

Abstract
Multi-organ microdevices can mimic tissue-tissue interactions that occur as a result of metabolite travel from one tissue to other tissues in vitro. These systems are capable of simulating human metabolism, including the conversion of a pro-drug to its effective metabolite as well as its subsequent therapeutic actions and toxic side effects. Since tissue-tissue interactions in the human body can play a significant role in determining the success of new pharmaceuticals, the development and use of multi-organ microdevices presents an opportunity to improve the drug development process. The goals are to predict potential toxic side effects with higher accuracy before a drug enters the expensive phase of clinical trials as well as to estimate efficacy and dose response. Multi-organ microdevices also have the potential to aid in the development of new therapeutic strategies by providing a platform for testing in the context of human metabolism (as opposed to animal models). Further, when operated with human biopsy samples, the devices could be a gateway for the development of individualized medicine. Here we review studies in which multi-organ microdevices have been developed and used in a ways that demonstrate how the devices’ capabilities can present unique opportunities for the study of drug action. We also discuss the challenges that are inherent in the development of multi-organ microdevices. Among these are how to design the devices, and how to create devices that mimic the human metabolism with high authenticity. Since single organ devices are testing platforms for tissues that can later be combined with other tissues within multi-organ devices, we will also mention single organ devices where appropriate in the discussion.
Introduction
1) Limitations of the Current Drug Development Process
Modern drug development requires implementation of extensive pre-clinical testing and validation protocols before potential therapeutic compounds are approved to progress to clinical evaluation. This process is costly and time-consuming, as well as inefficient as for every ten drugs entering clinical trials, only one or two will typically be licensed for eventual use in humans (Dimasi, 2001; Dimasi & Grabowski, 2007).
One of the major factors influencing this poor success rate is the lack of preclinical model systems capable of providing accurate predictions of human responses to novel therapeutic drugs. The current gold standard for laboratory based preclinical evaluation is a combination of in vitro cell culture assay and in vivo animal model experimentation and assessment. Cell culture assays are advantageous since they provide controlled environments where cellular maturation and activity are easily observed and tested. However, cultures of single cell types, or even co-cultures of 2 or 3 complimentary cell types, lack the complexity of living systems and are incapable of modeling situations where organ-organ or tissue-tissue communication are important. This simplicity is a major drawback in drug development studies since it is difficult to predict the oftentimes complex drug metabolism and the effect of metabolite activity on non-target tissues. Moreover, cells maintained in standard in vitro culture conditions often suffer from incomplete maturation, or are held in a configuration that prevents their full functional development, making predictions of in vivo tissue function more difficult to extrapolate.
Animal models maintain the intricacy of living systems, making assessment of organ-organ crosstalk and non-target organ toxicity possible. However, the inherent complexity of interconnected tissues can make specific modes of action difficult to elucidate and therefore confound observations. Furthermore, animal models have, on multiple occasions, been demonstrated to be poor predictors of human responses to drug treatment. The assumption that beneficial outcomes observed in animals will translate to human patients has led to clinical situations where treatments have proved ineffective or even harmful to patient wellbeing and recovery (Greek & Menache, 2013).
Most of the current in vitro models used by the pharmaceutical industry consist mainly of isolated single cells from a single organ. This simplification does not reflect the complexity of the organ’s interaction that occurs with the rest of the body in vivo. Indeed, it is well recognized that toxicity phenomena are a consequence of a complex series of events that may involve several organs. For example, bioactivation of a drug by specific liver enzymes may result in toxic events at a different organ. The current limitations of experimental methods confirms the need of an intermediate human in vitro model in the early stage of drug development that could efficiently reproduce multi-organ interactions to better predict the side effects observed in clinical trials.
The development of more appropriate and informative human models for preclinical drug screening is necessary to improve the success rate of clinical trials. Models that provide predictions with higher accuracy would reduce the cost of therapeutic development and improve the speed at which new drugs are approved for patients, as well as reducing (or ultimately eliminating) ethical concerns regarding the use of animals in experimentation. To this end, recent research efforts have focused on the establishment of physiologically relevant, multi-organ, functional in vitro models utilizing human cell sources. Such models are currently being designed to promote full functionality and molecular maturation of human cell types in configurations that facilitate the measurement and assessment of cell performance in real-time and in a high-throughput manner.
2) The Concept of Multi-Organ Microdevices
Multi-organ microdevices are microfluidic devices that mimic key aspects of human metabolism by connecting the fluidic streams from several on-chip in vitro tissue cultures with each other in a physiologically relevant manner so that metabolites are consumed, produced, and exchanged (via recirculation) between all tissues at physiologically relevant concentrations. The devices have been referred to as micro-cell culture analogs (μCCA), microphysiological systems, or body-on-a-chip systems. The combination of several tissues allows one to represent the function of several organs and observe their individual response to a drug as well as the influence this response exerts on other organs. Multi-organ devices can be used to simulate the conversion of a pro-drug to an effective compound that acts on another tissue as well as the compound’s toxicity at tissues that are not the intended target tissue (Sung & Shuler, 2009a). The ability of some tissues to modulate drug toxicity - for example, fat tissue, which modulates the toxicity by storing compounds and thereby reducing its concentration within the fluidic stream – can be observed (Viravaidya & Shuler, 2004a). The devices can also shed light on the quantitative influence of barrier tissues such as skin, lung epithelium, gastrointestinal tract epithelium, and endothelium impose on the bioavailability of a drug at the intended target tissue (Mahler, Esch, Glahn, & Shuler, 2009a). Within the device any number of tissues can be connected and the circulation of soluble metabolites between them enables the simulation and prediction of tissue-tissue interactions that are important in drug development (Baker, 2011; Esch, King, & Shuler, 2011).
While tissue-tissue interactions can be simulated with conventional, static cell culture systems (Li, 2009), the use of microtechnology allows chamber sizes and fluidic circuitry to be designed in a way that makes the simulation more physiologically relevant. For example, organ chamber sizes can be designed so that fluid residence times within them relate to each other as they do in vivo. According to this approach, a kidney chamber would be a fraction of the size of a liver chamber. In addition, the sizes of the connecting fluid channels can be designed to distribute the blood surrogate according to the in vivo blood distribution. For example, the circulation in the body delivers similar amounts of blood to the kidney and the liver despite their difference in tissue volume (Davies & Morris, 1993). Other advantages of microfabrication include the ability to fabricate many devices in a cost-effective manner. In addition, many cells/tissues respond to mechanical forces, such as those derived from fluid flow, and the cell’s response to mechanical forces can be mimicked in a microfabricated microfluidic system. The redesign and implementation of changes/improvements of the devices can be done fairly rapidly. Microfabrication techniques allow for the precise implementation of design principles while allowing for flexibility at a relatively low cost.
The principles guiding the development of physiologically based pharmacokinetic (PBPK) models provide guidance for designing multi-organ microdevices. In PBPK models, every organ is represented as a compartment. The uptake, distribution, metabolism, and excretion (ADME) of a drug is described with a set of differential equations. Multi-organ microdevices can be seen as physical representations of PBPK models, in which the organs are represented by a physical compartment, and the equations that govern the ADME of a drug are physically carried out by the tissues and fluidic channels that connect the organ chambers. This strategy led to classification of such devices as cell culture analogs of a PBPK, and later as the micro-cell culture analog of a PBPK (μCCA). Using this idea, one may design a device by first building a PBPK model that contains the organs of interest and then build a physical device that corresponds to the mathematical model. For example, figure 1 shows visually how the PBPK of the human body can be simplified by considering organs of interest explicitly but combining slowly perfused tissues and rapidly perfused tissues into two separate compartments. Combining organs is a valid approach when these organs neither react with nor absorb the drug or its metabolites and hence the organs are not important to capturing the full dynamics of drug and metabolite distribution. In the example given in figure 1, the gastrointestinal tissue, bone marrow, adipose tissue, kidney, and liver tissue were considered explicitly, while the other tissues were combined. Additionally, depending on the question that is to be addressed with the device, fluid pathways mimicking blood recirculation (expressed in the model as arrows) can be simplified. In a setup in which the fluid is split passively, the dimensions of the fluid channels determine how much fluid flows to each organ chamber. While the organ chambers volumes are scaled by a factor (typical scaling ranges from 40000–250000), the percentage of fluid flow that reaches each organ chamber should be comparable to the percent blood flow the respective organ receives in vivo. Through adjusting the pressure drop across each of the channels by adjusting the channels dimension, the needed fluid flow rate can be achieved. Of course, the fewer simplifications are made to the PBPK, the more authentic will be the device’s response. The complexity of the device should be sufficient to answer the question of interest, but the full model of the human body is not always necessary. An advantage that arises from this approach is that the results of the PBPK and the physical device can be compared and any discrepancies may lead to opportunities of further investigation and a deeper understanding of the biological system. PBPKs provide a powerful approach to designing multi-organ devices in a physiologically relevant manner.
Figure 1.
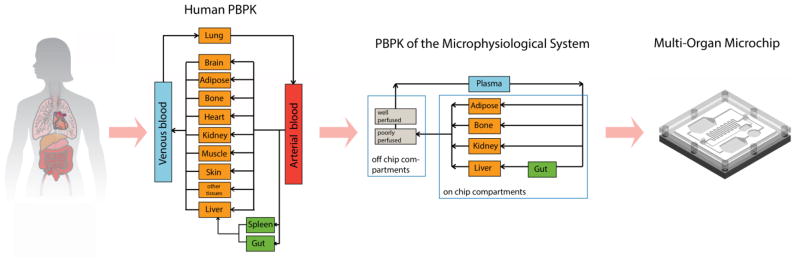
Illustration of how simplified PBPK models can guide the physiologic design of multi-organ microdevices: Organs that are of interest for a particular study are incorporated as chamber on the device. Organs that are not of interest can be combined within one volume (filled with liquid for well-perfused organs or filled with hydrogel for poorly perfused organs). Fluidic circuitry are the physical equivalent of the equations that express the transport of a drug in a in the PBPK model, and the cells that are cultured within each organ chamber are an equivalent for the equations that express the absorption and metabolism of drugs. The device that is illustrated here was used for the simulation of the first pass metabolism of acetaminophen.
3) Multi-Organ In Vitro Systems Versus Animal Models
Multi-organ microdevices have several advantages over the use of animal models for drug testing, with the most important one being that the devices can be operated with human cells and hence they are capable of mimicking the human metabolism instead of animal metabolism. When scaled appropriately (according to organ volumes and considering the activity of the available in vitro tissue constructs) and when operated with a physiologic ratio of blood surrogate to cells, the metabolite concentrations in the device are the same as in the human body and predictions of effective and toxic concentrations can be made. This is not always possible when extrapolating data from animal models to humans.
