

Abstract
The bacterium Myxococcus xanthus has two motility systems: S motility, which is powered by type IV pilus retraction, and A motility, which is powered by unknown mechanism(s). We found that A motility involved transient adhesion complexes that remained at fixed positions relative to the substratum as cells moved forward. Complexes assembled at leading cell poles and dispersed at the rear of the cells. When cells reversed direction, the A-motility clusters relocalized to the new leading poles together with S-motility proteins. The Frz chemosensory system coordinated the two motility systems. The dynamics of protein cluster localization suggest that intracellular motors and force transmission by dynamic focal adhesions can power bacterial motility.
During the exhibition of gliding motility, bacteria move across solid surfaces without the use of flagella (1). Glidingmotility is important for biofilm formation and bacterial virulence. Motility in Myxococcus xanthus, a Gram-negative rod-shaped bacterium, relies on two separate but coordinated motility engines. S motility is powered by type IV pili that are assembled at the leading cell pole; movement is produced as the pili bind to surface exopolysaccharides and are retracted, thereby pulling the cell forward (2). A motility, on the other hand, is not associated with pili or other obvious structures and is not well understood.
To investigate the A-motility system, we studied AglZ, a protein that is essential for A motility but dispensable for S motility (fig. S1, A and B) (3). AglZ is similar to FrzS, an S-motility protein that oscillates from one cell pole to the other when cells reverse direction (4) (fig. S1A). To track the localization of AglZ in moving cells, we constructed an M. xanthus strain containing a chimeric aglZ-yfp gene in place of the endogenous aglZ gene (fig. S2A). This chimeric gene encodes an AglZ–yellow fluorescent protein (YFP) fusion protein that was stable and functional (fig. S2, B and C). We followed AglZ-YFP localization using time-lapse video microscopy: In fully motile cells, AglZ-YFP was localized in ordered clusters spanning the cell length; in stalled cells, it was localized at the leading cell pole (fig. S3).
To study the link between the localization of AglZ-YFP and motility, we focused our observations on AglZ-YFP in fully motile cells. These cells showed an ordered array of AglZ-YFP clusters spanning the cell body (Fig. 1A). As cells moved forward, AglZ-YFP clusters maintained fixed positions with respect to the agar substrate rather than to their relative positions in the cell (Fig. 1A). We analyzed the clusters by taking line scans of the fluorescence intensity along motility paths for successive movie frames (Fig. 1B). To identify the position of peaks in multiple frames, we calculated a thresholded line-scan average (Fig. 1B) (5). This analysis not only located the positions of clusters shown in Fig. 1A, but also had the sensitivity to find other common peaks that were difficult to identify when viewing images with the eye. In every cell that was examined (n = 30), the AglZ-YFP clusters remained fixed relative to the substratum. The only AglZ-YFP clusters that moved relative to the cell body were located at the leading pole, which suggests that new sites were assembled at that pole. The number of clusters per cell was dependent on cell length; clusters were disassembled when they reached the rear half of the cell (fig. S4).
Fig. 1.
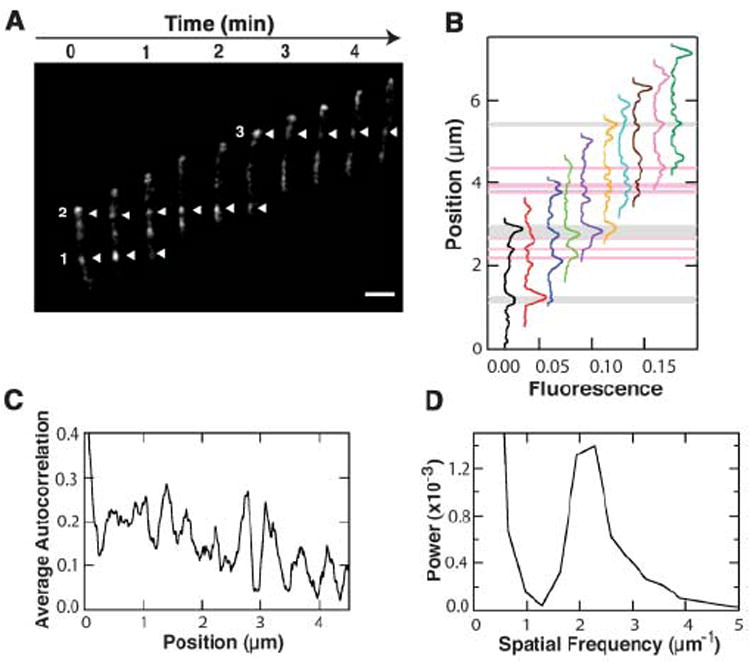
AglZ-YFP localizes to periodic sites that remain fixed relative to the substratum. (A) AglZ-YFP localization in a cell moving at constant velocity. Fluorescence micrographs captured every 30 s are shown. Numbered arrowheads highlight selected bright fluorescence clusters. Scale bar, 1 μm. (B) Line scans of fluorescence intensity as a function of position are shown for each movie frame in (A). For display purposes, individual scans have been shifted horizontally with time. Gray bars represent the three highest peaks in the average line scan, matching the positions of clusters 1 to 3 in (A). Pink bars denote all additional peaks found in the average scan. (C) Quantitative analysis of the AglZ-YFP fluorescence distribution in moving cells. The auto-correlation function of the thresholded line scan from six moving cells was averaged and displayed. (D) Power spectral density of the autocorrelation function.
