

1. Introduction
1.1. Uncharacterized Protein Segments Are a Source of Functional Novelty
Over the past decade, we have observed a massive increase in the amount of information describing protein sequences from a variety of organisms.1,2 While this may reflect the diversity in sequence space, and possibly also in function space,3 a large proportion of the sequences lacks any useful function annotation.4,5 Often these sequences are annotated as putative or hypothetical proteins, and for the majority their functions still remain unknown.6,7 Suggestions about potential protein function, primarily molecular function, often come from computational analysis of their sequences. For instance, homology detection allows for the transfer of information from well-characterized protein segments to those with similar sequences that lack annotation of molecular function.8−10 Other aspects of function, such as the biological processes proteins participate in, may come from genetic- and disease-association studies, expression and interaction network data, and comparative genomics approaches that investigate genomic context.11−17 Characterization of unannotated and uncharacterized protein segments is expected to lead to the discovery of novel functions as well as provide important insights into existing biological processes. In addition, it is likely to shed new light on molecular mechanisms of diseases that are not yet fully understood. Thus, uncharacterized protein segments are likely to be a large source of functional novelty relevant for discovering new biology.
1.2. Structure–Function Paradigm Enhances Function Prediction
Traditionally, protein function has been viewed as critically dependent on the well-defined and folded three-dimensional structure of the polypeptide chain. This classical structure–function paradigm (Figure 1; left panel) has mainly been based on concepts explaining the specificity of enzymes, and on structures of folded proteins that have been determined primarily using X-ray diffraction on protein crystals. The classical concept implies that protein sequence defines structure, which in turn determines function; that is, function can be inferred from the sequence and its structure. Even when protein sequences diverge during evolution, for example, after gene duplication, the overall fold of their structures remains roughly the same. Therefore, structural similarity between proteins can reveal distant evolutionary relationships that are not easily detectable using sequence-based methods.18,19 Structural genomics efforts such as the Protein Structure Initiative (PSI) have been set up to enlarge the space of known protein folds and their functions, thereby complementing sequence-based methods in an attempt to fill the gap of sequences for which there is no function annotation.20,21 Specifically, phase two of the PSI aimed to structurally characterize proteins and protein domains of unknown function, often providing the first hypothesis about their function and serving as a starting point for their further characterization.
Figure 1.
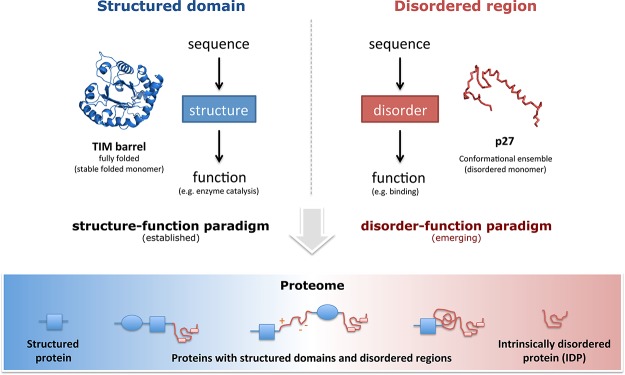
Structured domains and intrinsically disordered regions (IDRs) are two fundamental classes of functional building blocks of proteins. The synergy between disordered regions and structured domains increases the functional versatility of proteins. Adapted with permission from ref (50). Copyright 2012 American Association for the Advancement of Science.
1.3. Classification Further Facilitates Function Prediction
Classification schemes provide a guideline for systematic function assignment to proteins. Generally, proteins are made up of a single or multiple domains that can have distinct molecular functions. These domains, which are referred as structured domains, often fold independently, make precise tertiary contacts, and adopt a specific three-dimensional structure to carry out their function. The sequences that compose structured domains can be organized into families of homologous sequences, whose members are likely to share common evolutionary relationship and molecular function. The Pfam database classifies known protein sequences and contains almost 15 000 such families, for most of which there is some understanding about the function.22 Nevertheless, Pfam also contains more than 3000 families annotated as domains of unknown function, or DUFs.23 These families are largely made up of hypothetical proteins and await function annotation. Another powerful example of a protein classification scheme is the Structural Classification of Proteins (SCOP), which provides a means of grouping proteins with known structure together, based on their structural and evolutionary relationships.24,25 SCOP utilizes a hierarchical classification consisting of four levels, (i) family, (ii) superfamily, (iii) fold, and (iv) class, with each level corresponding to different degrees of structural similarity and evolutionary relatedness between members. Using this scheme, function of newly solved structures or sequences can be inferred from their similarity with existing protein classes through structure or sequence comparisons, for instance, as available via the SUPERFAMILY database.10 In this direction, another major initiative is Genome3D, which is a collaborative project to annotate genomic sequences with predicted 3D structures based on CATH26 (Class, Architecture, Topology, Homology) and SCOP24,25 domains to infer protein function.27
1.4. Intrinsically Disordered Regions and Proteins
While many proteins need to adopt a well-defined structure to carry out their function, a large fraction of the proteome of any organism consists of polypeptide segments that are not likely to form a defined three-dimensional structure, but are nevertheless functional.28−42 These protein segments are referred to as intrinsically disordered regions (IDRs; Figure 1; right panel).43 Because IDRs generally lack bulky hydrophobic amino acids, they are unable to form the well-organized hydrophobic core that makes up a structured domain31,44 and hence their functionality arises in a different manner as compared to the classical structure–function view of globular, structured proteins. In this framework, protein sequences in a genome can be viewed as modular because they are made up of combinations of structured and disordered regions (Figure 1; bottom panel). Proteins without IDRs are called structured proteins, and proteins with entirely disordered sequences that do not adopt any tertiary structure are referred to as intrinsically disordered proteins (IDPs). The majority of eukaryotic proteins are made up of both structured and disordered regions, and both are important for the repertoire of functions that a protein can have in a variety of cellular contexts.43 Traditionally, IDRs were considered to be passive segments in protein sequences that “linked” structured domains. However, it is now well established that IDRs actively participate in diverse functions mediated by proteins. For instance, disordered regions are frequently subjected to post-translational modifications (PTMs) that increase the functional states in which a protein can exist in the cell.45,46 In addition, they expose short linear peptide motifs of about 3–10 amino acids that permit interaction with structured domains in other proteins.47,48 These two features in isolation or in combination permit the interaction and recruitment of diverse proteins in space and time, thereby facilitating regulation of virtually all cellular processes.47 The prevalence of IDRs in any genome (see, for example, the D2P2 database,49 Box 1) in combination with their unique characteristics means that these regions extend the classical view of the structure–function paradigm and hence that of protein function. Thus, functional regions in proteins can either be structured or disordered, and these need to be considered as two fundamental classes of functional building blocks of proteins.50
Box 1. Databases of Intrinsically Disordered Regions and Proteins.
Several resources exist that collect experimental or computational information on disordered regions in proteins. The Database of Protein Disorder (DisProt, http://www.disprot.org/) was developed to facilitate research on protein disorder by organizing the rapidly increasing knowledge about the experimental characterization and the functionalities of IDRs and IDPs.203,400 The database includes the location of the experimentally determined disordered region(s) in a protein and the methods used for disorder characterization. Additionally, where known, entries list the biological function of an IDR and how it performs this function. As of the latest release (6.02, May 24, 2013), DisProt contained 694 IDP entries and 1 539 IDRs.
The IDEAL database (http://www.ideal.force.cs.is.nagoya-u.ac.jp/IDEAL/) also collects annotations of experimentally verified IDPs.388 This database focuses on regions that undergo coupled folding and binding upon interaction with other proteins (regions for which there is evidence for both a disordered isolated state and an ordered bound state), such as MoRFs and certain linear motifs (see section 3). It also suggests putative sequences for which there is only evidence of an ordered bound state, but that are thought to undergo induced folding based on, for example, the presence of a verified folding-upon-binding element in a homologue. The latest version (30 August 2013) contained 340 proteins with annotated IDRs of which 148 contain verified or putative elements that undergo folding upon binding.