Additionally, the devices are more cost effective and allow for the testing of combinations of compounds at varying concentrations when operating many of them in parallel. The number of animals that would be needed for such screening would be rather high and add to the costs of drug development as well as to the ethical concerns that accompany drug testing with animals.
Another important distinction from animal models is that multi-organ microdevices can be operated with physiologic and non-physiologic conditions. The use of physiologic conditions for predicting drug action is obvious, but the use of non-physiologic conditions can be of advantage as well. For example, if the origin of a toxic metabolite is unclear, some of the organs within the device could be set up with smaller or larger volumes than would be physiologic (or they could be left out entirely). The dose response information resulting from experiments with such devices can give important information about drug action.
Experimental results from experiments with multi-organ microdevices can be compared to predictions obtained with mathematical PBPK models. Mathematical models can be tailored to reflect the constellation of the on-chip situation. Since mathematical models rely on our knowledge of metabolic pathways, any discrepancies between the data from the models and experiments point to a gap in our understanding of the human metabolism. Additional further investigations will likely expand our knowledge and understanding, which can in turn be incorporated into the model.
Multi-organ microdevices open up paths to individualized medicine when operated with patient biopsy samples, and to investigating new therapeutic strategies that could bear a high risk, but have high potential impact.
4) Single Organ Microdevices
Microfluidic single organ models provide advantages over static cultures, and have been used to investigate disease states and drug actions. Since the liver is the most important organ for drug metabolism and clearance, many efforts has been dedicated to developing liver models that function well within microfluidic devices. (Chao, Maguire, Novik, & Cheng, 2009; Domansky et al., 2010; Griffith et al., 2006; Hattersley, Greenman, & Haswell, 2011; Kidambi et al., 2009; S.-A. Lee, Kang, Ju, Kim, & Lee, 2013; Ma, Zhang, Qin, & Lin, 2009; Novik, Maguire, Chao, Cheng, & Yarmush, 2010; Powers, Domansky, et al., 2002a; Powers, Janigian, et al., 2002b; Schütte et al., 2011; C. Zhang, Chia, Ong, Zhang, Toh, van Noort, et al., 2009a). On chip tissues of the gastrointestinal tract, (HwanáSung, 2011; Imura, Asano, Sato, & Yoshimura, 2009; Pusch et al., 2011) skin, (Ataç et al., 2013; Brauchle, Johannsen, Nolan, Thude, & Schenke-Layland, 2013) lung, (Hinderer et al., 2012; Huh, Matthews, Mammoto, & Montoya-Zavala, 2010), heart, (M. B. Chen, Srigunapalan, Wheeler, & Simmons, 2013) microvasculature, (Esch, Post, Shuler, & Stokol, 2011b; Schimek et al., 2013; Wong, Chan, & Kamm, 2012; Zheng et al., 2012) kidney (Ferrell et al., 2010; Ferrell, Ricci, Groszek, Marmerstein, & Fissell, 2011; Jang & Suh, 2010) have also been developed. Since we are focusing our attention on multi-organ devices that demonstrate a successful operation with at least two organs or tissues within one device, the discussion of single organ devices will not be exhaustive. For a more complete review of single organ tissues, we refer the reader to review articles that focus on single organ microdevices (Huh, Hamilton, & Ingber, 2011; Shintu et al., 2012), (Baker, 2011; Esch et al., 2011a; Ghaemmaghami, Hancock, Harrington, Kaji, & Khademhosseini, 2012; Neuži, Giselbrecht, Länge, Huang, & Manz, 2012; Sung, Esch, & Shuler, 2010; Sung et al., 2013).
Examples of Multi-Organ Microdevices and Ways in which They Can Contribute to the Drug Development Process
1) Lowering the Cost of Drug Discovery
The drug development process is expensive, especially in the phases of clinical trials, which can cost millions of dollars. However, despite extensive animal testing of drugs before starting a clinical trial with humans, many cancer drugs fail because of low efficacy or unexpected toxic side effects. Both low efficacy and toxic side effects were not predicted accurately and this highlights the fact that animal and human metabolism are different. The most promising advantage of body-on-a-chip devices is that the devices can mimic both animal and human metabolism and predict differences between them. This will allow for a higher level of accuracy when predicting the outcome of clinical trials. Accurately predicting toxic side effects can prevent unsuitable drug candidates from entering the expensive phase of clinical trials and limit costs and unrealistic expectations.
Body-on-a-chip devices can also reduce the cost of drug testing because they provides low cost platforms for the testing of many chemicals and combinations of chemicals. Using the devices, it is possible to test more drug combinations at a low cost and to eliminate ineffective or toxic concentrations from the parameter space that would be tested with animals. In order to predict effective drug concentrations, the devices are designed in a way that produces the same concentrations of metabolites as in the human body. This is not impossible but can be challenging considering that cellular activity and density of an in vitro construct can differ considerably from those of in vivo tissue.
Predicting Drug Efficacy and Toxic Side Effects for Humans
The first step to demonstrating that multi-organ microdevices can predict efficacy and drug toxicity is to build devices with a limited number of key organs and to challenge them with a drug whose efficacy and toxicity are known and can be measured in the device. Here we discuss two studies in which this has been achieved with known chemotherapeutics.
Liver and kidney tissues are of great interest to drug developers due to their predominant role during the absorption, distribution, metabolism, and excretion (ADME) process of a drug. Physiologically, the liver is the main organ in which the metabolism of drugs occurs, while the kidney is involved in their elimination. These critical processes make these two organs highly susceptible to drug injury. Experiments with the anti-cancer drug ifosfamide illustrate the importance of the liver-kidney interaction. Ifosfamide is a prodrug that is bioactivated by CYP450 enzymes in the liver. The generated metabolites have efficient anti-tumor effects but some of them, such as chloracetaldehyde, are recognized to be nephrotoxic. This mechanism was simulated in the first liver-kidney co-culture microchip. (Choucha-Snouber et al., 2012) When the authors used highly differentiated liver cells models (HepaRG), they were able to measure a perturbation of cell proliferation and calcium release in the kidney tissue. These results were not observed when the device did not contain liver cells or when the liver cell line HepG2/C3a (known to express lower levels of CYP450 enzymes than hepatocytes in the liver) was used. This contribution demonstrates the relevance of the multi-organ interactions in drug testing and also highlights the importance of the cell source when conducting toxicological studies.
Another system that demonstrated the ability of multi-organ microdevices to simulate the exchange of metabolites between organs is a system that contained liver cells (HepG2/C3A), colon cancer cells (HCT-116), and myeloblasts (Kasumi-1). (Sung & Shuler, 2009b) The device was subjected to Tegafur, an oral prodrug of the anti-cancer drug, 5-fluourouracil (5-FU). Upon oral uptake of 5-FU the enzyme dihydropyrimidine dehydrogenase (DPD) rapidly degrades the drug, resulting in a low effective concentration. (Malet-Martino, 2002) Administration of the prodrug Tegafur, which is converted to 5-FU by the P450 1A2, 2A6, and 2C8 enzymes, (Komatsu, Yamazaki, Shimada, Nakajima, & Yokoi, 2000) instead of administering 5-FU directly to patients has advantages because the uptake of Tegafur results in more stable concentrations of the effective component 5-FU. Challenging the multi-organ device with Tegafur, the metabolism of Tegafur, i.e. its conversion to 5-FU in the liver cell compartment was reproduced. 5-FU travelled through the microfluidic channels to the cancer cell compartment, where it caused a dose-dependent decrease in cell viability. Clinically Tegafur is often given with uracil, a competitive inhibitor of 5-FU, which slows its degradation and thereby increases the circulation time of 5-FU. The multi-organ system demonstrated that uracil addition did result in increased effectiveness of Tegafur (increased cell killing) and that the optimal ratio in vitro (about 4 moles of uracil to 1 mole of Tegafur) corresponds to the clinically determined optimum.
Predicting the Bioavailability of Drugs
The rate of first pass metabolism of ingested drugs (due to the direct transport to the liver via the portal circulation) and the distribution of its metabolites via the systemic circulation determines the drug’s bioavailability. Predicting the bioavailability of a drug accurately can be difficult with animal models. Multi-organ microdevices that contain a combination of the gastrointestinal tract epithelium and the liver at the appropriate sizes and with realistic liquid to cell ratios have the potential to predict the bioavailability of ingested drugs. To simulate the first pass metabolism of ingested drugs in conventional static models, Caco-2 cells are typically grown on porous membranes. The cells are in contact with hepatocytes that are grown in a wells beneath the membrane via a common medium. (Hyung Choi, Nishikawa, Sakoda, & Sakai, 2004; Lau, Chen, Liu, Li, & Cui, 2004) Substances that are transported across the Caco-2 cell layer and metabolites that are generated in the Caco-2 culture can reach the liver cell culture and vice versa. Microfluidic models of the liver (Chao et al., 2009; Novik et al., 2010; Powers, Domansky, et al., 2002a; C. Zhang, Chia, Ong, Zhang, Toh, van Noort, et al., 2009a) have the potential to be combined with models of the GI-tract epithelium. In fluidic models of the first pass metabolism, fluidic circuitry transports any substances that crossed the epithelial cell layer or that were generated by it from the basolateral side of the GI-tract cell culture to the liver cells that are located downstream (Brand, Hannah, Mueller, & Cetin, 2000). Re-circulation of medium between the basolateral side of the GI-tract cell culture and the liver cell culture has also been implemented (Choi, Fukuda, Sakoda, & Sakai, 2004; Mahler, Esch, Glahn, & Shuler, 2009a). Using such models, the first pass metabolism of drugs such as acetaminophen has been simulated (Mahler, Esch, Glahn, & Shuler, 2009b). The results show that liver cell damage occurs in a dose-dependant manner. Since acetaminophen is a small non-ionized molecule that diffuses passively across the GI-tract epithelium, the presence of the GI-tract tissue presents a barrier to the drug, which in turn modulates the effects of drug concentration in the liver. In addition, the GI-tract epithelium exhibited modest P450 activity and converted a portion of the acetaminophen into non-toxic metabolites. These metabolites were detected in the re-circulated medium using high-performance liquid chromatography (HPLC). The result obtained with the first pass metabolism model was consistent with those obtained with acetaminophen challenges in mice (Gujral, Knight, Farhood, Bajt, & Jaeschke, 2002). Models of the first pass metabolism are capable of estimating its bioavailability and its considerable effect on a drug’s toxicity to the liver.
