

Abstract
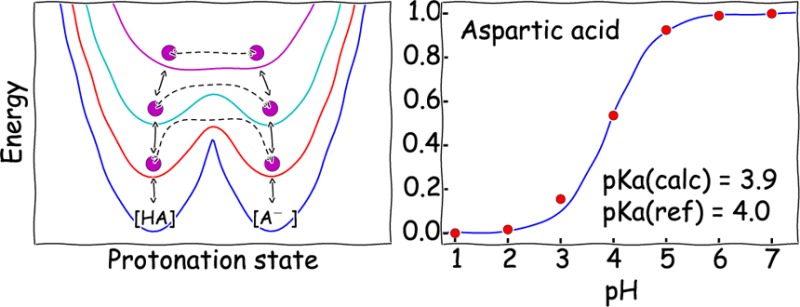
We present a new computational approach for constant pH simulations in explicit solvent based on the combination of the enveloping distribution sampling (EDS) and Hamiltonian replica exchange (HREX) methods. Unlike constant pH methods based on variable and continuous charge models, our method is based on discrete protonation states. EDS generates a hybrid Hamiltonian of different protonation states. A smoothness parameter s is used to control the heights of energy barriers of the hybrid-state energy landscape. A small s value facilitates state transitions by lowering energy barriers. Replica exchange between EDS potentials with different s values allows us to readily obtain a thermodynamically accurate ensemble of multiple protonation states with frequent state transitions. The analysis is performed with an ensemble obtained from an EDS Hamiltonian without smoothing, s = ∞, which strictly follows the minimum energy surface of the end states. The accuracy and efficiency of this method is tested on aspartic acid, lysine, and glutamic acid, which have two protonation states, a histidine with three states, a four-residue peptide with four states, and snake cardiotoxin with eight states. The pKa values estimated with the EDS-HREX method agree well with the experimental pKa values. The mean absolute errors of small benchmark systems range from 0.03 to 0.17 pKa units, and those of three titratable groups of snake cardiotoxin range from 0.2 to 1.6 pKa units. This study demonstrates that EDS-HREX is a potent theoretical framework, which gives the correct description of multiple protonation states and good calculated pKa values.
1. Introduction
Solution pH is one of the most important environmental variables that affects the structural and dynamic properties of biomolecules.1 Various biological events such as protein folding/unfolding,2 ligand binding,3−5 and enzyme activity6,7 heavily depend on solution pH. Solution pH affects protein denaturation,8 aggregation,9 and regulates many pH-dependent membrane proteins and channels.10−13 In cells, the pH in different compartments varies significantly; for example, the pH in the cytoplasm and nucleus is neutral (around 7.2), in vacuoles and Golgi, it is acidic (between 4.8 and 6.5), and in mitochondria, it is basic (around 8.0).14,15 At the same time, the pH of each compartment is tightly regulated, and a small pH change can lead to serious diseases.16
Solution pH affects proteins by changing the protonation states of ionizable/titratable residues. The protonation state of an ionizable residue is a function of its pKa value. However, the pKa values of ionizable residues in protein environments can be shifted from the standard pKa values experienced in water. The shift is especially large for groups found in protein interiors,17−19 and as a result, such buried groups can sometimes change protonation state even at physiological pH. Changes in the protonation state can be exploited for function, for example, when they are coupled to conformational reorganization, such as in the case of ATP synthase,12 bacteriorhodopsin,20 cytochrome c oxidase,21 or the photoactive yellow protein.22 To understand the mechanisms of such proteins, it is essential to know the pKa values of functionally important residues.
Experimental determination of pKa values of such residues can be a challenge, and carefully calibrated computational methods offer a possibility to obtain them. However, current computational methods have limitations when large scale structural reorganization is coupled to a change in protonation state. A widely used methodology for pKa calculation is based on the solutions of the Poisson–Boltzmann (PB) equation.23−26 The limitation of PB-based methods is that they may not properly represent the reorganization/response of protein induced by the titration of ionizable groups.27,28 Most PB-based methods use only a single conformation or allow perturbation of side-chain or hydrogen atoms.25,29 Also, the conformational response of a protein is modeled by a single value of a dielectric constant, which is dubious considering the inhomogeneous environment of the interior and surface of a protein. This approximation can also be problematic when water molecules are tightly coupled with ionizable groups of interest,30−32 which is commonly observed in many transmembrane proteins.10−13 A more accurate description of protein environment and polarizability can be achieved through quantum mechanical (QM) or mixed quantum mechanics/molecular mechanics (QM/MM) approaches.33−39 However, such calculations are computationally very expensive, and proper description of conformational changes triggered by protonation/deprotonation may not be achieved on a relevant time scale.
Protein conformational changes triggered by ionization can be considered explicitly using constant pH molecular dynamics simulations. Constant pH simulation methods can be categorized into two groups: (1) discrete protonation state models, and (2) continuous protonation state models. Constant pH methods based on discrete protonation state models generally use a hybrid approach combining molecular dynamics (MD) and Monte Carlo (MC). In the hybrid MC/MD methods, a MC procedure is performed to determine the protonation states of ionizable groups at a regular interval over the course of a MD simulation with either implicit or explicit water. During the MC procedure, a random walk between different protonation states is performed, and a state is determined based on the estimated free energy difference and Metropolis criteria. With a continuum electrostatic model,23,40 Dlugosz and Antosiewicz41,42 developed a MC/MD method based on the analytic continuum electrostatic method.43 Mongan et al. used a generalized Born (GB) solvation model to calculate (de)protonation free energies.44
In contrast to the implicit solvent methods above, the MD/MC method with explicit water requires a more sophisticated MC move due to solvent reorganization.45,46 Without solvent reorganization, a sudden change of charge distribution of a side-chain is likely to result in a large electrostatic penalty, which leads to an extremely low MC acceptance ratio. To increase the acceptance ratio of MC moves, various methods have been suggested. For example, Baptista et al. evaluated (de)protonation free energy by PB and performed a short MD run to relax the solvent.47,48 Bürgi et al.49 performed short thermodynamic integration (TI) calculations for MC moves, which is extremely expensive. Stern50 proposed a similar approach where MC moves consist of short MD simulations (not free energy calculations) using a time-dependent Hamiltonian that interpolates two protonation states. Most of these types of approaches require approximations that result in a loss of rigor that distorts the final ensemble.
In the continuous protonation state models, a protonation state is represented as a titration variable considered as an independent dynamic variable. Mertz and Pettitt developed an extended Hamiltonian approach.51 Baptista et al. performed MD simulations with the average charges of ionizable groups obtained by a mean field approximation.52 Börjesson and Hünenberger devised a model in which the extent of (de)protonation is equilibrated by weak coupling to a proton bath.53 Lee et al.54 applied, and Khandoghin et al. improved55 the λ dynamics approach56 to constant-pH simulation with the GB solvation model. Here, the extent of protonation is parametrized by a fictitious λ particle in the Hamiltonian, whose value fluctuates between 0 and 1. During the postprocessing of trajectories, conformations whose λ value is higher or lower than a threshold value are assigned to a protonation state, and other unphysical conformations are discarded. To avoid sampling of unphysical states, a barrier potential centered at λ = 0.5 is used. This potential has to be carefully adjusted to maximize the number of transitions and minimize sampling of unphysical states, however, even with this approach, most of the sampled conformations are different from physical states. Recently this approach has been extended to perform constant-pH MD simulations in explicit water.57−59
To enhance the accuracy of simulations that depend heavily on the accuracy of conformational sampling, constant pH methods with both discrete and continuous protonation states have been coupled with various enhanced sampling methods. The GB-based constant pH methods have been coupled with the temperature replica exchange60,61 and accelerated MD methods.62 A pH based replica exchange scheme has also been implemented63−66 where pH values are exchanged between replicas. To further increase sampling efficiency, the pH-exchange approach has been combined with reservoirs of conformations and protonation states within the framework of the double reservoir pH replica exchange method (work submitted for publication).
In this study, we develop a new constant-pH method that yields the correct description of protonation states by combining the enveloping distribution sampling (EDS)67,68 and Hamiltonian replica exchange (HREX)69,70 methods. The EDS method was devised to allow sampling of multiple end states from a single MD simulation of a hybrid Hamiltonian. Similar mixing schemes have been proposed by others.71−74 In the context of a constant pH simulation, the hybrid Hamiltonian is the sum of Boltzmann factors of multiple Hamiltonians, each corresponding to a different protonation state. The effect of solution pH is considered by the relative free energy differences between protonation states. To ensure proper sampling of all protonation states, a smoothness parameter, s, that controls the height of energy barriers is applied to the hybrid EDS Hamiltonian. When s → 0, the hybrid Hamiltonian becomes highly smoothed, which makes energy barriers disappear. On the other hand, as s → ∞, no smoothing is applied to the hybrid Hamiltonian, and it follows the minimum energy surface among the multiple end states, which corresponds to the physical Hamiltonian.
