARE components, VAMP7 and SYT7 (Martinez-Arca et al., 2000; Braun et al., 2004; Rao et al., 2004; Logan et al., 2006; Mollinedo et al., 2006; Flannery et al., 2010). Fig. 6A shows that VAMP7 siRNA reduced VAMP7 mRNA to 27.76%±3.99 of control siRNA levels (mean±s.e.m., n = 4, P<0.001), whereas Fig. 7A shows that SYT7 siRNA reduced SYT7 mRNA to 30.12%±4.11 of control siRNA levels (n = 3, P<0.001). VAMP7 mRNA levels were not altered by either a 3- or 48-hour exposure to 100 µM ZnCl2 (I. K., unpublished observation; SYT7 dependence on Zn2+ was not tested). VAMP7 KD was confirmed by examining protein levels through western blotting confirming that VAMP7 siRNA reduced VAMP7 protein expression to 30.21%±6.82 of control siRNA levels (n = 5, P<0.001) (Fig. 6B). VAMP7 and SYT7 KD were associated with changes in the lysosomal numbers and organization. There was an increase in total lysosomal numbers in VAMP7 KD cells (supplementary material Fig. S2), although there was a high degree of cell-to-cell variation with that metric. Both VAMP7 and SYT7 KD also caused clustering of lysosomes in large structures, which is compatible with the role of these SNARE proteins in membrane traffic. Examples of such clustering and its statistical analysis are shown in Figs 6, 7; supplementary material Fig. S2.
Fig. 6.

Inhibition of lysosomal exocytosis through VAMP7 KD inhibits Zn2+ secretion. (A) qRT-PCR results confirming VAMP7 KD by siRNA. (B) Quantification of western blot results confirming VAMP7 KD. Insert shows a representative blot (of five) of endogenous VAMP7 and actin levels under with either control or VAMP7 siRNA. (C) β-hexosaminidase activity assay for lysosomal exocytosis. Results are shown as the percentage of total β-hexosaminidase secreted after either 10 or 30 minutes; 1 µM ionomycin was used to stimulate lysosomal exocytosis. (D) Zn2+ secretion assay using cell-impermeant FluoZin-3 tetrapotassium salt. Results are shown as the percentage of the maximum fluorescence value recorded in the control+Zn2+ samples after 30 minutes, which were set at 100%. Results are mean±s.e.m.; *P<0.05, **P<0.001.
Fig. 7.

Inhibition of lysosomal exocytosis through SYT7 KD inhibits Zn2+ secretion. (A) qRT-PCR results confirming SYT7 KD by siRNA. (B) β-hexosaminidase activity assay for lysosomal exocytosis. Results are shown as the percentage of total β-hexosaminidase secreted over 10 or 30 minutes; 1 µM ionomycin was used to stimulate lysosomal exocytosis. (C) Zn2+ secretion assay using cell-impermeant FluoZin-3 tetrapotassium salt. Results are shown as percent of the maximum fluorescence value recorded in the control+Zn2+ samples at 30 minutes, which were taken as 100%. Results are mean±s.e.m.; *P<0.05, **P<0.001. (D) Confocal images of control, VAMP7 and SYT7 KD cells untreated or treated for 3 hours with 100 µM ZnCl2 and loaded with FluoZin-3,AM (green) and LysoTracker (red). Scale bars: 20 µm.
VAMP7 and SYT7 KD were functionally significant as they caused a loss of secreted activity of the lysosomal enzyme β-hexosaminidase (β-hex), a common marker of lysosomal exocytosis. In these experiments, lysosomal secretion was initiated by treatment with 1 µM of the Ca2+ ionophore ionomycin (Ion) (Fig. 6C; Fig. 7B). After 30 minutes, control KD cells secreted 18.80%±0.66 of their total β-hex content without stimulation, whereas VAMP7 KD cells secreted 14.09%±1.60 of their total β-hex content (mean±s.e.m., Fig. 6C). Once stimulated with ionomycin, however, control KD cells secreted 32.57%±2.36 of their total β-hex content, whereas VAMP7 KD cells only secreted 18.87%±1.57 of their total β-hex content (n = 3, P<0.001) (Fig. 6C). Similarly, SYT7 KD was also functionally significant, decreasing lysosomal exocytosis compared to control KD. Fig. 7B shows that after 30 minutes, ionomycin-treated control KD cells secreted 35.57%±1.36 of their total β-hex content, whereas SYT7 KD cells secreted 20.29%±1.65 of their total β-hex content (n = 3, P<0.001).