Besides the GI-tract epithelium, the body contains a number of other barrier tissues that limit bioavailability. Among these are the skin, the lung epithelium, the blood placental barrier, the blood brain barrier, and the endothelial lining of the microvasculature. Barrier tissues can be incorporated within multi-organ microdevices. For example, Brand et al. modified their model of the first pass metabolism to include a model of the skin instead of the GI-tract epithelium (Brand et al., 2000). With this model, the topical application of drugs can be simulated. Lee et al. (P. J. Lee, Hung, & Lee, 2007) have also been developing models of the liver that contain features that provide some functions of the microvasculature. Models that include a microvascular component will allow for the simulation of drugs that were intravenously administered in a more realistic way. (Zervantonakis et al., 2012)
Another tissue that modulates the concentrations of substances that circulate within the blood stream is adipose tissue. This tissue can store and release chemicals (depending on the degree of their hydrophobicity) and this feature was demonstrated in 2004 with a multi-organ microdevice. The authors showed that the presence of a fat compartment altered the dynamics of naphthalene toxicity. (Viravaidya & Shuler, 2004b; Viravaidya, Sin, & Shuler, 2004a) A later addition of differentiated 3T3-L1 adipocytes to the device suggested that storage of naphthalene and naphthoquinone in fat tissue reduces glutathione depletion in the lung compartment reducing the toxic effect. Generally, adipose tissue is an important tissue in modulating the concentration of a drug in the blood stream, but it is not often considered explicitly.
Testing Combinations of Drugs to Elucidate Synergistic Drug Action
Since microdevices are relatively inexpensive, and many such devices can be operated in parallel, it is possible to test many drugs and combinations of drugs at different concentrations with the devices. Testing combinations of drugs is useful when drug interactions may occur. Synergistic interactions are of particular interest. Several drugs that have similar functions, but different side effects could potentially be combined at reduced dosages to achieve the needed tissue response. To give an example that shows that synergistic drug action can be observed with multi-organ microdevices, we discuss a study that investigated a hypothesis that postulated the synergy of multiple drugs. The hypothesis was tested with a device that contained liver tissue, tumor tissues, and bone marrow tissue (Baker, 2011; Esch et al., 2011a; Tatosian & Shuler, 2009a). Using this device, Tatosian et al. tested a combination of three drugs: a chemotherapeutic drug, doxorubicin, and two drugs that suppress multidrug resistance (MDR), nicardipine and cyclosporine (A. P. Li, 2009; Tatosian & Shuler, 2009b). The two MDR drugs were chemical modulators that inhibit the action of the membrane transporter P-glycoprotein (P-gp) that pumps drugs out of the cell and prevents anti-cancer drugs from reaching sufficiently high intracellular concentrations to be effective as toxins for tumor cells. (Borowski, Bontemps-Gracz, & Piwkowska, 2005; Davies & Morris, 1993) Experiments with the uterine cancer cell line MES-SA, and an MDR variant of uterine cancer, MES-SA/DX-5, that overexpresses P-gp, indicated that combining the chemotherapeutic doxorubicin with the MDR modulators cyclosporine and nicardipine was more effective in inhibiting cancer cell proliferation than using doxorubicin alone or with only one of the two modulators, where the total dose of all modulators was kept constant (Tatosian & Shuler, 2009b). The device also contained megakaryoblast cells that form platelets (MEG-01) and liver cells (HepG2/C3A). Liver cells and bone marrow cells (i.e. MEG-01) were growth-inhibited when a single MDR modulator was used, but not when the combination of two modulators was used. This result was contrasted with those obtained in 96-well plates that suggested an additive effect rather than a synergistic effect as seen with the multi-organ microdevice. In clinical studies, when administered as single drugs, the high modulator concentration caused toxic side effects. The hypothesis that a combination of reduced doses of multiple modulators could be effectively combined with chemotherapeutics to reverse the growth of multi-drug resistant tumors with reduced side effects, (Lehnert, Dalton, Roe, Emerson, & Salmon, 1991; Pascaud, Garrigos, & Orlowski, 1998) was tested successfully with the multi-organ microdevice. Multi-drug combinations could potentially also be tested for particular patients that do not respond to routinely used drug combinations. Using biopsy cells within the devices and testing drugs directly with these devices could be one way to find individualized treatment options for these patients, estimating both drug efficacy and side effects. The possibility for individualized medicine is discussed under the paragraph that outlines the challenges of using multi-organ microdevices.
2) Experimenting with Non-Physiologic Versions of the Human Body
Body-on-a-chip systems also present opportunities that animal models do not. For example, body-on-a-chip devices can be operated with the entire set of organs of the human body, or with a subset. Leaving out an organ for a particular experiment or increasing its volume or activity beyond physiologic values can confirm or disprove hypotheses that aim to identify the origin of a toxic metabolite. Further, it is possible to modify the devices to simulate disease conditions such as limited activity of cells, and limited blood supply to a particular organ. Below we describe studies that benefited from this approach.
Low efficacy and the occurrence of toxic side effects are among the main reasons for drug attrition. Toxic metabolites can be generated from an initially non-toxic substance and these metabolites can circulate to other organs where they can cause substantial disturbances. Since multi-organ microdevices contain several tissues, the generation of toxic metabolites in any of these tissues and their actions on any of the other tissues within the device can be tested. Well-designed experimental sequences can even be used to test hypotheses about mechanisms of drug and chemical actions and this approach has been demonstrated with several designs. For example, a system that contained three tissues: liver, lung, and fat tissue was challenged with naphthalene and the response showed which organ was responsible for the generation of toxic metabolites and which metabolite was responsible for the cell death in another tissue. (Dimasi, 2001; Dimasi & Grabowski, 2007; Sweeney, Shuler, Babish, & Ghanem, 1995; Viravaidya & Shuler, 2004b; Viravaidya, Sin, & Shuler, 2004a) Naphthalene is an environmental toxin, but we discuss it in this review, because it illustrates how multi-organ microdevices could be used to clarify mechanisms of drug action and toxicity. After naphthalene addition to the system, the liver formed reactive metabolites, which were released subsequently into the recirculating blood surrogate. The medium stream delivered the metabolites to the lung compartment, causing dose dependent lung cell death. Removing the liver cells and replacing them with lung cells or no cells at all, allowed the authors to make the conclusion that the reactive metabolites of naphthalene were formed in the liver and not in the lung tissue. Increasing liver cell numbers caused an increase in toxicity in the lung compartment (Greek & Menache, 2013; Sweeney et al., 1995). This result was consistent with the hypothesis that the reactive product of naphthalene metabolism in the liver was the cause of lung cell death. The result also suggests that the toxic compounds had a sufficient lifetime in the medium to be excreted and circulate to the lung compartment. Subsequent work using a multi-organ system demonstrated that the reactive metabolite was likely naphthoquinone rather than the naphthalene epoxide that had been previously proposed as the toxic compound (Ghanem & Shuler, 2000; Sung & Shuler, 2009a). This finding was confirmed using a microscale system with lung, liver and fat compartments (Viravaidya & Shuler, 2004a; 2004b; Viravaidya, Sin, & Shuler, 2004a). This example suggests that several well-designed experiments with multi-organ microdevices can be used to determine which metabolite is responsible for a beneficial result within the target tissue, or which metabolite causes tissue damage.
3) Determining Parameters for Physiologically Based Pharmacokinetic Models
Physiologically based pharmacokinetic models (PBPKs) are mathematical models that are used to extrapolate data from animal experiments and predict human response to a drug. The models rely on our understanding and knowledge of a drug’s metabolism in order to give accurate predictions. Missing information means that the equations used in a PBPK are not complete and the model’s predictive power is not as high as it could be. Multi-organ microdevices can be modeled relatively precisely with PBPKs and discrepancies between the model’s prediction and experimental data obtained with the devices can point to gaps in our understanding. In fact, it might be possible to determine data for parameters for PBPK models from observations using the microdevices. The resulting models can be used to predict human response to a wide variety of combination of inputs with higher accuracy than before.
4) Individualized Medicine
Development of the pharmacogenetic and pharmacogenomic sciences, which focus on the analysis of patient-specific responses to drugs based on variations in genotype, has since given rise to the concept of personalized medicine (Dienstmann, Rodon, & Tabernero, 2013; Kalow, 2006). The knowledge of an individual’s genotype and epigenome could help modulate applicable therapies towards their personalized profile (Duffy, O’Donovan, & Crown, 2011). Similarly, the use of primary or induced pluripotent stem (iPS) cells, derived from patient tissue, could theoretically be used in personalized assays, recapitulating patient-specific responses in vitro. Since multi-organ microdevices can be operated with small numbers of cells, they could potentially be used to model multiple relevant organ functions for the assessment of drug responses in the context of a patient’s individual disease state (Romano, Morales, Marino, & Giordano, 2013; Rubin & Haston, 2011). However, iPS cells take time to develop so the need for the drug treatment must not be acute.
Challenges
1) Device Development
In order to benefit from experimenting with body-on-a-chip devices, the devices must reliably replicate human metabolism or at least a subset of the human metabolism. While several multi-organ devices have been developed for the purpose of demonstrating their usefulness in the drug development process, there are practical challenges that must be overcome if the devices are to be used by the pharmaceutical industry. In regards to device development, current efforts to overcome these challenges aim at improving the usability of the devices and the authenticity with which the human metabolism is mimicked. Currently, in the US, the Defense Advanced Research Projects Agency (DARPA) and the National Institute of Health are substantially funding research efforts (Microphysiological Systems) towards this goal. In particular, the funded research focuses on developing the devices so that they support the use of primary cells and stem cells for an extended period of time. The efforts also include the development of a common blood surrogate (cell culture medium). Below we discuss these challenges in more detail.
Device Design
Designing multi-organ microdevices in a physiologically relevant manner increases the predictive power of data obtained with them. Designing organ compartments in a non-physiologic manner can lead to an overproduction or underproduction of relevant metabolites. For example, if the liver compartment is larger than it should be according to physiological scaling and has a cellular construct of biological activity similar to natural tissue, toxic metabolites that are generated in the liver compartment will reach other tissues at a higher concentration and cause proportionately more damage than would be the case if the liver was appropriately scaled. One approach to scaling that has been used is calculating on-chip organ chamber sizes according to the needed fluid residence time within each organ chamber. In vivo, the blood residence time within organs depends on the size of the organ, the composition of the tissue, and the rate of perfusion. On the device the fluid flow rate, the organ chamber volume, and the composition of the tissue within determine the fluid residence time within each organ chamber. The fluid flow rate within each organ is a percentage of the overall flow or recirculation rate and relates to the fraction of blood distribution to each organ in vivo. Since fluid residence times per organ and the percent of total blood that reaches each organ (i.e. percent of total flow) are given by in vivo values, the chamber volume can be calculated so that a given fluid residence time is achieved for a particular tissue model under a particular flow rate. The volume of free liquid to cells in the device should be similar to that in the body. Data that were obtained with devices that were scaled according to fluid residence times are most accurate if the chambers contain 3D tissue-like constructs and there is less than 200 μm of distance between any cells and the medium supply to insure oxygen availability throughout the tissue for metabolically active tissue. Further, the liquid to cell ratio must be similar to that in the body. Although other design approaches are possible, these principles were considered when designing several devices that were discussed earlier (Mahler, Esch, Glahn, & Shuler, 2009b; Sung & Shuler, 2009a; Tatosian & Shuler, 2009a; Viravaidya, Sin, & Shuler, 2004b). The above approach ensures that the tissue construct is an accurate model of the tissue in the body. If the construct has a different activity than the native tissue (for example in a disease model), other metabolic processes such as matching the degree of conversion of a major nutrient may be used as a design criterion.