To obtain the correct ensemble of multiple protonation states while enhancing sampling efficiency, we coupled the EDS simulations with different smoothness parameter through the HREX method. The HREX method, often called the bias-exchange method, facilitates diverse conformational sampling by modifying a physical Hamiltonian or introducing various biasing potentials. To enhance conformational sampling efficiency, various Hamiltonian modification schemes have been suggested: scaling hydrophobic,70 long-range,75,76 solvent-related77,78 interactions, or biasing backbone dihedral angles.79,80 In the context of the EDS-HREX method, we use multiple EDS potentials with different smoothness parameter, including an EDS potential without smoothing that follows the minimum energy of multiple end states, and perform exchanges between them periodically. We assessed the performance of our method with titratable amino acid monomers: aspartic acid, glutamic acid, lysine, and histidine. We also tested out method with a small four-residue peptide (KAAE) and snake cardiotoxin. All systems were solvated with explicit water molecules. The results show that our method can successfully and efficiently reproduce the correct distribution of different protonation states at a given pH.
2. Theory
We briefly review the enveloping distribution sampling (EDS) and Hamiltonian replica exchange (HREX) methods and subsequently describe how these two methods are combined to sample the correct ensemble of different protonation states at a given pH.
2.1. Enveloping Distribution Sampling
The free energy difference between two states A and B is given by
| 1 |
where Z is the partition function of state and β is the inverse of the thermodynamic temperature.
In the EDS approach, a hybrid Hamiltonian enveloping both states is defined as follows,67
| 2 |
where EA and EB are the Hamiltonians of states A and B. In principle, a simulation performed on Eh allows sampling of the important phase space of both state A and B, and their free energy difference can be estimated by81
| 3 |
where ⟨...⟩h denotes an ensemble average of the hybrid state. However, if the energy difference between the minima of EA and EB is too large, the simulation will be trapped in the lowest energy basin of a single Hamiltonian. Additionally, if the energy barrier between minima is too high, transitions between the two states will be observed rarely, which can lead to large errors in the free energy result. To alleviate these problems, a modified EDS scheme with smoothing was suggested as follows,82
| 4 |
where N is the number of end states (e.g., in the case of two states, N = 2), s is a smoothness parameter, and Eioffset are energy offset parameters.
The schematic representations of mixing two protonation states, HA and A–, with different s values are illustrated in Figure 1. The relative energy difference between state HA and A– depends on the solution pH, which can be adjusted by the energy offset parameters. The pH dependence of energy offset values will be discussed in more detail in the following section. A lower s value leads to a lower energy barrier in the hybrid Hamiltonian, which can facilitate spontaneous state transitions. However, if s becomes too small, Eh adopts a single energy minimum, which deviates from the original energy minima of EA and EB. A simulation on Eh with such a small s value results in an ensemble comprising of only unphysical intermediate conformations. An iterative parameter optimization procedure was suggested to determine appropriate parameters for an efficient EDS simulation.82 In this paper, we address this problem by performing simulations at multiple s values and performing Hamiltonian replica between these simulations to enhance sampling of conformational transitions in the physically realistic Hamiltonian.
Figure 1.
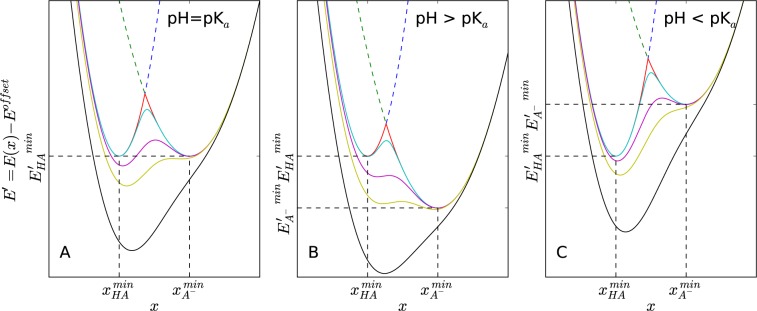
Schematic representation of EDS Hamiltonians (solid lines) mixing protonated (HA, blue dashed line) and deprotonated (A–, green dashed line) states for constant pH simulations under various pH conditions: (A) pH = pKa, (B) pH > pKa, and (C) pH < pKa. The difference between energy minima of each protonation state is determined by eq 8. Five EDS Hamiltonians constructed with different smoothness parameters are illustrated: s = ∞ (red), s = 0.7 (cyan), s = 0.22 (purple), s = 0.15 (yellow), s = 0.08 (black). Note that a smaller s value leads to a smoother EDS Hamiltonian with a lower energy barrier. If s is small enough, an EDS Hamiltonian has a single energy minimum, which is different from the energy minima of either original end state.
2.2. Constant pH Simulations with EDS
One goal of constant-pH simulation is to sample the equilibrium distribution of the protonated (HA) and deprotonated (A–) states of a titratable group of a biomolecule at a given pH. The equilibrium distribution of the two protonation states is determined by their free energy difference. However, this free energy difference cannot be calculated by a conventional molecular mechanics (MM) approach because it cannot account for two factors: (1) the quantum mechanical energy of bond breaking and formation and (2) the contribution of proton solvation, which is affected by external pH. Following Mongan et al.,44 we assume that the total protonation free energy of a titratable group in a protein (ΔGprotein) consists of the molecular mechanics (ΔGprotienMM) and nonmolecular mechanics (ΔGprotien) contributions:
| 5 |
The non-MM component can be estimated by introducing the model compound, which has the same titratable group as the protein but with a known experimental pKa value. In this study, a model compound is defined as a solvated amino acid monomer with capped termini. Based on the known pKa of the model compound (pKa,model), its protonation free energy is
Therefore, the non-MM component of the protonation free energy of the model compound is
| 8 |
With the assumption that the non-MM component of the model compound is identical with that of the protein, ΔGprotiennon-MM = ΔGmodel, eq 5 can be expressed as
| 9 |
where ΔGmodelMM can be readily obtained by conventional free energy calculation methods.
For a single ionizable group, when no other ionizable groups are titrated, ΔGproteinMM can be calculated by performing an EDS simulation with Hamiltonians of two protonation states, Eprotein (HA) and EproteinMM (A–). The ΔGprotein – ΔGmodelMM part in eq 9 can be viewed as a shift in free energy of the model compound that is due to the change in the environment of nonbonded interactions of the ionizable group when it is transferred from the solvent to the protein environment.54,83 By using eq 9, the sampling of different protonation states of a titratable group in a protein environment is performed in a pH dependent manner. Equation 9 is practically implemented such that the protonated state is considered the reference state and is not experiencing any energy offset, while the deprotonated state is experiencing the pH dependent energy offset −ΔGmodel + kBT ln 10 (pH – pKa,model), as also depicted in Figure 1.
When multiple ionizable groups in a protein are titrated, the number of states considered has to be increased, e.g. for two ionizable groups, four different states need to be considered, and for three ionizable groups, eight different states need to be considered. The advantage of this method, as opposed to free energy methods such as thermodynamic integration in which only two states are considered, is that it can estimate the pH dependent populations of multiple states in a single EDS simulation.
For example, in the case of two titratable groups, four states need to be considered. While one of the states (say the state in which both titratable groups are protonated) can be considered to be the model state, the pH dependent offset will be applied to the three other states. For a state in which titratable group 1 is deprotonated and group 2 is protonated, the pH dependent energy offset is −ΔGmodel1MM + kBT ln 10 (pH – pKa,model1); for a state in which titratable group 1 is protonated and group 2 is deprotonated, the pH dependent energy offset is −ΔGmodel2 + kBT ln 10 (pH – pKa,model2); for a state in which both groups are deprotonated, the offset is a sum of the two offsets. If the two titratable groups are of different nature, say Lys and Glu, the two model compound free energies ΔGmodelMM and pKa,model values will be different, but if two groups are the same, these energies and pKa,model values will be the same.
2.3. Hamiltonian Replica Exchange Method
In Hamiltonian replica exchange (HREX), replicas are swapped between different Hamiltonians periodically. Each Hamiltonian corresponds to a different environmental condition or representation of a system, such as external fields in Ising spin system or the strength of hydrophobic interaction in protein folding simulation. Generally, a proper exchange between different Hamiltonians can enhance the sampling efficiency while preserving the Boltzmann distribution.69 If the mth replica follows the Hamiltonian Em (x), its Boltzmann distribution is
| 10 |
where Zm is the partition function of Em. Because replicas are noninteracting, the joint probability of having configuration x in in the mth replica and configuration x′ in the nth replica is defined as
| 11 |
We define the probability of exchanging x in mth replica with x′ in nth replica as W(x,Em;x′,En), and the probability of the reverse process is W(x′,Em;x,En). To satisfy the detailed balance condition, the exchange probability between the two replica must follow the relation:
| 12 |
Combining eq 10 with eq 12 leads to
| 13 |
where
| 14 |
This condition can be satisfied by using the Metropolis-type criteria for exchanges,
2.4. Constant pH Simulation by Combination of EDS and HREX
The original EDS method with smoothing (eq 4) allows sampling of parts of the important phase space of multiple states in a single simulation, and it may lead to poor sampling of physical states. The conformations sampled in the middle of potential energy minima correspond to virtual intermediates between physical states, which may be similar to conformations with fractional charges (e.g., λ ∼ 0.5) in λ-dynamics.54,55,58 However, these mixed states are never included in analysis of the ensemble. To increase the efficiency of simulation, residence time at intermediate states should be reduced.
