

Abstract

Sedoheptulose 7-phosphate cyclases (SH7PCs) encompass three enzymes involved in producing the core cyclitol structures of pseudoglycosides and similar bioactive natural products. One such enzyme is ValA from Streptomyces hygroscopicus subsp. jinggangensis 5008, which makes 2-epi-5-epi-valiolone as part of the biosynthesis of the agricultural antifungal agent validamycin A. We present, as the first SH7PC structure, the 2.1 Å resolution crystal structure of ValA in complex with NAD+ and Zn2+ cofactors. ValA has a fold and active site organization resembling those of the sugar phosphate cyclase dehydroquinate synthase (DHQS) and contains two notable, previously unrecognized interactions between NAD+ and Asp side chains conserved in all sugar phosphate cyclases that may influence catalysis. Because the domains of ValA adopt a nearly closed conformation even though no sugar substrate is present, comparisons with a ligand-bound DHQS provide a model for aspects of substrate binding. One striking active site difference is a loop that adopts a distinct conformation as a result of an Asp → Asn change with respect to DHQS and alters the identity and orientation of a key Arg residue. This and other active site differences in ValA are mostly localized to areas where the ValA substrate differs from that of DHQS. Sequence comparisons with a second SH7PC making a product with distinct stereochemistry lead us to postulate that the product stereochemistry of a given SH7PC is not the result of events taking place during catalysis but is accomplished by selective binding of either the α or β pyranose anomer of the substrate.
Natural products have served as a major source of pharmaceuticals and bioactive molecules for centuries and continue to play key roles in guiding the development of new therapeutics today. Among these are pseudooligosaccharides,1 such as the antidiabetic drug acarbose, the crop protectant validamycin A, the antitumor agent cetoniacytone A, and the sunscreen mycosporin-like amino acids that have similar core cyclitol structures (Figure 1A). The core cyclitols of these molecules are generated from the pentose phosphate pathway intermediate sedoheptulose 7-phosphate (SH7P) by one of three enzymes present in some bacteria and fungi that allow SH7P to be used in secondary metabolism. The enzymes 2-epi-5-epi-valiolone synthase (EEVS), 2-epi-valiolone synthase (EVS), and desmethyl-4-deoxygadosol synthase (DDGS) each catalyze the cyclization of SH7P to a distinct C7-cyclitol product (Figure 1B). These enzymes, the first of which was identified ∼15 years ago2,3 are known as SH7P cyclases (SH7PCs) and are a part of the sugar phosphate cyclase (SPC) family of enzymes, all of which require NAD+ and a metal ion, either cobalt or zinc, as prosthetic groups.4−7
Figure 1.

Reactions catalyzed by known sugar phosphate cyclases. (A) Four cyclitol-containing natural products are shown and labeled by name, with their C7-cyclitol units made by SH7PCs highlighted in bold. (B) The substrates (above) and products (below) of five sugar phosphate cyclases are shown. A divalent metal cation is drawn next to the two substrate hydroxyls seen (for DHQS and DOIS) to coordinate it. SH7PCs and DHQS may utilize Zn2+ or Co2+ as the divalent metal cation6,40 but are shown here with Zn2+, the metal present in this structure of ValA and in structures of AnDHQS. DOIS uses only Co2+ for catalysis41 and is depicted as such. Abbreviations for the substrates and products are introduced, with each enzyme abbreviation being that of its product followed by an additional S for “synthase”. Because of resonance, DDG has an internal symmetry so the stereoconfiguration at C5 of the product after it is released into solution is not uniquely defined. We draw it here with the same stereochemistry as EEV, anticipating the proposal we make in this work that the cyclization products of EEVS and DDGS have the same C5 configuration.
The SH7PCs are structurally uncharacterized, and our current understanding of their enzyme mechanisms is based mostly on studies of two other sugar phosphate cyclases: dehydroquinate synthase (DHQS) and 2-deoxy-scyllo-inosose synthase (DOIS). DHQS acts on the substrate 3-deoxy-d-arabinoheptulosonate 7-phosphate [DAHP (Figure 1B)] as part of aromatic amino acid biosynthesis and has been well-studied because it is a target for antimicrobial drug development.4 Numerous structures of DHQS from bacteria, fungi, and plants have been determined under a variety of conditions, including in the presence and absence of carbaphosphonate (CBP), a substrate analogue inhibitor, and in the presence or absence of NAD+.4,8−12 DOIS acts on the substrate glucose 6-phosphate [G6P (Figure 1B)] and is not as well studied, but structures of it have been determined in the presence and absence of carbaglucose 6-phosphate (CG6P), a mechanism-based inhibitor.5
Both DHQS and DOIS form homodimers. Each individual subunit is composed of two domains connected by a hinge, and in DHQS, the domains move closer together upon substrate binding. The two domains are an N-terminal NAD+-binding α/β-sandwich and a C-terminal metal-binding α-helical domain. These same two domains are found in class III metal-dependent polyol dehydrogenases (polyol-DHs), which use NAD(P)+ and a bivalent metal to conduct chemistry that is distinct but related to that of SPCs.13 Altogether, these enzymes form what is known as the DHQS-like superfamily.14 In descriptions of SPC structure, the NAD+-binding domain has been identified as a Rossmann fold,4,5,15 but it has been noted that this assignment is not certain.13
Extensive studies4,16 have established that in converting DAHP to dehydroquinate (DHQ), the DHQS active site coordinates the substrate at its active site metal via two hydroxyls (Figure 1B) and then catalyzes a remarkable five reactions: alcohol oxidation by NAD+, phosphate β-elimination, carbonyl reduction by the earlier formed NADH, ring opening, and intramolecular aldol condensation. As is common among homologous enzyme pairs with distinct but related chemistry, the first step of the reaction in DHQS and the polyol-DH enzymes, NAD+-promoted oxidation of an alcohol, is conserved.17 On the basis of its crystal structures with and without substrate analogues, DOIS is proposed to have a mechanism similar to that of DHQS,5 and analogous mechanisms involving the same five steps have been proposed for EEVS, EVS, and DDGS.6 On the basis of comparisons of SH7PC sequences with those of DHQS and DOIS, 14 putative active site residues were identified in EEVS, EVS, and DDGS sequences that were mostly identical but showed characteristic variations in each of the three SH7PC types.6 These patterns of variation, however, provided no insight into how the active sites produce distinct products from the same substrate, especially regarding the differing stereochemistry at the C2 position of the EEVS and EVS products (Figure 1B); this remains a major unanswered question. Developing a better understanding of the structure and function of SH7PCs will provide a foundation for their use in generating new bioactive compounds through synthetic biology and semisynthetic production.18
Here, we present a crystal structure of ValA, the EEVS from Streptomyces hygroscopicus 5008, that is involved in the biosynthetic pathway of the agricultural antifungal agent validamycin A.3 This first structure of a SH7PC fortuitously includes tightly bound Zn2+ and NAD+ cofactors and provides an informative view of the residues lining the active site. We combine sequence comparisons with the various SH7PC sequences and structural comparisons with DHQS and DOIS substrate analogue complexes and develop an unexpected hypothesis for how these different SH7PCs can use the same substrate to generate different products.