MobiDB (http://mobidb.bio.unipd.it/) collects experimental data on IDRs from DisProt,203 IDEAL,388 and the Protein Data Bank147 (missing residues in crystal structures and structurally mobile regions in NMR ensembles).401 It also stores disorder prediction data from three methods. The total of disorder information is summarized in a weighted consensus. The latest version (1.2.1, August 28, 2012) contained 26 933 proteins for which there is experimental data on the presence or absence of disorder and disorder predictions for 4 662 776 proteins from 297 proteomes.
pE-DB (http://pedb.vib.be/) is the first database for the deposition of structural ensembles (see section 4.2) of intrinsically disordered proteins.398 Entries contain the primary experimental data (mainly NMR and SAXS, Box 2), the algorithms used in their calculation, and the coordinates of the structural ensembles, which are provided as a set of models in Protein Data Bank147 format. Development of pE-DB is intended to support the evolution of new methodologies for the structural descriptions of the disordered state. pE-DB stored 45 ensembles in 10 entries as of 17 January 2014.
Finally, the Database of Disordered Protein Prediction (D2P2, http://d2p2.pro/) stores disorder predictions (Box 3) made by nine different predictors for proteins from completely sequenced genomes.49 Alongside the disorder predictions, it contains information on MoRFs (ANCHOR386), PTM sites (PhosphoSitePlus402), and domains (SCOP24 and Pfam22). As of January 2014, D2P2 contained disorder predictions for 10 429 761 sequences in 1 765 genomes from 1 256 distinct species.
1.5. The Need for Classification of Intrinsically Disordered Regions and Proteins
IDRs and IDPs are prevalent in eukaryotic genomes. For instance, 44% of human protein-coding genes contain disordered segments of >30 amino acids in length49 (similar data shown in Figure 2A). In the human genome, 6.4% of all protein-coding genes do not have any function annotation in their description in Ensembl1 (Figure 2B). Further investigation using the D2P2 database of disorder in genomes49 revealed that most of these genes with no function annotation encode at least some disorder (Figure 2B) and that genes with no annotation contain proportionally more IDRs (Figure 2C). Given the absence of structural constraints, IDRs tend to evolve more rapidly than protein domains that adopt defined structures.51−56 As a result, identifying homologous regions is harder for IDRs and IDPs than it is for structured domains. This complicates the transfer of information about function between homologues and thus the prediction of function of IDRs and IDPs. Furthermore, much of protein annotation is based on information on sequence families and structured domains. However, less than one-half of all residues in the human proteome fall within such domains (Figure 3). Not only do most residues of human proteins fall outside domains, a large fraction of these residues are also disordered (Figure 3A and B, right bars). Moreover, although it is expected that SUPERFAMILY domains based on known protein structures have very little disorder (Figure 3A, left bar), Pfam domains based on sequence clustering do not contain much more (Figure 3B, left bar). These observations suggest that there is a large pool of protein segments that are not considered by conventional protein annotation methods, because the sequences of disordered regions are difficult to align, or because the methods do not explicitly consider disordered and nondomain regions of the protein sequence. Taken together, these considerations raise the need to devise a classification scheme specifically for disordered regions in proteins that may enhance the function prediction and annotation for this important class of protein segments.
Figure 2.
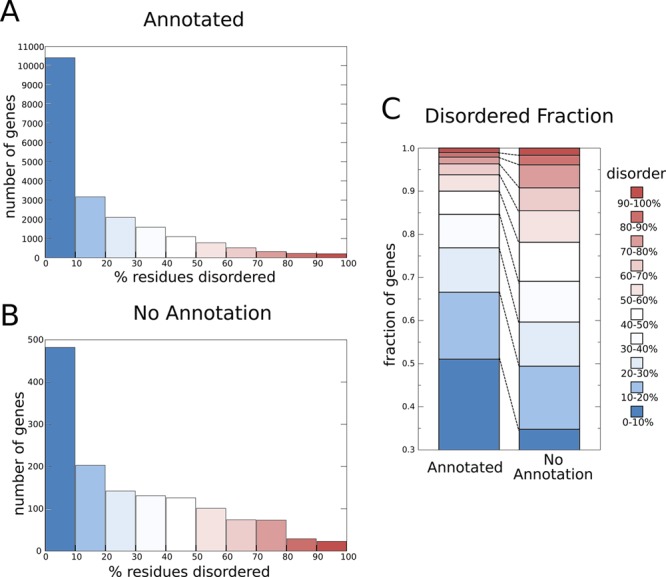
The number of protein-coding genes in the human genome with various amounts of disorder. Histograms of the numbers of human genes with annotation (A) and without annotation (B), grouped by the percentage of disordered residues. (C) A comparison of the fraction of annotated and unannotated human genes with different amounts of disorder. Residues in each protein are defined as disordered when there is a consensus between >75% of the predictors in the D2P2 database49 at that position. The set of human genes was taken from Ensembl release 63,1 and the representative protein coded for by the longest transcript was used in each case. The annotation was taken from the description field with “open reading frame”, “hypothetical”, “uncharacterized”, and “putative protein” treated as no annotation.
Figure 3.
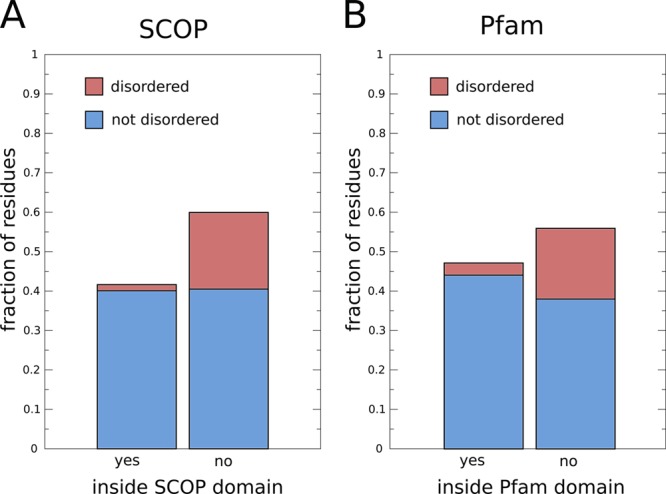
The fraction of disordered residues located in domains in human protein-coding genes: (A) residues inside (left) and outside (right) of SCOP domains,24 and (B) residues inside (left) and outside (right) of Pfam domains (only curated Pfam domains were considered, i.e., Pfam-A).22 The SCOP domains in human proteins are defined by the SUPERFAMILY database.10 Disordered residues were taken from the D2P2 database49 (when there is a consensus between >75% of the disorder predictors). The set of human genes was taken from Ensembl release 63.1
In this Review, we synthesize and provide an overview of the various classifications of intrinsically disordered regions and proteins that have been put forward in the literature since the start of systematic studies into their function some 15 years ago. We discuss approaches based on function, functional elements, structure, sequence, protein interactions, evolution, regulation, and biophysical properties (Table 1). Finally, we discuss resources that are currently available for gaining insight into IDR function (Table 2), we suggest areas where increased efforts are likely to advance our understanding of the functions of protein disorder, and we speculate how combinations of multiple existing classification schemes could achieve high quality function prediction for IDRs, which should ultimately lead to improved function coverage and a deeper understanding of protein function.
Table 1. Classifications of Intrinsically Disordered Regions and Proteins.
