

Abstract
Although acid-sensing ion channels (ASICs) have been studied in a variety of mammalian cells, little is currently known about their presence in intestinal epithelial cells. Therefore, the aims of the present study were to investigate the expression and function of ASIC isoforms in intestinal epithelial cells, particularly their physiological role in the acid-stimulated duodenal mucosal bicarbonate secretion (DMBS). RT-PCR and digital Ca2+ imaging were used to determine the expression and function of ASICs in HT29 cells, a human intestinal epithelial cell line. The acid-stimulated DMBS was measured in C57 black mice in vivo to study the role of ASICs in this physiological process. mRNA expression of ASIC1a was detected in the duodenal mucosa stripped from mice and HT29 cells, in which cytoplasmic free Ca2+ ([Ca2+]cyt) in response to extracellular acidosis was also measured. In Ca2+-containing solutions, acidosis raised [Ca2+]cyt in a pH-dependent manner with a half-maximal effect (pH0.5) at approximately 5.9. The acidosis-induced increase in [Ca2+]cyt was markedly inhibited by amiloride (an ASIC blocker), SK&F96365 (a blocker for non-selective cation channels), or in Ca2+-free solutions; but slightly affected by U73122, or nifedipine. After acidosis raised [Ca2+]cyt, stimulation of purinergic receptors with ATP further increased [Ca2+]cyt. Moreover, the acid-stimulated murine DMBS was significantly attenuated by amiloride. Therefore, ASIC1a is functionally expressed in intestinal epithelial cells, and may play an important role in the regulation of [Ca2+]cyt homeostasis in these cells and the acid-stimulated DMBS.
Keywords: Acid-sensing ion channels, cytosolic free Ca2+ concentration, intestinal epithelial cells, duodenal mucosal bicarbonate secretion
Introduction
Acid-sensing ion channels (ASICs) are members of the voltage-insensitive, amiloride-sensitive degenerin/epithelial Na+ channel (Deg/ENaC) family of cation channels1, 2. Although six ASIC subunits have been cloned so far, only five of these are sensitive to protons (ASIC1a, ASIC1b, ASIC2a, ASIC2b, ASIC3)2, 3. These subunits combine into homo- and heteromultimeric complexes to form functional channels, with the signature feature of activation by extracellular protons/acidosis but inhibition by amiloride1, 2. However, the pH of half-maximal activation (pH0.5) varies for each subunit: ASIC1a can be stimulated once extracellular pH drops below 6.9, with pH0.5 = 6.0–6.4; ASIC1b requires a more acidic pH for activation, pH0.5 = 5.9; ASIC2a requires even more acidic conditions as pH0.5 ~ 4.9; and ASIC3 is perhaps the most sensitive, pH0.5 = 6.5–6.72, 3. While ASICs are preferentially permeable to Na+, certain subunits also allow for the passage of Ca2+ (ASIC1a) and K+ (ASIC1b).
ASICs have been found in a variety of mammalian organs and systems, including the central and afferent nervous systems1–4, the cardiovascular system2, 5 and the gastrointestinal tract2, 6–8. Since acid-base regulation is a vital issue for cell and tissue homeostasis, there exists a multitude of pH sensors in the body. However, these acid sensors can also be upregulated and overactive under diseased states, where they have been shown to cause less than beneficial effects2. It has recently been shown that ischemic brain injury under acidosis occurs via the opening of Ca2+-permeable ASICs, leading to Ca2+ toxicity9–13. This acidosis induced rise in cytosolic free Ca2+ ([Ca2+]cyt) was shown to be absent in cells lacking ASIC1a, but present in the same cell type after transfection of ASIC1a, suggesting an important role of ASIC1a in the regulation of [Ca2+]cyt homeostasis13.
Interestingly, despite the gastrointestinal tract being the greatest source of acid production in the human body, the presence and function of ASICs in the gastrointestinal tract is still largely unknown. ASICs have been studied in gastrointestinal acid-sensing afferent neurons7, 8, 14, 15, but their expression and function in intestinal epithelial cells are poorly understood. Acid sensing is of paramount importance to gastrointestinal homeostasis, because there are huge variations in the intraluminal pH along the alimentary canal and the survival of epithelial cells requires maintenance of intracellular pH in a narrow physiological range6. This situation is met by multiple mechanisms controlling extra- and intracellular pH, in which several molecular acid sensors survey a wide pH range from acidic to alkaline environments. Given that the expression of ASICs has been found in human small intestine6, 8, the present study aims to investigate the expression and function of ASIC isoforms in intestinal epithelial cells, particularly their physiological role in acid-stimulated duodenal mucosal bicarbonate secretion (DMBS). After conducting molecular biology and functional studies, we provided evidence to support the notion that ASIC1a is functionally expressed in intestinal epithelial cells and may play an important role in the regulation of acid-stimulated DMBS.
Materials and Methods
Animals
This study was approved by the University of California, San Diego Animal Subjects Committee. Adult Harlan C-57 black mice were housed in an animal care room with a 12:12-hour light-dark cycle and were allowed free access to food and water. Before each experiment, mice were deprived of food and water for at least 1 hour.