Downscaling the organ chamber volumes as much as possible is an advantage if the cell sources are expensive. There must be, however, a minimum number of cells in the smallest organ chamber that is represented on the device. The smallest organ chamber should contain enough cells so that a meaningful metabolite concentration can be generated. From a practical point of view, handling very small numbers of cells (e.g. less than 100) might make an accurate device setup very difficult. Another consideration for establishing a minimum size of the device is the potential need to take medium from the device for subsequent metabolite analysis. The overall amount of medium within the device needs to be large enough so that a minimum of medium could be withdrawn without significantly perturbing the system (e.g. 25%). At the same time, some organs of the human body are very small (such as the pituitary glands) and would lead to a relatively large system if included in the device. Including such small organs implicitly in an “other organ” compartment rather than explicitly as an organ chamber that is populated with cells might be more practical for drug applications in which these organs do not play an important role.
More broadly, all tissues should be included either explicitly or implicitly if the devices are to be physiologic. For any organ or tissue that does not metabolize, absorb, or respond to the test compound or its metabolites, that organ/tissue can be included into an “other tissues” compartment. Such compartments do not have cells, but emulate the hold up of the fluid within that tissue (blood and interstitial fluid). These compartments may be divided into “rapidly perfused” and “poorly perfused” compartments. Such tissue compartments, even without cells, are included in PBPK type models to capture the appropriate dynamics and must be included in these microphysiological or body-on-a-chip devices to mimic the appropriate distribution and dynamics of the body’s response to a drug or chemical compound.
The Development of a Common Cell Culture Medium
In vitro cell-cultures are designed to mimic the relevant in vivo environment. A temperature of 37 °C, and a controlled humidified gas mixture of 5% CO2 and 95% O2 are the standard physical conditions, while a blood surrogate medium with appropriate micro and macronutrients is used to recreate the chemical milieu. Cell culture media have evolved from a salt solution to preserve tissue, to more complex compositions, able to maintain cells and tissues in an active state for extended periods of time. Different cell types often require the use of different cell culture media. That these different requirements can present a challenge for multi-organ microdevices was demonstrated with a device that showed tissue specific responses to a stimulant The device combined tissues of the liver, lung, kidney, and adipose within one platform (Mahler, Esch, Glahn, & Shuler, 2009a; C. Zhang, Zhao, Abdul Rahim, van Noort, & Yu, 2009b). The tissues were stimulated with TGF-β1, showing a dose-dependent response. TGF-β1 supported the growth of A549 lung cells, but inhibited the growth of HepG2/C3A liver cells. This response highlights the difficulty of finding a common medium with growth factors that support the growth of all cell types. This is a challenge for the development of multi-organ microdevices that must be solved in order for multi-organ microdevices to move forward. In this particular study the authors present an approach that uses gelatin microspheres to release TGF-β1 locally to support the lung compartment while maintaining low levels of TGF-β1in the circulation. This system demonstrated that the four tissues could remain viable within one device and that differential tissue response to TGF-β1 could be emulated.
Human or animal sera, the most commonly used being fetal bovine serum, are often used to supplement basic medium since they contain essential compounds for the growth and maintenance of cells and mimic many of the transport properties of blood. Recently, developing a serum-free medium has been a goal of many investigators, to improve the quality, consistency and definition of the culture medium, since variation in the serum composition has been known to affect cell culture maintenance and subsequent experimental data. (van der Valk et al., 2010) Serum-free medium formulations are based on the addition of cell-specific growth factors and supplements to a common base medium to facilitate the correct maintenance of specific cell cultures (Edwards, Das, Molnar, & Hickman, 2010). For example, the first serum-free defined culture system for hippocampal neurons was published by Hickman, (Schaffner, Barker, Stenger, & Hickman, 1995) and this model has since been adapted to facilitate the maintenance of cardiomyocytes, (Natarajan et al., 2011) motoneurons, (Das, Molnar, Devaraj, Poeta, & Hickman, 2003) sensory neurons, (Rumsey, Das, Bhalkikar, Stancescu, & Hickman, 2010) and skeletal muscle cells (Das et al., 2006) in defined in vitro conditions.
The recent development of novel multi-organ microdevices has required an advancement in medium formulations. One of the challenges for this new technology is to identify a common formulation that is capable of preserving each cell type’s morphology and functionality in co-culture. Recent attempts to find a common medium formulation have been published: i) Davis and co-workers have observed that oligodendrocyte precursors can be differentiated into mature oligodendrocytes that express myelin basic protein, using a serum-free medium in co-culture with rat embryonic motoneurons on a non-biological substrate. (Davis et al., 2012) ii) Guo et al. used a commercialized medium containing Neurobasal, B27, creatine, estrogen and cholesterol to promote neuromuscular junction formation between human stem cell-derived motoneurons and human skeletal muscle. (Guo, Gonzalez, Stancescu, Vandenburgh, & Hickman, 2011) iii) Zhang and co-workers optimized a common basal culture medium for enhancing the functions of four human cell types (liver, lung, kidney and adipose) in a multi-channel 3D micro uidic cell culture system (C. Zhang, Zhao, Abdul Rahim, van Noort, & Yu, 2009b). Despite this progress, successful co-culture of more than four cell types in a common serum-free medium is difficult, and further investigations are needed to increase the number of cell types that can be maintained within a single platform.
Cell Sources
Animal models are typically the primary source for most cell types utilized in experimental cultures. However, as already stated, their low predictive power, with regards to human responses to novel therapeutic treatment, makes them a poor candidate for use in next generation micro-devices for drug development protocols. Certain primary human cell types, such as skin, (Sakai et al., 2013) skeletal muscle, (Scherp et al., 2012) and blood, (Welin et al., 2013) are relatively easy to obtain. Acquisition of others, such as neurons, is more problematic due to the trauma caused by extraction, and in such cases investigators are often limited to cadaver tissue as a cell source. (Fakunle & Loring, 2012) As a result, either embryonic or induced pluripotent stem cells have become an attractive alternative for investigators seeking to model human tissue function in vitro. (Fakunle & Loring, 2012)
Stem cell technologies are attractive to investigators developing micro-devices for drug development applications, since they facilitate the production of cell lines maintaining stable transfections. (Gerrard, Zhao, Clark, & Cui, 2005) Such genetic manipulation can be used to produce functional human cell types carrying a fluorescent reporter gene, conjugated to a specific promoter, to allow optical assessment of metabolic activity in response to therapeutic treatment. (Xu, Kraus, & Shuler, 2008) Furthermore, the application of induced pluripotent stem cell technology from specific patients makes the concept of personalized medicine and patient specific disease models a possibility.
Widespread adoption of stem cells, particularly induced pluripotent stem cells, for in vitro applications has been questioned due to the reliability of the available cell lines. Such cell’s ability to successfully differentiate into specific lineages has been found to vary based on differences in donor genotype and tissue of origin (W. Wagner & Ho, 2007). Moreover, since stem cells are incredibly susceptible to differentiation induction based on their physical and chemical micro-environments, different labs have occasionally produced conflicting data, or been unable to recreate the work of others, calling into question the validity of certain differentiation protocols. (Morshead, Benveniste, Iscove, & van der Kooy, 2002; Raedt et al., 2007) Likewise, human embryonic stem cell lines, while sharing certain gene expression profiles, have been found to possess differences in the expression of several lineage markers. (Adewumi et al., 2007) Consequently, although numerous commercially available stem cell lineages are available, some investigators continue to focus on the use of primary tissue from human and animal models as a more consistent and reliable alternative. Although primary cells vary from donor to donor they typically maintain full, differentiated function while stem cell based constructs may not display the full adult phenotype. Efforts are underway to generate comprehensive selection criteria and more universal preparation standards for stem cell production, (W. Wagner & Ho, 2007) but what remains clear is that a more complete understanding of stem cell development and functional capacity is necessary to advance the development of next-generation human in vitro assays.
Such systems would be of tremendous benefit to the study of human genetic conditions for which no animal model exists or in instances when animal models fail to wholly recapitulate the complexity of the human condition. (Romano et al., 2013) For example, induced pluripotent stem cells have recently been used to model the electrophysiological profile of cardiomyocytes from a patient with type 1 long QT syndrome in vitro. (Itzhaki et al., 2011) Since substantial differences in cardiac physiology exist between humans and rodents, the use of mice or rats is unsuitable for investigating this condition, and highlights the importance of novel in vitro platforms for developing new therapies. Similarly, studies have been performed using induced pluripotent stem cells derived from patients with familial Parkinson’s disease, (Devine et al., 2011) as well as familial and sporadic Alzheimer’s disease, (Israel et al., 2012) to investigate specific cellular responses and physiological differences in cells possessing these common aberrant genotypes.
As mentioned previously, given the variability in performance of stem cell lines, development of stringent selection criteria and culture parameters are necessary to facilitate the widespread adoption of such cells into high throughput assays and screening systems. Use of such cells in multi-organ micro-devices is a goal yet to be realized. On a routine basis however, it remains an exciting prospect for improving patient care and the understanding of specific disease states, as well as their responses to novel therapeutics.
Authenticity of Cellular Behavior
Once an appropriate cell type is obtained, a further problem is the method of maintaining these cells within a housing that permits full functionality and correct emulation of in vivo behavior. Here single tissue models can be used to optimize tissue behavior. Three-dimensionality has been shown to create more authentic tissue responses than two-dimensional tissues (Weaver et al., 1997). Three-dimensional tissues and multi-cell type tissues that were cultured within microfluidic devices have been developed for the liver and other tissues (Chang et al., 2012; M. B. Chen et al., 2013; Domansky et al., 2010; HwanáSung, 2011; Kostadinova et al., 2013; Powers, Domansky, et al., 2002a; Pusch et al., 2011). While traditional in vitro assays often focus on measurement of biomarkers as indicators of cell health and functionality, direct measurement of functional output is a more accurate method, and likely to yield data with stronger correlative power to clinical observations. To that end, a number of groups have recently focused on the development of “on-chip” in vitro assay systems capable of emulating selected functional responses of key organs and tissues. Examples of such technologies can be found for heart, (Agarwal, Goss, Cho, McCain, & Parker, 2013; Edwards et al., 2010) liver, (Antunes, Andrade, Araújo, Ferreira, & Sarmento, 2013; Kostadinova et al., 2013; I. Wagner et al., 2013) lung, (Huh et al., 2010; Long et al., 2012) kidney, (Subramanian et al., 2010) skeletal muscle, (Guo et al., 2011; Wilson, Das, Wahl, Colton, & Hickman, 2010) hippocampus, (Natarajan, DeMarse, Molnar, & Hickman, 2013) gastro-intestinal tract, (Esch et al., 2012; Mahler, Esch, Glahn, & Shuler, 2009b; Mahler, Shuler, & Glahn, 2009c) and skin (I. Wagner et al., 2013) among others.