Materials and Methods
Expression, Purification, and Crystallization
Recombinant ValA was expressed as previously described.6 For purification, at 4 °C, cell pellets from 100 mL cultures were each resuspended in ∼5 mL of 40 mM HEPES and 300 mM NaCl (pH 8.0) (buffer A) with 10 mM imidazole, sonicated (13 W, 4 × 1 min), and centrifuged (14500 rpm for 30 min). The supernatant was loaded onto a Ni-NTA resin column (5 mL of resin, 0.8 mL/min). After being washed with 100 mL of buffer A with 20 mM imidazole, the protein was eluted using a 200 mL gradient from 20 to 500 mM imidazole in buffer A. Fractions (∼6 mL each) containing protein were combined and dialyzed overnight against 2 L of 10 mM Tris-HCl, 300 mM NaCl, and 5 mM imidazole (pH 8.0). A second phase of purification was conducted similarly using a TALON column (∼40 mL run at a rate of 0.3 mL/min) in buffer B [20 mM Tris-HCl and 300 mM NaCl (pH 8.0)] with 5 mM imidazole for column equilibration, 10 mM imidazole for washing, and a 200 mL gradient from 10 to 200 mM imidazole for elution. Fractions (∼4 mL each) containing pure ValA as judged by sodium dodecyl sulfate gel electrophoresis were combined and dialyzed against 2 L of 10 mM Tris-HCl (pH 7.5) (3 × 3 h). The protein solution was concentrated by ultrafiltration (10K cutoff membrane) to 10 mg/mL, flash-frozen in liquid nitrogen, and stored at −80 °C.
The enzyme was crystallized at 4 °C in hanging drops formed from 4 μL of the protein stock and 1 μL of a 0.6 M succinic acid reservoir solution (pH 6.5). The resulting crystals were rodlike with dimensions of ∼50 μm × 50 μm × 200 μm.
X-ray Diffraction Data Collection
For diffraction data collection (at −170 °C), crystals were briefly passed through a solution containing 20% PEG 400 and then flash-frozen in loops by being plunged into liquid nitrogen. Data were collected from two crystals using λ = 1.0 Å X-rays and Δφ = 1° steps at beamline 5.0.2 at the Advanced Light Source (Berkeley, CA). From both crystals, 120 2.0 s images were collected at a detector distance (d) of 250 mm, and from the second, an additional 200 2.0 s images were collected at a d of 350 mm. All these images were integrated using Mosflm19 and merged using the CCP4 suite of programs20,21 to obtain the data set used for structure solution and refinement. The merged data set was usable out to 2.1 Å using a CC1/2 of ∼0.2 as the cutoff criterion (Table 1), and a random 5% of reflections were marked for cross-validation. In addition, a third crystal was used for a fluorescence scan and to collect a data set at beamline 5.0.2 using λ = 1.282 Å X-rays to maximize the anomalous signal from the bound zinc. This data set included two sets of 60 Δφ = 1°, 4.0 s images offset by Δφ = 90° to collect the bijovet pairs and yielded data useful to 3.5 Å resolution (data not shown).
Table 1. Data Collection and Refinement Statisticsa.
| (A) Data | |
| resolution limits (Å) | 66.9–2.10 (2.21–2.10) |
| no. of unique observations | 20232 (2875) |
| multiplicity | 26.8 (19.3) |
| completeness (%) | 99.4 (98.9) |
| average I/σ | 11.2 (0.9) |
| Rmeas (%) | 38 (676) |
| CC1/2 (%) | 0.99 (0.22) |
| (B) Refinement | |
| no. of residues | 360 |
| no. of solvent atoms | 188 |
| total no. of atoms | 3004 |
| ⟨B⟩ for protein (Å2) | 48 |
| ⟨B⟩ for solvent (Å2) | 56 |
| Rcryst (%) | 17.9 (28.1) |
| Rfree (%) | 26.2 (34.7) |
| rmsd for bonds (Å) | 0.010 |
| rmsd for angles (deg) | 1.28 |
Numbers in parentheses correspond to values in the highest-resolution bin.
Structure Determination
The phase problem was initially solved by molecular replacement using MR-Rosetta with default settings.22 As search models, we tried both chain A of Vibrio cholerae DHQS [Protein Data Bank (PDB) entry 3OKF] and chain A of Helicobacter pylori DHQS (PDB entry 3CLH) that were the known structures that a BLAST search of the PDB showed as having sequences most similar to that of ValA (33 and 29% identical, respectively). V. cholerae DHQS did not yield a solution, but H. pylori DHQS gave a result with R and Rfree values of 0.25 and 0.31, respectively, at 2.1 Å resolution and 327 residues built. The electron density map from this solution allowed us to build almost all the side chains, the active site Zn2+, and the NAD+ prosthetic group. In contrast, conventional molecular replacement approaches yielded models with Rfree values near 50% and maps that were very difficult to interpret (data not shown). All manual model building was conducted in Coot.23 Refinements at various stages were conducted using Phenix24 or Buster25 with TLS refinement, with the final rounds being conducted using Phenix. Water molecules were manually placed on the basis of typical criteria: electron density of ≥3ρrms in Fo – Fc maps and ≥0.8ρrms in 2Fo – Fc maps and a reasonable potential H-bond partner. Three regions at or near crystallographic 2-fold axes were challenging to interpret. The first was a five-residue stretch, residues 46–50, that was near and crossing over a crystallographic 2-fold axis that was a nonphysiological crystal packing interaction. Into this weak helix-like main chain electron density we eventually modeled a portion, residues 48–50 and the side chain of Gln41, at 50% occupancy sharing the space with the same segments from its symmetry mate. As the program would not ignore contact of the side chain of Gln41 with itself, this side chain position was not allowed to move in the final refinement calculations. We conclude that the segment of only one of the monomers is ordered at a time and that in solution this part of the protein would be fairly dynamic. The second challenging region centered on a few residues N-terminal to residue 26. These residues had some positive density, but the electron density extended across the same crystallographic 2-fold axis noted above and then weakened, and we left this small section of density uninterpreted. The third challenging region was a β-hairpin turn (residues 32 and 33) located at the 2-fold axis generating the expected physiological dimer interaction. This turn showed weak electron density, while the associated β-strands showed strong and clear density. To follow the path of the electron density with a single conformation, we modeled the turn with an unfavorable cis-peptide bond before Lys32. This model also has a very short nonbonded collision (<2.5 Å) with its own symmetry mate, so we suspect that it does not represent a true conformation but only approximates the average chain path associated with a set of multiple conformations that allow reasonable packing at the interface. The final R and Rfree values were 0.179 and 0.262, respectively, with reasonable geometry (Table 1).