Measurement of duodenal bicarbonate secretion in mice
In vivo experiments were performed using a well-validated technique, as described previously16, with the HCO3− concentration of samples measured via a CO2-sensitive electrode. Female C57 mice were first anesthetized by intraperitoneal injection of a Ketamine/Midazolam cocktail (⅙ Ketamine, ⅙ Midazolam, 10 mg/kg). Respiratory rate and response to toe-pinch of the animals were carefully monitored. After initiation of anesthesia, the abdomen was opened, and the duodenum accessed through two small incisions: one just below the ribcage on the left side, and the other just below the sternum. Through the first incision, the stomach was located and another small incision was made proximal to the pyloric sphincter. A soft polyethylene catheter was then inserted into the stomach, gently pushed through the pyloric sphincter, and tied firmly with silk suture thread around the outside of the pyloric sphincter, isolating the proximal duodenum (5–10 mm) from the stomach. Through the second incision, the junction between the pancreatic duct and the duodenum was located and another incision was made in the duodenum. A second polyethylene catheter was inserted into the duodenum and secured into place just proximal to the junction with the pancreatic duct but distal to the duodenal blood supply. Thus, pancreatic secretions were excluded from the isolated duodenal segment while the blood supply remained intact. Throughout the duration of the experiment, the duodenum maintained a healthy pink color and was kept moist within the abdominal cavity.
After surgery, the proximal duodenum was perfused (0.15 cc/min) with isotonic saline for 20 minutes. After this initial washout and recovery period, basal HCO3− secretion was measured for 20 minutes. This was followed with a luminal pretreatment of 100 μM amiloride or simply more saline, and HCO3− secretion was measured for 6 minutes. The duodenal segment was then perfused with 10 mM HCl in isotonic saline for 5 minutes, after which a 5-min washout period was allowed to flush out any residual acid. HCO3− secretion was then measured for an additional 42 minutes. After each experiment, the length of the duodenal test segment was measured in situ to the nearest millimeter. Animals could be sustained under these experimental conditions for at least 2 hours. Sample volumes were measured by weight to the nearest 0.01 mg. The amount of HCO3− in the effluents was quantitated by used of a CO2-sensitive electrode (Thermo Orion, Beverly, MA). The electrode was calibrated before each day’s use by constructing a semilogarithmic standard curve using known HCO3− concentrations. A 1-mL aliquot from each 6-min perfusion period was individually sampled, yielding a reading in millivolts. This reading was then converted back to an HCO3− concentration, as dictated by the previously generated standard curve. In this way, HCO3− outputs were determined for each 6-min period and expressed as micromoles per centimeter per hour. Stimulated HCO3− outputs are presented as HCO3− output over time and as net HCO3− output (peak minus average basal output).
HT29 cell culture
HT29cl.19A is a human intestinal epithelial cell line17. Cells were fed with fresh Dulbecco’s modified Eagle medium supplemented with 10% FBS, L-glutamine, and streptomycin, and grown to confluence (~5 days) in 75-cm2 flasks. Before use, the cells were replated onto 10 mm round coverslips (Warner Instruments, Hamden, CT) for at least 24 h before use.
[Ca2+]cyt measurement by digital Ca2+ imaging
[Ca2+]cyt measurements in HT29 cells were measured by Fura 2 fluorescence ratio digital imaging under the room temperature (~22 °C), as described previously16. Briefly, HT29 cells were grown on coverslips and loaded with 5 μM Fura 2-acetoxymethyl ester (AM) dissolved in 0.01% Pluronic F-127 plus 0.1% DMSO in physiological salt solution, in the dark at room temperature for 50 min, then washed in physiological salt solution for at least 30 min to remove extracellular dye and allow intracellular esterase to cleave cytoplasmic Fura 2-AM into active Fura 2. The coverslips with HT29 cells were then mounted in a perfusion chamber on a Nikon microscope stage. Cells were initially superfused with normal physiological salt solution with pH 7.4 for 3–5 minutes and then switched to the solutions with different pH values in the absence or the presence of different drugs. In some experiments, Ca2+-free solutions were used. The ratio of Fura 2 fluorescence (510-nm light emission excited by 340- and 380-nm illuminations) from the cells, as well as background fluorescence, was collected at room temperature with the use of a 20x Nikon UV-Fluor objective and an intensified CCD camera (ICCD200). The fluorescence signals emitted from the cells were monitored continuously using a MetaFluor Imaging System (Universal Imaging, Downingtown, PA) and were recorded in an IBM-compatible computer for later analysis.
RT-PCR analysis
Total RNA from HT29 cells or mouse duodenal mucosa was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions. Five micrograms of total RNA were converted into cDNA with reverse transcriptase. After inactivation at 70°C for 10 min, 1 μl of the reaction mixture was incubated in buffer containing 0.2 mM dATP, dCTP, dGTP, and dTTP, 0.2 μM oligonucleotide primers, as shown below, 3 mM MgCl2, 500 mM KCl, and a 10x buffer consisting of 200 mM Tris·HCl (pH 8.0), together with 1 unit of Taq polymerase (Invitrogen). Primers were synthesized by Integrated DNA Technologies (Coralville, IA). ASIC1a-specific sense and antisense primers (GenBank accession number is NM_020039) were 5′-GCTATGGCAAAGAGCTGTCC-3′ and 5′-AAGGGGACAGGT-GTCATCAG-3′, respectively. GAPDH sense and antisense primers, as previously used, were 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′ respectively. The samples were amplified in an automated thermal cycler (GeneAmp 2400; Applied Biosystems). DNA amplification conditions included an initial 3-min denaturation step at 94°C, 35 cycles of 30 s at 94°C, 30 s at 57°C, 40 s at 72°C, and a final elongation step of 10 min at 72°C. The products were electrophoresed on a 1.5% agarose gel, stained with ethidium bromide (0.5 μg/ml), and then photographed under UV light.