We calculated the autocorrelation function of the line-scan averages from six different moving cells (Fig. 1C). Autocorrelations are useful for finding repeating patterns in a signal, such as the determination of the presence of periodicities buried under noise. The average of all six auto-correlation functions displayed a clear periodicity (Fig. 1C), represented by a single large peak at a spatial frequency of 2.15 ± 0.03 μm−1 in its power spectral density (Fig. 1D). This frequency corresponded to a spatial periodicity of 466 ± 7 nm, which is very similar to the helical pitch of bacterial actin MreB as seen in both Bacillus subtilis [470 nm (6)] and Escherichia coli [460 ± 80 nm (7)].
Because AglZ-YFP clusters remained fixed relative to the substratum as cells moved forward, they must be moving in the opposite direction to that of the cell. These dynamics suggest the formation of cytoskeleton-anchored transient surface adhesions that could power cellular movement. We analyzed cells that bend while in motion to investigate whether AglZ-YFP clusters could indicate sites of transient adhesion between the cell surface and the substratum. First, in a moving cell that was progressively bending, two distinct sites of curvature were observed (Fig. 2A). If AglZ-YFP localizes at sites that mediate adhesion with the substratum, then clusters should accumulate at sites of maximum bending. As expected, accumulation of AglZ-YFP was specifically observed at each site. Second, cells sometimes became stuck at their leading end, which prevented forward movement; these cells bent into U or S shapes as the active A engine pushed against the flexible cell wall. It has been proposed that these flailing motions are due to a motor pushing from the lagging end of the cell (8). However, if multiple motor-coupled adhesion complexes pushed cells forward, flailing would also occur. In that situation, cell bends would form between adhesion complexes. In a flailing cell expressing AglZ-YFP (Fig. 2B), as the front of the cell became fixed, the still-motile cell adopted a right-handed U shape and then relaxed to adopt a left-handed U shape that transitioned into an S shape to become a right-handed U shape again (neighboring cells probably block full relaxation between 2 to 3 min and 8 to 9 min). Cell shape correlated with the pattern of AglZ-YFP clusters. At each bend, enlarged AglZ-YFP fluorescence clusters were not observed at the maximum inflection points, but rather between the bends. The fluorescent clusters maintained relatively fixed positions with respect to the agar substrate, whereas the cell body appeared to move through them (Fig. 2, B and C). Furthermore, as the AglZ-YFP clusters faded and dispersed at the lagging cell pole, terminal cell bends relaxed and a spike in velocity was observed, which suggest that dispersal of the AglZ-YFP clusters indeed removed adhesion constraints (Fig. 2C). Thus, the cell body appears to move through “focal adhesion” sites where AglZ-YFP accumulates.
Fig. 2.