Assessing authentic tissue behavior in multi-organ micro-devices requires the real-time recording of primary functional outputs of different cell types. Physical movement of contractile cell types, such as skeletal muscle myotubes, cardiomyocytes and smooth muscle cells can be evaluated through measurement of substrate deflection, either by use of cantilevers (Wilson et al., 2010) or flexible posts (Vandenburgh et al., 2008). Electrical activity of neurons is usually assessed in vitro using electrophysiological patch clamp recordings, (Guo, Johe, Molnar, Davis, & Hickman, 2010) however, such techniques are invasive and difficult to scale up for high throughput applications. Use of microelectrode arrays facilitate the high throughput interrogation of cultured neuronal networks in a high throughput manner, and are far more amenable to integration with multi-organ platforms. (Natarajan et al., 2013) Moreover, the use of microelectrodes can also be used as a means to assess the concentrations of a wide variety of functionally relevant analytes such as superoxide radicals (X. J. Chen, West, Cropek, & Banta, 2008) and lactate. (Ges & Baudenbacher, 2010) Optical techniques can also be used to assess functional metabolism of drug compounds and their effect on cell viability in microdevices (Ma et al., 2009; Sung & Shuler, 2009b; Tatosian & Shuler, 2009a; Viravaidya & Shuler, 2004b; Viravaidya, Sin, & Shuler, 2004a).
Without the means to measure and assess the biomimicity of cultured tissue analogs, very little of the data required for accurate predictions of in vivo drug responses can be obtained. Development of appropriate analytical techniques for application within novel multi-organ micro-devices is essential as a means to assess the appropriate real time physiological and functional effects of drug treatment in vitro.
2) Commercialization
Commercial development of multi-organ devices for drug testing is currently underway. For example, Hurel corporation, discusses a microfluidic platform for testing two fluidically interconnected chambers with cells/tissues, a medium reservoir and in situ “pumps” for moving fluid. This device is currently in beta testing and will presumably be commercially available soon.
In order for the pharmaceutical industry to adopt new devices, they must be easy to use and provide a profit by being lower in cost than conventional approaches. Low cost of the physical devices can be achieved by utilizing polymeric materials for device fabrication and device designs that are pumpless and valveless, or have low cost strategies for moving fluids in a controllable manner. However, there are other challenges that must be overcome in order to fulfill the ease of use and low cost requirement. Some of the these challenges are discussed below.
Longevity
The need to maintain in vitro models for extended periods of time is of great importance for drug development applications that aim to predict the effects of chronic drug exposure. Prolonged metabolite exposure and waste build-up within the organ compartments limits the lifetime of multi-organ microdvices. In addition, long in vitro culture periods tend to lead to cellular senescence or induction of apoptotic pathways, which can confound data from toxicity studies. Furthermore, typically cell maturation takes a few hours up to two or three weeks in culture. This requires coordinating the cell seeding process so that all cells are mature and functional at the time of the experiment. Complex culture environments are often necessary to produce accurate models of in vivo tissues. However, the incorporation of increasing numbers of disparate cell types into common culture conditions makes the maintenance of such platforms problematic since the conservation of optimized parameters for all cell types becomes more and more difficult. One solution to this problem may be to culture each cell type in its separate medium until the cells reach maturation and operate the device with a common medium for the duration of the experiment. Using this procedure, devices have been operated for 24–72 hours without any media exchange.
Relatively long culture periods have been established for certain individual cell types. For example, skeletal muscle cultures have been shown to survive in vitro for up to 90 days, during which time they promote phenotype maturation, as evidenced by quantifiable changes in myosin heavy chain isoform composition. (Das, Rumsey, Bhargava, Stancescu, & Hickman, 2009) This system was subsequently modified to support the long-term (30+ days) co-culture and functional interaction of skeletal muscle myotubes and motoneurons (Das, Rumsey, Bhargava, Stancescu, & Hickman, 2010). Such data demonstrate that maintenance of co-cultures is possible over long periods provided careful consideration is given to culture variables such as surface chemistry and topography, medium formulation and correct temporal addition of exogenous stimuli. The described data were obtained using rodent cells, however, and similar studies using human cells were successful in co-cultures for 10 days (Guo et al., 2011). Such disparity is likely due to inherent differences in rodent and human cellular maturation and maintenance, and highlights the need for further assessment of optimal culture conditions to promote more long-term survival. It should be noted that data pertaining to the long-term survival and functional viability of human neuronal cell types has been reported, (Guo et al., 2010; Guo, Spradling, Stancescu, Lambert, & Hickman, 2013) so the possibility of functional nerve-muscle co-cultures using human cells in the near future seems possible.
Standardization
The exponential development of microfabricated devices has led to a variety of available tools dedicated to multiple cell cultures within one device. However, the approaches all differ by materials, configuration, and criteria of design. All of these variables have the potential to influence cellular behavior on chip. Proof of concept experiments with the described multi-organ devices have provided some evidence for their usefulness in early stage drug testing. However, if the devices are to be used in industry there is a real need for biological validation and standardization. The currently funded efforts by DARPA aim specifically to develop standard platforms that can be used by many different groups to utilize body-on-a-chip systems. The development of such biological platform requires finding a balance between complexity, required by the need to recreate the in vivo situation accurately, and the need for the devices to be inexpensive and easy to use. The approach used by cosmetic industry to replace animal testing encouraged by the ICCVAM program in the US could provide some direction for the development of such standards. A mutual endpoint toolbox could allow inter-laboratory and inter-platform evaluation. Quantitatively and qualitatively assessment of a platform’s functionality would increase our confidence in the results obtained with them. In this regard, a multi-organ platform dedicated to drug development should focus on the ADMET process with relevant biomarker of early toxicity for each organ. For example, a common method to evaluate the potential of a drug to induce various CYP450 enzyme expression, which can be an early response indicating drug toxicity, has been proposed by ECVAM (Gerin, Dell’Aiera, Richert, Smith, & Chanteux, 2013). A cocktail of specific probes were designed as a calibrator for the four main CYP enzymes involved in drug metabolism. Using the same cocktail of drugs and evaluation methods across laboratories will allow the validation of different models in a defined way. Working towards standardization will require a close collaboration between engineers, biologists, and the pharmaceutical industry.
Conclusions
Multi-organ microdevices have the potential to contribute to the early stage drug development process in ways that are not possible with conventional in vitro models. Because the devices are designed to mimic the physiologic relationship of organs and their interaction via soluble metabolites, they can capture inter-organ effects in vitro. The low cost of the devices permits testing a large number of drugs and drug combinations with human tissues instead of animal tissues. This bears the potential advantage of providing a higher degree of accuracy when predicting toxic side effects for humans. Further, the devices can easily be modified to mimick disease conditions or entirely un-physiologic conditions that allow for an increase in flexibility when experimenting with new drug candidates. Here we have reviewed and discussed studies that have demonstrated the devices’ capability to simulate the first pass metabolism, the conversion of anti-cancer prodrugs and their subsequent effects on tumor tissue, the synergistic actions of two MDR modulators, and the modulation of bioavailability and toxicity via barrier tissues and tissues that absorb and store chemical compounds. Most importantly, we have discussed how the devices can be used to test hypotheses about mechanistic models of drug toxicity. Experimenting with multi-organ microdevices gives us the ability to obtain useful, and often non-obvious information, on biological mechanism and the exchange of metabolites between tissues.
However, significant challenges must be overcome in order for the devices to become relevant for the pharmaceutical industry. Currently, DARPA and NIH are funding efforts in the US that will likely result in several platforms that can be used by investigators. The efforts also have the potential to solve some of the most important questions regarding the authenticity of the mimicked metabolism since all developments are required to utilize primary or stem cell sources. In addition, the resulting collaborations are a strong catalyst for the development and combining of ideas. In general, we can expect to see an increase of the total number of organs that are explicitly included in body-on-a-chip devices.
Table 1.
Examples of multi-organ microdevices and their demonstrated capabilities for drug testing applications
| Organ combination | Tested Drug/Toxin | Demonstrated capability | Endpoints | References |
|---|---|---|---|---|
| GI-tract-liver | Acetaminophen, 7-ethoxycoumarin (7-EC), 7-hydroxycoumar in (7-HC) and lidocaine | Metabolism of acetaminophen in the GI-tract and liver, modulated acetaminophen toxicity in the liver due to the presence of the GI-tract, Modulation of liver toxicity due to addition of bile acids to GI-tract model | Acetaminophe n metabolite detection via HPLC, live/dead assay, P450 7A1 (CYP7A1) activity in the liver | (Mahler, Esch, Glahn, & Shuler, 2009a; van Midwoud, Merema, Verpoorte, & Groothuis, 2010) |
| Microvasculature-liver | Not challenged with any drugs | Increased albumin and urea production as a result of co-culture | Consumption of glucose by liver cells, albumin synthesis, urea production | (Chang et al., 2012; Guzzardi. Vozzi, & Ahluwalia, 2009; P. J. Lee et al., 2007: Vozzi, Heinrich, Bader, & Ahluwalia, 2008) |
| Liver-tumor-bone marrow | tegafur | Pro-drug conversion, synergy of anti-cancer drugs | Live/dead assay | (Sung & Shuler, 2009a) |
| Liver-lung-fat | naphthalene | Identification of toxic metabolite, modulation of toxicity by fat tissue | Live/dead assay | (Viravaidya & Shuler, 2004a; Viravaidya, Sin, & Shuler, 2004a) |
| Liver-kidney | ifosfamide | Pro-drug conversion and nephrotoxicity | Metabolite detection via mass spectrometry, cell proliferation, calcium release | (Choucha-Snouber et al., 2012) |
| Liver-lung-kidney-fat | TGF-β1 | Dose-dependent response of each cell type to TGF-β1 and tissue-specific support through localization of TGF-β1 release | Albumin secretion, PROD activity, GGT activity, adiponectin secretion | (C. Zhang, Zhao, Abdul Rahim, van Noort, & Yu, 2009b) |
| Skin-liver | bpV(phen) | Uptake of bpV(phen) through the skin and subsequent stimulation of glucose consumption | Glucose consumption | (Brand et al., 2000) |
Acknowledgments
The authors acknowledge support from the National Institute of Health (NIH) through Grant No. UH2TR000516. MBE and MLS also acknowledge support from the National Science Foundation (NSF) under grant No. CBET-1106153. MLS and MBE we also supported by the Cornell Center on the Microenvironment & Metastasis through Award Number U54CA143876 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. AS, COS, and JH acknowledge support from the National Institute of Health (NIH) through Grant No. R01NS050452, Grant No. R01EB009429, and Grant No. UH2TR000516.