Structural Comparisons and Analyses
Secondary structure assignments were made using DSSP,26,27 and structure-based sequence alignments were generated using the Dali server.28
Results and Discussion
Overall Structure
The structure of recombinant ValA from S. hygroscopicus 5008 presented a challenging molecular replacement problem, as the most similar known structures were only ∼30% identical in sequence. The structure determination was greatly facilitated by the MR-Rosetta algorithm,22 which yielded solutions of a quality much higher than the quality of conventional molecular replacement (see Materials and Methods). In addition to being aided by MR-Rosetta, the quality of the solution was also enhanced by the inclusion of weak high-resolution data that would have been discarded on the basis of conventional high-resolution cutoff criteria. For the data set used here, the conventional high-resolution cutoff criterion of an Rmeas of ∼60% or an ⟨I/σ⟩ of ∼2 would lead to a limit of 2.85 or 2.3 Å, respectively, whereas the more generous criterion (CC1/2 of ∼0.2), shown in recent work to produce better refined models,29−31 leads to a limit of 2.1 Å (Table 1). To test how the inclusion of weak high-resolution data impacted the molecular replacement calculations, we conducted MR-Rosetta runs using these three justifiable resolution cutoffs. On the basis of Rfree values, using the 2.1 Å resolution cutoff yielded the best solution, with the 2.3 and 2.85 Å cutoffs being slightly worse and much worse, respectively (Table 2). This example thus shows that weak high-resolution data (out to CC1/2 ∼ 0.2 and ⟨I/σ⟩ ∼ 0.9 in this case) can help with challenging molecular replacement solutions as well as produce better refined models.
Table 2. Resolution Dependence of MR-Rosetta Results.
| resolution (Å) | R | Rfree | no. of residues builta |
|---|---|---|---|
| 2.85 | 0.30 | 0.40 | 306 (221) |
| 2.30 | 0.26 | 0.33 | 334 (318) |
| 2.10 | 0.25 | 0.31 | 327 (298) |
The total number of residues built in backbone segments and, in parentheses, the number of these modeled as specific residues in the sequence of the target structure.
Further refinement of the molecular replacement solution yielded a model for the one chain in the asymmetric unit with final R and Rfree values of 17.9 and 26.2%, respectively, to 2.1 Å resolution (Table 1). The large majority of the main chain as well as an active site NAD+ and Zn2+ are well ordered with strong and clear density, and an absorption scan and anomalous difference map clearly confirm the presence and placement of the active site Zn2+ (Figure 2). The final structure includes 360 of the 414 expected residues, 188 waters, one PEG, one Zn2+, and one NAD+. The missing residues (1–25, 46, 47, 58–62, 244–249, and 399–414) are not modeled because of weak or unclear electron density. Additionally, three sections, including the residues just N-terminal to residue 26, a β-hairpin turn at residues 32 and 33, and a weakly ordered helix at residues 46–50, laid on or near crystallographic 2-fold axes and had weak, ill-formed density, making them challenging to model (see Materials and Methods). A crystallographic 2-fold axis brings two ValA chains together to form a dimer that, according to the PISA server,32 buries 4220 Å2 of surface area (i.e., 2110 Å2 per monomer). This dimer (Figure 3A) is equivalent to those observed for the homologous enzymes DHQS and DOIS,4,5,11,12,16,33 and the dimer interface is well-conserved, implying that it is the physiological form of ValA.
Figure 2.
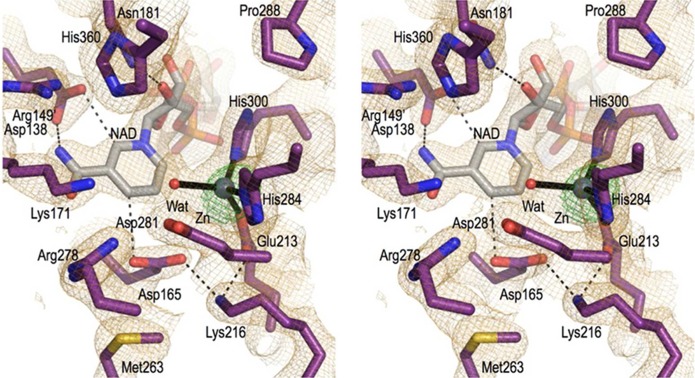
Electron density map quality and active site structure. Stereoview of the ValA active site residues (purple carbons) and a water (red sphere) that are near the NAD+ (gray carbons) and the Zn2+ (silver sphere) cofactors. Coordination bonds (black lines) and select H-bonds (black dashes) are shown along with the 2Fo – Fc electron density (orange, contoured at 1ρrms) and an anomalous difference map (green, contoured at 12ρrms).
Figure 3.
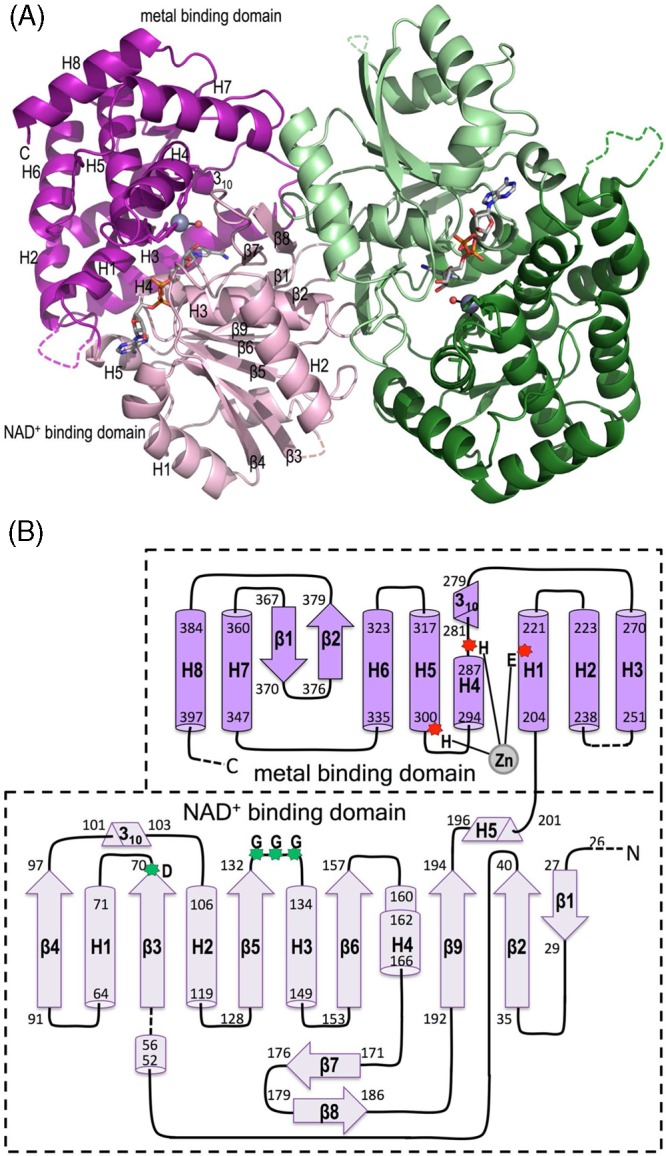
Overall structure and topology of ValA. (A) Ribbon diagrams of the two chains of the ValA dimer are shown in purple and green tones, respectively, with the N-terminal NAD+-binding domains in light hues and the C-terminal metal-binding domains in dark hues. Dashed lines indicate internal unmodeled backbone segments. The NAD+ and the Zn2+ with its coordinating ligands are shown (colored as in Figure 1). Secondary structural elements in each domain of one monomer are labeled. (B) Topology diagram showing α-helices (cylinders), β-stands (arrows), 310-helices (triangular prisms), and π-helices (wider cylinder) with their first and last residues given. The minimal length α- and 310-helices (five and three residues, respectively) are left out of the family secondary structure nomenclature. The domains are colored light and dark purple as indicated, and helices (H) and strands (β) common to the SPCs are named sequentially within each domain. Dashed lines denote unmodeled backbone segments. The three Zn2+-binding residues (red asterisks) and the glycine-rich turn and acidic residues (green asterisks) important for NAD+ binding are indicated.