To measure the effect of suppressing lysosomal exocytosis on Zn2+ secretion, we incubated control and VAMP7, or SYT7 KD cells with 100 µM ZnCl2 for 3 hours. Next, cells were placed in normal medium (no Zn2+) and Zn2+ secretion was analyzed using FluoZin-3 fluorescence as described previously (Kukic et al., 2013). Both VAMP7 (Fig. 6D) and SYT7 (Fig. 7C) KD significantly reduced Zn2+ secretion. Within 30 minutes of secretion, VAMP7 KD cells were only able to secrete 51.79%±19.26 (n = 3, P<0.05) of the amount of Zn2+ secreted by the control KD cells (100%) (Fig. 6D), indicating that VAMP7 is necessary for Zn2+ secretion. Similarly, SYT7 KD cells were only able to secrete 58.90±2.87% (n = 3, P<0.05) of the amount of Zn2+ secreted by the control KD cells at 30 minutes, which was taken as 100% (Fig. 7C). This inhibition of Zn2+ secretion in VAMP7 and SYT7 KD cells was also corroborated by the elevated cellular Zn2+ levels, as seen by assessing FluoZin-3,AM staining using confocal microscopy (Fig. 7D).
The effects of suppressing lysosomal secretion on Zn2+-induced cell death were analyzed using the AnnV and PI assay described above. This assay revealed significant upregulation of Zn2+-induced cell death in VAMP7 KD cells treated with 100 µM ZnCl2 for 48 hours, compared to control KD cells undergoing the same treatment (Fig. 8; supplementary material Fig. S1). In summary, the data described here show that the Zn2+ sink integrates Zn2+ absorption from the cytoplasm with its secretion through lysosomal exocytosis. Both aspects of the Zn2+ sink function are new and necessary for Zn2+ detoxification.
Fig. 8.
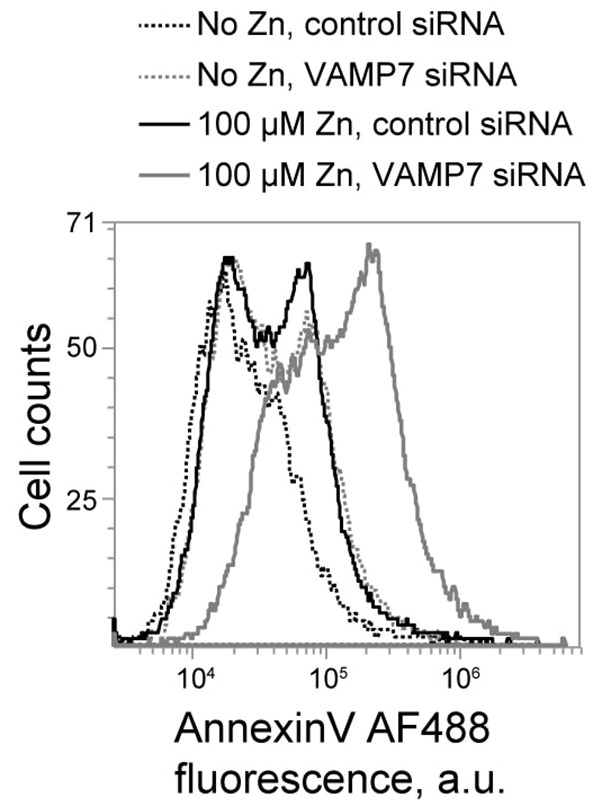
Inhibition of lysosomal exocytosis through VAMP7 KD increases cell death in Zn2+-treated cells. Flow cytometry data of AnnV- and PI-stained control and VAMP7-KD cells treated for 48 hours with 100 µM ZnCl2, showing increased Annexin V fluorescence in VAMP7- Zn2+ treated cells (gray solid line) compared to control- Zn2+ treated cells (solid black line).
Finally, because lysosomal biogenesis and exocytosis are regulated by TFEB and related factors (Sardiello et al., 2009; Martina et al., 2014), TFEB should have an impact on lysosomal Zn2+ clearance. Indeed, Fig. 9 shows that TFEB overexpression, a common protocol used to study its function, increases both lysosomal exocytosis and Zn2+ secretion. Fig. 9A shows that after 30 minutes, mock (empty vector)-transfected cells secreted 13.95%±0.67 of their total β-hex content, while TFEB-transfected cells secreted 23.11%±3.75 (mean±s.e.m.; n = 3, P<0.05) of their total β-hex content. Once stimulated with ionomycin, mock-transfected cells secreted 20.30%±1.47 (n = 3, P<0.01) of their total β-hex content, whereas TFEB-transfected cells secreted 38.65%±1.98 (n = 3, P<0.001) of their total β-hex content. TFEB also increased Zn2+ secretion. Fig. 9B shows that after 15 minutes, control KD cells were able to secrete 76.70%±1.37 of secretable Zn2+ in our assay, whereas TFEB-transfected cells were able to secrete 104.52%±10.12 (n = 3, P<0.05) of secretable Zn2+. This increase in Zn2+ secretion was dependent on the lysosomal fusion machinery as TFEB cDNA and SYT7 siRNA transfected cells had similar levels of secretion to control cells (71.70%±3.09). To our knowledge, this is the first evidence linking increased Zn2+ secretion by TFEB and lysosomal exocytosis.
Fig. 9.

Enhancing lysosomal exocytosis though TFEB overexpression increases Zn2+ secretion. (A) β-hexosaminidase activity assay for lysosomal exocytosis. Results are shown as the percentage of total β-hexosaminidase secreted over 10 or 30 minutes; 1 µM ionomycin was used to stimulate lysosomal exocytosis. (B) Zn2+ secretion assay using cell-impermeant FluoZin-3 tetrapotassium salt. Results are shown as the percentage of the maximum fluorescence value recorded in the control+Zn2+ samples at 30 minutes, which were taken as 100%. The results shown above are after 15 minutes of secretion time (chase). Results are mean±s.e.m.; *P<0.05; **P<0.001.