| basis for classification | classes | description | examples | |
|---|---|---|---|---|
| function | (33,39,57,58) | •entropic chains | IDRs carrying out functions that benefit directly from their conformational disorder, e.g., flexible linkers and spacers | MAP2 projection domain, titin PEVK domain, RPA70, MDA5 |
| •display sites | flexibility of IDRs facilitates exposure of motifs and easy access for proteins that introduce and read PTMs | p53, histone tails, p27, CREB kinase-inducible domain | ||
| •chaperones | their binding properties (many different partners, rapid association/disassociation, and folding upon binding) make IDPs suitable for chaperone functions | hnRNP A1, GroEL, α-crystallin, Hsp33 | ||
| •effectors | folding upon binding mechanics allow effectors to modify the activity of their partner proteins | p21, p27, calpastatin, WASP GTPase-binding domain | ||
| •assemblers | assembling IDRs have large binding interfaces that scaffold multiple binding partners and promote the formation of higher-order protein complexes | ribosomal proteins L5, L7, L12, L20, Tcf 3/4, CREB transactivator domain, Axin | ||
| •scavengers | disordered scavengers store and neutralize small ligands | chromogranin A, Pro-rich glycoproteins, caseins and other SCPPs | ||
| functional features | linear motifs47,125 | •structural modification | sites of conformational alteration of a peptide backbone | peptidylprolyl cis–trans isomerase Pin1 sites |
| •proteolytic cleavage | sites of post-translational processing events or proteolytic cleavage scission sites | Caspase-3/-7, separase, taspase1 scission sites | ||
| •PTM removal/addition | specific binding sequences that recruit enzymes catalyzing PTM moiety addition or removal | cyclin-dependent kinase phosphorylation site, SUMOylation site, N-glycosylation site | ||
| •complex promoting | motifs that mediate protein–protein interactions important for complex formation; often associated with signal transduction | proline-rich SH3-binding motif, cyclin box, pY SH2-binding motif, PDZ-binding motif, TRAF-binding motifs in MAVS | ||
| •docking | motifs that increase the specificity and efficiency of modification events by providing an additional binding surface | KEN box degron, MAPK docking sites | ||
| •targeting or trafficking | signal sites that localize proteins within particular subcellular organelles or act to traffic proteins | nuclear localization signal, clathrin box motif, endocytosis adaptor trafficking motifs | ||
| molecular recognition features (MoRFs)121 | •alpha | disordered motifs that form α-helices upon target binding | p53 ∼ Mdm2, p53 ∼ RPA70, p53 ∼ S100B(ββ), RNase E ∼ enolase, inhibitor IA3 ∼ proteinase A | |
| •beta | disordered motifs that form β-strands upon target binding | RNase E ∼ polynucleotide phosphorylase, Grim ∼ DIAP1, pVIc ∼ adenovirus 2 proteinase | ||
| •iota | disordered motifs that form irregular secondary structure upon target binding | p53 ∼ Cdk2-cyclin A, amphiphysin ∼ α-adaptin C | ||
| •complex | disordered motifs that contain combinations of different types of secondary structure upon target binding | amyloid β A4 ∼ X11, WASP ∼ Cdc42 | ||
| intrinsically disordered domains (IDDs)158,159 | some protein domains identified using sequence-based approaches are fully or largely disordered | WH2, RPEL, BH3, KID domains | ||
| co-occurrence of protein domains with disordered regions161,162 | particular disordered regions frequently co-occur in the same sequence with specific protein domains | |||
| structure | structural continuum37 | proteins function within a continuum of differently disordered conformations, extending from fully structured to completely disordered, with everything in between and no strict boundaries between the states | ||
| protein quartet32,34,166 | •intrinsic coil | flexible regions of extended conformation with hardly any secondary structure; high net charge differentiates these from disordered globules | ribosomal proteins L22, L27, 30S, S19, prothymosin α | |
| •pre-molten globule | disordered protein regions with residual secondary structure, often poised for folding upon binding events; lower net charge makes them more compact than coils | Max, ribosomal proteins S12, S18, L23, L32, calsequestrin | ||
| •molten globule | globally collapsed conformation with regions of fluctuating secondary structure | nuclear coactivator binding domain of CREB binding protein | ||
| •folded | structured proteins with a defined three-dimensional structure | most enzymes, transmembrane domains, hemoglobin, actin | ||
| sequence | sequence–structural ensemble relationships166,204 | •polar tracts | sequence stretches enriched in polar amino acids often form globules that are generally devoid of significant secondary structure preferences | Asn- and Gly-rich sequences, Gln-rich linkers in transcription factors and RNA-binding proteins |
| •polyelectrolytes | amino acid compositions biased toward charged residues of one type; strong polyelectrolytes (high net charge) form expanded coils | Arg-rich protamines, Glu/Asp-rich prothymosin α | ||
| •polyampholytes | sequences with roughly equal numbers of positive and negative charges; conformations of polyampholytes are governed by the linear distribution of oppositely charged residues, with segregation of opposite charges leading to globules, while well-mixed charged sequences adopt random-coil or globular conformations, depending on the total charge | RNA chaperones, splicing factors, titin PEVK domain, yeast prion Sup35 | ||
| prediction flavors205 | •V | predicted best by the VL-2V predictor, for which the hydrophobic amino acids are the most influential attributes | E. coli ribosomal proteins | |
| •C | VL-2C is the best predictor for flavor C, which has more histidine, methionine, and alanine residues than the other flavors | poly- and oligosaccharide binding domains | ||
| •S | flavor with less histidine than the others, best predicted by predictor VL-2S, which has a measure of sequence complexity as the most important attribute | proteins that facilitate binding and interaction | ||
| disorder–sequence complexity206 | IDPs from different functional classes show distinct disorder–sequence complexity distributions | proteins with disordered linkers between structured domains populate compact and disordered DC regions | ||
| overall degree of disorder35,51,68,161,208,209 | •fraction | categorization of proteins based on the fraction of residues predicted to be disordered | 0–10/10–30/30–100% disorder | |
| •overall score | overall disorder scores for the whole protein | minimum average disorder score depending on the predictor | ||
| •continuous stretches | presence or absence of continuous stretches of disordered residues | typically >30 residues | ||
| length of disordered regions211 | •>500 residues | proteins that contain disordered regions of different lengths are enriched for different types of functions | transcription | |
| •300–500 residues | kinase and phosphatase functions | |||
| •<50 residues | (metal) ion binding, ion channels, GTPase regulatory activity | |||
| position of disordered regions211 | •N-terminal | proteins that contain disordered regions at different locations in the sequence are enriched for different types of functions | DNA-binding, ion channel | |
| •internal | transcription regulator, DNA-binding | |||
| •C-terminal | transcription repressor/activator, ion channel | |||
| tandem repeats217,218 | •Q/N | glutamine- and asparagine-rich proteins regions are both important for normal cellular function and prone to cause harmful aggregation | huntingtin, Sup35p, Ure2p, Ccr4, Pop2 | |
| •S/R | tandem repeats composed of arginine and serine residues are phosphorylated and disordered, and play a role in spliceosome assembly | ASF/SF2, SRp75, SRSF1 | ||
| •K/A/P | tandem repeats composed of lysine, alanine, and proline function in binding nucleosome linker DNA | histone H1 | ||
| •F/G | disordered domains with phenylalanine-glycine repeats influence NPC gating behavior | nucleoporins | ||
| •P/T/S | extensively glycosylated regions rich in proline, threonine, and serine residues are involved in mucus formation | mucins | ||
| •others | ||||
| protein interactions | fuzzy complexes by topology242 | •polymorphic | a form of static disorder, with alternative bound conformations serving distinct functions by having different effects on the binding partner | β-catenin ∼ Tcf4, NLS ∼ importin-α, actin ∼ WH2 domain |
| •clamp | complex formation through folding upon binding of two disordered protein segments, connected by a linker that remains disordered | Ste5 ∼ Fus3, myosin VI ∼ actin filament, Oct-1 ∼ DNA | ||
| •flanking | complex formation through folding upon binding of a central disordered protein segment, flanked by two regions that remain disordered | SF1 splicing factor ∼ U2AF, proline-rich peptides ∼ SH3 domains, p27Kip1 ∼ cyclin-Cdk2 | ||
| •random | disordered regions that remain highly dynamic even in the bound state | elastin self-assembly, Sic1 ∼ Cdc4 | ||
| fuzzy complexes by mechanism176,251 | •conformational selection | the fuzzy region facilitates the formation of the binding-competent form by shifting the conformational equilibrium | Max ∼ DNA, MeCP2 ∼ DNA | |