Each chain of ValA encompasses the expected N-terminal NAD+-binding domain and C-terminal metal-binding domain common to the DHQS-like superfamily. We describe here the domain topologies (Figure 3B) using a secondary structure nomenclature that takes into account which elements are conserved among the SPCs (Figure 4). The NAD+-binding domain has a core seven-strand β-sheet (with a 1-2-9-6-5-3-4 strand order) surrounded by five α-helices, one β-hairpin (β7 and β8), and two short 310-helices. The metal-binding domain is mainly α-helical and includes eight α-helices, one 310-helix, and one β-hairpin. This domain contains not only the Zn2+-binding residues but also, on the basis of what has been seen in DHQS and DOIS, the majority of the residues involved in substrate recognition and so has also been called the substrate-binding domain.5 However, the sugar phosphate substrate actually binds in a cleft between the two domains, and its recognition involves residues from both domains.
Figure 4.

Sequence alignment of ValA with representative related enzymes. The sequence of ValA is listed first, and its secondary structure elements are schematically shown above the sequence. Other sequences in descending order are AvDDGS (A. variabilis DDGS, Ava_3858), AmEVS (Ac. mirum EVS, Amir_2000), PDB entry 1DQS (As. nidulans DHQS), PDB entry 2D2X (B. circulans DOIS), and PDB entry 1JQ5 (B. stearothermophilus glycerol dehydrogenase). For the structurally known proteins, the residues in β-strands (yellow), α-helices (teal), 310-helices (blue), and π-helices (orange) are highlighted. Residues involved in metal binding (m), NAD+ binding (n), and substrate binding and/or catalysis (∗) are denoted below the sequences, and active site residues with notable variation (↓) are denoted above the sequences.
Relationships to Other Structurally Known Proteins
A structural similarity search performed using the DALI server28 showed that ValA is most similar to assorted DHQSs (rmsd of ∼2.2–2.6 Å, Z scores of ∼35–45), followed by DOIS (rmsd of ∼2.2 Å, Z score of 34) and then various polyol-DHs (rmsd of ∼2.7–3.6 Å, Z scores of ∼23–28). Interestingly, although the structures used as search models in molecular replacement were the two PDB structures (PDB entries 3OKF and 3CLH) with sequences most similar to that of ValA, in terms of structural similarity these rank only eighth (Z score of 41) and twenty-first (Z score of 36), respectively. This underscores why it can be useful to try all potential homologues in molecular replacement rather than just the ones most similar in sequence.34 The DALI search further showed that there are no known protein structures outside of the DHQS-like superfamily that share noteworthy structural similarity to ValA or either of its individual domains.
On the basis of these results, representative enzymes were chosen for a structure-based sequence alignment (Figure 4): Aspergillus nidulans DHQS (AnDHQS, PDB entry 1DQS), the most well-studied DHQS;4Bacillus circulans DOIS (BcDOIS, PDB entry 2D2X), the only structurally known DOIS;5 and Bacillus stearothermophilus glycerol dehydrogenase (BsGlyDH, PDB entry 1JQ5), the most structurally similar member of the polyol-DH family.13 Representatives from the two structurally unknown types of the SH7PCs were also included in this sequence alignment: a DDGS from Anabaena variabilis (AvDDGS) and an EVS from Actinosynnema mirum (AmEVS). The structure-based alignment between ValA and DHQS is largely consistent with alignments that led to the previously proposed putative active site residues in ValA.6,7 The only change is that Lys356 in AnDHQS had been previously aligned with Pro370 in ValA, but the structure-based alignment identifies the equivalent residue as His360.
Zinc and NAD+ Binding
Although zinc and NAD+ were not added during sample preparation or crystallization, the electron density maps showed their unambiguous presence in the crystal structure (Figure 2), presumably meaning that they were bound by ValA already in the E. coli cytosol and carried along during the purification. The zinc and NAD+ are both bound in a manner quite similar to what has been described for DHQS4 and DOIS,5 so aside from some novel observations, we will here only briefly summarize the features of the binding. All residues in direct contact with zinc and NAD+ are designated in Figure 4, and most are conserved among the SPCs. The Zn2+ ion is coordinated by Glu213, His284, and His300, all from the C-terminal metal-binding domain, and a water. As inferred from the liganded structures of DHQS and DOIS, this active site water will be displaced upon substrate binding.
The binding of NAD+ includes characteristic residues conserved among the SPCs such as Asp70 at the end of strand β3 that H-bonds with the adenosine ribose O2′ hydroxyl, the glycine-rich turn connecting β5 and H3 providing backbone amides that H-bond to the pyrophosphate oxygens, and Glu101, Lys104, Lys180, and Asn181 that H-bond with the nicotinamide ribose hydroxyls. The nicotinamide amide nitrogen donates H-bonds to the Asp138 side chain and the Lys171 backbone oxygen, and the oxygen forms H-bonds with surrounding waters in this structure. A fascinating pair of interactions that is conserved in known DHQS-like superfamily structures but has not been described before places carboxylate oxygens from Asp138 and Asp165 each roughly in the plane of the nicotinamide ring where they are in position to accept weakly polar H-bonds from the nicotinamide C2 and C4 atoms, respectively (see Figure 2). We expect that these interactions will preferentially stabilize the positive charge on the oxidized form of the nicotinamide ring, which is distributed among ring carbon atoms 2, 4, and 6 via resonance forms. These interactions may help explain the high affinity of these enzymes for NAD+, and in particular, the interaction with the reactive nicotinamide C4 position may play a role in modulating the nicotinamide redox properties during the catalytic cycle.