DISCUSSION
Although lysosomes are most commonly discussed in terms of their role in endocytic digestion and absorption, mounting evidence points towards their role in cell death and in signaling (Settembre et al., 2013). Recent evidence indicates that under oxidative stress conditions, Zn2+ accumulates in the lysosomes and can lead to LMP (Hwang et al., 2008). Furthermore, this Zn2+ dysregulation is mechanistically linked to autophagy (Lee et al., 2009; Yoon et al., 2010; Cho et al., 2012). Considering that autophagy and oxidative stress play a crucial role in cell death and neurodegenerative pathologies, it is likely that Zn2+ represents a new target for modulating diseases and a key step in understanding neuronal cell death. These recent developments emphasize the role of lysosomes in metal toxicity. Thus, the accumulation of Zn2+ and other metals in lysosomes has been shown to lead to LMP, to the release of lysosomal enzymes into the cytoplasm, and to cell death. Although pathological aspects of Zn2+ buildup in the lysosomes are undisputable, its physiological role is unclear.
Our recent data suggest that lysosomes play a role of a ‘Zn2+ sink’, working in parallel with the transcriptional regulation of Zn2+ chelation and transport proteins. Cytoplasmic Zn2+ is gauged by the transcription factor MTF-1, which, upon Zn2+ binding, induces transcription of Zn2+ chelators, such as metallothioneins, and Zn2+ transporters, such as ZnT1. The ability to absorb Zn2+ through active transport involving ZnT transporters makes lysosomes a good candidate for absorbing rapid cytoplasmic Zn2+ spikes. The recently published data on the lysosomal absorption of Zn2+ released from metallothioneins by H2O2 highlight such a function of lysosomes (Tang et al., 2002; Suntres and Lui, 2006; Hwang et al., 2008; Lee et al., 2009).
Although ZIP transporters such as ZIP8 have been shown to exist in the lysosomes (Aydemir et al., 2009), their impact on the function of the lysosomal Zn2+ sink has not been shown. At the same time, the ion channel TRPML1, whose dysfunction causes the lysosomal storage disease mucolipidosis IV (MLIV) (Slaugenhaupt et al., 1999), has been shown to be a component of the lysosomal Zn2+ transport machinery. Its loss has been shown to cause Zn2+ buildup in the lysosomes in studies by two groups (Eichelsdoerfer et al., 2010; Kukic et al., 2013) and its permeability to Zn2+ by another group (Dong et al., 2010). It seems reasonable to conclude that TRPML1 is involved in trafficking Zn2+ from lysosomes into the cytoplasm. We proposed that upon entering the cell, Zn2+ is registered by MTF-1, which leads to an eventual increase in transcription and translation of Zn2+ chelators and exporters. However, Zn2+ must be rapidly eliminated from the cytoplasm in order to protect against toxicity. Thus, in parallel, Zn2+ is scavenged by the lysosomes, to be later released through a TRPML1-dependent mechanism.
The data in Fig. 6D, Fig. 7C and Fig. 9B show that lysosomal secretion is also involved in Zn2+ clearance from the cell. What is the relationship between the two mechanisms of Zn2+ clearance? We think that their function reflects their ability to respond to changes in Zn2+ flow. Such changes can be caused by increased autophagy of Zn2+-rich organelles such as mitochondria or proteins. Interestingly, TRPML1 is upregulated in response to increased endocytic load in a manner that requires the transcription factor TFEB, whereas VAMP7 remains unchanged (I.K., unpublished observation). Based on this, we propose that TRPML1-driven Zn2+ release is a dynamic response to increased Zn2+ load, whereas VAMP7- and SYT7-dependent secretion is a constitutive mechanism. We believe that our data clearly show that Zn2+ absorption into the lysosomes, followed by its secretion, is an important detoxification mechanism. Furthermore, the fact that suppression of the lysosomal function causes Zn2+ redistribution into Golgi and mitochondria shows that the lysosomal Zn2+ sink has a major impact on the cellular Zn2+ handling.
LMP following Zn2+ exposure has clearly been shown to be a cell death pathway (Hwang et al., 2008; Chung et al., 2009; Lee et al., 2009; Hwang et al., 2010). Why would cells employ a Zn2+ detoxification mechanism that effectively kills them? We think that the answer to this question lies in the relation between the lysosomal Zn2+ absorption rates and the rate of its secretion and release. Such a ratio might depend on the tissue, developmental stage and other factors. We propose that under normal conditions, or upon moderate Zn2+ exposure, the Zn2+ sink limits cytoplasmic Zn2+ by absorbing it (model in Fig. 5). Zn2+ is later released into the cytoplasm (as discussed above), or secreted across the plasma membrane. However, during the exposure to high (200 µM) levels of Zn2+, Zn2+ extraction lags, leading to LMP and cell death (supplementary material Fig. S3). Our data show a role of lysosomes in transition metal toxicity and identify a novel detoxification mechanism.