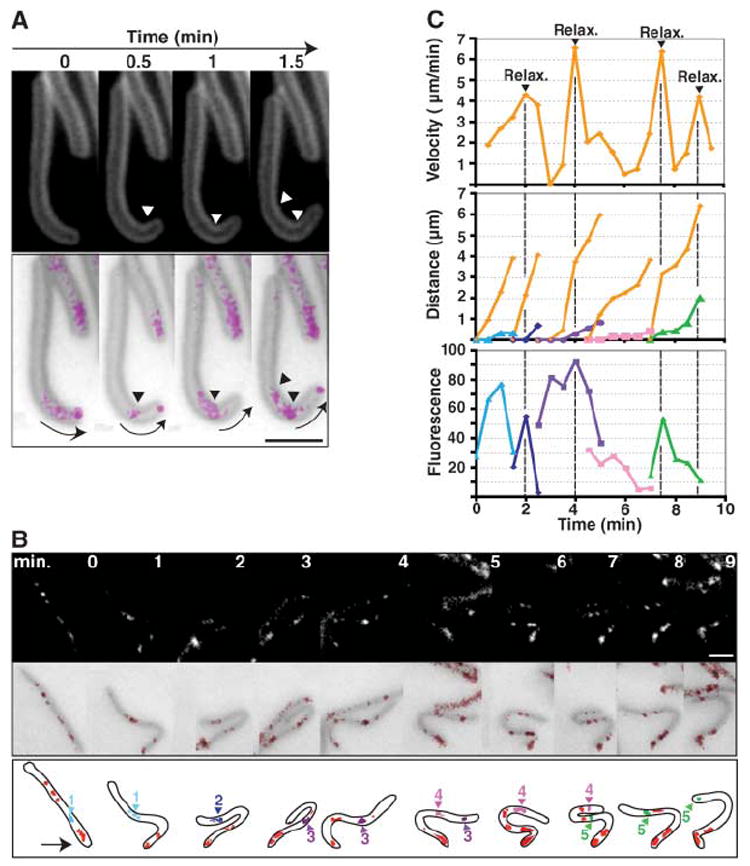
AglZ-YFP localizes to transient adhesion sites. (A) AglZ-YFP fluorescence clusters in a cell that bends while in motion. (Top) Cells stained with the membrane dye FM4-64 are shown. (Bottom) An overlay of the membrane signal (gray) and the AglZ-YFP signal (magenta), which is artificially colored for better clarity, are shown. White and black arrowheads point to regions of cell-body curvature and localization of the YFP signal, respectively. Arrows indicate the direction of movement. Scale bar, 1 μm. (B) AglZ-YFP fluorescence clusters in a cell undergoing flailing motion. Fluorescence and overlaid phase micrographs (top and middle rows, respectively) are shown. Time intervals, 1 min. A cartoon representation (bottom row) shows the clusters numbered and color-coded for the analysis shown in (C). The arrow indicates the stuck leading pole. Scale bar, 2 μm. (C) Dynamic behavior of the AglZ-YFP fluorescence clusters in the cell shown in (B). Time intervals, 30 s. (Top) The velocity of the lagging pole over time is shown. Dotted lines mark the times where relaxation of the terminal bend (Relax.) is observed. The leading pole remained immobilized for the entire duration of the time lapse. (Middle) The distance traveled by the AglZ-YFP clusters, color-coded and numbered as in (B), over time is shown. 1, blue triangles; 2, blue diamonds; 3, purple squares; 4, pink squares; 5, green triangles. For each cluster, the distance traveled by the lagging pole (orange diamonds) during the same time interval was plotted to show that the clusters remain mostly fixed relative to the substratum. (Bottom) The relative fluorescence intensity of each cluster over time. The same color code as that used in the middle panel applies.
The dynamics of AglZ-YFP localization (Fig. 1A) suggest that A-engine clusters are assembled at the leading cell pole and disassembled at the lagging end. It follows then that, during cellular reversals, proteins of the A-motility system should be shifted to the new leading pole along with S-motility components (4). Indeed, upon cellular reversal, AlgZ-YFP localized rapidly to the new leading pole (Fig. 3A and table S2). This was a two-step process: Immediately before reversal, the cell paused for 10 s, andAglZ-YFP became diffuse (Fig. 3B). The cell then reversed, and AglZ-YFP localized to the new leading pole after a 10-s delay. Thus, AglZ may not trigger polar switching, but rather might be assembled at the new leading pole along with other motility components.
Fig. 3.