Chemicals and solutions
ATP, SK&F96365, U-73122, nifedipine, amiloride, ruthenium red were purchased from Sigma. Fura 2-AM was from Molecular Probes (Eugene, OR). The other chemicals were obtained from Fisher Scientific (Santa Clara, CA). The normal physiological salt solution used in digital Ca2+ measurement contained the following (in mM): 140 Na+, 5.0 K+, 2 Ca2+, 147 Cl−, 10 HEPES, and 10 glucose. pH was adjusted to 7.4 under the room temperature. The solutions with different pH values were adjusted by adding HCl. For the Ca2+ -free solution, Ca2+ was omitted, and 0.5 mM EGT A was added prevent possible Ca2+ contamination. The osmolalities for all solutions were ~284 mosmol/kgH2O.
Statistical analysis
Results are expressed as means ± SE. Differences between means were considered to be statistically significant at P < 0.05 using Student’s t-test or one-way ANOVA followed by Newman-Keuls post hoc test, as appropriate.
Results
Expression of ASIC1a in mouse duodenal epithelia and HT29 cells
Although a total of six ASIC subunits have been well characterized in the mammalian cells, ASIC1a is the focus of the present study because it has been extensively studied and is widely distributed in the different systems, including the gastrointestinal tract8, 13. RT-PCR analysis was performed to detect mRNA expression of ASIC1a in the intestinal epithelium. Since expression of ASIC1a has been confirmed in the mammalian central nervous system9,13, rat brain tissue was used as a positive control. As shown in Fig. 1, expression of ASIC1a transcripts was clearly detected in mouse duodenal epithelia and HT29 cells, a human intestinal epithelial cell line. Therefore, we confirmed the expression of ASIC1a mRNA in murine and human intestinal epithelial cells.
Figure 1. Expression of ASIC1a in mouse duodenal epithelia and HT29 cells.
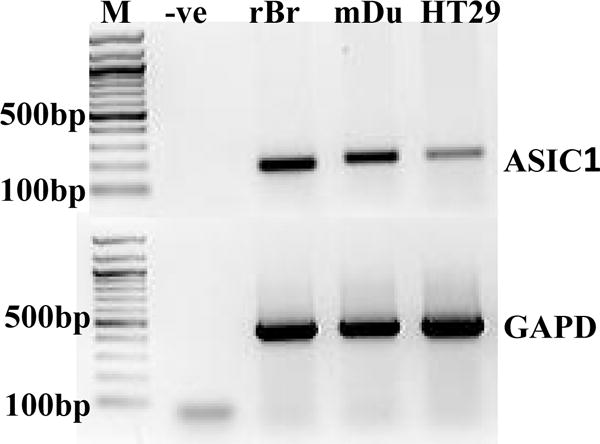
RNA was isolated from mouse duodenal epithelia (mDu) and HT29 cells, in which ASIC1a transcripts were detected by RT-PCR analysis. Rat brain tissue (rBr) was used as a positive control, while a negative control contained all the reaction ingredients except cDNA (-ve), and M is the molecular weight marker. GAPDH was used as an internal loading control. The data shows one representative of the similar results obtained from three different experiments.
Extracellular acidity triggers Ca2+ mobilization in HT29 cells
Although gastrointestinal lumen contains the highest acidity in the human body6, the functional study of ASICs in intestinal epithelia has been unexpectedly neglected. Thus, after detecting the expression of ASIC1a, we sought to investigate its function in intestinal epithelial cells. Since ASIC1a has be reported to show a significant permeability to extracellular Ca2+ 13, we used a digital Ca2+ imaging system to study the kinetics of [Ca2+]cyt in HT29 cells. After the cells were superfused with normal physiological salt solution at pH 7.4 to record the baseline for ~3 min, the superfusion was switched to solutions with varying pH values (6.0–4.0). As Figure 2 illustrates, acidosis induced a pH-dependent increase in [Ca2+]cyt with a half-maximal effect (pH0.5) at approximately 5.9 and peak at pH 5.0 (p<0.01, n=52) (Figure 2A&B). This rise in [Ca2+]cyt was relatively quick and peaked within a few minutes, then gradually returned to approximately baseline levels without additional stimulation (Figure 2A). Therefore, ASIC1a may be functionally expressed in human intestinal epithelial cells and likely play an important role in the regulation of [Ca2+]cyt homeostasis as in neurons13.
Figure 2. Extracellular acidity triggers Ca2+ mobilization in HT29 cells in Ca2+-containing solutions.
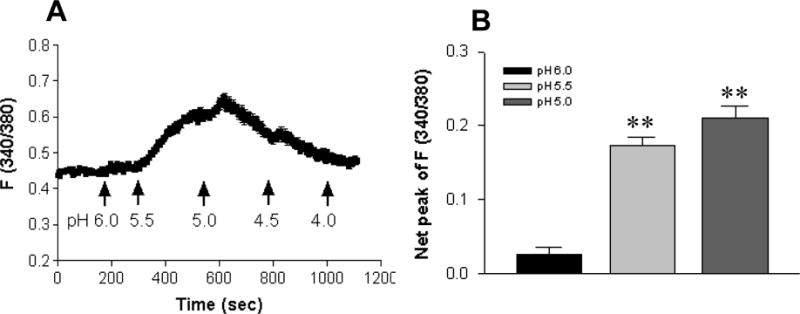
A: Normal physiological salt solutions with varying pH values (6.0–4.0) adjusted by HCl triggered a rise in [Ca2+]cyt that was peaked at pH 5.0, then gradually returned to the baseline levels even with additional extracellular acidosis. B: Summarized data showing that extracellular acidosis caused a pH (6.0–5.0)-dependent rise in net peak of [Ca2+]cyt with a pH0.5 of approximate 5.9. Values are mean ± SE, n = 52 cells for each tracing or bar. **P < 0.01 vs. pH 7.4.