Footnotes
Disclosure: MLS is on the Hurel scientific advisory board.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adewumi O, Aflatoonian B, Ahrlund-Richter L, Amit M, Andrews PW, Beighton G, et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nature Biotechnology. 2007;25(7):803–816. doi: 10.1038/nbt1318. [DOI] [PubMed] [Google Scholar]
- Agarwal A, Goss JA, Cho A, McCain ML, Parker KK. Microfluidic heart on a chip for higher throughput pharmacological studies. Lab on a Chip. 2013 doi: 10.1039/c3lc50350j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes F, Andrade F, Araújo F, Ferreira D, Sarmento B. Establishment of a triple co-culture in vitro cell models to study intestinal absorption of peptide drugs. European Journal of Pharmaceutics and Biopharmaceutics. 2013;83:427–235. doi: 10.1016/j.ejpb.2012.10.003. [DOI] [PubMed] [Google Scholar]
- Ataç B, Wagner I, Horland R, Lauster R, Marx U, Tonevitsky AG, et al. Skin and hair on-a-chip: in vitro skin models versus ex vivo tissue maintenance with dynamic perfusion. Lab on a Chip. 2013;13(18):3555–3561. doi: 10.1039/C3LC50227A. [DOI] [PubMed] [Google Scholar]
- Baker M. A living system on a chip. Nature. 2011;471(7340):661–665. doi: 10.1038/471661a. [DOI] [PubMed] [Google Scholar]
- Borowski E, Bontemps-Gracz MM, Piwkowska A. Strategies for overcoming ABC-transporters-mediated multidrug resistance (MDR) of tumor cells. Acta Biochimica Polonica. 2005;52(3):609–627. [PubMed] [Google Scholar]
- Brand R, Hannah T, Mueller C, Cetin Y. A novel system to study the impact of epithelial barriers on cellular metabolism. Annals of Biomedical Engineering. 2000;28:1210–1217. doi: 10.1114/1.1318926. [DOI] [PubMed] [Google Scholar]
- Brauchle E, Johannsen H, Nolan S, Thude S, Schenke-Layland K. Design and analysis of a squamous cell carcinoma in vitro model system. Biomaterials. 2013;34(30):7401–7407. doi: 10.1016/j.biomaterials.2013.06.016. [DOI] [PubMed] [Google Scholar]
- Chang K-W, Lee C-T, Tushar Harishchandra P, Chen H-P, Yueh T-R, Valagerahally Puttaswamy S, et al. 3D biomimetic chip integrated with microvascular system for studying the liver specific functions. Presented at the 2012 7th IEEE International Conference on Nano/Micro Engineered and Molecular Systems (NEMS), IEEE; 2012. pp. 218–221. [DOI] [Google Scholar]
- Chao P, Maguire T, Novik E, Cheng K. Evaluation of a microfluidic based cell culture platform with primary human hepatocytes for the prediction of hepatic clearance in human. Biochemical Pharmacology. 2009;78(6):625–632. doi: 10.1016/j.bcp.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MB, Srigunapalan S, Wheeler AR, Simmons CA. A 3D microfluidic platform incorporating methacrylated gelatin hydrogels to study physiological cardiovascular cell–cell interactions. Lab on a Chip. 2013;13(13):2591–2598. doi: 10.1039/c3lc00051f. [DOI] [PubMed] [Google Scholar]
- Chen XJ, West AC, Cropek DM, Banta S. Detection of the superoxide radical anion using various alkanethiol monolayers and immobilized cytochrome c. Anal Chem. 2008;80(24):9622–9629. doi: 10.1021/ac800796b. [DOI] [PubMed] [Google Scholar]
- Choi SH, Fukuda O, Sakoda A, Sakai Y. Enhanced cytochrome P450 capacities of Caco-2 and Hep G2 cells in new coculture system under the static and perfused conditions: evidence for possible organ-to-organ interactions against exogenous stimuli. Materials Science & Engineering C. 2004;24(3):333–339. [Google Scholar]
- Choucha-Snouber L, Aninat C, Grsicom L, Madalinski G, Brochot C, Poleni PE, et al. Investigation of ifosfamide nephrotoxicity induced in a liver-kidney co-culture biochip. Biotechnology and bioengineering. 2012;110(2):597–608. doi: 10.1002/bit.24707. [DOI] [PubMed] [Google Scholar]
- Das M, Gregory CA, Molnar P, Riedel LM, Wilson K, Hickman JJ. A defined system to allow skeletal muscle differentiation and subsequent integration with silicon microstructures. Biomaterials. 2006;27(24):4374–4380. doi: 10.1016/j.biomaterials.2006.03.046. [DOI] [PubMed] [Google Scholar]
- Das M, Molnar P, Devaraj H, Poeta M, Hickman JJ. Electrophysiological and morphological characterization of rat embryonic motoneurons in a defined system. Biotechnology Progress. 2003;19(6):1756–1761. doi: 10.1021/bp034076l. [DOI] [PubMed] [Google Scholar]
- Das M, Rumsey JW, Bhargava N, Stancescu M, Hickman JJ. Skeletal muscle tissue engineering: A maturation model promoting long-term survival of myotubes, structural development of the excitation-contraction coupling apparatus and neonatal myosin heavy chain expression. Biomaterials. 2009;30(29):5392–5402. doi: 10.1016/j.biomaterials.2009.05.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das M, Rumsey JW, Bhargava N, Stancescu M, Hickman JJ. A defined long-term in vitro tissue engineered model of neuromuscular junctions. Biomaterials. 2010;31(18):4880–4888. doi: 10.1016/j.biomaterials.2010.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharmaceutical Research. 1993;10(7):1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- Davis H, Gonzalez M, Bhargava N, Stancescu M, Hickman JJ, Lambert S. Rat Cortical Oligodendrocyte-Embryonic Motoneuron Co-Culture: An Axon-Oligodendrocyte Interaction Model. J Biomater Tissue Eng. 2012;2(3):206–214. doi: 10.1166/jbt.2012.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, et al. Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat Commun. 2011;2:440. doi: 10.1038/ncomms1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienstmann R, Rodon J, Tabernero J. Biomarker-driven patient selection for early clinical trials. Curr Opin Oncol. 2013;25(3):305–312. doi: 10.1097/CCO.0b013e32835ff3cb. [DOI] [PubMed] [Google Scholar]
- Dimasi JA. New drug development in the United States from 1963 to 1999. Clin Pharmacol Ther. 2001;69(5):286–296. doi: 10.1067/mcp.2001.115132. [DOI] [PubMed] [Google Scholar]
- Dimasi JA, Grabowski H. The Cost of Biopharmaceutical R&D: Is Biotech Different? Managerial and Decision Economics. 2007;28(469):469–479. [Google Scholar]
- Domansky K, Inman W, Serdy J, Dash A, Lim MHM, Griffith LG. Perfused multiwell plate for 3D liver tissue engineering. Lab on a Chip. 2010;10(1):51–58. doi: 10.1039/B913221J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy MJ, O’Donovan N, Crown J. Use of molecular markers for predicting therapy response in cancer patients. Cancer Treat Rev. 2011;37(2):151–159. doi: 10.1016/j.ctrv.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Edwards D, Das M, Molnar P, Hickman JJ. Addition of glutamate to serum-free culture promotes recovery of electrical activity in adult hippocampal neurons in vitro. J Neurosci Methods. 2010;190(2):155–163. doi: 10.1016/j.jneumeth.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esch MB, King TL, Shuler ML. The role of body-on-a-chip devices in drug and toxicity studies. Annual Review of Biomedical Engineering. 2011a;13:55–72. doi: 10.1146/annurev-bioeng-071910-124629. [DOI] [PubMed] [Google Scholar]
- Esch MB, Post DJ, Shuler ML, Stokol T. Characterization of in vitro endothelial linings grown within microfluidic channels. Tissue Engineering Part A. 2011b;17(23–24):2965–2971. doi: 10.1089/ten.tea.2010.0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esch MB, Sung JH, Yang J, Yu C, Yu J, March JC, Shuler ML. On chip porous polymer membranes for integration of gastrointestinal tract epithelium with microfluidic “body-on-a-chip” devices. Biomedical Microdevices. 2012;14(5):895–906. doi: 10.1007/s10544-012-9669-0. [DOI] [PubMed] [Google Scholar]
- Fakunle ES, Loring JF. Ethnically diverse pluripotent stem cells for drug development. Trends Mol Med. 2012;18(12):709–716. doi: 10.1016/j.molmed.2012.10.007. [DOI] [PubMed] [Google Scholar]
- Ferrell N, Desai RR, Fleischman AJ, Roy S, Humes HD, Fissell WH. A microfluidic bioreactor with integrated transepithelial electrical resistance (TEER) measurement electrodes for evaluation of renal epithelial cells. Biotechnology and bioengineering. 2010;107(4):707–716. doi: 10.1002/bit.22835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell N, Ricci KB, Groszek J, Marmerstein JT, Fissell WH. Albumin handling by renal tubular epithelial cells in a microfluidic bioreactor. Biotechnology and bioengineering. 2011;109(3):797–803. doi: 10.1002/bit.24339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerin B, Dell’Aiera S, Richert L, Smith S, Chanteux H. Assessment of cytochrome P450 (1A2, 2B6, 2C9 and 3A4) induction in cryopreserved human hepatocytes cultured in 48-well plates using the cocktail strategy. Xenobiotica. 2013;43(4):320–335. doi: 10.3109/00498254.2012.719088. [DOI] [PubMed] [Google Scholar]
- Gerrard L, Zhao D, Clark AJ, Cui W. Stably transfected human embryonic stem cell clones express OCT4-specific green fluorescent protein and maintain self-renewal and pluripotency. Stem Cells. 2005;23(1):124–133. doi: 10.1634/stemcells.2004-0102. [DOI] [PubMed] [Google Scholar]
- Ges IA, Baudenbacher F. Enzyme-coated microelectrodes to monitor lactate production in a nanoliter microfluidic cell culture device. Biosens Bioelectron. 2010;26(2):828–833. doi: 10.1016/j.bios.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami AM, Hancock MJ, Harrington H, Kaji H, Khademhosseini A. Biomimetic tissues on a chip for drug discovery. Drug discovery today. 2012;17(3–4):173–181. doi: 10.1016/j.drudis.2011.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem A, Shuler ML. Combining Cell Culture Analogue Reactor Designs and PBPK Models to Probe Mechanisms of Naphthalene Toxicity. Biotechnology Progress. 2000;16(3):334–345. doi: 10.1021/bp9901522. [DOI] [PubMed] [Google Scholar]