| •flexibility modulation | the fuzzy region modulates the flexibility of the binding interface and changes binding entropy | Ets-1 ∼ DNA, SSB ∼ DNA | ||
| •competitive binding | the fuzzy region serves as an intramolecular competitive partner for the binding surface. | HMGB1 ∼ DNA, RNase1 ∼ RNase inhibitor | ||
| •tethering | the fuzzy region increases the local concentration of a weak-affinity binding domain near the target, or anchors it via transient interactions | RPA ∼ DNA, UPF1 ∼ UPF2, PC4 ∼ VP16 | ||
| binding plasticity257 | •static | mono-/polyvalent complexes, chameleons, penetrators, huggers | for examples, see Figure 12 | |
| •coiled-coil based | intertwined strings, long cylindrical containers, connectors, armature, tweezers and forceps, grabbers, tentacles, pullers, stackers | |||
| •dynamic | cloud contacts and protein interaction ensembles | |||
| evolution | sequence conservation54 | •flexible | regions that require the property of disorder for functionality regardless of the exact sequence | signaling and regulatory proteins (Sky1, Bur1) |
| •constrained | regions of conserved disorder that also have highly conserved amino acid sequences | ribosomal proteins (Rpl5), protein chaperones (Hsp90) | ||
| •nonconserved | no conservation of the disorder, nor of the underlying sequence; no clear functional hallmarks | yeast Ty1 retrotransposon domains A and B | ||
| conservation of amino acid composition260 | •HR | IDRs with high residue conservation | transcription regulation and DNA binding | |
| •LRHT | IDRs with low residue conservation but high conservation of the amino acid composition of the region | ATPase and nuclease activities | ||
| •LRLT | IDRs with neither conservation of sequence nor conservation of amino acid composition | (metal) ion binding proteins | ||
| lineage and species specificity159 | •prokaryotes | species from different kingdoms of life seem to use disorder for different types of functions | longer lasting interactions involved in complex formation | |
| •eukaryotes and viruses | transient interactions in signaling and regulation | |||
| evolutionary history and mechanism of repeat expansion61 | •Type I | repeats that showed no function diversification after expansion | titin PEVK domain, salivary proline-rich proteins | |
| •Type II | repeats that acquired diverse functions through mutation or differential location within the sequence | RNA polymerase II (CTD) | ||
| •Type III | repeats that gained new functions as a consequence of their expansion | prion protein octarepeats | ||
| regulation | expression patterns208 | •constitutive | IDPs encoded by constitutively highly expressed transcripts are almost entirely disordered and often ribosomal proteins | ribosomal L proteins |
| •high | IDP-encoding transcripts showing high expression levels in most tissues and little tissue specificity | protease inhibitors, splicing factors, complex assemblers | ||
| •medium | these IDP-encoding transcripts are expressed at medium levels, with some tissue-specificity | DNA binding, transcription regulation | ||
| •tissue-specific | IDP-encoding transcripts with highly tissue-specific expression | cell organization regulators, complex disassemblers | ||
| •low or transient | IDP-encoding transcripts that are present in undetectable amounts; more than one-half of analyzed IDPs | variety of functions | ||
| alternative splicing304,305,309,312,313 | regulation and evolutionary patterns of inclusion and exclusion of IDR-encoding exons can provide insights into whether the encoded IDR functions in protein regulation and interactions | a tissue-specific region with a phosphosite in the TJP1 protein in mouse, a mammalian-specific region in the PTB1 splicing regulator | ||
| degradation kinetics315,316,318,320,321 | •degradation accelerators | IDRs that can influence and accelerate proteasomal degradation of the protein containing it | ||
| •others | IDRs that have no influence on protein half-life or increase it, e.g., because of sequence compositions that impede proteasome processivity | low complexity sequences such as glycine-alanine repeats and polyglutamine repeats | ||
| post-translational processing and secretion337,340 | secreted proteins are depleted for IDPs, but structural disorder is important in, e.g., prohormones, the extracellular matrix, and biomineralization | pre-pro-opiomelanocortin, elastic fiber proteins, SIBLINGs, mucins | ||
| biophysical properties | solubility209 | the sequence features of IDPs are generally associated with aqueous solubility, although some IDPs are thermostable, while others are not; this is likely modulated by sequence–structural ensemble relationships, such as the degree of compaction | 4E-BP1, calpastatin, CREB, p21, p27, Sp1, stathmin, WASP | |
| phase transition137,353 | certain IDRs (such as those that contain specific low-complexity regions or interaction motifs) can undergo phase transitions like the formation of protein-based droplets or hydrogels | multivalent SH3-binding motifs in phase separation, granule-like assemblies of RNA-binding proteins containing low-complexity IDRs, mucins | ||
| biomineralization117,341 | structural disorder is common in proteins with roles in biomineralization, such as the formation of bone and teeth | caseins, osteopontin, bone sialoprotein 2, dentin sialophosphoprotein | ||
Table 2. Current Methods for Function Prediction of Intrinsically Disordered Regions and Proteins.
| basis for method | description | method | Web site |
|---|---|---|---|
| linear motifs | annotation of well-characterized linear motifs, which can be mapped onto other protein sequences | ELM125 | http://elm.eu.org/ |
| MiniMotif126 | http://mnm.engr.uconn.edu/ | ||
| identification of putative uncharacterized motifs in protein sequences | SLiMPrints372 | http://bioware.ucd.ie/slimprints.html | |
| phylo-HMM373 | http://www.moseslab.csb.utoronto.ca/phylo_HMM/ | ||
| DiliMot374 | http://dilimot.russelllab.org/ | ||
| SLiMFinder375 | http://bioware.ucd.ie/slimfinder.html | ||
| PTM sites | resources of experimentally verified PTM sites, mostly phosphorylation | Phospho.ELM268 | http://phospho.elm.eu.org/ |
| PhosphoSite376 | http://www.phosphosite.org/ | ||
| PHOSIDA377 | http://www.phosida.com/ | ||
| identification and collection of peptide motifs that direct post-translational modifications | ScanSite380 | http://scansite.mit.edu/ | |
| NetPhorest381 | http://netphorest.info/ | ||
| NetworKIN382 | http://networkin.info/ | ||
| PhosphoNET383 | http://www.phosphonet.ca/ | ||
| molecular recognition features | collection of verified sequence elements that undergo coupled folding and binding | IDEAL388 | http://www.ideal.force.cs.is.nagoya-u.ac.jp/IDEAL/ |
| prediction of sequences that undergo disorder-to-order transitions | MoRFpred385 | http://biomine.ece.ualberta.ca/MoRFpred/ | |
| ANCHOR386 | http://anchor.enzim.hu/ | ||
| intrinsically disordered domains | annotation of disordered protein domains, which can be detected by sequence profiles | Pfam22 | http://pfam.sanger.ac.uk/ |
| other | prediction of gene ontology functions using protein sequence features such as intrinsic disorder | FFPred391 | http://bioinf.cs.ucl.ac.uk/psipred/ |
| function annotation of experimentally verified disordered protein regions | DisProt203 | http://www.disprot.org/ | |
| predictions of disordered regions combined with information on MoRFs, PTM sites, and domains | D2P2 49 | http://d2p2.pro/ |
2. Function
Dunker and co-workers57 distinguished 28 separate functions for disordered regions, based on literature analysis of 150 proteins containing disordered regions of 30 residues or longer. These functionalities can be summarized as molecular recognition, molecular assembly, protein modification, and entropic chains. Further development of this scheme resulted in one comprising six different functional classes of disordered protein regions: entropic chains, display sites, chaperones, effectors, assemblers, and scavengers (Figure 4).33,58 In another classification scheme, Gsponer and Babu classified IDR function into three broad functional categories: (i) facilitated regulation via diverse post-translational modifications, (ii) scaffolding and recruitment of different binding partners, and (iii) conformational variability and adaptability (Figure 5).39 A single protein may consist of several disordered regions that belong to different functional classes.59 The following section will address and exemplify the six functionalities of disordered regions.
Figure 4.
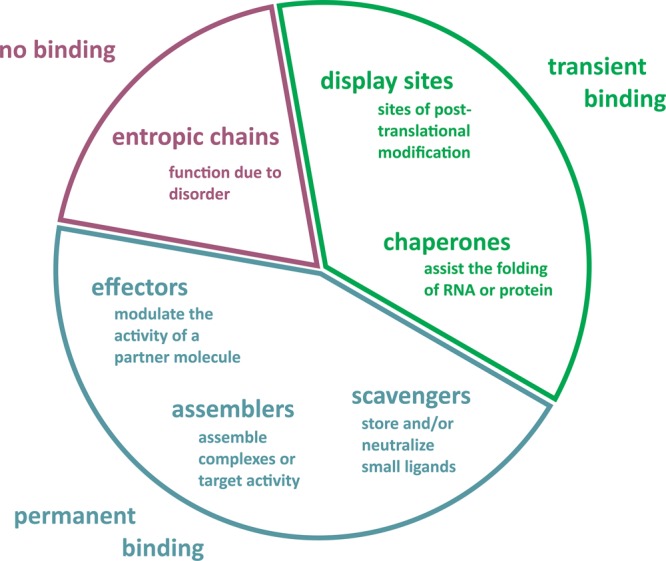
Functional classification scheme of IDRs. The function of disordered regions can stem directly from their highly flexible nature, when they fulfill entropic chain functions (such as linkers and spacers, indicated in dark-tone red), or from their ability to bind to partner molecules (proteins, other macromolecules, or small molecules). In the latter case, they bind either transiently as display sites of post-translational modifications or as chaperones (indicated in green), or they bind permanently as effectors, assemblers, or scavengers (indicated in dark-tone blue). More extensive descriptions and examples are found in the main text. Adapted with permission from ref (58). Copyright 2005 Elsevier.