MATERIALS AND METHODS
Cell culture
HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, St Louis, MO) supplemented with 10% FBS. For siRNA and cDNA transfection, medium was changed after 24 hours. 100 µM ZnCl2 was added directly to the medium, containing serum, 24 hours after transfection, for the indicated times. Bafilomycin A1 (#196000, EMD MIllipore, Darmstadt, Germany) was used at 1 µM for the indicated amount of time.
siRNA-mediated KD and plasmid transfection
The VAMP7 siRNA (cat. number SASI_Hs01_00197188), SYT7 (cat. number SASI_Hs01_0047888), ZnT2 siRNA (Cat number SASI_Hs01_00055662), ZnT4 siRNA (cat. number SASI_Hs00225995), and MTF-1 siRNA (cat. number SASI_Hs01_00177112) were from Sigma-Aldrich. Non-targeting control siRNA#1 (Sigma) was used as a negative control. Cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbag, CA) as described by the manufacturer's protocol using 600 nM siRNA per 35-mm well (1200 nM siRNA per 35-mm well for efficient VAMP7 and SYT7 KD). All KDs were confirmed using SYBR-green based qPCR. For DNA transfections, 2 µg of GalT–mCherry and TFEB was transfected per 35-mm dish.
Microscopy
Cells were seeded on glass coverslips and loaded with dyes for 15 minutes at 37°C in buffer containing, in mM: 150 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES pH 7.4, 1 g/l glucose. The loading was followed by a 15-minute washout in all cases. Lysotracker Red DND-99 and FluoZin-3,AM (F-24195, Invitrogen, Carlsbag, CA) were used at 0.1 µM. Confocal microscopy was performed using Leica TCS SP5 and BioRad 3000 confocal microscopes. Live cells were treated as above. Images were analyzed using ImageJ (Bethesda, MD).
Reverse transcriptase and quantitative qPCR
RNA was isolated from cells using Trizol (Invitrogen, Carlsbag, CA) according to the manufacturer's protocol. cDNA was synthesized using the GeneAmp RNA PCR system (Applied Biosystems, Carlsbad, CA) with oligo(dT) priming. qPCR was performed using SYBR green (Fermentas, Glen Burnie, MD). The amount of cDNA loaded was normalized to starting RNA concentrations, with a final concentration of 9 ng (40 ng in ZnT experiments) of RNA loaded per experimental well. Six-point standard curves were generated for each primer using 1∶2 dilutions of cDNA. cDNA for the following genes were amplified using the indicated primers (IDT, Coralville, IA). MT2a, forward, 5′AAGTCCCAGCGAACCCGCGT-3′, reverse, 5′-CAGCAGCTGCACTTGTCCGACGC-3′; VAMP7, forward, 5′-CCGGACAGACTGAAGCCAT-3′, reverse, 5′-ATCTGCTCTGTCACCTCCAG-3′; SYT7, forward, 5′-AAGCGGGTGGAGAAGAAGAA-3′, reverse, 5′-CGAAGGCGAAGGACTCATTG-3′; ZnT1 (SLC30A1), forward, 5′-GGGAGCAGCGACATCAACGT-3′, reverse, 5′-GGGTCTGCGGGGTCCAATT-3′; ZnT2 (SLC30A2), forward, 5′-GCAATCCGGTCATACACGGGAT-3′, reverse, 5′-CAGCTCAATGGCCTGCAAGT-3; ZnT4 (SLC30A4), forward, 5′-CACATACAGCTAATTCCTGGAAGTTCATCT-3′, reverse, 5′-GCCTGTAACTCTGAAGCTGAATAGTACAT-3′; and LAMP1, forward, 5′-GGACAACACGACGGTGACAAG-3′, reverse, 5′-GAACTTGCATTCATCCCGAACTGGA-3′. All primers were designed to span exons and reverse-transcriptase-negative controls were tested to ensure amplification of cDNA only. qPCR was performed using the standard curve method on the 7300 Real Time System (Applied Biosystems, Carlsbad, CA). Reactions were run on the following parameters: 2 minutes at 50°C, 10 minutes at 95°C, and 40 cycles at 95°C for 15 seconds followed by 60°C for 1 minute. All experimental samples were run in triplicate and normalized to an RPL32 endogenous control.
β-Hexosamindase activity assay
Untreated control and transfected cells were washed with 37°C PBS, and 300 µl 37°C PBS with 1 mM CaCl2 was added to each 35-mm dish. For each sample, 100 µl of the supernatant was incubated with 300 µl 3 mM 4-nitrophenyl N-acetyl-β-D-glucosaminide (N9376, Sigma-Aldrich) for 30 minutes at 37°C in 0.1 M citrate buffer (pH 4.5) (C2488, Sigma-Aldrich). This volume was replaced with fresh 100 µl of 37°C of PBS with 1 mM CaCl2 to the culture dish. Samples were collected every after 0, 10 and 30 minutes. Reactions were stopped by adding 650 µl borate buffer (100 mM boric acid, 75 mM NaCl, 25 mM sodium borate pH 9.8) and the absorbance was measured in a spectrophotometer at 405 nm. To determine total cellular content of β-hexosamindase, cells were lysed with 300 µl 1% Triton X-100 in PBS and, after a 10,000 g spin for 5 minutes at 4°C, 10 µl of the cell extracts were used for the enzyme activity reaction. Enzyme activity was determined as the amount of 4-nitrophenol produced per mg of protein (Bradford method). Absorbance was calibrated with different amounts of 4-nitrophenol (N7660, Sigma-Aldrich) in 0.1 M citrate buffer.