Acidosis-induced Ca2+ mobilization via ASIC1a in HT29 cells
To test if acidosis-induced Ca2+ mobilization is via ASIC1a, we used an extracellular pH value of 5.0 as a control to perform further experiments in HT29 cells. As shown in Figure 3A, acidosis induced a marked Ca2+ mobilization (ΔF340/380 0.295 ± 0.022, n=47 cells). However, after pretreatment with amiloride (100 μM), a widely used ASIC blocker3, 13, acidosis-induced increase in Ca2+ mobilization was significantly attenuated (ΔF340/380 0.175 ± 0.019, n=47 cells, p<0.01). The net peak of acidosis-induced Ca2+ signaling was reduced by 41% in the presence of amiloride (Figure 3B). To test if acidosis induces extracellular Ca2+ entry, we compared the acidosis-induced Ca2+ signaling in the Ca2+ -free and Ca2+ -containing solutions. Our results show that the Ca2+ signaling was significantly attenuated in the Ca2+ -free solutions (ΔF340/380 0.130 ± 0.016 vs. 0.280 ± 0.020, n=50 cells, p<0.01), in which the net peak of acidosis-induced Ca2+ signaling was reduced by 54% (Figure 3C). Since ASIC1a is the nonselective cation channels1, 13, we further tested the effect of SK&F96365 (30μM), a widely used blocker for the nonselective cation channels18. As shown in Figure 3, the acidosis-induced Ca2+ signaling in HT29 cells was significantly attenuated by SK&F96365 (n=52 cells, p<0.01). The net peak of acidosis-induced Ca2+ signaling was reduced by 31% in the presence of SK&F96365 (Figure 3D). This data provided further evidence for the functional expression of ASIC1a in human intestinal epithelial cells and its important role in the regulation of [Ca2+]cyt homeostasis in these cells.
Figure 3. The major component of acidosis-induced Ca2+ mobilization is via Ca2+ entry through ASICs in HT29 cells.
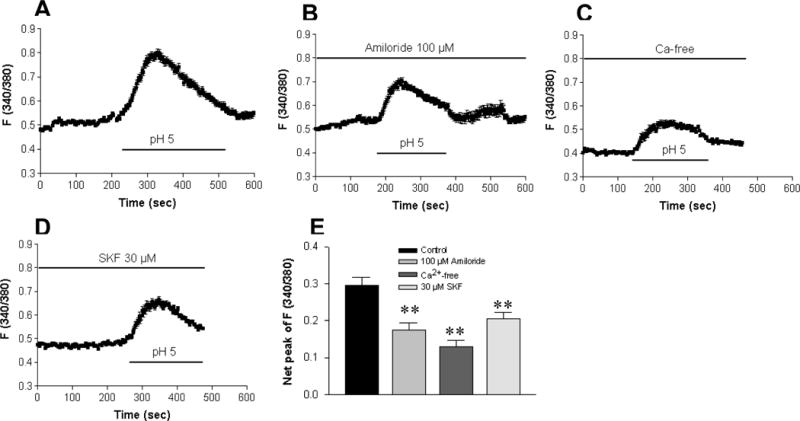
A: Superperfusion with Ca2+-containing solution (pH 5.0) induced a rise in [Ca2+]cyt in a control experiment. The acidosis-induced rise in [Ca2+]cyt was significantly reduced by amiloride (100 μM, B), removal of extracellular Ca2+ (C), or SK&F96365 (30 μM, D). E: Summarized data showing net peak of [Ca2+]cyt rise induced by pH 5.0 in the cells exposed to different treatments. Values are mean ± SE, n =47–50 cells for each tracing or bar. **P < 0.01 vs. control with Ca2+-containing solution in the absence of inhibitors.
Specificity of Acidosis-induced Ca2+ mobilization in HT29cells
As there may exist a variety of pathways for Ca2+ mobilization in HT29 cells, we performed further experiments to determine the possible involvement of other mechanisms responsible for this acidosis-induced rise in [Ca2+]cyt. Again, an extracellular pH of 5.0 was used as a control (Figure 4A). To test if acidosis-induced Ca2+ mobilization is via voltage-gated Ca2+ channels2, 19, HT29 cells were pretreated with nifedepine (10 μM), a widely used blocker for voltage-gated Ca2+ channels. As shown in Figure 4, acidosis-induced Ca2+ mobilization was slightly altered by nifedepine (n=49, p<0.05), which reduced the Ca2+ signaling by 19% (Figure 4B). To test if G protein coupled receptor (GPCR)-mediated phospholipase C (PLC) pathway is involved2, 20, U73122 (10 μM), a PLC inhibitor, was used. U73122 slightly attenuated acidosis-induced Ca2+ mobilization (n=52, p<0.05), and reduced the Ca2+ signaling by 18% (Figure 4C). Since experimental evidence suggests that transient receptor potential ion channels of vanilloid (TRPV) plays a role as a acid sensor for signaling mucosal hyperemia in the duodenum in response to luminal acidification2, 6, we used ruthenium red (100 μM), a TRPV channel blocker, to test the possible involvement of TRPV in HT29 cells. As shown in Figure 4, acidosis-induced Ca2+ mobilization was not significantly altered by ruthenium red (n=52, p>0.05), which reduced the Ca2+ signaling by 5% only (Figure 4D). Therefore, the major component of Ca2+ entry into HT29 cells seemed to be through ASIC1a.
Figure 4. The minor component of acidosis-induced Ca2+ mobilization is via the other Ca2+ entry pathways in HT29 cells.