As shown in Figure 1, when s is small, the corresponding EDS simulation will mainly reside in an intermediate region in the phase space, which deviates substantially from the physical energy minima. To address this problem while preserving the sampling efficiency of EDS, we combined the EDS method with the Hamiltonian exchange method (Figure 2). In this study, we introduce the baseline EDS Hamiltonian without smoothing for conformational sampling, E0(x;s = ∞), which follows the minimum potential surface among the original states. Assume that state i with Hamiltonian Ei′ has the lowest energy at x0 among {E1,...,EN′},
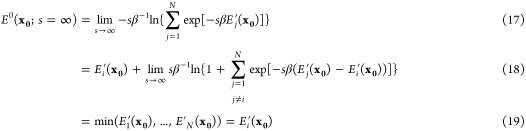 |
All exponential terms in eq 18 vanish as s → ∞ because −sβ(Ej′ – Ei) becomes a large negative number. Therefore, all conformations sampled with E0 exactly correspond to one of the original end states, and we denote the corresponding ensemble Γ0. In other words, E0 connects multiple Hamiltonians in a way that maximizes the correspondence to the original end states. This effect is more prominent near transition state regions, where the original potential energy surfaces are distorted most by a positive s (Figure 1). In addition, E0 enables a more accurate sampling of equilibrium ensembles of given Hamiltonians than the original EDS method, which uses an effective s = 1.0 and slightly deviates from the original Hamiltonians near the transition state region. The contribution of a given conformation x in Γ0 to each partition function can be obtained by simply calculating a corresponding Boltzmann factor. These properties make this minimum energy surface the logical choice for collecting the accurate constant pH ensemble.
Figure 2.
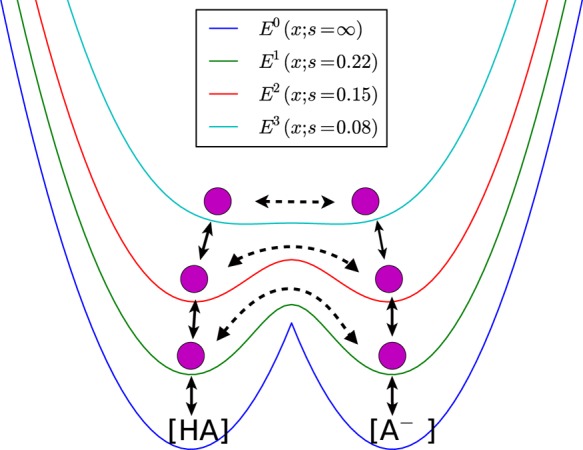
Schematic representation of the combination of EDS and Hamiltonian exchange methods for constant pH simulation. An EDS Hamiltonian with s = ∞, E0(x;s = ∞), follows the minimum of either Hamiltonian of protonated or deprotonated state, min(Eprot(x),Edeprot(x)). The other hybrid Hamiltonians with positive s values have lower energy barriers, which enhances protonation state transitions.
Generally, E0 has a high energy barrier in explicit solvent simulations due to solvent reorganization, which makes it impossible to observe spontaneous protonation state transitions within a computationally accessible time scale. Thus, to accelerate transitions, we introduce additional hybrid Hamiltonians with smaller s, which lowers the energy barriers in transition regions (Figure 2) and perform exchanges between the Hamiltonians at a regular interval. At the potential with the smallest s value, there is virtually no energy barrier, thus sampling mostly nonphysical conformations. These nonphysical conformations will be filtered through successive exchanges with more physical Hamiltonians, and eventually, only physically accessible conformations will be collected in the E0 trajectory. The exchange criterion between Hamiltonian is defined in eq 16.
3. Methods
3.1. Preparing Initial Structures
In this study, we used five test systems: an aspartic acid, glutamic acid, lysine monomers, a KAAE peptide, and snake cardiotoxin V from Naja naja atra (CTX A5, PDB ID: 1CVO).86 The input structures of all test systems were generated from the CHARMM22 topology files87 using the CHARMMing server.88 The N-termini and C-termini of all test systems are capped with the neutral acetyl and N-methyl groups, respectively. The amino acid monomers and the KAAE peptide are solvated in a 30 Å cubic box with explicit TIP3P water molecules, and the snake cardiotoxin is solvated in a 60 Å cubic box. The protonated and deprotonated states only differ in the partial charges of their side chains. The deprotonated state has a dummy nonzero mass hydrogen atom without charge while keeping the bond, angle, and van der Waals interactions. As discussed in the previous section, the contributions of these terms cancel out because the non-MM free energy components of the model compound and protein environment are almost identical. The charges of titratable amino acids are adopted from the CHARMM22 parameter set.54
3.2. MD Simulations
The combination of EDS and HREX is implemented in the CHARMM program.89 The EDS calculation is performed with the EDS command90 of the MSCALE facility,91 which can run simulations of multiple independent but connected systems. Each MSCALE subsystem is constructed to represent a protonation state. Their potential energy values are calculated in subprocesses and are used to calculate the EDS energy and associated gradients67 in the main processes. The HREX calculation is performed by the REPD facility.92 For all MD simulations in this study, exchanges between replicas are attempted every 1000 MD steps, and the SHAKE algorithm is used to constrain the bond length of hydrogen atoms. A time step of 1 fs is used, and the Nosé–Hoover thermostat93,94 is employed to maintain a temperature of 300 K. A nonbonded cutoff of 15 Å is used, electrostatic interactions are truncated by the force shift method, and van der Waals interactions are truncated with a switching function between 10 and 12 Å. All initial solvated systems are minimized by 200 steps of steepest descent followed by 200 steps with the adopted basis Newton–Raphson method.95 After minimization, the systems are equilibrated for 1 ns with constant pressure simulations at 1 atm. The last snapshot of the equilibration run is used as the initial structure for the constant pH simulations, and the size of water box is kept constant.
3.3. Calculation of ΔGmodelMM
To carry out a constant pH simulation, the MM contribution to the protonation free energy of the model compound is required, which effectively arises from the change of electrostatic interactions. In this study, a model compound is defined as a solvated amino acid monomer with capped termini. The ΔGmodelMM values of model compounds are obtained by TI,
| 20 |
where λ represents a coupling parameter between the protonated and deprotonated states. Eele (λ) is defined as
| 21 |
where Eele,prot and Eele,deprot are the electrostatic potentials of protonated and deprotonated states, respectively. The TI calculations are performed using the PERT facility in CHARMM. The λ value changes from 0 to 1 in increments of 0.05 at every 120 ps, using 20 ps of equilibration and 100 ps for gathering statistics. The calculated ΔGmodelMM values are summarized in Table 1.
Table 1. Experimental pKa Values84,85 and Calculated Free Energy Differences ΔFelec,w of Titratable Residues in Explicit Water.
| titratable residue | pKa,w | ΔFelec,w (kcal/mol) |
|---|---|---|
| Asp | 4.0 | –43.60 |
| Glu | 4.4 | –46.15 |
| Lys | 10.4 | 22.40 |
| His-δ | 6.5 | –4.39 |
| His-ε | 7.1 | –13.12 |
3.4. Calculation of pKa Values
With a given pKa value, the fraction of deprotonated samples depends on the pH value and can be obtained with the Hill equation:
| 22 |
where fd (pH) is the fraction of deprotonated states at a given pH, and n is the Hill coefficient. In our method, the fd value is estimated by comparing the Boltzmann factors of the protonated and deprotonated states of the conformations in Γ0 sampled with the baseline Hamiltonian E0 (eq 19). The fd value can be obtained with
| 23 |
where N is the number of configurations in Γ0, xi ∈ Γ0, and E′ = E – Eoffset.
4. Results and Discussion
4.1. Titration of Two-State Systems
To assess the accuracy of EDS-HREX constant-pH simulation, we estimated the pKa values of several two-state systems, including aspartic acid, glutamic acid, and lysine monomers in explicit water. For each system, we performed three independent constant-pH simulations for 1 ns at different pH values. Aspartic acid was run for 5 ns to investigate the convergence of a small system with our method. Each EDS-HREX simulation consists of 4 EDS replicas, E0 to E3, using s values of ∞, 0.027, 0.020, and 0.01. The energy offset value of the deprotonated state is determined by eq 8. The pKa values were estimated by fitting the baseline ensembles Γ0 to the Hill equation. The average pKa values and standard deviations of all benchmark systems are summarized in Table 2.