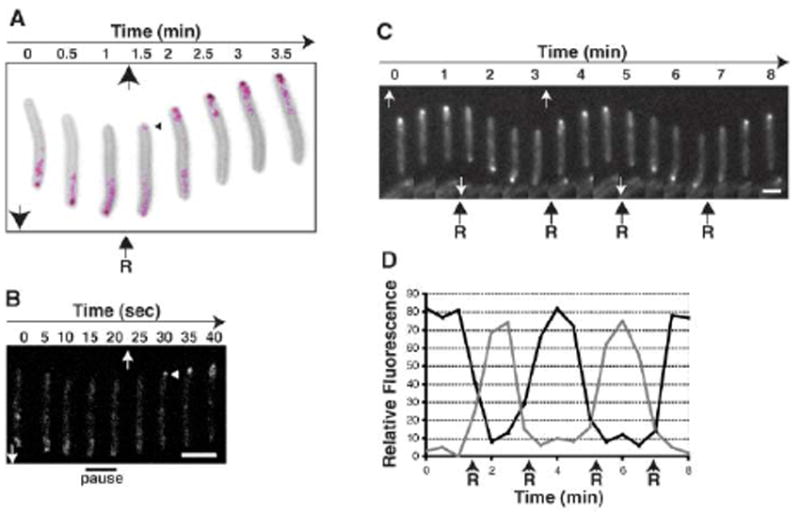
AglZ-YFP oscillates from pole to pole upon cellular reversals. (A) AglZ-YFP localized to the new leading pole upon cellular reversals. Fluorescent micrographs of AglZ-YFP (magenta) and a representative reversing cell stained with FM4-64 (gray) were overlaid to show AglZ-YFP dynamics every 30 s. The black arrows inside the panel indicate the direction of movement. The arrowheads in (A) and (B) show the relocalization of AglZ-YFP at the new leading pole. R, reversal. (B) AglZ-YFP dynamics at the time of reversal. Fluorescence micrographs of a reversing cell captured every 5 s are shown. The 10-s delay is indicated by “pause.” Scale bar, 2 μm. (C) AglZ-YFP oscillations in hyper-reversing cells. Fluorescent micrographs of a frzCDc cell that expresses AglZ-YFP captured every 30 s are shown. The white arrows in (B) and (C) indicate the direction of movement. Scale bar, 2 μm. (D) Quantitative fluorescence analysis of the cell presented in (C). The relative fluorescence intensities of each cell pole were measured in arbitrary units and plotted over time. The black line indicates the initial leading pole, and the gray line indicates the initial trailing pole.
The Frz chemosensory pathway, which regulates cell reversals, controls FrzS translocation to the leading pole when cells reverse direction (4). In a nonreversing frzE mutant, AglZ-YFP localization never switched poles. In contrast, a hyper-reversing frzCDc mutant showed very frequent reversals that were always followed by AglZ-YFP polar switching (Fig. 3, C and D, and table S2). In this frzCDc strain, AglZ-YFP was mostly polar in distribution, and ordered intracellular fluorescence clusters were only observed transiently. Thus, cellular reversals result from the concerted switching of both A- and S-motility components to the new leading pole (4). Coordination of these two engines must be achieved through the signaling activity of a common pathway.
Although AglZ-YFP localized to transient adhesion sites, it is unclear whether the force that produces locomotion is generated at those sites. To address this question, we investigated the motility of cells treated with the antibiotic cephalexin. Cephalexin-treated cells, which elongate up to 10 times their natural length, showed almost normal A motility but greatly reduced S motility, which suggests that the A engine is distributed along the cell body whereas the S engine is polar (9). We also observed that 10- to 30-μm-long A+S−-motile cephalexin-treated cells moved with velocities that were independent of cell length. The localization of AglZ-YFP was also correlated with the activity of the A engine in these cells (Fig. 4A). In these moving filaments, AglZ-YFP was localized in clusters that were distributed in the front part of the cell, whereas the back of the cell was largely depleted of clusters (Fig. 4A); consequently, the number of clusters per cell was largely independent of cell length (Fig. 4B). Thus, in the filaments, we could test whether force was produced at the sites where AglZ-YFP accumulates by analyzing the relationship between the number of sites and the “drag force overcome” [i.e., the force necessary to power the motility of a cell of given cell length and velocity (5)]. Indeed, the drag force overcome was proportional to the number of clusters in filamentous cells (Fig. 4C), indicating that motility force seems to be produced at the adhesion sites; these characteristics are similar to eukaryotic focal adhesions where both adhesion and force are generated (10).
Fig. 4.
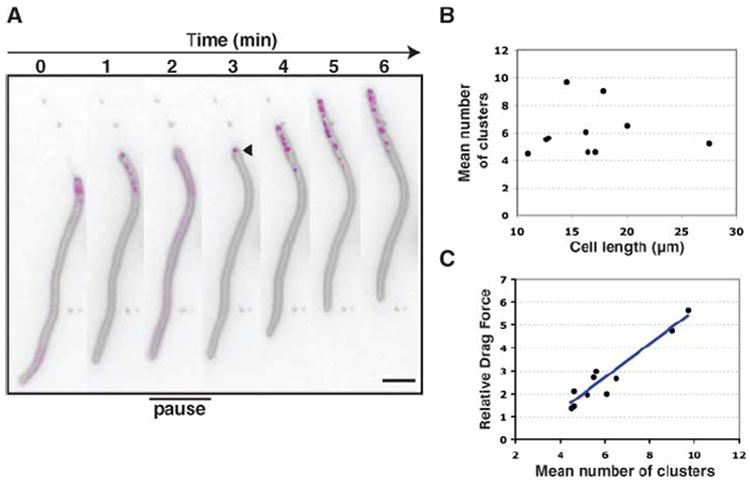
Dynamics of AglZ-YFP clusters in artificially elongated cells. (A) AglZ-YFP dynamics in A+S−-motile filamentous cells. Fluorescent micrographs of a representative 20-μm-long cephalexin-treated cell stained with FM4-64 (gray) expressing AglZ-YFP (magenta). AglZ-YFP is only found distributed over the front part of the cell when the cell is in motion. The arrowhead indicates polar condensation of AglZ-YFP. “Pause” indicates times when the cell motion is stopped. Scale bar, 2 μm. (B) Relationship between cluster number and filamentous cell length. (C) Relationship between relative drag force overcome and cluster number in filamentous cells.