Figure 5.

Functional classification of IDRs according to their interaction features. (A) The flexibility of IDRs facilitates access to enzymes that catalyze post-translational modifications and effectors that bind these PTMs. This permits combinatorial regulation and reuse of the same components in multiple biological processes. (B) The availability of molecular recognition features and linear motifs within the IDRs enables the fishing for (“fly casting”) and gathering of different partners. (C) Conformational variability enables a nearly perfect molding to fit the binding interfaces of very diverse interaction partners. Context-dependent folding of an IDR can activate signaling processes in one case or inhibit them in another, resulting in completely different outcomes. Adapted with permission from ref (39). Copyright 2009 Elsevier.
2.1. Entropic Chains
Entropic chains carry out functions that benefit directly from their conformational disorder; that is, they function without ever becoming structured. Examples of entropic chains include flexible linkers, which allow movement of domains positioned on either ends of the linker relative to each other, and spacers that regulate the distances between domains. Evidence that flexibility is a functional characteristic that needs to be maintained came from studies on a family of flexible linkers in the 70 kDa subunit of replication protein A (RPA70), which display conserved dynamic behavior in the face of negligible sequence conservation.60 The microtubule-associated protein 2 (MAP2) projection domain exemplifies spacer behavior as it repels molecules that approach microtubules, thereby providing spacing in the cytoskeleton. Another subcategory of entropic chains are entropic springs, such as those present in the titin protein, which contains repeat regions rich in PEVK amino acids that generate force upon overstretching to help restore muscle cells to their relaxed length.61,62
2.2. Display Sites
Post-translational modifications (PTMs) affect the stability, turnover, interaction potential, and localization of proteins within the cell.63 These aspects of PTMs are particularly relevant for proteins involved in regulation and signaling, as are many IDPs.35,37,39,64,65 The conformational flexibility of disordered protein regions as display sites provides advantages over structured regions. (i) Flexibility facilitates the deposition of PTMs by enabling transient but specific interaction with catalytic sites of modifying enzymes.47,66 This is because, upon binding, a flexible, disordered region loses more conformational freedom (i.e., entropy), which reduces the overall free energy of binding, leading to weaker and more transient binding as compared to a folded protein region that interacts with equal strength (i.e., the same binding enthalpy, or, equal specificity).28,30,37 (ii) The flexibility of IDRs also allows for easy access and recognition of the PTMs within the IDR by effector proteins that mediate downstream outcomes upon binding.47,66 Indeed, experimental and computational approaches have shown that disordered regions are enriched for sites that can be phosphorylated,45,46,67 and suggest that IDPs are likely to be substrates of a large number of kinases and other modifying enzymes as they are heavily post-translationally modified.46,68,69 Furthermore, PTM sites are often located within short peptide motifs, modification of which influences the affinity for interaction with diverse binding partners (see section 3.1).70,71 In turn, disordered protein regions are strongly enriched for these motifs,47,72−74 underlining the importance of intrinsic disorder as PTM display sites. Well-characterized examples of IDPs in which PTMs are key to function and regulation include, among others, histones, p53, and the cyclin-dependent kinase regulator p27.75−77
2.3. Chaperones
Chaperones are proteins that assist RNA and protein molecules to reach their functionally folded states.78,79 Disordered regions make up over one-half of the sequences of RNA chaperones and over one-third of the sequences of protein chaperones.80,81 The versatility of disordered segments seems well suited for chaperone function, although mechanistic evidence is still scarce.82 First, their capacity to structurally adapt to many different binding partners matches the need for chaperones to bind a wide range of proteins. Second, disordered segments enable fast macromolecular interactions. This is because the highly dynamic nature of IDRs prolongs the lifetime of the encounter complex of the binding event due to rapid sampling of many different conformations, thereby increasing the number of nonspecific interactions as compared to an encounter of a structured protein. In turn, this results in a higher probability to sample the specific conformation that results in the stable interaction complex and increases the association rate of the interaction.83,84 The quick binding of misfolded proteins by disordered chaperones could, for example, prevent the formation of toxic aggregates by providing a solubilizing effect (see section 9.1). Finally, the binding thermodynamics of disordered regions are well suited for the cycles of repeated chaperone binding and release that enable substrate folding. It has been proposed that transient binding of disordered chaperone regions to misfolded substrates induces local folding of the disordered chaperone, and promotes unfolding of the substrate, thereby providing the substrate with a chance to refold correctly.80 This reversible exchange of entropy represents a distinct type of chaperone function that relies on disordered regions and does not require ATP. Loss of flexibility of disordered regions upon substrate binding has been demonstrated for the chaperones GroEL85 and α-crystallin.86,87 This mechanism can even be switched on and off at need by regulated transitions between folded and disordered states,88 as reported in the case of the redox-regulated chaperone Hsp33.89,90
2.4. Effectors
Another functional class of disordered regions is that of the effectors, which interact with other proteins and modify their activity. Upon binding their interaction partners, IDRs often undergo a disorder-to-order transition, also known as coupled folding and binding.91,92 Examples of two effectors that fold upon binding are p21 and p27, which regulate different cyclin-dependent kinases (Cdk) that are responsible for the control of cell-cycle progression in mammals.66 p21 and p27 exhibit functional diversity by achieving opposite effects on different Cdk–cyclin complexes, promoting the assembly and catalytic activity of some (e.g., Cdk4 paired with D-type cyclins), and inhibiting others (e.g., Cdk2 paired with A- and E-type cyclins).66 Another effector IDP is calpastatin, which undergoes significant folding upon binding calpain, thereby achieving specific and reversible inhibition.93
IDRs can also affect the activity of other parts within the same protein, either through competitive interactions or through allosteric modulation. The intrinsically disordered GTPase-binding domain (GBD) of the Wiskott–Aldrich syndrome protein (WASP) illustrates competitive binding that controls autoinhibition.94 Binding of the GBD to the Cdc42 protein promotes the interaction of WASP with the actin cytoskeleton regulatory machinery. However, GDB adopts a different structure when it folds back on other parts of WASP to inhibit actin interaction. Indeed, autoinhibitory regions are generally enriched for intrinsic disorder and often have different structures in the inhibitory and functionally active states of the protein.95 A striking example of allosteric coupling in a disordered protein was revealed between different binding sites in the adenovirus E1A oncoprotein.96 Complexes of E1A with the TAZ2 domain of CREB-binding protein (CBP) and the retinoblastoma protein (pRb) can have either positive or negative cooperativity, depending on the available E1A interaction sites (i.e., binding of either pRb or CBP to E1A increases or decreases, respectively, the probability that the other one will also bind). These findings support earlier studies that suggest allosteric coupling does not always require a well-defined structural route to propagate through the protein, but can also be determined by the stabilities of individual conformations of the protein that change upon binding their interaction partners.97−99 Such a mechanism could be one explanation for how the availability of different binding partners regulates the outcomes of multiple binding events involving disordered proteins in a cellular context.96
2.5. Assemblers
Disordered assemblers bring together multiple binding partners to promote the formation of higher-order protein complexes,100,101 such as the ribosome (many ribosomal proteins are disordered102), activated T-cell receptor complexes,58 the RIP1/RIP3 necrosome,103 and the transcription preinitiation complex.104 The presence of different functional regions within the disordered segments, such as molecular recognition features (MoRFs) and short linear peptide motifs (SLiMs), enables binding and can bring together different partners (see sections 3.1 and 3.2). Indeed, larger complexes are assembled from proteins that tend to be more disordered,105 and intrinsic disorder is a common feature of hubs in protein interaction networks.106,107 The open structure of disordered assemblers is largely preserved upon scaffolding their partner proteins, resulting in a large binding interface that enables multiple proteins to be bound by a single IDR.108,109 Furthermore, disordered regions largely avoid the steric hindrance that prevents the formation of comparably large complexes from structured proteins.