Table 2. Calculated pKa Values of Amino Acids with Two Protonation States from EDS-HREX Constant pH Simulation in Explicit Water.
| titratable residue | estimated pKa | std. dev. | experimental pKa |
|---|---|---|---|
| Asp (1 ns) | 3.92 | 0.094 | 4.0 |
| Asp (5 ns) | 3.94 | 0.046 | 4.0 |
| Glu | 4.33 | 0.094 | 4.4 |
| Lys | 10.43 | 0.034 | 10.4 |
For aspartic acid, we carried out 3 independent sets of 6 EDS-HREX simulations with pH values ranging from 2 to 7 with an interval of 1. Figure 3A illustrates the average deprotonated fraction of the aspartic acid at each pH condition obtained from 3 independent EDS-HREX simulations, along with the corresponding Hill equation. The average pKa value of the aspartic acids is estimated to be 3.92, which agrees well with the experimental pKa value of 4.0.
Figure 3.
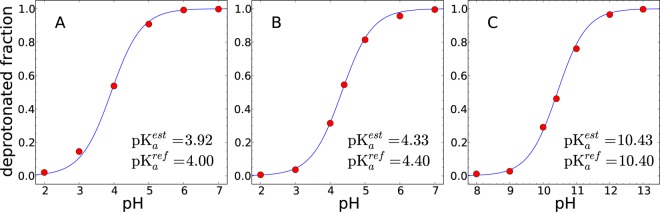
Deprotonated fractions of (A) aspartic acid, (B) glutamic acid, and (C) lysine by EDS-HREX constant pH simulation with explicit water molecules. The average deprotonated fractions of three independent 1 ns simulations are shown as red dots. The fitted titration curves are shown as solid lines.
To test the convergence and accuracy of the EDS-HREX method, we performed constant pH simulations of aspartic acid for 5 ns at 6 different pH conditions: 18 constant pH simulations in total. The average estimated pKa values and standard deviations are calculated for 1 ns time windows (Table 3). The results show that a deviation from experiment of only 0.08 pKa units, corresponding to 0.11 kcal/mol, can be achieved even with a 1 ns simulation. In addition, the estimated pKa value remains stable for 5 ns. Starting from a standard deviation of 0.094 pKa unit, the value decreases to 0.032 after 2 ns. Little change in the standard deviation after 2 ns indicates that the simulations are converged within 2 ns. These results demonstrate that the EDS-HREX method can give a reliable pKa estimate.
Table 3. Averages of estimated pKa Values, Standard Deviations, and Absolute Errors of the Blocked Aspartic Acid from 18 EDS-HREX Constant pH Simulations by 1 ns Time Window Along 5 ns Trajectories.
| time (ns) | pKa | std. dev. |
|---|---|---|
| 0–1 | 3.92 | 0.094 |
| 0–2 | 3.90 | 0.032 |
| 0–3 | 3.89 | 0.022 |
| 0–4 | 3.93 | 0.014 |
| 0–5 | 3.94 | 0.046 |
To verify that the exchange with smoothed EDS potentials enhances the state transitions, we traced the protonation state transition of replica 0 of the aspartic acid from one simulation over time (Figure 4). To determine the state of a conformation x in the smoothed EDS Hamiltonians, we define the likelihood of state i, θi (x), that is, being protonated or deprotonated, as follows:
| 24 |
where E′ = E – Eoffset, which is adopted from the Zwanzig equation (eq 3). In Figure 4A, if θi (x) is larger than 0.9999, x is considered to be the state i. If both θi (x) and θj (x) are less than 0.9999, x is considered as an intermediate state, which could be unphysical. It can be observed that, after multiple exchanges between the EDS potentials, replica 0 of aspartic acid returns to E0 and its protonation state is changed from the protonated to the deprotonated state at 350 ps. After 80 ps, replica 0 reaches the EDS Hamiltonian with the smallest s, E3. At E3, intermediate states are dominantly sampled due to a lowered energy barrier. In addition, fast spontaneous protonation state transitions without Hamiltonian exchange are readily observed. These results demonstrate that an EDS potential with a small s facilitates state transitions through nonphysical intermediate states, as expected.
Figure 4.
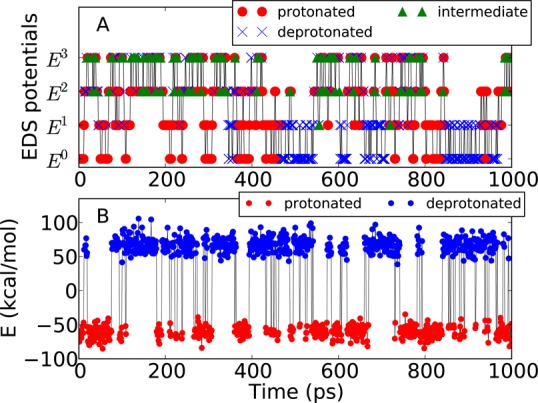
(Top) Protonation states and the visited EDS potentials of replica 0 during 1 ns EDS-HREX simulation at pH = 4. Based on the state likelihood θi, the protonated (red circle), deprotonated (blue × marks), and intermediate (green triangle) states are assigned. (Bottom) Difference between the adjusted potential energies of two protonation states, ΔE = (Eprot – Eprotoffset) – (Edeprot – Edeprot). Because E0 follows the lower energy between (Eprot – Eprotoffset) and (Edeprot – Edeprot), if ΔE is negative, a configuration corresponds to the protonated state (red). Otherwise, the configuration corresponds to the deprotonated state (blue).
The sampling efficiency of EDS-HREX can be estimated from the number of protonation state transitions observed in the production ensemble Γ0 (Figure 4B). During the 1 ns simulation of aspartic acid at pH 4, we observe an average of 83 protonation state transitions, which is comparable to previous λ-dynamics based approaches. Donnini et al.57 observed ∼100 transitions during 20 ns of the titration of imidazole, corresponding to ∼5 transitions per ns, and Goh et al.58 achieved ∼50 transitions per ns for the titrations of adenine and cytosine.
To verify that the ensemble Γ0 consists only of physical states, we compare the radial distribution functions (RDFs) of water molecules around the OD atoms of aspartic acid in each protonation state obtained by our constant-pH simulation with those produced by conventional MD simulations with fixed charges (Figure 5). The RDFs obtained by our constant-pH simulations are almost identical with those from conventional fixed-charge MD simulations, which demonstrates that our method samples physical states rather than approximate states with noninteger charges obtained by λ-dynamics. The RDFs between the water hydrogen atoms and the OD atoms of aspartic acids are significantly different depending on the protonation state. In the deprotonated state, due to the negatively charged OD atoms, the water hydrogen atoms form a sharp peak of the first solvation shell at 1.8 Å, and the second solvation shell is observed at 3.1 Å. However, in the protonated state, a hydrogen atom with +0.44e charge is bonded to an OD atom, which repels water hydrogen atoms and reduces the peak height of first solvation shell substantially. The RDFs of water oxygens display similar differences. In the deprotonated state, the first and second solvation shells are clearly observed at 2.7 and 4.8 Å, while only the first solvation shell is observed in the protonated state.
Figure 5.
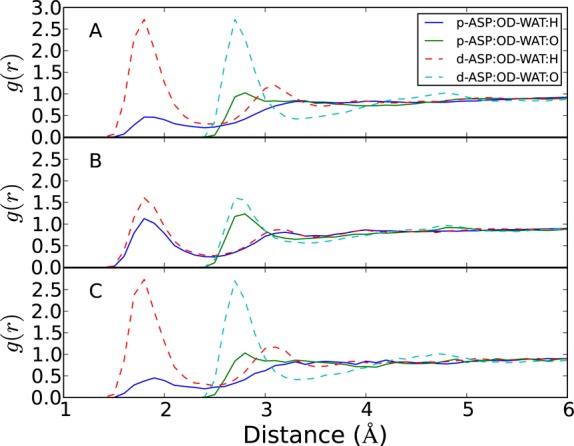
Radial distribution functions (RDFs) between the OD atoms of aspartic acid and water molecules obtained by EDS-HREX constant pH simulation at (A) E0(x;s = ∞) and (B) E3(x;s = 0.01) with a state likelihood threshold of 0.99. The subplot C is obtained from 2 ns MD simulations with the fixed charges for the protonated and deprotonated states. The RDFs of protonated and deprotonated states are shown as solid and dashed lines, respectively.
The RDFs obtained from the ensemble sampled with E3 (x;s = 0.01) show much less difference between the protonation states. The protonation state is determined based on the state likelihood criterion, θ (x) > 0.99. The peaks of the deprotonated aspartic acid become lower, and those of the protonated state become higher, which converges to the average of the RDFs of the two protonation states. This shows that the EDS sampling with a positive s leads to a significant deviation from the original states. Thus, one should be cautious when interpreting an ensemble obtained with a modified Hamiltonian, such as λ-dynamics or an EDS potential with s > 0, because the estimated thermodynamic properties can significantly diverge from the true values.