Previously, a “slime gun” model for gliding motility was proposed because, in several bacterial species, motility is correlated with the secretion of slime through pores (nozzles) located in the outer membrane (11, 12), and a biophysical model suggested that the hydration of slime within the nozzles could generate sufficient force to propel bacteria forward (11). Our results are consistent with an alternate model, whereby intracellular motor complexes that connect to both membrane-spanning adhesion complexes and to the cytoskeleton power motility by pushing against the substratum and moving the cell body forward, much like focal adhesion–based traction or apicomplexan gliding motility in eukaryotic organisms (fig. S5) (10, 13). The periodicity of the AglZ-YFP clusters strongly implies the existence of a continuous helical filament that spans the length of the cell. Thus, the action of the motor complexes may induce the cell body to rotate as it pulls the cell forward (fig. S5). Rotation of the cell body as cells move has been shown for Cytophaga sp., an organism that also secretes slime during motility (14). Slime secretion may be part of the motility system, by supplying additional power for movement and/or adhesion or lubrication of the interface between the cell body and the substratum (10). The dynamics of AglZ-YFP and FrzS–green fluorescent protein (GFP) fusion protein (4) suggest how the A and S engines might be coordinated: A and S complexes oscillate so that they are targeted together to the new leading pole upon cellular reversal, which is synchronized by the Frz chemosensory system.
Supplementary Material
Acknowledgments
We thank Y. Inclàn for the construction of pZeroΩaglZ and comments on the manuscript; and J. Merlie, R. Losick, and G. Oster for helpful discussions and comments. This research was supported by a grant from NIH to D.R.Z. (GM20509).
Footnotes
www.sciencemag.org/cgi/content/full/315/5813/853/DC1
Materials and Methods
SOM Text
Figs. S1 to S5
Tables S1 and S2
References
References and Notes
- 1.Spormann AM. Microbiol Mol Biol Rev. 1999;63:621. doi: 10.1128/mmbr.63.3.621-641.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Y, et al. Proc Natl Acad Sci U S A. 2003;100:5443. doi: 10.1073/pnas.0836639100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang R, et al. J Bacteriol. 2004;186:6168. doi: 10.1128/JB.186.18.6168-6178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mignot T, Merlie JP, Zusman DR. Science. 2005;310:855. doi: 10.1126/science.1119052. [DOI] [PubMed] [Google Scholar]
- 5.Materials and methods are available as supporting material on Science Online
- 6.Defeu Soufo HJ, Graumann PL. BMC Cell Biol. 2005;6:10. doi: 10.1186/1471-2121-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kruse T, Moller-Jensen J, Lobner-Olesen A, Gerdes K. EMBO J. 2003;22:5283. doi: 10.1093/emboj/cdg504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolgemuth CW. Biophys J. 2005;89:945. doi: 10.1529/biophysj.105.062513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun H, Yang Z, Shi W. Proc Natl Acad Sci U S A. 1999;96:15178. doi: 10.1073/pnas.96.26.15178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Biochim Biophys Acta. 2004;1692:103. doi: 10.1016/j.bbamcr.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 11.Wolgemuth C, Hoiczyk E, Kaiser D, Oster G. Curr Biol. 2002;12:369. doi: 10.1016/s0960-9822(02)00716-9. [DOI] [PubMed] [Google Scholar]
- 12.Hoiczyk E, Baumeister W. Curr Biol. 1998;8:1161. doi: 10.1016/s0960-9822(07)00487-3. [DOI] [PubMed] [Google Scholar]
- 13.Baum J, Papenfuss AT, Baum B, Speed TP, Cowman AF. Nat Rev Microbiol. 2006;4:621. doi: 10.1038/nrmicro1465. [DOI] [PubMed] [Google Scholar]
- 14.Godwin SL, Fletcher M, Burchard RP. J Bacteriol. 1989;171:4589. doi: 10.1128/jb.171.9.4589-4594.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.