Zinc secretion assay
Cells were plated on a 12-well plate and 48 hours after, transfection pulsed with 100 µM ZnCl2 for 3 hours, washed twice with warm PBS, and chased in 1 ml DMEM per well. Duplicate time-points were collected for 0, 5, 15, 30 or 60 minutes and replaced with new 50 µl of DMEM. For each sample, 50 µl of supernatant was placed in a 96-well plate. Zn2+ content was measured by incubating the supernatants with 10 µM cell-impermeant FluoZin-3 tetrapotassium salt (F-24194, Molecular Probes, Invitrogen, Carlsbag, CA) for 15 minutes at 37°C. The 96-well plate was read using a FujiFilm FLA-5100 fluorescent image analyzer. After the last time point, cells were washed with PBS, 200 µl detergent solution was added to lyse the cells and fluorescence was normalized to total protein in each sample.
Caspase-3 activity assay
Cells were prepared and measured using the EnzChek Caspase-3 Assay Kit #1 (E13183, Invitrogen, Carlsbag, CA) following the manufacturer's instructions. AMC substrate fluorescence was measured using a fluorometer at an excitation wavelength of 342 nm and an emission wavelength of 441 nm.
Western blot analysis
Cells were solubilized for 10 min at room temperature in either a 1× detergent solution (0.5 M EDTA, pH 8.0, 1 M Tris, pH 8.0, 0.4% deoxycholate, 1% Nonidet P-40 substitute) for (LAMP1) or a 1% CHAPS in PBS solution (for VAMP7) containing protease inhibitor mixture III (Calbiochem, Gibbstown, NJ) and centrifuged at 16,000 g for 5 minutes. The supernatant was collected and protein concentrations were determined using a Bradford assay. Protein was incubated at 100°C for 5 minutes in sample buffer containing 14% β-mercaptoethanol. Equal amounts of protein were loaded on a 10% precast Tris-HCl polyacrylamide gel (Bio-Rad) for each experimental sample. Proteins were transferred onto PVDF membrane (EMD Millipore, Darmstadt, Germany) and blocked in 10% nonfat dried milk for 1 hour. The following primary antibodies were used: monoclonal LAMP1 (sc-20011, Santa Cruz, Santa Cruz, CA) at 1∶1000 dilution, monoclonal VAMP7 (ab36195, Abcam, Cambridge, US) at 1∶500, and monoclonal β-actin (ab6276, Abcam, Cambridge, US) at 1∶5000 dilution. Horseradish peroxidase (HRP)-conjugated goat anti-mouse Ig secondary antibodies (Amersham Biosciences) were used at 1∶20,000 dilutions. Immunodetection was performed with the Luminata Forte HRP substrate (EMD Millipore, Darmstadt, Germany). Band densities were measured using ImageJ (Bethesda, MD).
Flow cytometry
Transfected HeLa cells were treated with either 100 µM ZnCl2 for 48 hours for Baf and VAMP7 experiments, or 200 µM ZnCl2 for 12 hours for TFEB experiments. Cells were then washed with PBS, trypsinized, and counted. 2×106 cells were pelleted for each sample, washed with PBS and then resuspended in Annexin Binding Buffer from the Vybrant Apoptosis Assay Kit #3 (V.13242, Molecular Probes). Cells were then loaded with 1 µl propidium iodide and 2 µl Annexin V 488 and sorted on the Accuri (BD) C6 at the University of Pittsburgh Cancer Institute Cytometry Facility. Cell sorting was gated to include healthy and apoptotic cells while excluding debris.
Statistics
Statistical significance was calculated using a one-tailed, unpaired Student's t-test with P≤0.05 considered significant. Data are presented as mean±s.e.m.
Supplementary Material
Acknowledgments
This project used the University of Pittsburgh Cancer Institute Cytometry Facility that is supported in part by award P30CA047904. We thank Michael Meyer and Bratislav Janjic for help with flow cytometry data analysis. The GalT mcherry construct was kindly provided by Ora Weisz and the TFEB construct was kindly provided by Rosa Puertollano.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
I.K. conceptualized, designed, executed and interpreted the data, and prepared the article. S.L.K. conceptualized and interpreted the data. K.K. conceptualized, designed and interpreted the data, and prepared the article.