Assembler function can be imagined in two ways. (i) The first is structural mortar, which helps to bring together proteins by stabilizing the complexes they form. A well-studied example of this behavior is the assembly of the ribosome, which relies on a sequence of cooperative binding steps of protein and RNA.110 Although the initial stages of rRNA folding are probably driven by the RNA itself,111 ribosomal proteins subsequently fold upon binding the rRNAs,112,113 which induces structural changes in both the RNA and the protein, and guides the complex toward its native state.110 (ii) The second is scaffolds that serve as backbones for the spatiotemporally regulated assembly of different signaling partners. An example of this mechanism is the Axin scaffold protein, which colocalizes β-catenin, casein kinase Iα, and glycogen synthetase kinase 3β by their binding to Axin’s long intrinsically disordered region, thereby effectively yielding a complex of structured domains with flexible linkers.114 The assembly of all four proteins accelerates interactions between them by raising their local concentrations and leads to the efficient phosphorylation and subsequent destruction of β-catenin. Scaffolding regions have one of the highest degrees of disorder of all functional categories.109,115
2.6. Scavengers
The final distinct functional class of IDRs and IDPs are scavengers, which store and neutralize small ligands. Chromogranin A, one of the earliest examples of an IDP, functions as a scavenger by storing ATP and adrenaline in the medulla of the adrenal gland.116 NMR studies showed that chromogranin is a random coil in both the isolated form and in its cellular environment in the intact adrenal gland.116 Caseins and other calcium-binding phosphoproteins (SCPPs) are highly disordered proteins that solubilize clusters of calcium phosphate in milk and other biofluids (see section 9.3).117 Finally, salivary proline-rich glycoproteins are scavenger IDPs that bind tannin molecules in the digestive tract.33
3. Functional Features
Different types of functional regions in intrinsically disordered proteins have been uncovered by investigations aimed both directly at increasing the understanding of IDRs and indirectly by linking previously studied functionality of proteins to disordered regions. First, the majority of linear motifs (such as the SH2 domain interaction motif) have been found as enriched in IDRs.48,72,118 Second, the development of disorder prediction methods (Box 3) has led to the identification of segments that promote disorder-to-order transitions called molecular recognition features (MoRFs),119−123 which have been verified using known crystal structures. Third, some interaction domains identified using crystallography, by sequence analysis, and by other techniques, turn out to be intrinsically disordered in solution (e.g., the BH3 domain124). The following section discusses these three interaction features separately and points out the underlying connections between them.
Box 3. Prediction of Intrinsically Disordered Regions and Proteins.
Predicting disordered regions from amino acid sequence allows the analysis of disordered proteins at a genome-wide scale and provides initial hypotheses about the presence of structural disorder in individual proteins.38,406 A large number of prediction methods have been developed and are regularly benchmarked as part of the Critical Assessment of Techniques for Protein Structure Prediction (CASP).407,408 Excellent overviews of disorder prediction methods are given elsewhere,406,409,410 and nonexhaustive lists of publicly available prediction software and webservers can be found at http://en.wikipedia.org/wiki/List_of_disorder_prediction_software and http://www.disprot.org/predictors.php.
Three general prediction strategies currently exist:
-
•
Disorder prediction based directly on sequence properties. For instance, IUPred is a physicochemical sequence-based method that estimates residue interaction energies.411 Sequences with lower predicted pairwise interaction energies are considered more likely to be disordered due to a lack of stabilizing contacts. Similarly, FoldIndex considers weakly hydrophobic regions of high net charge. Such regions are likely to be disordered due to their low energy benefit when adopting a compact conformation.31,412
-
•
Machine learning is used in the majority of predictors, for example, by using unresolved residues in X-ray structures as a training set.410 For example, DISOPRED2 uses linear support vector machines (SVMs) trained on PSI-BLAST sequence profiles surrounding unresolved residues.35 Similarly, PONDR XL1 employs a feed-forward neural network trained on sequence attributes found associated with unresolved residues.271
-
•
Meta-predictors that combine several individually successful disorder prediction methods have been developed more recently, resulting in increases in prediction accuracy.407 For instance, metaPrDOS413 and MFDp414 both apply SVM-based machine learning to the results of a number of individual prediction methods to arrive at a final score. Similarly, the MobiDB401 and D2P2 databases49 (Box 1) provide a consensus overview of several independent prediction methods.
Curated databases containing experimentally determined disordered regions, such as DisProt203 and IDEAL388 (Box 1), provide a gold standard for assessing disorder prediction methods. Overall, the quality of the predictions appears to have reached a reasonable plateau of accuracy, with modest recent progress.407,408 Additional data on biologically relevant long disordered regions may lead to future improvements in predicting IDRs and IDPs.408
3.1. Linear Motifs
A common functional module within IDRs is the linear motif,47,48,72 also known as LMs, short linear motifs (SLiMs),125 or MiniMotifs.126 By regulating low-affinity interactions, these short sequence motifs (annotated instances are usually 3–10 amino acids long48) can target proteins to a particular subcellular location, recruit enzymes that alter the chemical state of the motif by post-translational modifications (PTMs), control the stability of a protein, and promote recruitment of binding factors to facilitate complex formation.47,48 Linear motifs, helped by the flexible nature of the disordered regions that surround them,71 primarily bind onto the surfaces of globular domains,127,128 and their compact binding surface promotes them to occur multiple times within one protein.47,48 Moreover, the short nature of many linear motifs means they have a high propensity to convergently evolve and emerge in unrelated proteins.47,48 A consequence of these properties is that pathogenic viruses and bacteria have evolved to mimic these linear motifs, allowing them to manipulate regulation of cellular processes.129,130
Linear motifs can be broadly divided into two major families: those that act as modification sites and those that act as ligands, with each having numerous subgroups (Figure 6).131 The first major family, the enzyme binding or modification motifs, can be divided into three groups. (i) The first is post-translational processing events or proteolytic cleavage. A well-known example is the motif recognized by Caspase-3 and -7, which has an [ED]xxD[AGS] consensus sequence. Caspases are a family of proteases that promote apoptosis and inflammation by cleaving such motifs in their substrate proteins.132 Hundreds of proteins have convergently evolved the Caspase-3/-7 motif, and thereby have come under the regulation of the apoptotic pathway.133 (ii) The second is PTM moiety removal and addition. Many enzymes that catalyze post-translational modifications recognize a specific binding sequence on the substrate. For example, the cyclin-dependent kinase recognition motif [ST]Px[KR] is present in many mitotic proteins, and its phosphorylation is key for regulating cell cycle progression.134 (iii) The third is structural modifications. This group of motifs is involved in the catalyzed conformational alteration of a peptide backbone. The classic example is the peptidylprolyl cis–trans isomerase (PPIase) Pin1, which binds [ST]P motifs in a phosphorylation dependent manner to catalyze the cis–trans isomerization of the proline peptide bond. This modification can regulate the recognition of phosphorylated [ST]P sites by phosphatases.135
Figure 6.

Functional classification of linear motifs. Linear motifs can be divided into two major families, which each have three further subgroups. The modification class motifs all act as recognition sites for enzyme active sites, whereas the ligand class motifs are always recognized by the binding surface of a protein partner. More detailed classification beyond the graph shown here is possible. For example, an important subgroup of docking motifs are the degrons, which regulate protein stability by recruiting members of the ubiquitin–proteasome system. In the regular expressions, x corresponds to any amino acid, while other letters represent single letter codes of amino acids; letters within square brackets mean either residue is allowed in that position.