- Greek R, Menache A. Systematic Reviews of Animal Models: Methodology versus Epistemology. Int J Med Sci. 2013;10(3):206–221. doi: 10.7150/ijms.5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith LG, WU B, CIMA MJ, Powers MJ, CHAIGNAUD B, Vacanti JP. In Vitro Organogenesis of Liver Tissuea. Annals of the New York Academy of Sciences. 2006;831(1):382–397. doi: 10.1111/j.1749-6632.1997.tb52212.x. [DOI] [PubMed] [Google Scholar]
- Gujral JS, Knight TR, Farhood A, Bajt ML, Jaeschke H. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicological sciences. 2002;67(2):322–328. doi: 10.1093/toxsci/67.2.322. [DOI] [PubMed] [Google Scholar]
- Guo X, Gonzalez M, Stancescu M, Vandenburgh HH, Hickman JJ. Neuromuscular junction formation between human stem cell-derived motoneurons and human skeletal muscle in a defined system. Biomaterials. 2011;32(36):9602–9611. doi: 10.1016/j.biomaterials.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Johe K, Molnar P, Davis H, Hickman J. Characterization of a human fetal spinal cord stem cell line, NSI-566RSC, and its induction to functional motoneurons. Journal of Tissue Engineering and Regenerative Medicine. 2010;4(3):181–193. doi: 10.1002/term.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Spradling S, Stancescu M, Lambert S, Hickman JJ. Derivation of sensory neurons and neural crest stem cells from human neural progenitor hNP1. Biomaterials. 2013;34(18):4418–4427. doi: 10.1016/j.biomaterials.2013.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattersley SM, Greenman J, Haswell SJ. Study of ethanol induced toxicity in liver explants using microfluidic devices. Biomedical Microdevices. 2011;13(6):1005–1014. doi: 10.1007/s10544-011-9570-2. [DOI] [PubMed] [Google Scholar]
- Hinderer S, Schesny M, Bayrak A, Ibold B, Hampel M, Walles T, et al. Engineering of fibrillar decorin matrices for a tissue-engineered trachea. Biomaterials. 2012;33(21):5259–5266. doi: 10.1016/j.biomaterials.2012.03.075. [DOI] [PubMed] [Google Scholar]
- Huh D, Hamilton GA, Ingber DE. From 3D cell culture to organs-on-chips. Trends in Cell Biology. 2011;21(12):745–754. doi: 10.1016/j.tcb.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh D, Matthews B, Mammoto A, Montoya-Zavala M. Reconstituting organ-level lung functions on a chip. Science. 2010;328:1662–1668. doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HwanáSung J. Microscale 3-D hydrogel scaffold for biomimetic gastrointestinal (GI) tract model. Lab on a Chip. 2011;11(3):389–392. doi: 10.1039/c0lc00273a. [DOI] [PubMed] [Google Scholar]
- Hyung Choi S, Nishikawa M, Sakoda A, Sakai Y. Feasibility of a simple double-layered coculture system incorporating metabolic processes of the intestine and liver tissue: application to the analysis of benzo [a] pyrene toxicity. Toxicology in vitro. 2004;18(3):393–402. doi: 10.1016/j.tiv.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Imura Y, Asano Y, Sato K, Yoshimura E. A microfluidic system to evaluate intestinal absorption. Analytical Sciences. 2009;25(12):1403–1407. doi: 10.2116/analsci.25.1403. [DOI] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482(7384):216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471(7337):225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- Jang KJ, Suh KY. A multi-layer microfluidic device for efficient culture and analysis of renal tubular cells. Lab on a Chip. 2010;10(1):36–42. doi: 10.1039/B907515A. [DOI] [PubMed] [Google Scholar]
- Kalow W. Pharmacogenetics and pharmacogenomics: origin, status, and the hope for personalized medicine. Pharmacogenomics J. 2006;6(3):162–165. doi: 10.1038/sj.tpj.6500361. [DOI] [PubMed] [Google Scholar]
- Kidambi S, Yarmush RS, Novik E, Chao P, Yarmush ML, Nahmias Y. Oxygen-mediated enhancement of primary hepatocyte metabolism, functional polarization, gene expression, and drug clearance. Proceedings of the National Academy of Sciences. 2009;106(37):15714–15719. doi: 10.1073/iti0937106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu T, Yamazaki H, Shimada N, Nakajima M, Yokoi T. Roles of cytochromes P450 1A2, 2A6, and 2C8 in 5-fluorouracil formation from tegafur, an anticancer prodrug, in human liver microsomes. Drug Metabolism and Disposition. 2000;28(12):1457–1463. [PubMed] [Google Scholar]
- Kostadinova R, Boess F, Applegate D, Suter L, Weiser T, Singer T, et al. A long-term three dimensional liver co-culture system for improved prediction of clinically relevant drug-induced hepatotoxicity. Toxicology and Applied Pharmacology. 2013;268(1):1–16. doi: 10.1016/j.taap.2013.01.012. [DOI] [PubMed] [Google Scholar]
- Lau Y, Chen Y, Liu T, Li C, Cui X. Evaluation of a novel in vitro Caco-2 hepatocyte hybrid system for predicting in vivo oral bioavailability. Drug Metabolism and Disposition. 2004;32(9):937–942. [PubMed] [Google Scholar]
- Lee PJ, Hung PJ, Lee LP. An artificial liver sinusoid with a microfluidic endothelial-like barrier for primary hepatocyte culture. Biotechnology and bioengineering. 2007;97(5):1340–1346. doi: 10.1002/bit.21360. [DOI] [PubMed] [Google Scholar]
- Lee SA, Kang E, Ju J, Kim DS, Lee S. Spheroid-based three-dimensional liver-on-a-chip to investigate hepatocyte–hepatic stellate cell interactions and flow effects. Lab on a Chip. 2013;13(18):3435–3766. doi: 10.1039/c3lc50197c. [DOI] [PubMed] [Google Scholar]
- Lehnert M, Dalton WS, Roe D, Emerson S, Salmon SE. Synergistic inhibition by verapamil and quinine of P-glycoprotein-mediated multidrug resistance in a human myeloma cell line model. Blood. 1991;77(2):348–354. [PubMed] [Google Scholar]
- Li AP. The use of the integrated discrete multiple organ co-culture (IdMOC (R)) system for the evaluation of multiple organ toxicity. ATLA-Alternatives to Laboratory Animals. 2009;37(4):377. doi: 10.1177/026119290903700408. [DOI] [PubMed] [Google Scholar]
- Long C, Finch C, Esch M, Anderson W, Shuler M, Hickman J. Design optimization of liquid-phase flow patterns for microfabricated lung on a chip. Annals of Biomedical Engineering. 2012;40(6):1255–1267. doi: 10.1007/s10439-012-0513-8. [DOI] [PubMed] [Google Scholar]
- Ma B, Zhang G, Qin J, Lin B. Characterization of drug metabolites and cytotoxicity assay simultaneously using an integrated microfluidic device. Lab on a Chip. 2009;9(2):232–238. doi: 10.1039/b809117j. [DOI] [PubMed] [Google Scholar]
- Mahler GJ, Esch MB, Glahn RP, Shuler ML. Characterization of a gastrointestinal tract microscale cell culture analog used to predict drug toxicity. Biotechnology and bioengineering. 2009a;104(1):193–205. doi: 10.1002/bit.22366. [DOI] [PubMed] [Google Scholar]
- Mahler GJ, Esch MB, Glahn RP, Shuler ML. Characterization of a gastrointestinal tract microscale cell culture analog used to predict drug toxicity. Biotechnology and bioengineering. 2009b;104(1):193–205. doi: 10.1002/bit.22366. [DOI] [PubMed] [Google Scholar]
- Mahler GJ, Shuler ML, Glahn RP. Characterization of Caco-2 and HT29-MTX cocultures in an in vitro digestion/cell culture model used to predict iron bioavailability. The Journal of nutritional biochemistry. 2009c;20(7):494–502. doi: 10.1016/j.jnutbio.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Malet-Martino M. Clinical Studies of Three Oral Prodrugs of 5-Fluorouracil (Capecitabine, UFT, S-1): A Review. The Oncologist. 2002;7(4):288–323. doi: 10.1634/theoncologist.7-4-288. [DOI] [PubMed] [Google Scholar]
- Morshead CM, Benveniste P, Iscove NN, van der Kooy D. Hematopoietic competence is a rare property of neural stem cells that may depend on genetic and epigenetic alterations. Nature Medicine. 2002;8(3):268–273. doi: 10.1038/nm0302-268. [DOI] [PubMed] [Google Scholar]
- Natarajan A, DeMarse TB, Molnar P, Hickman JJ. Engineered In Vitro Feed-Forward Networks. J Biotechnol Biomater. 2013;3(153):2. [Google Scholar]
- Natarajan A, Stancescu M, Dhir V, Armstrong C, Sommerhage F, Hickman JJ, Molnar P. Patterned cardiomyocytes on microelectrode arrays as a functional, high information content drug screening platform. Biomaterials. 2011;32(18):4267–4274. doi: 10.1016/j.biomaterials.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuži P, Giselbrecht S, Länge K, Huang TJ, Manz A. Revisiting lab-on-a-chip technology for drug discovery. Nature Reviews Drug Discovery. 2012;11(8):620–632. doi: 10.1038/nrd3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novik E, Maguire TJ, Chao P, Cheng KC, Yarmush ML. A microfluidic hepatic coculture platform for cell-based drug metabolism studies. Biochemical pharmacology. 2010;79(7):1036–1044. doi: 10.1016/j.bcp.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascaud C, Garrigos M, Orlowski S. Multidrug resistance transporter P-glycoprotein has distinct but interacting binding sites for cytotoxic drugs and reversing agents. Biochem J. 1998;333:351–358. doi: 10.1042/bj3330351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers MJ, Domansky K, Kaazempur-Mofrad MR, Kalezi A, Capitano A, Upadhyaya A, et al. A microfabricated array bioreactor for perfused 3D liver culture. Biotechnology and bioengineering. 2002a;78(3):257–269. doi: 10.1002/bit.10143. [DOI] [PubMed] [Google Scholar]