Funding
This work was supported by National Institutes of Health [grant numbers HD058577 and ES01678 to K.K.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.145318/-/DC1
References
- Andrews G. K. (2001). Cellular zinc sensors: MTF-1 regulation of gene expression. Biometals 14, 223–237 10.1023/A:1012932712483 [DOI] [PubMed] [Google Scholar]
- Aydemir T. B., Liuzzi J. P., McClellan S., Cousins R. J. (2009). Zinc transporter ZIP8 (SLC39A8) and zinc influence IFN-gamma expression in activated human T cells. J. Leukoc. Biol. 86, 337–348 10.1189/jlb.1208759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun V., Fraisier V., Raposo G., Hurbain I., Sibarita J. B., Chavrier P., Galli T., Niedergang F. (2004). TI-VAMP/VAMP7 is required for optimal phagocytosis of opsonised particles in macrophages. EMBO J. 23, 4166–4176 10.1038/sj.emboj.7600427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao Y., Fu D. (2004). Kinetic study of the antiport mechanism of an Escherichia coli zinc transporter, ZitB. J. Biol. Chem. 279, 12043–12050 10.1074/jbc.M313510200 [DOI] [PubMed] [Google Scholar]
- Cho K. S., Yoon Y. H., Choi J. A., Lee S. J., Koh J. Y. (2012). Induction of autophagy and cell death by tamoxifen in cultured retinal pigment epithelial and photoreceptor cells. Invest. Ophthalmol. Vis. Sci. 53, 5344–5353 10.1167/iovs.12--9827 [DOI] [PubMed] [Google Scholar]
- Chung H., Yoon Y. H., Hwang J. J., Cho K. S., Koh J. Y., Kim J. G. (2009). Ethambutol-induced toxicity is mediated by zinc and lysosomal membrane permeabilization in cultured retinal cells. Toxicol. Appl. Pharmacol. 235, 163–170 10.1016/j.taap.2008.11.006 [DOI] [PubMed] [Google Scholar]
- Decressac M., Mattsson B., Weikop P., Lundblad M., Jakobsson J., Björklund A. (2013). TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. USA 110, E1817–E1826 10.1073/pnas.1305623110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X. P., Shen D., Wang X., Dawson T., Li X., Zhang Q., Cheng X., Zhang Y., Weisman L. S., Delling M. et al. (2010). PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat. Commun. 1, 38 10.1038/ncomms1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichelsdoerfer J. L., Evans J. A., Slaugenhaupt S. A., Cuajungco M. P. (2010). Zinc dyshomeostasis is linked with the loss of mucolipidosis IV-associated TRPML1 ion channel. J. Biol. Chem. 285, 34304–34308 10.1074/jbc.C110.165480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcón-Pérez J. M., Dell'Angelica E. C. (2007). Zinc transporter 2 (SLC30A2) can suppress the vesicular zinc defect of adaptor protein 3-depleted fibroblasts by promoting zinc accumulation in lysosomes. Exp. Cell Res. 313, 1473–1483 10.1016/j.yexcr.2007.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery A. R., Czibener C., Andrews N. W. (2010). Palmitoylation-dependent association with CD63 targets the Ca2+ sensor synaptotagmin VII to lysosomes. J. Cell Biol. 191, 599–613 10.1083/jcb.201003021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsleff L., Schauss A. G., Bier I. D., Stuart S. (1999). Evidence of functional zinc deficiency in Parkinson's disease. J. Altern. Complement. Med. 5, 57–64 10.1089/acm.1999.5.57 [DOI] [PubMed] [Google Scholar]
- Fraldi A., Annunziata F., Lombardi A., Kaiser H. J., Medina D. L., Spampanato C., Fedele A. O., Polishchuk R., Sorrentino N. C., Simons K. et al. (2010). Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J. 29, 3607–3620 10.1038/emboj.2010.237 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Frederickson C. J., Bush A. I. (2001). Synaptically released zinc: physiological functions and pathological effects. Biometals 14, 353–366 10.1023/A:1012934207456 [DOI] [PubMed] [Google Scholar]
- Heming T. A., Traber D. L., Hinder F., Bidani A. (1995). Effects of bafilomycin A1 on cytosolic pH of sheep alveolar and peritoneal macrophages: evaluation of the pH-regulatory role of plasma membrane V-ATPases. J. Exp. Biol. 198, 1711–1715 [DOI] [PubMed] [Google Scholar]
- Huang L., Gitschier J. (1997). A novel gene involved in zinc transport is deficient in the lethal milk mouse. Nat. Genet. 17, 292–297 10.1038/ng1197--292 [DOI] [PubMed] [Google Scholar]
- Huang L., Kirschke C. P., Gitschier J. (2002). Functional characterization of a novel mammalian zinc transporter, ZnT6. J. Biol. Chem. 277, 26389–26395 10.1074/jbc.M200462200 [DOI] [PubMed] [Google Scholar]
- Hwang J. J., Lee S. J., Kim T. Y., Cho J. H., Koh J. Y. (2008). Zinc and 4-hydroxy-2-nonenal mediate lysosomal membrane permeabilization induced by H2O2 in cultured hippocampal neurons. J. Neurosci. 28, 3114–3122 10.1523/JNEUROSCI.0199--08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J. J., Kim H. N., Kim J., Cho D. H., Kim M. J., Kim Y. S., Kim Y., Park S. J., Koh J. Y. (2010). Zinc(II) ion mediates tamoxifen-induced autophagy and cell death in MCF-7 breast cancer cell line. Biometals 23, 997–1013 10.1007/s10534--010--9346--9 [DOI] [PubMed] [Google Scholar]