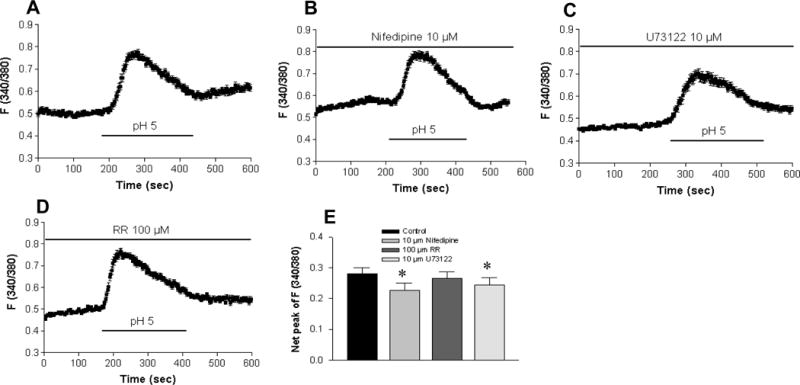
A: Superperfusion with Ca2+-containing solution (pH 5.0) induced a rise in [Ca2+]cyt in a control experiment. The acidosis-induced rise in [Ca2+]cyt was slightly reduced by nifidepine (10 μM; B), U73122 (10 μM; C). However, ruthenium red (100 μM; D) did not significantly affect this acidosis-induced rise in [Ca2+]cyt. E: Summarized data showing net peak of [Ca2+]cyt rise induced by pH 5.0 in the cells exposed to different treatments. Values are mean ± SE, n =49–50 cells for each tracing or bar. *P < 0.05 vs. control with Ca2+-containing solution in the absence of inhibitors.
There remains the possibility that the rise in [Ca2+]cyt might be due to acidosis-induced ATP release from HT29 cells and through activation of the P2 purinoceptors2. However, when an increase in [Ca2+]cyt (ΔF340/380 0.163 ± 0.026, n=50 cells, p<0.05) was first induced by acidosis (pH of 5.5), ATP (10 μM) induced an additional rise in [Ca2+]cyt with an immediate peak (ΔF340/380 0.228 ± 0.042, n=50 cells, p<0.01). Acidosis and ATP caused different time courses of [Ca2+]cyt in HT29 cells (Figure 5). These data suggest that the acidosis-induced increase in [Ca2+]cyt does not depend on ATP release.
Figure 5. Acidosis and ATP causes different time courses of [Ca2+]cyt in HT29 cells.
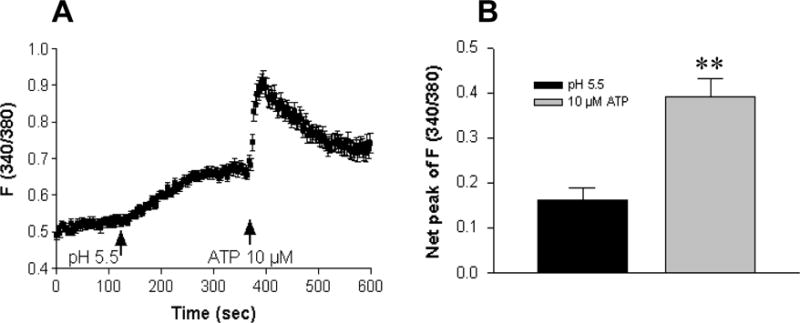
A: A slow rise in [Ca2+]cyt was first induced by extracellular acidosis of pH 5.5, and further induced by ATP (10 μM) with an immediate peak. B: Summarized data showing net peak of [Ca2+]cyt rise induced by pH 5.5 or ATP (10 μM). Values are mean ± SE, n = 50 cells for each tracing or bar. **P < 0.01 vs. pH 5.5.
Amiloride attenuation of acid-stimulated duodenal bicarbonate secretion in vivo
After the expression and function of ASIC1a in intestinal epithelial cells have been identified, we sought to test if ASIC1a is involved in the acid-stimulated duodenal mucosal bicarbonate secretion (DMBS). Female C57 mice were anesthetized and the proximal duodenum were isolated followed by luminal perfusion with either saline as a control or acid as a physiological stimulant. Figure 6A shows a time course study of acid-stimulated murine DMBS in vivo. Duodenal luminal perfusion of HCl (10 mM) resulted in a robust increase in DMBS, which reached a maximal level at about 30 min after acid stimulation and declined thereafter to the baseline level. Net peak HCO3− secretion, calculated from the difference between the baseline and the peak value, was used to assess acid-stimulated HCO3− secretion (Figure 6B). Luminal pretreatment with amiloride (100 μM), a widely used ASIC blocker2, 3, did not affect basal murine DMBS in vivo, but significantly attenuated the acid-stimulated murine DMBS (Δpeak 10.63 ± 2.18 μmol/cm-h with HCl vs. 0.05 ± 0.56 μmol/cm-h with HCl plus amiloride, n=5, p<0.01). Amiloride reduced net peak of the acid-stimulated DMBS by 95% (Figure 6). These data suggest that acid may act on duodenal ASIC1a to stimulate murine DMBS in vivo.
Figure 6. Acid-stimulated murine duodenal mucosal bicarbonate secretion (DMBS) in vivo is significantly inhibited by amiloride.