ValA Sugar Phosphate-Binding Site
Although no substrate or substrate analogue is bound in this crystal form of ValA, we can still gain insight into its substrate binding by comparisons with the ligand-bound structures of AnDHQS and BcDOIS (henceforth termed DHQS and DOIS, respectively). For convenience, we refer to sequence differences between ValA and DHQS as mutations or changes with respect to DHQS, even though ValA did not evolve from a modern DHQS. As noted in the introductory section, DHQS undergoes a conformational change from “open” in the absence of a sugar phosphate ligand to “closed” upon binding the substrate analogue CBP via a domain rotation of ∼12° that brings the N- and C-terminal domains closer together.8 A recent structure of DHQS from Actinidia chinensis reinforces the relevance of the closed conformation seen, as the same closed conformation appears to be stabilized by the binding of inorganic phosphate and glycine in a way that mimics that of the substrate.35 DOIS, in contrast, was reported to not undergo such a domain closure based on comparisons of its structures with or without a substrate analogue.5
A set of overlays of ValA with representative liganded and unliganded forms of DHQS and DOIS show that our ValA structure has a conformation between the open and closed DHQS forms but much closer to the closed form, varying by only ∼2° (Figure 5). They further show that the unliganded and liganded DOIS structures do indeed have minimal differences in their domain orientations, but that the DOIS conformation is ∼10° more open in chain A and ∼7.5° more open in chain B than the DHQS closed structure (data not shown), suggesting that it might not accurately represent the ligand-bound structure. Also supporting this possibility is the fact that the DOIS-liganded structure was obtained by soaking crystals of the unliganded enzyme with inhibitor, during which some crystal cracking was observed.5 This implies that the enzyme could not undergo a complete domain closure without compromising the integrity of the crystal. For this reason, we focus in the following comparisons solely on the DHQS·CBP complex, which on the basis of the all the evidence accurately represents a true inhibitor-bound conformation.
Figure 5.

ValA overlay with closed and open DHQS structures. Shown are ribbon diagrams of an unliganded, open DHQS (blue, PDB entry 1NRX), a CBP-bound, closed DHQS (cyan, PDB entry 1DQS), and ValA (purple), all overlaid on the basis of their NAD+-binding domains. Pale hues are used for the NAD+-binding domains and NAD+ and dark hues for the metal-binding domains. For the sake of clarity, only select secondary structure elements of the metal-binding domain are shown along with the three zinc-ligating residues (sticks) and the zinc (gray sphere). The active site side chain that does not align well between DHQS (Arg264) and ValA (Arg277) is colored green for both structures, and the alternate equivalent ValA residue (Arg278) is colored orange. The fact that other open DHQS structures, such as those of TtDHQS (PDB entry 1UJN) and HpDHQS (PDB entry 3CLH), are up to 5° different in domain orientation compared with AnDHQS (PDB entry 1NRX) does not alter the conclusions of this analysis.
With only this unliganded structure of ValA available, we cannot make any claims about what ligand-induced conformational changes may occur. However, the similarity of ValA to the closed conformation of DHQS is fortunate as it means that the NAD+, the zinc, and nearly all of the ValA residues equivalent to DHQS active site residues align rather well (Figure 6), giving us confidence that this comparison provides an informative picture of which ValA residues will play a role in substrate binding. Of the ligand-binding residues in the DHQS complex, only one, Arg264, is not in the proximity of its corresponding residue in ValA. The equivalent residue in ValA is Arg277 (Figure 4), and it points in the opposite direction (see the green side chains in Figure 5). Interestingly, Arg277 is not conserved among EEVSs (data not shown), suggesting it is not a key residue for this enzyme. Even more interestingly, because of a different nearby loop conformation, the following residue in ValA, Arg278, has its side chain close to that of DHQS Arg264 (Figure 6) and is conserved among EEVSs, suggesting that it may be the functionally equivalent residue. An important question then becomes whether the different loop conformation is a robust difference between ValA and DHQS or whether it may be simply due to the ValA structure not having a ligand bound.
Figure 6.
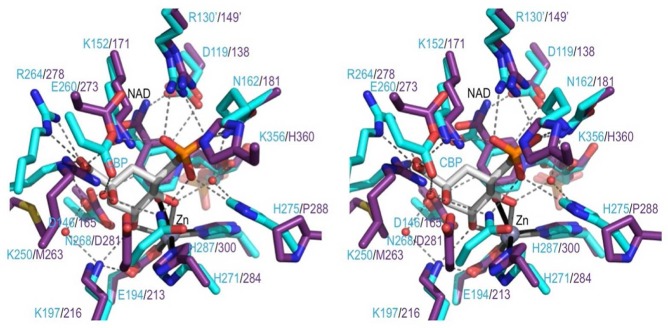
Comparing the ValA active site region with the DHQS·CBP complex. Stereoview of select active site residues in ValA (purple) overlaid on the DHQS (cyan) in complex with CBP (white) shown in roughly the same orientation as DAHP in Figure 1. H-Bonding interactions in the DHQS active site (dashed lines) and coordination bonds with Zn2+ (solid lines) are shown. A prime on a residue number means it is from the other subunit of the dimer.
A closer look at the loop (residues 257–264 in DHQS and residues 270–278 in ValA) identifies another key active site position and confirms that the difference in loops is robust (Figure 7). In both unliganded and liganded DHQS structures, the loop wraps around the side chain of Asp257 that accepts multiple backbone amide H-bonds to stabilize the conformation. The equivalent residue in ValA, Asn270, is not compatible with the DHQS loop conformation but plays an equally central role in stabilizing the alternate less compact loop path (Figure 7).
Figure 7.