- Jansen J., Karges W., Rink L. (2009). Zinc and diabetes – clinical links and molecular mechanisms. J. Nutr. Biochem. 20, 399–417 10.1016/j.jnutbio.2009.01.009 [DOI] [PubMed] [Google Scholar]
- Jiang L. J., Vasák M., Vallee B. L., Maret W. (2000). Zinc transfer potentials of the alpha- and beta-clusters of metallothionein are affected by domain interactions in the whole molecule. Proc. Natl. Acad. Sci. USA 97, 2503–2508 10.1073/pnas.97.6.2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanninen K. M., Grubman A., Caragounis A., Duncan C., Parker S. J., Lidgerwood G. E., Volitakis I., Ganio G., Crouch P. J., White A. R. (2013). Altered biometal homeostasis is associated with CLN6 mRNA loss in mouse neuronal ceroid lipofuscinosis. Biol. Open 2, 635–646 10.1242/bio.20134804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher S. L., Seo Y. A., Lopez V. (2009). Mammary gland zinc metabolism: regulation and dysregulation. Genes Nutr. 4, 83–94 10.1007/s12263--009--0119--4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher S. L., McCormick N. H., Velasquez V., Lopez V. (2011). Zinc in specialized secretory tissues: roles in the pancreas, prostate, and mammary gland. Adv. Nutr. 2, 101–111 10.3945/an.110.000232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiedrowski L. (2012). Cytosolic acidification and intracellular zinc release in hippocampal neurons. J. Neurochem. 121, 438–450 10.1111/j.1471--4159.2012.07695.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukic I., Lee J. K., Coblentz J., Kelleher S. L., Kiselyov K. (2013). Zinc-dependent lysosomal enlargement in TRPML1-deficient cells involves MTF-1 transcription factor and ZnT4 (Slc30a4) transporter. Biochem. J. 451, 155–163 10.1042/BJ20121506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. J., Koh J. Y. (2010). Roles of zinc and metallothionein-3 in oxidative stress-induced lysosomal dysfunction, cell death, and autophagy in neurons and astrocytes. Mol. Brain 3, 30 10.1186/1756--6606--3--30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. Y., Cole T. B., Palmiter R. D., Suh S. W., Koh J. Y. (2002). Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. USA 99, 7705–7710 10.1073/pnas.092034699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. J., Cho K. S., Koh J. Y. (2009). Oxidative injury triggers autophagy in astrocytes: the role of endogenous zinc. Glia 57, 1351–1361 10.1002/glia.20854 [DOI] [PubMed] [Google Scholar]
- Logan M. R., Lacy P., Odemuyiwa S. O., Steward M., Davoine F., Kita H., Moqbel R. (2006). A critical role for vesicle-associated membrane protein-7 in exocytosis from human eosinophils and neutrophils. Allergy 61, 777–784 10.1111/j.1398--9995.2006.01089.x [DOI] [PubMed] [Google Scholar]
- Lopez V., Foolad F., Kelleher S. L. (2011). ZnT2-overexpression represses the cytotoxic effects of zinc hyper-accumulation in malignant metallothionein-null T47D breast tumor cells. Cancer Lett. 304, 41–51 10.1016/j.canlet.2011.01.027 [DOI] [PubMed] [Google Scholar]
- Martina J. A., Diab H. I., Lishu L., Jeong-A L., Patange S., Raben N., Puertollano R. (2014). The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal. 7, ra9 10.1126/scisignal.2004754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Arca S., Alberts P., Zahraoui A., Louvard D., Galli T. (2000). Role of tetanus neurotoxin insensitive vesicle-associated membrane protein (TI-VAMP) in vesicular transport mediating neurite outgrowth. J. Cell Biol. 149, 889–900 10.1083/jcb.149.4.889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick N. H., Kelleher S. L. (2012). ZnT4 provides zinc to zinc-dependent proteins in the trans-Golgi network critical for cell function and Zn export in mammary epithelial cells. Am. J. Physiol. 303, C291–C297 10.1152/ajpcell.00443.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina D. L., Fraldi A., Bouche V., Annunziata F., Mansueto G., Spampanato C., Puri C., Pignata A., Martina J. A., Sardiello M. et al. (2011). Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 21, 421–430 10.1016/j.devcel.2011.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedeva Y. V., Lin B., Shuttleworth C. W., Weiss J. H. (2009). Intracellular Zn2+ accumulation contributes to synaptic failure, mitochondrial depolarization, and cell death in an acute slice oxygen-glucose deprivation model of ischemia. J. Neurosci. 29, 1105–1114 10.1523/JNEUROSCI.4604--08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollinedo F., Calafat J., Janssen H., Martín-Martín B., Canchado J., Nabokina S. M., Gajate C. (2006). Combinatorial SNARE complexes modulate the secretion of cytoplasmic granules in human neutrophils. J. Immunol. 177, 2831–2841 10.4049/jimmunol.177.5.2831 [DOI] [PubMed] [Google Scholar]