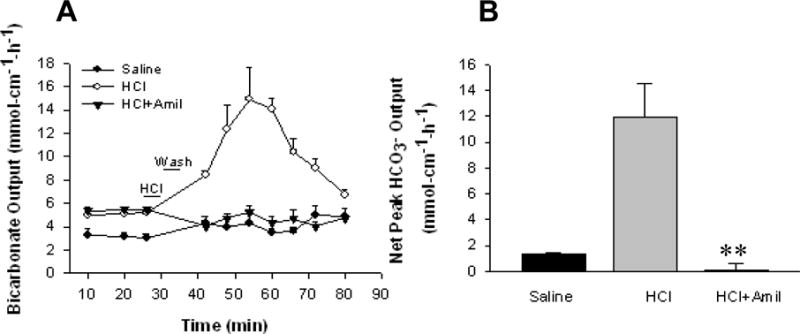
A: Time course of murine DMBS induced by luminal perfusion with saline, HCl (10 mM) alone, or HCl plus amiloride (100 μM). B: Summarized data showing net peak HCO3− secretion calculated from the difference between the baseline and the peak value at 30 min after HCl perfusion. Values are mean ± SE, n = 5 animals for each tracing or bar. **P < 0.01 vs. HCl alone.
Discussion
Although acid-sensing ion channels (ASICs) have been studied in various types of mammalian cells, their presence and function in intestinal epithelial cells are poorly understood. ASIC proteins co-assemble to form homo and heteromeric cation channels, typically with Ca2+ and Na+ as the major charge carriers1–3. Most ASICs are gated by external H+ with pH between 7.2 – 4.0 and are sensitive to amiloride2. Previous studies support the presence of ASICs in human gastrointestinal tract2, 6–8, but it is currently unknown about the expression and function of ASIC isoforms in intestinal epithelial cells. The major findings of this study are that: 1) ASIC1a is expressed in human intestinal epithelial cells and murine duodenal mucosa, 2) ASIC1a plays an important role in the regulation of [Ca2+]cyt homeostasis in human intestinal epithelial cells, and 3) acid may stimulate murine DMBS via ASIC1a-mediated Ca2+ signaling.
Expression of ASICs have been detected in both the nervous and vascular systems, and their functional role have been intensively studied in neurons and cerebral vascular smooth muscle cells1–3. In the nervous system, ASIC1a was found to be expressed in synapses of central neurons and to provide a non-voltage-gated pathway for Ca2+ entry into neurons13. Activation of ASIC1a may lead to neuronal injury9, 10, 21. In the vascular system, ASIC1b was found to predominantly expressed in cerebral vascular smooth muscle cells and to provide an inward Na+ current5, 9, 10, 21. However, in the gastrointestinal tract, the expression and function of ASICs were mainly studied in primary afferent neurons2, 6, 7. ASIC1-3 are expressed in dorsal root ganglion cells and may contribute to abdominal hyperalgesia and pain during the gastrointestinal luminal acidosis resulted from inflamation, ischemia, or malignant tumor growth2, 6. Although ASICs have been molecularly cloned from the human small intestine8, little is currently known about the expression and function of ASICs in intestinal epithelial cells. We readily detected mRNA expression of ASIC1a in human intestinal epithelial cells and murine duodenal mucosal epithelia, which laid a foundation for the functional role of ASIC1a in intestinal epithelial cells.
Digital Ca2+ imaging was utilized to study the function of ASIC1a in human intestinal epithelial cells since ASIC1a shows a significant Ca2+ permeability13. We found that acidosis induced a pH-dependent increase in [Ca2+]cyt with a pH0.5 of 5.9 in HT29 cells, which is same to the pH0.5 in COS cells expressing ASIC1a13. Furthermore, acidosis-induced increase in [Ca2+]cyt was attenuated in the absence of extracellular Ca2+. Both the ASIC channel blocker amiloride and the nonselective cation channel blocker SK&F96365 significantly reduced the amplitude of the [Ca2+]cyt response. These findings provide strong evidence for the functional expression of ASIC1a in human intestinal epithelial cells and its important role in the regulation of [Ca2+]cyt homeostasis in these cells. We also determined the possible involvement of other mechanisms responsible for this acidosis-induced rise in [Ca2+]cyt. Although experimental evidence indicates that TRPV plays a role as a acid sensor in the duodenum2, 6, a TRPV channel blocker ruthenium red failed to affect acidosis-induced Ca2+ mobilization, excluding its involvement in HT29 cells. However, voltage-gated Ca2+ channels and PLC pathway may play a minor role in the acidosis-induced Ca2+ mobilization in HT29 cells as in other types of mammalian cells2, 19, 20. Taken together, three mechanisms may be responsible for the acidosis-induced Ca2+ mobilization in intestinal epithelial cells: 1) ASIC1a that plays a predominate role (~50%), 2) voltage-gated Ca2+ channels that play a minor role (~20%), and 3) intracellular Ca2+ release through the PLC pathway that also plays a minor role (~20%). Moreover, ATP induced an additional rise in [Ca2+]cyt after acidosis-induced Ca2+ mobilization in HT29 cells, causing a different time course of [Ca2+]cyt. This data suggests that acidosis may raise [Ca2+]cyt via a direct activation of ASIC1a rather than via ATP release-activated P2 receptors in intestinal epithelial cells.
While the gastrointestinal lumen is the greatest source of acid production in the human body, the presence and function of ASICs in the gastrointestinal epithelial cells are poorly understood. Acid sensing is of paramount importance to pH homeostasis in gastroduodenal lumen2, 6, 7. Acid and bicarbonate secretion are the major determinants of duodenal pH, making the average diurnal pH in the empty human duodenum at about 6.06, which is close to the pH0.5 of ASIC1a13. Since ASIC1a is expressed in human intestinal epithelial cells and murine duodenal epithelia and activation of ASIC1a induced a rise in [Ca2+]cyt, it is logical to speculate that ASIC1a plays a role in the regulation of acid-stimulated DMBS and duodenal pH homeostasis via Ca2+ signaling. Indeed, we found that ASIC blocker amiloride significantly attenuated the acid-stimulated murine DMBS in vivo, suggesting that acid may stimulate DMBS via ASIC1a-mediated Ca2+ signaling.