4.2. Histidine
To verify that our method can be extended to a chemically coupled multistate titration straightforwardly, we performed a constant pH simulation of histidine, which has two coupled titratable sites, ND1 and NE2, leading to four possible tautomeric states. The titration of histidine is one of the most important goals of constant pH simulations because its experimental pKa value is in the range of physiological conditions. In this work, we consider three protonation states defined in the CHARMM22 force field, the doubly protonated state (residue type HSP), the ND1-protonated state (HSD), and the HE2-protonated state (HSE). From the given microscopic equilibrium constants, k1 and k2, for the reactions HSP ↔ HSD and HSP ↔ HSE, the macroscopic equilibrium constant, k, for histidine can be derived as
| 25 |
By using the definition of pKa, the macroscopic experimental pKa value of histidine can be obtained from the microscopic pKa values, pKa,1 and pKa,2, as follows
| 26 |
From the given experimental pKa value of 6.5 for HSD and 7.1 for HSE,85 the macroscopic experimental pKa value is determined to be 6.4.
We carried out 1 ns constant pH simulations of histidine at 5 different pH values ranging from 5 to 9. Because histidine has three protonation states, more state transitions are required for convergence than for the two protonation state systems. We used 6 EDS potentials with s = ∞, 0.06, 0.05, 0.04, 0.024, and 0.01. Figure 6 shows the macroscopic and two microscopic titration curves of histidine, and the estimated pKa values are listed in Table 4. The estimated macroscopic pKa value is 6.24 with a standard deviation of 0.033, which agrees well with the experimental pKa. The estimated microscopic pKa values of HSD and HSE are 6.33 and 7.20, respectively, which also agree with the experimental pKa values. These results demonstrate that our method can successfully perform constant pH simulations of multiple titratable sites with chemical coupling.
Figure 6.
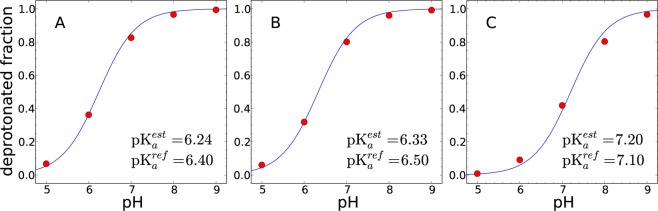
Titration curves of histidine obtained by EDS-HREX constant pH simulation with explicit water molecules. (A) The macroscopic, the total deprotonated fraction of Nδ and Nε, and two microscopic titration curves of (B) Nδ and (C) Nε are illustrated. The average deprotonated fractions of three 1 ns simulations are shown as red dots. The fitted titration curves are shown as solid lines.
Table 4. Calculated pKa Values of Histidine in Explicit Water.
| titratable residue | estimated pKa | std. dev. | experimental pKa |
|---|---|---|---|
| His-δ | 6.33 | 0.016 | 6.5 |
| His-ε | 7.20 | 0.132 | 7.1 |
| His-all | 6.24 | 0.033 | 6.4 |
4.3. KAAE Peptide
The KAAE peptide contains two titratable residues, Lys-1 and Glu-4. For this peptide, four different protonation states were considered: state 1, with both groups protonated, state 2 with Lys-1 deprotonated and Glu-4 protonated, state 3 with Lys-1 protonated and Glu-4 deprotonated, and state 4 with both groups deprotonated. Because the pKa of a Lys model compound is 10.4 and the pKa value of Glu model compound is 4.4, state 2 with deprotonated Lys and protonated Glu is improbable and could have been omitted. However, for consistency, we kept it in the calculations.
Simulations of the KAAE peptide were performed at pH values 2.4 to 13.4 in steps of 1 pH unit. Three different sets of simulations were performed, each with different initial velocities and using slightly different EDS potentials. The first set of simulations was performed at s = ∞, 0.027, 0.021, 0.016, and 0.012. The acceptance ratios in replica exchange simulations between replicas 3 and 4 were 45%, that is, larger than the target acceptance ratio of 20%. Thus, the second set of simulations was performed with the same s value of the highest replica decreased from 0.012 to 0.0086. The acceptance ratios for replica exchange between all replicas were still higher than 20% (the target acceptance ratio), except at a few pH values between replicas 0 and 1, and at one pH value between replicas 1 and 2. Thus, we performed the third set of simulations at s = ∞, 0.03, 0.022, 0.016, and 0.0086.
The titration curves for the peptide were determined by averaging the three simulations (Figure 7). The pKa values and Hill coefficients were determined from the Hill equation. The calculated pKa values and their standard deviations are shown in Table 5. Based on the standard deviations, we conclude that the change in distribution of EDS potentials had virtually no effect on the population of the four states and the calculated pKa values and Hill coefficients.
Figure 7.
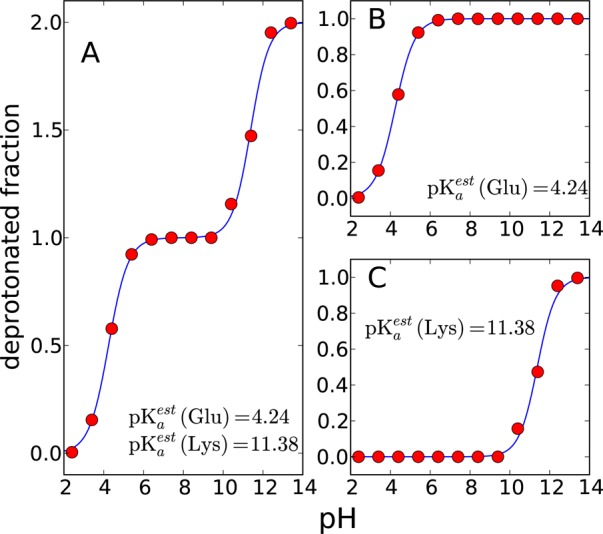
Titration curves of KAAE peptide in explicit water. (A) The macroscopic, the sum of deprotonated fractions of glutamic acid and lysine, and two microscopic titration curves of (B) glutamic acid and (C) lysine are illustrated. The average deprotonated fractions of three 1 ns simulations are shown as red dots. The fitted titration curves are shown as solid lines.
Table 5. Calculated pKa Values and Hill Coefficients of KAAE Peptide.
| estimated
pKa |
Hill
coefficient |
|||
|---|---|---|---|---|
| titratable residue | avg | std | avg | std |
| Glu | 4.23 | 0.15 | 0.94 | 0.27 |
| Lys | 11.38 | 0.09 | 0.96 | 0.08 |
The pKa value of Lys-1 is 11.38; that is, it is shifted by 1 pH unit from the model compound pKa value of 10.4, while the pKa of Glu-4 is 4.23, only 0.2 pH units lower than that of the model compound. Hydrogen bond analysis was performed with VMD96 for simulations at pH values 2.4, 7.4, and 13.4, which corresponded to states 1, 3, and 4, respectively, being predominately populated. As a measure of ion-pair interactions, we looked at the distance between Lys-1:NZ and Glu-4:CD. For pH values 2.4 and 13.4, these two atoms were never closer than 4 Å to each other, but at pH 7, they were within 4 Å 6% of the time. In terms of hydrogen bonding interactions with the rest of the peptide, Glu-4 did not engage in any, while Lys-1 was hydrogen bonded 8% of the time at pH 2.4, 20% of the time at pH 7.4, and 2% of the time at pH 13.4. These hydrogen bonding interactions may explain why the calculated pKa value of Lys was shifted more than that of Glu.
4.4. Snake Cardiotoxin
Finally, we performed a constant pH simulation of snake cardiotoxin V from Naja naja atra (CTX A5, PDB ID: 1CVO).86 CTX A5 has three titratable residues, Glu-17, Asp-42, and Asp-59 that affect the stability of the protein between pH values 2 and 5.97,98 We considered all possible protonation state combinations of these residues (8 total).
Three sets of 1 ns EDS-HREX simulations were performed at pH values ranging from 1 to 6 in steps of 1 pH unit. Each EDS-HREX simulation consists of 6 replicas with s values of ∞, 0.033, 0.027, 0.022, 0.018, and 0.01 corresponding to a total simulation time of 108 ns. The deprotonated fractions of titratable residues were obtained from the average of three sets of simulations, and the variables for the Hill equation were obtained by fitting the average data points to the Hill equation (Figure 8). The calculated pKa values and Hill coefficients of three titratable residues are listed in Table 6.
Figure 8.
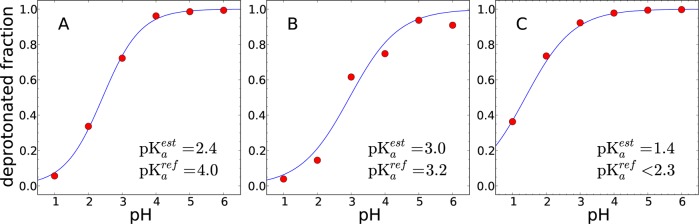
Titration curves of three titratable residues of snake cardiotoxin (CTX A5) are shown. The deprotonated fractions of (A) Glu-17, (B) Asp-42, and (C) Asp-59 are illustrated. The average deprotonated fractions of three 1 ns simulations are shown as red dots. The fitted titration curves are shown as solid lines.