- Ohana E., Hoch E., Keasar C., Kambe T., Yifrach O., Hershfinkel M., Sekler I. (2009). Identification of the Zn2+ binding site and mode of operation of a mammalian Zn2+ transporter. J. Biol. Chem. 284, 17677–17686 10.1074/jbc.M109.007203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri M., Impey S., Kang H., di Ronza A., Pelz C., Sardiello M., Ballabio A. (2011). Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 20, 3852–3866 10.1093/hmg/ddr306 [DOI] [PubMed] [Google Scholar]
- Palmiter R. D., Findley S. D. (1995). Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J. 14, 639–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiter R. D., Cole T. B., Findley S. D. (1996). ZnT-2, a mammalian protein that confers resistance to zinc by facilitating vesicular sequestration. EMBO J. 15, 1784–1791 [PMC free article] [PubMed] [Google Scholar]
- Pastore N., Ballabio A., Brunetti-Pierri N. (2013). Autophagy master regulator TFEB induces clearance of toxic SERPINA1/α-1-antitrypsin polymers. Autophagy 9, 1094–1096 10.4161/auto.24469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad A. S. (2013). Discovery of human zinc deficiency: its impact on human health and disease. Adv. Nutr. 4, 176–190 10.3945/an.112.003210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S. K., Huynh C., Proux-Gillardeaux V., Galli T., Andrews N. W. (2004). Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. J. Biol. Chem. 279, 20471–20479 10.1074/jbc.M400798200 [DOI] [PubMed] [Google Scholar]
- Rink L., Gabriel P. (2000). Zinc and the immune system. Proc. Nutr. Soc. 59, 541–552 10.1017/S0029665100000781 [DOI] [PubMed] [Google Scholar]
- Rulon L. L., Robertson J. D., Lovell M. A., Deibel M. A., Ehmann W. D., Markesber W. R. (2000). Serum zinc levels and Alzheimer's disease. Biol. Trace Elem. Res. 75, 79–85 10.1385/BTER:75:1--3:79 [DOI] [PubMed] [Google Scholar]
- Sardiello M., Palmieri M., di Ronza A., Medina D. L., Valenza M., Gennarino V. A., Di Malta C., Donaudy F., Embrione V., Polishchuk R. S. et al. (2009). A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 [DOI] [PubMed] [Google Scholar]
- Saydam N., Adams T. K., Steiner F., Schaffner W., Freedman J. H. (2002). Regulation of metallothionein transcription by the metal-responsive transcription factor MTF-1: identification of signal transduction cascades that control metal-inducible transcription. J. Biol. Chem. 277, 20438–20445 10.1074/jbc.M110631200 [DOI] [PubMed] [Google Scholar]
- Schissel S. L., Schuchman E. H., Williams K. J., Tabas I. (1996). Zn2+-stimulated sphingomyelinase is secreted by many cell types and is a product of the acid sphingomyelinase gene. J. Biol. Chem. 271, 18431–18436 10.1074/jbc.271.31.18431 [DOI] [PubMed] [Google Scholar]
- Settembre C., Fraldi A., Medina D. L., Ballabio A. (2013). Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296 10.1038/nrm3565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaugenhaupt S. A., Acierno J. S., Jr, Helbling L. A., Bove C., Goldin E., Bach G., Schiffmann R., Gusella J. F. (1999). Mapping of the mucolipidosis type IV gene to chromosome 19p and definition of founder haplotypes. Am. J. Hum. Genet. 65, 773–778 10.1086/302549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suntres Z. E., Lui E. M. (2006). Antioxidant effect of zinc and zinc-metallothionein in the acute cytotoxicity of hydrogen peroxide in Ehrlich ascites tumour cells. Chem. Biol. Interact. 162, 11–23 10.1016/j.cbi.2006.04.007 [DOI] [PubMed] [Google Scholar]
- Tang Z. L., Wasserloos K. J., Liu X., Stitt M. S., Reynolds I. J., Pitt B. R., St Croix C. M. (2002). Nitric oxide decreases the sensitivity of pulmonary endothelial cells to LPS-induced apoptosis in a zinc-dependent fashion. Mol. Cell. Biochem. 234-235, 211–217 10.1023/A:1015930927407 [DOI] [PubMed] [Google Scholar]
- Vinceti M., Bergomi M., Nacci G., Pietrini V., Ferrari A., Fortini K., Guidetti D., Sola P., Rocchi E., Mancia D. et al. (2002). Erythrocyte zinc, copper, and copper/zinc superoxide dismutase and risk of sporadic amyotrophic lateral sclerosis: a population-based case-control study. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 3, 208–214 [DOI] [PubMed] [Google Scholar]
- Yoon Y. H., Cho K. S., Hwang J. J., Lee S. J., Choi J. A., Koh J. Y. (2010). Induction of lysosomal dilatation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Invest. Ophthalmol. Vis. Sci. 51, 6030–6037 10.1167/iovs.10--5278 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.