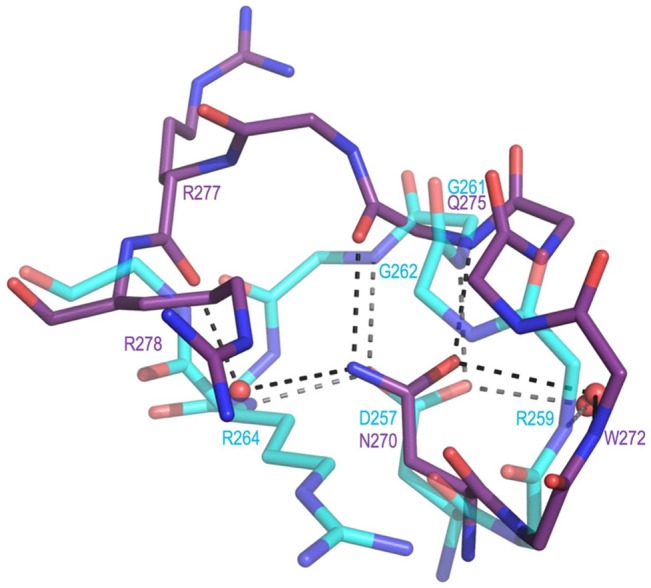
Active site loop difference that relates to the presence of Asp257 in DHQS vs Asn270 in ValA. Shown are residues 269–278 of ValA (purple) and residues 256–264 of DHQS (cyan, PDB entry 1DQS) after the proteins have been overlaid as in Figure 5. H-Bonding interactions (dashed lines) involving the loop residues and associated waters are shown. In DHQS, the Asp257 carboxylate receives H-bonds directly or indirectly (via water) from four backbone nitrogens (from Arg259, Gly261, Gly262, and Arg264). In ValA, the Asn270 side chain amide directly or indirectly makes H-bonds with two backbone nitrogens (from Trp272 and Glu275) and two backbone oxygens (from Gln275 and Arg277).
If ValA Arg278 is taken to be the equivalent of DHQS Arg264, Figure 6 compares the DHQS residues surrounding the substrate analogue CBP with their ValA equivalents. Among these, just four ValA residues are different types: Met263 replaces a Lys, Asp281 replaces an Asn, Pro288 replaces a His, and His360 replaces a Lys. Using atom numbering for the substrate (see Figure 1) rather than the CBP inhibitor, the essential features of binding of CBP to DHQS (clockwise from the top of Figure 6) are a phosphate-binding pocket (at two o’clock), bidentate metal coordination by the C5 and C4 hydroxyls (at three to six o’clock), which also serves to point the C5 hydrogen at the nicotinamide C4 atom in good geometry for hydride transfer, and then a pocket for the C2 hydroxyl and carboxylate groups (around ten o’clock). In ValA, the metal and nicotinamide are nearly identically positioned as are key residues interacting with the phosphate (Lys171, Arg149′, and Asn181) and the metal-coordinating hydroxyls (Asp165 and Lys216). We take this to mean that the analogous parts of the ValA substrate will be bound similarly to CBP in DHQS. Having a high degree of spatial conservation of these parts of the substrate makes sense, as they are where most of the chemistry takes place.
In contrast, the significantly shifted or mutated residues (Arg278, Met263, and Asp281) are present at the pocket around the C2 hydroxyl and carboxylate groups where the ValA substrate has different substituents. In particular, the Lys → Met change makes sense with the absence of the substrate carboxylate. The two remaining changes, involving ValA residues Pro288 and His360, create more space around the phosphate group, but we do not understand why that might be. The DHQS residue replaced by Pro288 is His275, which has been proposed to serve as an acid/base during catalysis.4
On the basis of the findings mentioned above, we conclude that ValA will bind its substrate, SH7P, with the phosphate group and the C5 and C4 metal-coordinating hydroxyls in positions similar to those in the DHQS·CBP complex. However, other aspects of the binding mode such as ring conformation and/or orientation must differ from those of CBP, especially because the configuration of the C4 hydroxyl in SH7P differs (see Figure 1B) such that it and the C5 hydroxyl cannot simultaneously be equatorial. This difference makes predicting details of the binding mode of SH7P more challenging.
Variations among the SH7PCs and a Proposal for How They Catalyze Different Reactions
As noted in the introductory section, a major open question about SH7PC enzymes is how they bind the same substrate and produce different products (Figure 1B), with the most conceptually confusing aspect being how EEVS produces one stereochemistry at position C5 of the product (derived from the substrate C2 atom, as shown in Figure 1B) while EVS produces the other.6 Because of an internal symmetry in the DDGS product, it could be produced with either C5 stereochemistry (Figure 1B). Interestingly, EEVS and DDGS are more similar to each other in sequence than they are to EVS, with both being reported to vary from DHQS in the identities of four putative active site residues while EVS varied in only one6,7 (Figure 8). Our structural results for ValA (an EEVS) strengthen this pattern, in that the Asn residue associated with its distinct loop conformation and the alternate active site Arg residue (Figure 7) are also both present in DDGSs (Figure 4). In contrast, EVS, like DHQS, has an Asp in the loop and conserves the first Arg (Figure 4). This implies that these two subsets of the SH7PCs (EEVS and DDGS vs EVS) have distinct binding environments for the substituents of the substrate C2 atom and leads us to hypothesize that the discrimination related to the stereochemistry at C5 of the product actually occurs upon substrate binding rather than during catalysis.
Figure 8.

Variation in active site residues among sugar phosphate cyclases. Schematic drawing of residues lining the substrate-binding pocket in the DHQS·CBP complex shown in roughly the same orientation as in Figure 6. Each DHQS residue shown is labeled (cyan), and under that label are listed the corresponding residues found in the structures of DOIS (pink) and EEVS (purple) or, on the basis of the alignment in Figure 4, residues expected to be equivalent in DDGS and EVS (black). The CBP ligand is shown in bold. H-Bond interactions (dashed lines) and coordination bonds with Zn2+ (solid lines) are shown. The residue numbering corresponds to the representative proteins used in Figure 4 (AnDHQS, BcDOIS, ValA, AvDDGS, and AmEVS).
Specifically, each enzyme would selectively bind either the α- or β-pyranose form of SH7P, and the rapid interconversion between the various pyranose and furanose forms would allow these normally less populated SH7P forms (∼16 and <1%, respectively36) to accumulate in the enzyme active sites (Figure 9A). In this way, rather than the variation in aldol acceptor geometry between EEVS and EVS requiring a 180° rotation of the polar C5 substituents in the middle of the catalytic cycle, each enzyme would start with a substrate preorganized to generate the correct configuration at that position (Figure 9B). As EEV has the configuration at C5 that would be derived from α-pyranose SH7P, this is the form of substrate that EEVS and DDGS would bind, and as EV has the same configuration at C5 as would be derived from β-pyranose SH7P, the EVS active site would select for that form of the substrate. This hypothesis is completely consistent with the behavior of DHQS and DOIS, which both bind their substrate analogues with a fixed configuration corresponding to that of their product. In the case of DOIS, the enzyme’s selectivity is directly observed in its preferential binding from a racemic mixture of the inhibitor CG6P only the form that mimics the β-anomer of the substrate.5,37
Figure 9.