Since amiloride is not a specific blocker for ASICs, it also blocks Na+/H+ exchanger (NHE) and epithelial Na+ channels (ENaC). Caution must be taken to interpret this data. There exists the possibility that our results could be caused by the inhibition of NHE by amiloride; however, it should be noted that the inhibition of the NHE actually activates DMBS22, 23, therefore the decrease in DMBS must be caused by the inhibitive effect of amiloride on ASICs. Although it is an important pathway for Na+ entry into a variety of epithelial cells, expression of ENaC β-subunit is lacking in the duodenal epithelium, making the activity of duodenal ENaC unlikely24. Taken together, our data showing both expression and function of ASIC1a in intestinal epithelial cells have provided support for its important role in mediating acid-stimulated DMBS. However, we found that amiloride (100 μM) inhibits 10 mM acid (pH 2.0)-stimulated DMBS in vivo (by 95%) over two-fold stronger than blocks acidosis (pH 5.0)-induced Ca2+ signaling in HT29 cells in vitro (by 41%). This may be due to: 1) different acidosis stimulation (pH 2.0 vs.5.0), 2) different measurements (cell signaling vs. end-point function), and 3) different levels of the study (single cells vs. whole animals).
In the classical model, the acid-stimulated DMBS is a relatively long and indirect process25, 26: the discharge of gastric acid into the duodenum initially stimulates the release of multiple biological factors from the enteric nerve endings or enterochromaffin cells in the duodenum (such as ACh, PGE2, 5-HT, etc)27, 28, which subsequently act on duodenal epithelial cells to stimulate DMBS via [Ca2+]cyt, cAMP, and cGMP signaling pathways26, 28, 29. However, to the best of our knowledge, no evidence has yet been found of a relatively short and direct pathway on duodenal epithelial cells for the acid-stimulated DMBS. Although further studies using specific ASIC1a blockers are needed to confirm our hypothesis, the present study may provide the first evidence for this direct pathway, which has physiological implication for the acid-stimulated DMBS and duodenal protection against gastric acid25, 30.
Acknowledgments
This work was supported by the American Gastroenterological Association Student Research Fellowship Award to X. Dong, and partially supported by American Heart Association Beginning Grant-in-Aid Award (0565025Y) and American Lung Association to H. Dong, and by the University of California San Diego Digestive Diseases Research Development Center grant (DK-080506), in which H. Dong serves as director of cell imaging core.
References
- 1.Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A protongated cation channel involved in acid-sensing. Nature. 1997;386:173–7. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- 2.Holzer P. Acid-sensitive ion channels and receptors. Handb Exp Pharmacol. 2009:283–332. doi: 10.1007/978-3-540-79090-7_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lingueglia E. Acid-sensing ion channels in sensory perception. J Biol Chem. 2007;282:17325–9. doi: 10.1074/jbc.R700011200. [DOI] [PubMed] [Google Scholar]
- 4.Lingueglia E, de Weille JR, Bassilana F, Heurteaux C, Sakai H, Waldmann R, Lazdunski M. A modulatory subunit of acid sensing ion channels in brain and dorsal root ganglion cells. J Biol Chem. 1997;272:29778–83. doi: 10.1074/jbc.272.47.29778. [DOI] [PubMed] [Google Scholar]
- 5.Chung WS, Farley JM, Swenson A, Barnard JM, Hamilton G, Chiposi R, Drummond HA. Extracellular acidosis activates ASIC-like channels in freshly isolated cerebral artery smooth muscle cells. Am J Physiol Cell Physiol. doi: 10.1152/ajpcell.00511.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holzer P. Taste receptors in the gastrointestinal tract. V. Acid sensing in the gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol. 2007;292:G699–705. doi: 10.1152/ajpgi.00517.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holzer P. Acid-sensitive ion channels in gastrointestinal function. Curr Opin Pharmacol. 2003;3:618–25. doi: 10.1016/j.coph.2003.06.008. [DOI] [PubMed] [Google Scholar]
- 8.Schaefer L, Sakai H, Mattei M, Lazdunski M, Lingueglia E. Molecular cloning, functional expression and chromosomal localization of an amiloride-sensitive Na(+) channel from human small intestine. FEBS Lett. 2000;471:205–10. doi: 10.1016/s0014-5793(00)01403-4. [DOI] [PubMed] [Google Scholar]
- 9.Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–98. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 10.Xiong ZG, Chu XP, Simon RP. Ca2+ -permeable acid-sensing ion channels and ischemic brain injury. J Membr Biol. 2006;209:59–68. doi: 10.1007/s00232-005-0840-x. [DOI] [PubMed] [Google Scholar]
- 11.Wang WZ, Chu XP, Li MH, Seeds J, Simon RP, Xiong ZG. Modulation of acid-sensing ion channel currents, acid-induced increase of intracellular Ca2+, and acidosis-mediated neuronal injury by intracellular pH. J Biol Chem. 2006;281:29369–78. doi: 10.1074/jbc.M605122200. [DOI] [PubMed] [Google Scholar]