Table 6. Calculated pKa Values and Hill Coefficients of Titratable Residues of CTX A5.
| calculated
pKa |
Hill
coefficient |
||||
|---|---|---|---|---|---|
| titratable residue | avg | std | avg | std | exp |
| Glu-17 | 2.4 | 0.10 | 0.77 | 0.04 | 4.0 |
| Asp-42 | 3.0 | 0.37 | 0.60 | 0.08 | 3.2 |
| Asp-59 | 1.4 | 0.28 | 0.77 | 0.17 | <2.3 |
The calculated pKa values, directions of the pKa shifts, and Hill coefficients are in accordance with the experiment. It is known that Asp-59 strongly interacts with the adjacent Lys-2, which results in a large shift of pKa value of Asp-59, from 4.0 to less than 2.3.97,98 In our result, the pKa is calculated to be 1.4, which is consistent with this. The pKa of Asp-42 is calculated to be 3.0, which is close to the experimental value of 3.2. The largest error is observed in Glu-17, whose calculated pKa value is lower than the experiment by 1.6 pKa units.
One possible source of this error may be limited conformational sampling. To obtain accurate pKa estimates, multiple transitions between different protonation states should be sampled. Generally, the titrations of residues are strongly coupled with protein conformational changes. Therefore, sufficient conformational sampling is important to reproduce experimental results. To check the convergence of our simulations, we counted the average number of protonation transitions of replicas sampled with the s = ∞ Hamiltonian at pH 2 and pH 3 (Figure 9). The protonation states of three residues, Glu-17, Asp-42, and Asp-59, are denoted by three letters (e.g., PDD). P and D represent the protonated and deprotonated state, respectively, and thus PDD would correspond to Glu-17 protonated, Asp-42 deprotonated, and Asp-59 deprotonated.
Figure 9.
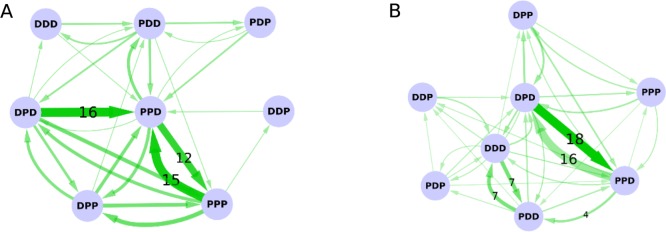
Summary of protonation state transitions at pH (A) 2 and (B) 3 are shown. Each node represents a protonation state and the width of the edge is proportional to the average number of transitions observed. The numbers of the most frequently observed transitions are displayed on the edges. The thinnest edge corresponds to only one transition from three sets of EDS-HREX simulations.
The majority of state transitions are observed between a subset of states, while the rest of the states are rarely visited. This indicates that the simulations are not fully converged, possibly due to limited conformational sampling. At pH 2, most protonation transitions occur between states PPP, PPD, DPP, and DPD, while the transition to state DDP is sampled only once during all 3 simulations (Figure 9A and B). When the external pH is 3, transitions between two pairs of states are mainly observed: state PPD–DPD and PDD–DDD.
The current EDS scheme lowers energy barriers caused by different energy terms between end states. In constant pH simulations, only electrostatic interactions are affected by the EDS mixing. In other words, the energy barriers originating from other energy terms (i.e., van der Waals or dihedral terms) are conserved after mixing by EDS, which can limit the conformational sampling of titratable groups. This sampling issue may be solved by combining the current EDS scheme with other accelerated sampling methods that preserve the canonical ensemble, such as self-guided Langevin dynamics with reweighting90,99,100 or orthogonal space random walk.101 In addition, introducing additional dimensions of Hamiltonian exchange to allow exchanges between pH values can also improve the convergence rate of simulations.61,63−66
Another source of error may originate from an imperfect representation of electrostatistics: using the classical MM model with fixed partial charges. For Glu-17, three positively charged lysines, Lys-2, Lys-13, and Lys-19, are located in the vicinity of Glu-17 and can affect the titration behavior. Lys-2 is considered to be especially important in controlling the stability of CTX A5 through interaction with Glu-17.97,98 If electrostatic interactions between Glu-17 and these three neighboring lysines are overestimated with the current MM force field, it may result in overpopulation of deprotonated Glu-17 leading to a lower calculated pKa value. This issue can be addressed by using polarizable force fields102,103 or QM/MM approaches,104,105 which can treat electrostatic interactions more accurately. Additionally, considering charge-leveling may improve the accuracy of simulations. Recently, Wallace and Shen have shown that a charge-leveling by simultaneous ionization or neutralization of a ion in solution can help to reproduce an experimental pKa value more accurately.106
4.5. Advantages of the EDS-HREX Method
The EDS-HREX method can be readily extended to titratable groups with chemically coupled moieties, such as histidine. This approach solves a problem inherent in λ-dynamic based approaches,54,55,57 where a new model or coordinate must be implemented to control the interconversion between such states. For example, histidine has two titratable sites, Nδ and Nε, and their atomic charges depend on each other. This dependence cannot be represented properly by a single titration coordinate. To address this issue, Khandogin and Brooks introduced a tautomeric state variable in addition to λ,55 and Donnini et al. performed linear-interpolations between all possible combinations of protonation states explicitly.57 In the EDS-HREX method, transitions between any pair of protonation states are automatically considered by performing MD simulation with the hybrid Hamiltonian.
The EDS-HREX method is compatible with any existing force field because the energy and forces of the hybrid Hamiltonian can be readily obtained from those of end states, which are calculated independently. In most current force fields, an atom type and its associated force field parameters depend on its protonation state. Our method can consider the change of atomic parameters other than charge, such as the van der Waals or Generalized Born solvation radius parameters. To apply the λ-dynamics approach to the change of general force field parameters rigorously, the parameters have to be interpolated linearly with respect to λ.55,57 Otherwise, this can be an inherent source of error as discussed in previous constant pH simulation with the GBSW implicit solvent model.54,55 The linear interpolation of parameters also requires the analytic derivatives of energy functions associated with λ, which can be highly complicated to compute.
The EDS-HREX method can be used for any free-energy calculation, not only those involving pH. It yields accurate free energy estimates because the E0 replica does not smooth the EDS Hamiltonian at all (s = ∞). In the original EDS method, free energies are calculated via eq 3. However, this equation only converges if energy differences between the original hybrid states are small.107−109 Therefore, a trade-off must be made between efficiency of sampling (small s) and convergence of the result (achievable with large s). To address this, an iterative parameter optimization scheme has been proposed to find the ideal s value, which optimizes accuracy.82 However, this method still samples some number of unphysical system states. In the EDS-HREX method, the E0 replica always has a s of ∞, while other replicas are used to explore different conformations. Therefore, the free energy differences between states can be directly calculated by comparing their Boltzmann factors from replica E0 because this replica only samples physical states. Another potentially significant advantage of EDS-HREX is that the resultant ensemble has only discrete protonation states, and these can be coupled to a high quality quantum mechanics (QM) or QM/MM surface using a non-Boltzmann Bennett approach.104,105
4.6. Performance Characteristics and Optimization
Determining the optimal replica distribution and smoothness parameters efficiently requires further investigation. In this study, the parameters were determined by trial-and-error, using a series of short simulations. We plan to devise an automatic procedure to determine the optimal parameter set for a given problem, which is similar to an iterative procedure that optimizes parameters for a single EDS simulation.82 As general guidelines to determine parameters, two conditions should be satisfied. First, spontaneous state transitions should be observed with highly smoothed EDS potentials, as shown in Figure 4A. This ensures that an EDS-HREX simulation actually samples important protonation states, which is essential in constant pH simulations. Second, an average exchange rate between replicas should be in the range 20–30% for an efficient sampling of various protonation state.
Our method requires more computational resources than λ-dynamics for the same simulation length. However, this increased cost is offset by the advantage that every time step may be used to collect the final ensemble. No steps need to be discarded. Currently, the EDS method is implemented via the MSCALE facility in CHARMM, which requires the independent energy evaluation of each end state. Therefore, the apparent cost of our method is simply proportional to the number of possible protonation states times the number of replicas used. For example, if there are x titratable groups with two protonation states, we need at most 2xNr times more computational resources than a single Hamiltonian simulation, where Nr is the number of replicas. However, in some cases, at any given pH, the number of states considered can be reduced. For example, a histidine residue has four possible protonation states but the only three of those states need to be considered under physiological conditions; the fully deprotonated state will not contribute. In the case of the KAAE peptide, the state in which Glu is protonated and Lys is deprotonated could have also been omitted, reducing the total number of states from four to three. Finally, CTX A5 has multiple lysines on its surface, which were assumed to be positive in this study because they are almost fully exposed and expected to experience little pKa shifts. Therefore, the cost may be reduced by eliminating rarely populated charge states.