Proposed enzyme-specific selection of forms of sedoheptulose 7-phosphate. (A) The pyranose and furanose α- and β-anomers of SH7P with their relative abundance as determined by NMR36 are shown along with the linear form via which they interconvert. Also shown is our proposal that EEVS and DDGS bind the α-pyranose anomer while EVS binds the β-pyranose anomer. (B) Ring opening and intramolecular aldol condensation steps of the proposed reaction mechanisms of EEVS and EVS, emphasizing how the α- and β-anomers of pyranose of SH7P are preorganized for the generation of the respective stereochemistries at C5 in the products. B represents an active site base that may aid ring opening.
In this way, rather than viewing the SH7PCs as a family of enzymes that use one substrate to specifically give two stereochemically distinct types of products, one can then view them as enzymes that bind distinct substrates, either α-pyranose or β-pyranose SH7P, to give products with stereochemistries matching the stereochemistries of those substrates. In terms of the evolutionary origin of the SH7PCs, because EVS sequences are more similar to those of DHQSs,6 we suggest that EVS evolved from DHQS with only slight changes to the active site being needed to allow binding of the β-pyranose anomer of SH7P. In contrast, the key differentiating step in EEVS evolution would have been the Asp → Asn mutation that changed which Arg pointed into the active site pocket, as this allowed binding of the α-pyranose anomer of SH7P leading to the production of EEV. Then further mutations, including two active site mutations of Asp → Ala and His → Thr (Figures 4 and 8), gave rise to the mechanistic difference that characterizes the DDGS enzymes. Although we do not understand this transition yet, the more minor nature of differences between the DDGSs and EEVSs is emphasized by phylogenetic trees showing that EEVS and DDGS are more closely related to each other than to the other SPCs.6
Although much remains to be learned about ValA and the SH7PCs in general, the ValA structure presented here sheds much light on this enzyme family. Especially seeing the spatial orientation of active site residues (Figure 8) has provided insight into how variation in the active site pocket allows for the different specificities found in the SPC superfamily. Nevertheless, the detailed roles of catalytic residues in the EEVS, DDGS, and EVS mechanisms still remain open questions. For example, the residue that takes the catalytic role of an essential histidine in DHQS (His275) and DOIS (His250) has not been identified in these enzymes. We expect that answering such questions for the SH7PCs will best be approached through kinetics and structural studies conducted with stereospecific carbacyclic phosphonate analogues of SH7P that do not yet exist but that are in concept similar to the DAHP analogues used in informative studies of DHQS.4,8,38,39 We are now initiating studies in this direction.
Acknowledgments
We thank Dale Tronrud and Camden Driggers for help with the crystallographic work and Murugesh Padmanarayan for growing and analyzing the first crystals.
Glossary
Abbreviations
- CG6P
carbaglucose 6-phosphate
- CBP
carbaphosphonate
- DAHP
3-deoxy-d-arabinoheptulosonate 7-phosphate
- DHQ(S)
dehydroquinate (synthase)
- DDG(S)
desmethyl-4-deoxygadusol (synthase)
- DOI(S)
2-deoxy-scyllo-inosose (synthase)
- EEV(S)
2-epi-5-epi-valiolone (synthase)
- EV(S)
2-epi-valiolone (synthase)
- G6P
glucose 6-phosphate
- SH7P
sedoheptulose 7-phosphate
- SH7PC
sedoheptulose 7-phosphate cyclase
- SPC
sugar phosphate cyclase.
Accession Codes
The coordinates and structure factors have been deposited as Protein Data Bank entry 4P53.
The authors declare no competing financial interest.
This work was supported in part by an Oregon State University Provost’s Distinguished Graduate Fellowship (to K.M.K.) and National Institute of Allergy and Infectious Diseases Grant R01AI061528 (to T.M.).
Funding Statement
National Institutes of Health, United States
References
- Mahmud T. (2003) The C7N aminocyclitol family of natural products. Nat. Prod. Rep. 20, 137–166. [DOI] [PubMed] [Google Scholar]
- Stratmann A.; Mahmud T.; Lee S.; Distler J.; Floss H. G.; Piepersberg W. (1999) The AcbC protein from Actinoplanes species is a C-7-cyclitol synthase related to 3-dehydroquinate synthases and is involved in the biosynthesis of the α-glucosidase inhibitor acarbose. J. Biol. Chem. 274, 10889–10896. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Bai L. Q.; Minagawa K.; Jian X. H.; Li L.; Li J. L.; Chen S. Y.; Cao E. H.; Mahmud T.; Floss H. G.; Zhou X. F.; Deng Z. X. (2005) Gene cluster responsible for validamycin biosynthesis in Streptomyces hygroscopicus subsp jinggangensis 5008. Appl. Environ. Microbiol. 71, 5066–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter E. P.; Hawkins A. R.; Frost J. W.; Brown K. A. (1998) Structure of dehydroquinate synthase reveals an active site capable of multistep catalysis. Nature 394, 299–302. [DOI] [PubMed] [Google Scholar]
- Nango E.; Kumasaka T.; Hirayama T.; Tanaka N.; Eguchi T. (2008) Structure of 2-deoxy-scyllo-inosose synthase, a key enzyme in the biosynthesis of 2-deoxystreptamine-containing aminoglycoside antibiotics, in complex with a mechanism-based inhibitor and NAD+. Proteins: Struct., Funct., Bioinf. 70, 517–527. [DOI] [PubMed] [Google Scholar]
- Asamizu S.; Xie P. F.; Brumsted C. J.; Platt P. M.; Mahmud T. (2012) Evolutionary Divergence of Sedoheptulose 7-Phosphate Cyclases Leads to Several Distinct Cyclic Products. J. Am. Chem. Soc. 134, 12219–12229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Flatt P. M.; Schlorke O.; Zeeck A.; Dairi T.; Mahmud T. (2007) A comparative analysis of the sugar phosphate cyclase superfamily involved in primary and secondary metabolism. ChemBioChem 8, 239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols C. E.; Ren J.; Lamb H. K.; Hawkins A. R.; Stammers D. K. (2003) Ligand-induced conformational changes and a mechanism for domain closure in Aspergillus nidulans dehydroquinate synthase. J. Mol. Biol. 327, 129–144. [DOI] [PubMed] [Google Scholar]