The second major family of motifs comprises ligand motifs, which can also be divided into three main groups (Figure 6). (i) Complex promoting motifs are the most well-known class of motifs and include the phosphorylated tyrosine motif recognized by SH2 (Src homology 2) domains, the C-terminal motifs that bind PDZ domains, and the proline-rich PxxP motifs that interact with SH3 (Src homology 3) domains.136 These motifs often function in protein scaffolding, and their multivalency (tendency to occur multiple times in one sequence) can increase the avidity of interactions and promote phase transition (see section 9.2).137 (ii) Docking motifs increase the specificity and efficiency of modification events (e.g., addition or removal of PTMs, see above) by providing additional binding surface. These docking motifs are distinct from the modification sites, but are usually in the same protein. Examples are the KEN box and D box degrons, which act as recognition surfaces for ubiquitin ligases that ubiquitinate the protein on a different position, leading to degradation of the protein by the 26S proteasome.138,139 The KEN box motif occurs in several key mitotic kinases to ensure their degradation or deactivation at mitotic exit.139 In some cases, the docking site is present in a protein different from that which contains the modification site, as exemplified by the F box motif. Another part of F box proteins recognizes post-translationally modified degradation motifs of substrates, while the F box itself docks the Skp1 components of SCF (Skp, Cullin, F box) E3 ligase complexes.140 (iii) Targeting motifs can localize proteins toward subcellular organelles. For example, importin proteins involved in nuclear transport recognize the nuclear localization signal (NLS), usually a motif containing a short cluster of lysines and arginines, and translocate NLS-containing proteins into the nucleus.141 Targeting motifs can also act to traffic proteins, as in the case of endocytic motifs. These are recognized by adaptor proteins at different stages of endocytosis to ensure that cargo proteins are packaged into vesicles and trafficked to the right location.142,143
An important feature of linear motifs is their propensity to act as molecular switches. This is for two major reasons. (i) Linear motif-mediated interactions are generally low affinity due to the limited binding surface. This means that large, bulky post-translational modifications have a big impact on their binding properties.71 (ii) Their small footprint (i.e., size) allows motifs to occur multiple times in the same protein, thereby promoting high avidity interactions and the recruitment of multiple factors (e.g., the LAT complex in T-cell receptor signaling144).99 This also means two different motifs can overlap, resulting in mutually exclusive binding of interaction partners.73 The ability of a motif to rapidly switch between binding partners and create multivalent complexes is crucial for the creation of dynamic signaling networks.71
3.2. Molecular Recognition Features
Disordered segments can also contain another type of peptide motif (10–70 amino acids) that promotes specific protein–protein interactions. These functional elements are called preformed structural elements (PSEs),119 molecular recognition features (MoRFs) or elements (MoREs),120−122 or prestructured motifs (PreSMos).123 Importantly, MoRFs undergo disorder-to-order transitions upon binding their interaction partners (i.e., folding upon binding),38,121,123 and often the unbound form of these preformed elements is biased toward the conformation that they adopt in the complex.119 Preformed structural elements and MoRFs may serve as initial contact points for interaction events, which have different kinetic and thermodynamic properties than interactions between structured protein regions as discussed before. Binding of preformed elements is one version of conformational selection (see section 6), suggested long ago for interactions with flexible ligands.145 At the other extreme is induced folding, in which structure formation and binding occur concomitantly after the formation of the initial encounter complex. Given the complexity of many complexes involving intrinsically disordered regions, interactions involving both conformational selection of preformed elements and induced folding likely occur.92,146
MoRFs occurring in the Protein Data Bank147 can be classified into subtypes according to the structures they adopt in the bound state: α-MoRFs, β-MoRFs, and ι-MoRFs (Figure 7A–C),121 which form α-helices, β-strands, and irregular (but rigid) secondary structure when bound, respectively. MoRFs that contain combinations of different types of secondary structure are called complex (Figure 7D).121 The p53 protein contains multiple MoRFs that are disordered in the absence of their interactors (Figure 7E).120,121 The first p53 MoRF is located near the N-terminus and undergoes a transition from a disordered to an α-helical state upon interaction with the Mdm2 protein. In fact, this region of p53 exemplifies the high potential of IDRs for multiple partner binding as it is known to bind more than 40 different partners. However, for most of these complexes, the 3D structures are not determined, and therefore the MoRF type is not always known. The region between p53 residues 40 and 60 features an α-MoRF that functions as a secondary binding site for Mdm2 as well as a primary binding site for RPA70.148 In the absence of any binding partner, this region shows evidence of minimal helical secondary structure,149 whereas when bound to either Mdm2150 or RPA70,151 a stronger helical structure is observed. The C-terminal region of p53 also contains a MoRF that interacts with multiple partners, giving rise to different bound structures. For example, the S100B(ββ) protein induces a helical structure, while interaction with the Cdk2–cyclin A complex leads to an irregular ι-MoRF. An example of the role of MoRFs in scaffolding proteins is RNase E, which assembles the RNA degradosome.152 The flexible C-terminal end of RNase E contains several recognition motifs that are central to its scaffolding function and serve as binding sites for other members of the degradosome.153 For example, an α-MoRF interacts with enolase,154 and a β-MoRF binds polynucleotide phosphorylase.155 The recognition features are connected by disordered segments that accommodate assembly of the multiprotein complex by providing the required space and flexibility. Lee and co-workers123 have annotated the secondary structure propensities of many other regions that display transient structural elements and undergo disorder-to-order transitions, all of which have been experimentally confirmed by NMR spectroscopy.
Figure 7.

Classification of molecular recognition features (MoRFs) based on the secondary structure of the bound state. MoRFs (red ribbons) undergo disorder-to-order transition upon binding their partners (blue surfaces). (A) α-MoRF. BH3 domain of BAD (MoRF) bound to bcl-xl (partner) (PDB ID: 1G5J). (B) β-MoRF. Inhibitor of apoptosis protein DIAP1 (partner) bound to N-terminus of cell death protein GRIM (MoRF) (PDB ID: 1JD5). (C) ι-MoRF. AP-2 (partner) bound to the recognition motif of amphiphysin (MoRF) (PDB ID: 1KY7). (D) Complex-MoRF. Phosphotyrosine-binding domain (PTB) of the X11 protein (partner) bound to amyloid β A4 protein (MoRF) (PDB ID: 1X11). Note that the PTB domain of X11 actually binds unphosphorylated peptides and is a PTB by sequence similarity. Panels A–D reprinted with permission from ref (122). Copyright 2007 American Chemical Society. (E) Promiscuity of disorder-controlled interactions illustrated by the p53 interaction network. A structure versus disorder prediction on the p53 amino acid sequence is shown in the center of the figure (up = disorder, down = order) along with the structures of various regions of p53 bound to 14 different partners. The predictions for a central region of structure, and the disordered amino and carbonyl termini have been confirmed experimentally for p53. The various regions of p53 are color coded to show their structures in the complex and to map the binding segments to the amino acid sequence. Starting with the p53–DNA complex (top, left, magenta protein, blue DNA), and moving in a clockwise direction, the Protein Data Bank147 IDs and partner names are given as follows for the 14 complexes: (1tsr – DNA), (1gzh – 53BP1), (1q2d – gcn5), (3sak – p53 (tetramerization domain)), (1xqh – set9), (1h26 – cyclin A), (1ma3 – sirtuin), (1jsp – CBP bromo domain), (1dt7 – s100ββ), (2h1l – sv40 Large T antigen), (1ycs – 53BP2), (2gs0 – PH), (1ycr – MDM2), and (2b3g – RPA70). Reprinted with permission from ref (40). Copyright 2010 Elsevier.
Sequence context can play an active role in modulating the degree of structural preorganization of a MoRF. An example pertains to the study of DNA binding motifs in the basic regions (bRs) of basic region leucine zipper transcription factors.156 The bRs are 28–30 residue long regions predicted to be highly disordered and include a strongly conserved 10-residue DNA binding motif (DBM). The α-helicity (i.e., preference for α-helical conformation) of the DBM in the unbound form is modulated by the sequence of the N-terminal segment that is directly in cis to the DBM.156 For example, the N-terminal sequence contexts of Gcn4 and Cys3 DBMs contribute to a higher level of helicity of the DBM than the same region in c-Fos and Fra1 (whose DBMs have a low helicity). Essentially, the N-terminal sequence contexts are helix caps, and these can be used in different ways to ensure different levels of structural preorganization within an α-MoRF, thereby suggesting that investigating sequence contexts can provide useful clues when classifying MoRFs and linear motifs.157
3.3. Intrinsically Disordered Domains
Most protein domains that are identified using sequence-based approaches are structured, but some can be fully or largely disordered158 or contain conserved disordered regions,159 known as intrinsically disordered domains (IDDs). For instance, about 14% of Pfam domains have more than 50% of their residues in predicted disordered regions. Many well-known domains, such as the kinase-inhibitory domain (KID) of Cdk inhibitors (e.g., p2766) and the Wiskott–Aldrich syndrome protein (WASP)-homology domain 2 (WH2) of actin-binding proteins,158 have been shown experimentally to be fully disordered in isolation and solution. Protein domains with conserved disordered regions have a variety of functions, but are most commonly involved in DNA, RNA, and protein binding.159 Furthermore, domains that were gained during evolution by the extension of existing exons contain the highest degree of disordered regions.160 This suggests that exonization of previously noncoding regions could be an important mechanism for the addition of disordered segments to proteins.