On a similar note, we point out that the scalability of λ-dynamics may not be completely linear with the number of titratable groups. When there are N independent titratable groups, and if we assume that a probability to obtain a physical charge state of a single titratable site, that is, λ > 0.8 or λ < 0.2, is p, the fraction of snapshots that all titratable groups are in physical charge states becomes pN.
Our method focuses on obtaining accurate configurations of different protonation states. As shown in Figure 5, even with a rather strict state likelihood value >0.99, the radial distribution functions between titratable sites and water molecules obtained from a smoothed EDS potential are significantly distorted. Therefore, the reliability of ensembles obtained with fluctuating charges is questionable. Thus, the major contribution of our method is in the improved quality of ensembles, because our method samples original end states with the s = ∞ Hamiltonian.
The computational cost can also be reduced with an improved EDS scheme when only a few atom charges change in a large system. The EDS equation only requires energy differences. Instead of calculating the full energy of each charge state, computing just the energy differences can greatly reduce the number of required floating point operations. In other words, instead of calculating E1, E2, E3, and E4, we can perform an EDS simulation by calculating E1, E2 – E1, E3 – E1, and E4 – E1. In constant pH simulations, the energies of different states differ only in electrostatic interactions between titratable groups and their neighboring atoms within a cutoff radius. Therefore, the energies and gradients of a subset of nonbonded pairs should be recalculated with different charge sets. This can significantly reduce computational cost compared to the current EDS implementation.
5. Conclusion
We devised a new computational approach for constant-pH simulations in explicit solvent by combining the EDS and Hamiltonian replica exchange algorithms. We showed that this method can reproduce the correct description of multiple protonation states with frequent state transitions. A comparison of radial distribution functions between aspartic acid and water molecules demonstrates that the ensemble obtained with the baseline EDS Hamiltonian agrees well with those of MD simulations with fixed charges. In terms of sampling efficiency, we observed over 80 protonation state transitions of blocked aspartic acid during 1 ns of simulation, which is comparable to the λ-dynamics based approaches. We also showed that the EDS-HREX method can be easily extended to multiple protonation state cases with the titration of histidine, KAAE peptide, and snake cardiotoxin. Due to the generality of the EDS-HREX method, it can be applied to free energy calculations in various problems that require frequent transitions between multiple states separated by large energy barriers.
Acknowledgments
This research was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute, National Institutes of Health. A.D. was partially supported by NIH Grant RO1 GM073838 to Bertrand Garcia-Moreno. The authors thank Gerhard König for helpful discussions.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Yang A.; Honig B. J. Mol. Biol. 1993, 231, 459. [DOI] [PubMed] [Google Scholar]
- Muñoz V.; Serrano L. J. Mol. Biol. 1995, 245, 297. [DOI] [PubMed] [Google Scholar]
- Doster W.; Beece D.; Bowne S. F.; DiIorio E. E.; Eisenstein L.; Frauenfelder H.; Reinisch L.; Shyamsunder E.; Winterhalter K. H.; Yue K. T. Biochemistry 1982, 21, 4831. [DOI] [PubMed] [Google Scholar]
- Tan A.; Young A. D.; Noble R. J. Biol. Chem. 1972, 247, 2493. [PubMed] [Google Scholar]
- Hünenberger P. H.; Helms V.; Narayana N.; Taylor S. S.; McCammon J. A. Biochemistry 1999, 38, 2358. [DOI] [PubMed] [Google Scholar]
- Davies R.; Neuberger A.; Wilson B. Biochim. Biophys. Acta, Enzymol. 1969, 178, 294. [DOI] [PubMed] [Google Scholar]
- Thomas P.; Russell A.; Fersht A. Nature 1985, 318, 375. [Google Scholar]
- Anderson D. E.; Becktel W. J.; Dahlquist F. W. Biochemistry 1990, 29, 2403. [DOI] [PubMed] [Google Scholar]
- Kelly J. W. Curr. Opin. Struct. Biol. 1996, 6, 11. [DOI] [PubMed] [Google Scholar]
- Schlesinger P. H.; Gross a.; Yin X. M.; Yamamoto K.; Saito M.; Waksman G.; Korsmeyer S. J. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuello L. G.; Romero J. G.; Cortes D. M.; Perozo E. Biochemistry 1998, 37, 3229. [DOI] [PubMed] [Google Scholar]
- Rastogi V. K.; Girvin M. E. Nature 1999, 402, 263. [DOI] [PubMed] [Google Scholar]
- Schnell J. R.; Chou J. J. Nature 2008, 451, 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seksek O.; Bolard J. J. Cell Sci. 1996, 109, 257. [DOI] [PubMed] [Google Scholar]
- Llopis J.; McCaffery J. M.; Miyawaki a.; Farquhar M. G.; Tsien R. Y. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Moreno E B. J. Biol. 2009, 8, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom D. G.; Castañeda C. A.; Cannon B. R.; Velu P. D.; García-Moreno E B. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 16096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom D. G.; Castañeda C. A.; Cannon B. R.; García-Moreno E B. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.; Lee J.; Seok C. Phys. Chem. Chem. Phys. 2013, 15, 5844. [DOI] [PubMed] [Google Scholar]
- Lanyi J. K. Annu. Rev. Physiol. 2004, 66, 665. [DOI] [PubMed] [Google Scholar]
- Yoshikawa S.; Shinzawa-Itoh K.; Nakashima R.; Yaono R.; Yamashita E.; Inoue N.; Yao M.; Fei M. J.; Libeu C. P.; Mizushima T.; Yamaguchi H.; Tomizaki T.; Tsukihara T. Science 1998, 280, 1723. [DOI] [PubMed] [Google Scholar]
- Hoff W. D.; Xie A.; Van Stokkum I. H. M.; Tang X.-J.; Gural J.; Kroon A. R.; Hellingwerf K. J. Biochemistry 1999, 38, 1009. [DOI] [PubMed] [Google Scholar]
- Tanford C.; Kirkwood J. J. Am. Chem. Soc. 1957, 79, 5333. [Google Scholar]
- Gilson M.; Honig B. Nature 1987, 330, 84. [DOI] [PubMed] [Google Scholar]
- Alexov E.; Mehler E. L.; Baker N.; Baptista A. M.; Huang Y.; Milletti F.; Nielsen J. E.; Farrell D.; Carstensen T.; Olsson M. H. M.; Shen J. K.; Warwicker J.; Williams S.; Word J. M. Proteins: Struct., Funct., Bioinf. 2011, 79, 3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warwicker J. Proteins: Struct., Funct., Bioinf. 2011, 79, 3374. [DOI] [PubMed] [Google Scholar]
- Damjanović A.; Wu X.; García-Moreno E B.; Brooks B. R. Biophys. J. 2008, 95, 4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damjanović A.; Brooks B. R.; García-Moreno E B. J. Phys. Chem. A 2011, 115, 4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu R. E.; Alexov E. G.; Gunner M. R. Biophys. J. 2002, 83, 1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch C. A.; Karp D. A.; Lee K. K.; Stites W. E.; Lattman E. E.; Garca-Moreno E B. Biophys. J. 2002, 82, 3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov V. P.; Schlessman J. L.; García-Moreno E B.; Halle B. Biophys. J. 2004, 87, 3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damjanović A.; García-Moreno E B.; Lattman E. E.; García A. E. Proteins: Struct., Funct., Bioinf. 2005, 60, 433. [DOI] [PubMed] [Google Scholar]
- Sagnella D. E.; Tuckerman M. E. J. Chem. Phys. 1998, 108, 2073. [Google Scholar]
- Kamerlin S. C. L.; Warshel A. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 30. [Google Scholar]
- Lill M. A.; Helms V. J. Chem. Phys. 2001, 115, 7993. [Google Scholar]
- Voth G. A. Acc. Chem. Res. 2006, 39, 143. [DOI] [PubMed] [Google Scholar]
- Riccardi D.; Schaefer P.; Yang Y.; Yu H.; Ghosh N.; Prat-Resina X.; König P.; Li G.; Xu D.; Guo H.; Elstner M.; Cui Q. J. Phys. Chem. B 2006, 110, 6458. [DOI] [PubMed] [Google Scholar]
- Jensen J. H.; Li H.; Robertson A. D.; Molina P. A. J. Phys. Chem. A 2005, 109, 6634. [DOI] [PubMed] [Google Scholar]
- Merz K. M. J. Am. Chem. Soc. 1991, 113, 3572. [Google Scholar]