- Nichols C. E.; Hawkins A. R.; Stammers D. K. (2004) Structure of the ‘open’ form of Aspergillus nidulans 3-dehydroquinate synthase at 1.7 Å resolution from crystals grown following enzyme turnover. Acta Crystallogr. D60, 971–973. [DOI] [PubMed] [Google Scholar]
- Nichols C. E.; Ren J.; Leslie K.; Dhaliwal B.; Lockyer M.; Charles I.; Hawkins A. R.; Stammers D. K. (2004) Comparison of ligand-induced conformational changes and domain closure mechanisms, between prokaryotic and eukaryotic dehydroquinate synthases. J. Mol. Biol. 343, 533–546. [DOI] [PubMed] [Google Scholar]
- Sugahara M.; Nodake Y.; Sugahara M.; Kunishima N. (2005) Crystal structure of dehydroquinate synthase from Thermus thermophilus HB8 showing functional importance of the dimeric state. Proteins: Struct., Funct., Bioinf. 58, 249–252. [DOI] [PubMed] [Google Scholar]
- Liu J. S.; Cheng W. C.; Wang H. J.; Chen Y. C.; Wang W. C. (2008) Structure-based inhibitor discovery of Helicobacter pylori dehydroquinate synthase. Biochem. Biophys. Res. Commun. 373, 1–7. [DOI] [PubMed] [Google Scholar]
- Ruzheinikov S. N.; Burke J.; Sedelnikova S.; Baker P. J.; Taylor R.; Bullough P. A.; Muir N. M.; Gore M. G.; Rice D. W. (2001) Glycerol dehydrogenase. Structure, specificity, and mechanism of a family III polyol dehydrogenase. Structure 9, 789–802. [DOI] [PubMed] [Google Scholar]
- Murzin A. G.; Brenner S. E.; Hubbard T.; Chothia C. (1995) SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247, 536–540. [DOI] [PubMed] [Google Scholar]
- Sillitoe I.; Cuff A. L.; Dessailly B. H.; Dawson N. L.; Furnham N.; Lee D.; Lees J. G.; Lewis T. E.; Studer R. A.; Rentzsch R.; Yeats C.; Thornton J. M.; Orengo C. A. (2013) New functional families (FunFams) in CATH to improve the mapping of conserved functional sites to 3D structures. Nucleic Acids Res. 41, D490–D498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park A.; Lamb H. K.; Nichols C.; Moore J. D.; Brown K. A.; Cooper A.; Charles I. G.; Stammers D. K.; Hawkins A. R. (2004) Biophysical and kinetic analysis of wild-type and site-directed mutants of the isolated and native dehydroquinate synthase domain of the AROM protein. Protein Sci. 13, 2108–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett G. J.; Borkakoti N.; Thornton J. M. (2003) Catalysing new reactions during evolution: Economy of residues and mechanism. J. Mol. Biol. 331, 829–860. [DOI] [PubMed] [Google Scholar]
- Winter J. M.; Tang Y. (2012) Synthetic biological approaches to natural product biosynthesis. Curr. Opin. Biotechnol. 23, 736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battye T. G.; Kontogiannis L.; Johnson O.; Powell H. R.; Leslie A. G. (2011) iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D62, 72–82. [DOI] [PubMed] [Google Scholar]
- Terwilliger T. C.; Dimaio F.; Read R. J.; Baker D.; Bunkoczi G.; Adams P. D.; Grosse-Kunstleve R. W.; Afonine P. V.; Echols N. (2012) phenix.mr_rosetta: Molecular replacement and model rebuilding with Phenix and Rosetta. J. Struct. Funct. Genomics 13, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O. S., Vonrhein C., and Womack T. O. (2011) BUSTER, version 2.11.2, Global Phasing Ltd., Cambridge, U.K. [Google Scholar]
- Kabsch W.; Sander C. (1983) Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- Joosten R. P.; te Beek T. A.; Krieger E.; Hekkelman M. L.; Hooft R. W.; Schneider R.; Sander C.; Vriend G. (2011) A series of PDB related databases for everyday needs. Nucleic Acids Res. 39, D411–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L.; Rosenstrom P. (2010) Dali server: Conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus P. A.; Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kershaw N. J.; Murphy J. M.; Liau N. P.; Varghese L. N.; Laktyushin A.; Whitlock E. L.; Lucet I. S.; Nicola N. A.; Babon J. J. (2013) SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat. Struct. Mol. Biol. 20, 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins A.; Gretes M. C.; Nelson K. J.; Poole L. B.; Karplus P. A. (2012) Mapping the active site helix-to-strand conversion of CxxxxC peroxiredoxin Q enzymes. Biochemistry 51, 7638–7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Brown K. A.; Carpenter E. P.; Watson K. A.; Coggins J. R.; Hawkins A. R.; Koch M. H. J.; Svergun D. I. (2003) Twists and turns: A tale of two shikimate-pathway enzymes. Biochem. Soc. Trans. 31, 543–547. [DOI] [PubMed] [Google Scholar]
- Stokes-Rees I.; Sliz P. (2010) Protein structure determination by exhaustive search of Protein Data Bank derived databases. Proc. Natl. Acad. Sci. U.S.A. 107, 21476–21481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelstadt G.; Negron L.; Schofield L. R.; Marsh K.; Parker E. J. (2013) Biochemical and structural characterisation of dehydroquinate synthase from the New Zealand kiwifruit Actinidia chinensis. Arch. Biochem. Biophys. 537, 185–191. [DOI] [PubMed] [Google Scholar]
- Charmantray F.; Helaine V.; Legeret B.; Hecquet L. (2009) Preparative scale enzymatic synthesis of d-sedoheptulose-7-phosphate from β-hydroxypyruvate and d-ribose-5-phosphate. J. Mol. Catal. B: Enzym. 57, 6–9. [Google Scholar]
- Nango E.; Eguchi T.; Kakinuma K. (2004) Active site mapping of 2-deoxy-scyllo-inosose synthase, the key starter enzyme for the biosynthesis of 2-deoxystreptamine. Mechanism-based inhibition and identification of lysine-141 as the entrapped nucleophile. J. Org. Chem. 69, 593–600. [DOI] [PubMed] [Google Scholar]
- Bender S. L.; Widlanski T.; Knowles J. R. (1989) Dehydroquinate synthase: The use of substrate analogues to probe the early steps of the catalyzed reaction. Biochemistry 28, 7560–7572. [DOI] [PubMed] [Google Scholar]
- Montchamp J. G. (1992) Mechanism-based carbocyclic inhibitors of dehydroquinate synthase. Ph.D. Dissertation, Purdue University, West Lafayette, IN. [Google Scholar]
- Moore J. D.; Skinner M. A.; Swatman D. R.; Hawkins A. R.; Brown K. A. (1998) Reactivation of 3-dehydroquinate synthase by lanthanide cations. J. Am. Chem. Soc. 120, 7105–7106. [Google Scholar]
- Kudo F.; Hosomi Y.; Tamegai H.; Kakinuma K. (1999) Purification and characterization of 2-deoxy-scyllo-inosose synthase derived from Bacillus circulans. A crucial carbocyclization enzyme in the biosynthesis of 2-deoxystreptarnine-containing aminoglycoside antibiotics. J. Antibiot. 52, 81–88. [DOI] [PubMed] [Google Scholar]