- 12.Pignataro G, Simon RP, Xiong ZG. Prolonged activation of ASIC1a and the time window for neuroprotection in cerebral ischaemia. Brain. 2007;130:151–8. doi: 10.1093/brain/awl325. [DOI] [PubMed] [Google Scholar]
- 13.Yermolaieva O, Leonard AS, Schnizler MK, Abboud FM, Welsh MJ. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci U S A. 2004;101:6752–7. doi: 10.1073/pnas.0308636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holm M, Johansson B, von Bothmer C, Jonson C, Pettersson A, Fandriks L. Acid-induced increase in duodenal mucosal alkaline secretion in the rat involves the Larginine/NO pathway. Acta Physiol Scand. 1997;161:527–32. doi: 10.1046/j.1365-201X.1997.00239.x. [DOI] [PubMed] [Google Scholar]
- 15.Page AJ, Brierley SM, Martin CM, Price MP, Symonds E, Butler R, Wemmie JA, Blackshaw LA. Different contributions of ASIC channels 1a, 2, and 3 in gastrointestinal mechanosensory function. Gut. 2005;54:1408–15. doi: 10.1136/gut.2005.071084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong X, Smoll EJ, Ko KH, Lee J, Chow JY, Kim HD, Insel PA, Dong H. P2Y receptors mediate Ca2+ signaling in duodenocytes and contribute to duodenal mucosal bicarbonate secretion. Am J Physiol Gastrointest Liver Physiol. 2009;296:G424–32. doi: 10.1152/ajpgi.90314.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van den Berghe N, Vaandrager AB, Bot AG, Parker PJ, de Jonge HR. Dual role for protein kinase C alpha as a regulator of ion secretion in the HT29cl.19A human colonic cell line. Biochem J. 1992;285(Pt 2):673–9. doi: 10.1042/bj2850673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 19.Roy A, Rozanov C, Iturriaga R, Mokashi A, Lahiri S. Acid-sensing by carotid body is inhibited by blockers of voltage-sensitive Ca2+ channels. Brain Res. 1997;769:396–9. doi: 10.1016/s0006-8993(97)00820-2. [DOI] [PubMed] [Google Scholar]
- 20.Christensen BN, Kochukov M, McNearney TA, Taglialatela G, Westlund KN. Proton-sensing G protein-coupled receptor mobilizes calcium in human synovial cells. Am J Physiol Cell Physiol. 2005;289:C601–8. doi: 10.1152/ajpcell.00039.2005. [DOI] [PubMed] [Google Scholar]
- 21.Xiong ZG, Chu XP, Simon RP. Acid sensing ion channels–novel therapeutic targets for ischemic brain injury. Front Biosci. 2007;12:1376–86. doi: 10.2741/2154. [DOI] [PubMed] [Google Scholar]
- 22.Furukawa O, Bi LC, Guth PH, Engel E, Hirokawa M, Kaunitz JD. NHE3 inhibition activates duodenal bicarbonate secretion in the rat. Am J Physiol Gastrointest Liver Physiol. 2004;286:G102–9. doi: 10.1152/ajpgi.00092.2003. [DOI] [PubMed] [Google Scholar]
- 23.Repishti M, Hogan DL, Pratha V, Davydova L, Donowitz M, Tse CM, Isenberg JI. Human duodenal mucosal brush border Na(+)/H(+) exchangers NHE2 and NHE3 alter net bicarbonate movement. Am J Physiol Gastrointest Liver Physiol. 2001;281:G159–63. doi: 10.1152/ajpgi.2001.281.1.G159. [DOI] [PubMed] [Google Scholar]
- 24.Odes HS, Smirnoff P, Guberman R, Pollak-Charcon S, Sperber AD, Fich A, Fraser GM, Schwartz B. Cystic fibrosis transmembrane conductance regulator and Na+ channel subunits mRNA transcripts, and Cl- efflux, show a different distribution in rat duodenum and colon. Acta Physiol Scand. 2003;178:231–40. doi: 10.1046/j.1365-201X.2003.01138.x. [DOI] [PubMed] [Google Scholar]
- 25.Flemstrom G, Isenberg JI. Gastroduodenal mucosal alkaline secretion and mucosal protection. News Physiol Sci. 2001;16:23–8. doi: 10.1152/physiologyonline.2001.16.1.23. [DOI] [PubMed] [Google Scholar]
- 26.Seidler U, Blumenstein I, Kretz A, Viellard-Baron D, Rossmann H, Colledge WH, Evans M, Ratcliff R, Gregor M. A functional CFTR protein is required for mouse intestinal cAMP-, cGMP- and Ca2+-dependent HCO3− secretion. J Physiol. 1997;505(Pt 2):411–23. doi: 10.1111/j.1469-7793.1997.411bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong H, Sellers ZM, Smith A, Chow JY, Barrett KE. Na+/Ca2+ exchange regulates Ca2+-dependent duodenal mucosal ion transport and HCO3− secretion in mice. Am J Physiol Gastrointest Liver Physiol. 2005;288:G457–65. doi: 10.1152/ajpgi.00381.2004. [DOI] [PubMed] [Google Scholar]
- 28.Smith AJ, Chappell AE, Buret AG, Barrett KE, Dong H. 5-Hydroxytryptamine contributes significantly to a reflex pathway by which the duodenal mucosa protects itself from gastric acid injury. Faseb J. 2006;20:2486–95. doi: 10.1096/fj.06-6391com. [DOI] [PubMed] [Google Scholar]
- 29.Sellers ZM, Mann E, Smith A, Ko KH, Giannella R, Cohen MB, Barrett KE, Dong H. Heat-stable enterotoxin of Escherichia coli (STa) can stimulate duodenal HCO3(−) secretion via a novel GC-C- and CFTR-independent pathway. Faseb J. 2008;22:1306–16. doi: 10.1096/fj.06-7540com. [DOI] [PubMed] [Google Scholar]
- 30.Ham M, Kaunitz JD. Gastroduodenal mucosal defense. Curr Opin Gastroenterol. 2008;24:665–73. doi: 10.1097/MOG.0b013e328311cd93. [DOI] [PubMed] [Google Scholar]