- Powers MJ, Janigian DM, Wack KE, Baker CS, Stolz DB, Griffith LG. Functional Behavior of Primary Rat Liver Cells in a Three-Dimensional Perfused Microarray Bioreactor. Tissue Engineering. 2002b;8(3):499–513. doi: 10.1089/107632702760184745. [DOI] [PubMed] [Google Scholar]
- Pusch J, Votteler M, Göhler S, Engl J, Hampel M, Walles H, Schenke-Layland K. The physiological performance of a three-dimensional model that mimics the microenvironment of the small intestine. Biomaterials. 2011;32(30):7469–7478. doi: 10.1016/j.biomaterials.2011.06.035. [DOI] [PubMed] [Google Scholar]
- Raedt R, Pinxteren J, Van Dycke A, Waeytens A, Craeye D, Timmermans F, et al. Differentiation assays of bone marrow-derived Multipotent Adult Progenitor Cell (MAPC)-like cells towards neural cells cannot depend on morphology and a limited set of neural markers. Exp Neurol. 2007;203(2):542–554. doi: 10.1016/j.expneurol.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Romano G, Morales F, Marino IR, Giordano A. A commentary on iPS cell biology: Potential applications in autologous tissue and cell transplantation, development of new models for the study of human pathological conditions and drug screening. Journal of cellular physiology. 2013 doi: 10.1002/jcp.24437. [DOI] [PubMed] [Google Scholar]
- Rubin LL, Haston KM. Stem cell biology and drug discovery. BMC Biol. 2011;9:42. doi: 10.1186/1741-7007-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumsey JW, Das M, Bhalkikar A, Stancescu M, Hickman JJ. Tissue engineering the mechanosensory circuit of the stretch reflex arc: Sensory neuron innervation of intrafusal muscle fibers. Biomaterials. 2010;31(32):8218–8227. doi: 10.1016/j.biomaterials.2010.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai Y, Koike M, Hasegawa H, Yamanouchi K, Soyama A, Takatsuki M, et al. Rapid fabricating technique for multi-layered human hepatic cell sheets by forceful contraction of the fibroblast monolayer. PloS one. 2013;8(7):e70970. doi: 10.1371/journal.pone.0070970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffner AE, Barker JL, Stenger DA, Hickman JJ. Investigation of the factors necessary for growth of hippocampal neurons in a defined system. J Neurosci Methods. 1995;62(1–2):111–119. doi: 10.1016/0165-0270(95)00063-1. [DOI] [PubMed] [Google Scholar]
- Scherp P, Putluri N, LeBlanc GJ, Wang ZQ, Zhang XH, Yu Y, et al. Proteomic analysis reveals cellular pathways regulating carbohydrate metabolism that are modulated in primary human skeletal muscle culture due to treatment with bioactives from Artemisia dracunculus L. J Proteomics. 2012;75(11):3199–3210. doi: 10.1016/j.jprot.2012.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimek K, Busek M, Brincker S, Groth B, Hoffmann S, Lauster R, et al. Integrating biological vasculature into a multi-organ-chip microsystem. Lab on a Chip. 2013;13(18):3588. doi: 10.1039/c3lc50217a. [DOI] [PubMed] [Google Scholar]
- Schütte J, Hagmeyer B, Holzner F, Kubon M, Werner S, Freudigmann C, et al. “Artificial micro organs”—a microfluidic device for dielectrophoretic assembly of liver sinusoids. Biomedical Microdevices. 2011;13(3):493–501. doi: 10.1007/s10544-011-9517-7. [DOI] [PubMed] [Google Scholar]
- Shintu L, Baudoin R, Navratil V, Prot JM, Pontoizeau C, Defernez M, et al. Metabolomics-on-a-Chip and Predictive Systems Toxicology in Microfluidic Bioartificial Organs. Anal Chem. 2012;84(4):1840–1848. doi: 10.1021/ac2011075. [DOI] [PubMed] [Google Scholar]
- Subramanian B, Rudym D, Cannizzaro C, Perrone R, Zhou J, Kaplan DL. Tissue-engineered three-dimensional in vitro models for normal and diseased kidney. Tissue Engineering Part A. 2010;16(9):2821–2831. doi: 10.1089/ten.TEA.2009.0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung JH, Shuler ML. A micro cell culture analog (μCCA) with 3-D hydrogel culture of multiple cell lines to assess metabolism-dependent cytotoxicity of anti-cancer drugs. Lab on a Chip. 2009a;9(10):1385–1394. doi: 10.1039/b901377f. [DOI] [PubMed] [Google Scholar]
- Sung JH, Shuler ML. A micro cell culture analog (microCCA) with 3-D hydrogel culture of multiple cell lines to assess metabolism-dependent cytotoxicity of anti-cancer drugs. Lab on a Chip. 2009b;9(10):1385–1394. doi: 10.1039/b901377f. [DOI] [PubMed] [Google Scholar]
- Sung JH, Esch MB, Shuler ML. Integration of in silico and in vitro platforms for pharmacokinetic-pharmacodynamic modeling. Expert Opinion on Drug Metabolism & Toxicology. 2010;6(9):1063–1081. doi: 10.1517/17425255.2010.496251. [DOI] [PubMed] [Google Scholar]
- Sung JH, Esch MB, Prot JM, Long CJ, Smith A, Hickman JJ, Shuler ML. Microfabricated mammalian organ systems and their integration into models of whole animals and humans. Lab on a Chip. 2013;13(7):1201–1212. doi: 10.1039/c3lc41017j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney LM, Shuler ML, Babish JG, Ghanem A. A cell culture analogue of rodent physiology: Application to naphthalene toxicology. Toxicology in vitro. 1995;9(3):307–316. doi: 10.1016/0887-2333(95)00007-U. [DOI] [PubMed] [Google Scholar]
- Tatosian DA, Shuler ML. A novel system for evaluation of drug mixtures for potential efficacy in treating multidrug resistant cancers. Biotechnology and bioengineering. 2009a;103(1):187–198. doi: 10.1002/bit.22219. [DOI] [PubMed] [Google Scholar]
- van der Valk J, Brunner D, De Smet K, Fex Svenningsen A, Honegger P, Knudsen LE, et al. Optimization of chemically defined cell culture media--replacing fetal bovine serum in mammalian in vitro methods. Toxicol In Vitro. 2010;24(4):1053–1063. doi: 10.1016/j.tiv.2010.03.016. [DOI] [PubMed] [Google Scholar]
- Vandenburgh H, Shansky J, Benesch-Lee F, Barbata V, Reid J, Thorrez L, et al. Drug-screening platform based on the contractility of tissue-engineered muscle. Muscle & Nerve. 2008;37(4):438–447. doi: 10.1002/mus.20931. [DOI] [PubMed] [Google Scholar]
- Viravaidya K, Shuler ML. Incorporation of 3T3 -L1 cells to mimic bioaccumulation in a microscale cell culture analog device for toxicity studies. Biotechnology Progress. 2004a;20(2):590–597. doi: 10.1021/bp034238d. [DOI] [PubMed] [Google Scholar]
- Viravaidya K, Shuler ML. Incorporation of 3T3-L1 cells to mimic bioaccumulation in a microscale cell culture analog device for toxicity studies. Biotechnology Progress. 2004b;20(2):590–597. doi: 10.1021/bp034238d. [DOI] [PubMed] [Google Scholar]
- Viravaidya K, Sin A, Shuler ML. Development of a microscale cell culture analog to probe naphthalene toxicity. Biotechnology Progress. 2004a;20(1):316–323. doi: 10.1021/bp0341996. [DOI] [PubMed] [Google Scholar]
- Viravaidya K, Sin A, Shuler ML. Development of a microscale cell culture analog to probe naphthalene toxicity. Biotechnology Progress. 2004b;20(1):316–323. doi: 10.1021/bp0341996. [DOI] [PubMed] [Google Scholar]
- Wagner I, Materne EM, Brincker S, Sussbier U, Fradrich C, Busek M, et al. A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture. Lab on a Chip. 2013 doi: 10.1039/c3lc50234a. [DOI] [PubMed] [Google Scholar]
- Wagner W, Ho AD. Mesenchymal stem cell preparations--comparing apples and oranges. Stem Cell Rev. 2007;3(4):239–248. doi: 10.1007/s12015-007-9001-1. [DOI] [PubMed] [Google Scholar]
- Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. Journal of Cell Biology. 1997;137(1):231–245. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welin A, Amirbeagi F, Christenson K, Bjorkman L, Bjornsdottir H, Forsman H, et al. The Human Neutrophil Subsets Defined by the Presence or Absence of OLFM4 Both Transmigrate into Tissue In Vivo and Give Rise to Distinct NETs In Vitro. PloS one. 2013;8(7):e69575. doi: 10.1371/journal.pone.0069575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson K, Das M, Wahl KJ, Colton RJ, Hickman J. Measurement of contractile stress generated by cultured rat muscle on silicon cantilevers for toxin detection and muscle performance enhancement. PloS one. 2010;5(6) doi: 10.1371/journal.pone.0011042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K, Chan JM, Kamm RD. Microfluidic models of vascular functions. Annual review of Biomedical Engineering. 2012;14(1):205–230. doi: 10.1146/annurev-bioeng-071811-150052. [DOI] [PubMed] [Google Scholar]
- Xu H, Kraus W, Shuler M. Development of a stable dual cell-line GFP expression system to study estrogenic endocrine disruptors. Biotechnology and Bioengineering. 2008;101(6):1276–1287. doi: 10.1002/bit.21991. [DOI] [PubMed] [Google Scholar]
- Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proceedings of the National Academy of Sciences. 2012;109(34):13515–13520. doi: 10.1073/pnas.1210182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Chia SM, Ong SM, Zhang S, Toh YC, van Noort D, YU H. The controlled presentation of TGF-β1 to hepatocytes in a 3D-microfluidic cell culture system. Biomaterials. 2009a;30(23):3847–3853. doi: 10.1016/j.biomaterials.2009.03.052. [DOI] [PubMed] [Google Scholar]
- Zhang C, Zhao Z, Abdul Rahim NA, van Noort D, Yu H. Towards a human-on-chip: Culturing multiple cell types on a chip with compartmentalized microenvironments. Lab on a Chip. 2009b;9(22):3185–3192. doi: 10.1039/b915147h. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Chen J, Craven M, Choi NW, Totorica S, Diaz-Santana A, et al. In vitro microvessels for the study of angiogenesis and thrombosis. Proceedings of the National Academy of Sciences. 2012;109(24):9342–9347. doi: 10.1073/pnas.1201240109. [DOI] [PMC free article] [PubMed] [Google Scholar]