- Matthew J. B. Annu. Rev. Biophys. Biophys. Chem. 1985, 14, 387. [DOI] [PubMed] [Google Scholar]
- Dlugosz M.; Antosiewicz J.; Robertson A. Phys. Rev. E 2004, 69, 021915. [DOI] [PubMed] [Google Scholar]
- Dlugosz M.; Antosiewicz J. M. Chem. Phys. 2004, 302, 161. [Google Scholar]
- Schaefer M.; Karplus M. J. Phys. Chem. 1996, 1578. [Google Scholar]
- Mongan J.; Case D. A.; McCammon J. A. J. Comput. Chem. 2004, 25, 2038. [DOI] [PubMed] [Google Scholar]
- Gennett T.; Milner D.; Weaver M. J. Phys. Chem. 1985, 89, 2787. [Google Scholar]
- Grunwald E.; Steel C. J. Am. Chem. Soc. 1995, 117, 5687. [Google Scholar]
- Baptista A. M.; Teixeira V. H.; Soares C. M. J. Chem. Phys. 2002, 117, 4184. [Google Scholar]
- Machuqueiro M.; Baptista A. M. J. Phys. Chem. B 2006, 110, 2927. [DOI] [PubMed] [Google Scholar]
- Bürgi R.; Kollman P. A.; Van Gunsteren W. F. Proteins: Struct., Funct., Bioinf. 2002, 47, 469. [DOI] [PubMed] [Google Scholar]
- Stern H. A. J. Chem. Phys. 2007, 126, 164112. [DOI] [PubMed] [Google Scholar]
- Mertz J. E.; Pettitt B. M. Int. J. High. Perform. C 1994, 8, 47. [Google Scholar]
- Baptista A. M.; Martel P. J.; Petersen S. B. Proteins: Struct., Funct., Bioinf. 1997, 27, 523. [PubMed] [Google Scholar]
- Börjesson U.; Hünenberger P. H. J. Chem. Phys. 2001, 114, 9706. [Google Scholar]
- Lee M. S.; Salsbury F. R.; Brooks C. L. Proteins: Struct., Funct., Bioinf. 2004, 56, 738. [DOI] [PubMed] [Google Scholar]
- Khandogin J.; Brooks C. L. Biophys. J. 2005, 89, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X.; Brooks C. L. J. Chem. Phys. 1996, 105, 2414. [Google Scholar]
- Donnini S.; Tegeler F.; Groenhof G.; Grubmüller H. J. Chem. Theory Comput. 2011, 7, 1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh G. B.; Knight J. L.; Brooks C. L. J. Chem. Theory Comput. 2012, 8, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh G. B.; Knight J. L.; Brooks C. L. J. Chem. Theory Comput. 2013, 9, 935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandogin J.; Brooks C. L. Biochemistry 2006, 45, 9363. [DOI] [PubMed] [Google Scholar]
- Meng Y.; Roitberg A. E. J. Chem. Theory Comput. 2010, 6, 1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S. L.; de Oliveira C. A. F.; McCammon J. A. J. Chem. Theory Comput. 2010, 6, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh S. G.; Damjanović A.; Brooks B. R. Proteins: Struct., Funct., Bioinf. 2011, 79, 3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace J. A.; Shen J. K. J. Chem. Theory Comput. 2011, 7, 2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabri Dashti D.; Meng Y.; Roitberg A. E. J. Phys. Chem. B 2012, 116, 8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swails J. M.; York D. M.; Roitberg A. E. J. Chem. Theory Comput. 2014, 10, 1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ C. D.; van Gunsteren W. F. J. Chem. Phys. 2007, 126, 184110. [DOI] [PubMed] [Google Scholar]
- Christ C. D.; van Gunsteren W. F. J. Chem. Phys. 2008, 128, 174112. [DOI] [PubMed] [Google Scholar]
- Sugita Y.; Kitao A.; Okamoto Y. J. Chem. Phys. 2000, 113, 6042. [Google Scholar]
- Fukunishi H.; Watanabe O.; Takada S. J. Chem. Phys. 2002, 116, 9058. [Google Scholar]
- Valleau J.; Card D. J. Chem. Phys. 1972, 57, 5457. [Google Scholar]
- Bennett C. H. J. Comput. Phys. 1976, 22, 245. [Google Scholar]
- Han K.-K. Phys. Lett. A 1992, 165, 28. [Google Scholar]
- Best R. B.; Chen Y.-G.; Hummer G. Structure 2005, 13, 1755. [DOI] [PubMed] [Google Scholar]
- Laghaei R.; Mousseau N.; Wei G. J. Phys. Chem. B 2011, 115, 3146. [DOI] [PubMed] [Google Scholar]
- Laghaei R.; Mousseau N. J. Phys. Chem. B 2010, 7071. [DOI] [PubMed] [Google Scholar]
- Wang L.; Friesner R. A.; Berne B. J. J. Phys. Chem. B 2011, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Kim B.; Friesner R. a.; Berne B. J. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 13749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan S.; Zacharias M. Proteins: Struct., Funct., Bioinf. 2009, 76, 448. [DOI] [PubMed] [Google Scholar]
- Curuksu J.; Zacharias M. J. Chem. Phys. 2009, 130, 104110. [DOI] [PubMed] [Google Scholar]
- Zwanzig R. W. J. Chem. Phys. 1954, 22, 1420. [Google Scholar]
- Christ C. D.; van Gunsteren W. F. J. Chem. Theory Comput. 2009, 5, 276. [DOI] [PubMed] [Google Scholar]
- Bashford D.; Gerwert K. J. Mol. Biol. 1992, 224, 473. [DOI] [PubMed] [Google Scholar]
- Nozaki Y.; Tanford C. Methods Enzymol. 1967, 69, 715. [Google Scholar]
- Kyte J.Structure in Protein Chemistry; Structure in Protein Chemistry Series; Garland Publishing, Inc.: New York, 1995. [Google Scholar]
- Singhal A. K.; Chien K. Y.; Wu W. G.; Rule G. S. Biochemistry 1993, 32, 8036–44. [DOI] [PubMed] [Google Scholar]
- MacKerell A.; Bashford D. J. Phys. Chem. B 1998, 5647, 3586. [DOI] [PubMed] [Google Scholar]
- Miller B. T.; Singh R. P.; Klauda J. B.; Hodoscek M.; Brooks B. R.; Woodcock H. L. J. Chem. Inf. Model 2008, 48, 1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L.; Mackerell A. D.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; Caflisch A.; Caves L.; Cui Q.; Dinner A. R.; Feig M.; Fischer S.; Gao J.; Hodoscek M.; Im W.; Kuczera K.; Lazaridis T.; Ma J.; Ovchinnikov V.; Paci E.; Pastor R. W.; Post C. B.; Pu J. Z.; Schaefer M.; Tidor B.; Venable R. M.; Woodcock H. L.; Wu X.; Yang W.; York D. M.; Karplus M. J. Comput. Chem. 2009, 30, 1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König G.; Miller B. T.; Boresch S.; Wu X.; Brooks B. R. J. Chem. Theory Comput. 2012, 8, 3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock H. L.; Miller B. T.; Hodoscek M.; Okur A.; Larkin J. D.; Ponder J. W.; Brooks B. R. J. Chem. Theory Comput. 2011, 7, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W.; Hodoscek M.; Roux B. J. Chem. Theory Comput. 2009, 5, 2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nose S. J. Chem. Phys. 1984, 81, 511. [Google Scholar]
- Hoover W. Phys. Rev. A 1985, 31, 1695. [DOI] [PubMed] [Google Scholar]
- Chu J.-W.; Trout B. L.; Brooks B. R. J. Chem. Phys. 2003, 119, 12708. [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. J. Mol. Graph. 1996, 14, 33. [DOI] [PubMed] [Google Scholar]
- Chiang C. M.; Chien K. Y.; Lin H. J.; Lin J. F.; Yeh H. C.; Ho P. L.; Wu W. G. Biochemistry 1996, 35, 9167. [DOI] [PubMed] [Google Scholar]
- Chiang C. M.; Chang S. L.; Lin H. J.; Wu W. G. Biochemistry 1996, 35, 9177. [DOI] [PubMed] [Google Scholar]
- Wu X.; Brooks B. R. Chem. Phys. Lett. 2003, 381, 512. [Google Scholar]
- Wu X.; Brooks B. R. J. Chem. Phys. 2011, 135, 204101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L.; Chen M.; Yang W. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 20227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes P. E. M.; Roux B.; Mackerell A. D. Theor. Chem. Acc. 2009, 124, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponder J. W.; Wu C.; Ren P.; Pande V. S.; Chodera J. D.; Schnieders M. J.; Haque I.; Mobley D. L.; Lambrecht D. S.; DiStasio R. a.; Head-Gordon M.; Clark G. N. I.; Johnson M. E.; Head-Gordon T. J. Phys. Chem. B 2010, 114, 2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König G.; Hudson P. S.; Boresch S.; Woodcock H. L. J. Chem. Theory Comput. 2014, 10, 1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König G.; Pickard F. C.; Mei Y.; Brooks B. R. J. Comput.-Aided Mol. Des. 2014, 28, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace J. A.; Shen J. K. J. Chem. Phys. 2012, 137, 184105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radmer R. J.; Kollman P. A. J. Comput. Chem. 1997, 18, 902. [Google Scholar]
- Lu N.; Kofke D. A. J. Chem. Phys. 2001, 114, 7303. [Google Scholar]
- Chipot C.; Pohorille A.. Free Energy Calculations: Theory and Applications in Chemistry and Biology; Springer Series in Chemical Physics; Springer: Dordrecht, 2007. [Google Scholar]