Interestingly, it has also been observed that particular disordered regions frequently co-occur in the same sequence with specific protein domains.161,162 Some domain families appear only to require the presence of disorder in their neighborhood for functioning, while others seem to rely on the occurrence of disordered regions in specific locations relative to the start or end of the protein domain.161 For example, particular combinations of domains, involved mainly in regulatory, binding, receptor, and ion-channel roles, only occur with a disordered region inserted between them, while others only occur without a disordered domain between them. These observations imply that short disordered regions in the vicinity of protein domains complement the function of a structured domain, and in some cases may comprise separate functional modules in their own right. Thus, the co-occurrence of IDRs and structured domains in the same protein might be useful to gain insight into unannotated disordered regions.
3.4. Continuum of Functional Features
A measure that is often used to distinguish the different types of disordered binding modules is length; however, this is likely to stem primarily from the different methodology used for their detection. Protein domain detection relies on hidden Markov models,22 which is not the best approach for identifying short sequences, and therefore domain annotation tends to focus on larger sequence regions. In contrast, linear motifs in the ELM database are biased toward short binding modules (∼3–10 amino acids48,125) as these are more straightforward to annotate. Finally, the tendency of MoRFs and preformed elements to undergo disorder-to-order transitions and the statistics used for their detection means that these features tend to be slightly longer than annotated linear motifs.
Thus, although there are differences in the definitions of linear motifs and MoRFs, they share many common features72,163 including a tendency to undergo disorder-to-order transition (all MoRFs by definition and ∼60% of LMs48), an enrichment in IDRs (MoRFs by definition and ∼80% of LMs are in IDRs48,72), and a tendency to promote complex formation.48,100,122 Intrinsically disordered domains (IDDs) can also have significant overlap with MoRFs and linear motifs. For example, the WH2 domain is considered an IDD158 and is also defined as a motif in the ELM database.125 One feature that is probably more common in IDDs is that some are not only capable of binding to well-folded, structured domains (a mechanism shared with motifs and MoRFs), but can also bind each other in a process of mutually induced folding. For example, the nuclear coactivator binding domain (NCBD) of CREB-binding protein (CBP) and the activator for thyroid hormone and retinoid receptors (ACTR) domain of p160 are both disordered on their own but upon interaction form a complex by mutual synergistic folding.164 The overlap between linear motifs and MoRFs especially, but also IDDs, suggests that these functional features are different states in the same continuum of binding mechanisms involving disordered regions.
4. Structure
Intrinsically disordered regions and proteins show a wide variety of structural subtypes. These different types of disorder can be characterized using an array of experimental techniques (Box 2), and several resources collect computationally identified and experimentally verified disordered regions (Box 1). The following section discusses classification schemes that are based on structural features of disordered proteins.
Box 2. Experimental Characterization of Intrinsically Disordered Regions and Proteins.
IDPs and IDRs have been studied using a variety of experimental techniques, including NMR, SAXS, and smFRET. Nuclear magnetic resonance (NMR) spectroscopy is the key method to characterize protein disorder, due to its ability to provide residue-level information on protein structure and dynamics in solution.403 Many aspects of structural disorder can be detected directly using NMR, including local disorder, folding upon binding, and disorder in complex. In contrast to NMR methods, detection of disorder using X-ray crystallography techniques is mainly indirect as it relies on missing electron density.32 Another powerful method for detecting and characterizing IDPs is small-angle X-ray scattering (SAXS), which assesses protein dimensions and shape by measuring the scattered X-ray intensity caused by a sample. SAXS can be used to determine hydrodynamic parameters and the degree of globularity of a protein, which are good indicators to determine whether a protein is compact or unfolded.183,404 Single-molecule methods are also emerging for the study of structural disorder.179−182 These techniques minimize averaging over the heterogeneous ensembles of conformations in which disordered proteins naturally exist and thus are able to measure dynamics of individual molecules. For example, single-molecule fluorescence resonance energy transfer (smFRET) can measure dynamics and individual conformations of the unbound ensemble, intermediates during induced folding, and internal friction in the folding process.180−182 Atomic force microscopy (AFM) is also useful for the characterization of the conformational heterogeneity of single proteins.182 High-throughput proteomic approaches are mainly used to identify IDPs. These techniques enrich cellular extracts for disordered proteins, and then separate structured from disordered proteins, followed by identification (e.g., by mass spectrometry). For example, heat treatment enriches cell extracts for IDPs and depletes for proteins containing folded domains (see section 9.1).209 IDPs can also be identified on the basis of their susceptibility to degradation by the 20S proteasome under conditions in which structured proteins are resistant (see section 8.3).332 The degradation assays can be used to identify binding partners of IDPs that provide protection against degradation. Finally, computational techniques such as molecular dynamics (MD) simulations complement experimental approaches and provide important insights into IDP behavior.196,405 The DisProt, IDEAL, MobiDB, and pE-DB databases collect experimentally verified disordered regions and proteins (Box 1).
4.1. Structural Continuum
Proteins have been proposed to function within a conformational continuum, ranging from fully structured to completely disordered.37 The spectrum covers tightly folded domains that display either no disorder or only local disorder in loops or tails, multidomain proteins linked by disordered regions, compact molten globules containing extensive secondary structure, collapsed globules formed by polar sequence tracts, unfolded states that transiently populate local elements of secondary structure, and highly extended states that resemble statistical coils (Figure 8). In this model, there are no boundaries between the described states and native proteins could appear anywhere within the continuous landscape. IDRs are highly dynamic and fluctuate rapidly over an ensemble of heterogeneous conformations (see section 4.2).165 Thus, an IDR may fluctuate stochastically between several different states, transiently sampling coil-like states, localized secondary structure, and more compact globular states. Transient localized elements of secondary structure (most often helices) are common in amphipathic regions of the sequence and potentially play a role in binding processes.92 The structural characteristics and populations of the individual states in the conformational ensemble and the degree of compaction of the polypeptide chain are determined by the nature of the amino acids and their distribution in the IDR sequence (see section 5.1).166−168 For example, low and high average charges typically lead to disordered globules and swollen coils, respectively.166,167
Figure 8.
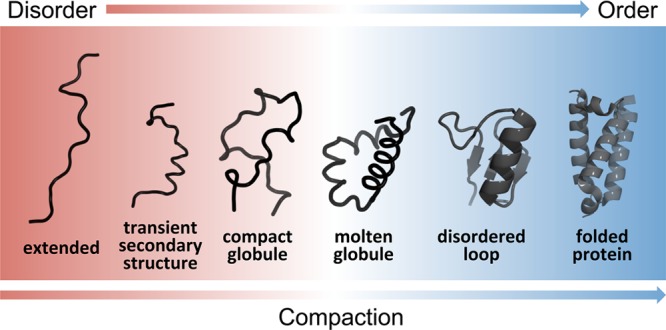
Schematic representation of the continuum model of protein structure. The color gradient represents a continuum of conformational states ranging from highly dynamic, expanded conformational ensembles (red) to compact, dynamically restricted, fully folded globular states (blue). Dynamically disordered states are represented by heavy lines, stably folded structures as cartoons. A characteristic of IDPs is that they rapidly interconvert between multiple states in the dynamic conformational ensemble. In the continuum model, the proteome would populate the entire spectrum of dynamics, disorder, and folded structure depicted.
4.2. Conformational Ensembles
Disordered regions in the native unbound state exist as dynamic ensembles of rapidly interconverting conformations,165,169,170 which can be described by relatively flat energy landscapes.99,171,172 Conditions, post-translational modifications, and binding events (see section 6) change the relative free energies of individual conformations as well as the energy differences between conformations.99,173−176 As a result, the populations of individual conformations within the ensemble change under different conditions. These individual states are often important for function. Thus, the dynamic nature of IDPs is best modeled by statistical approaches that describe the probabilities of individual conformations in the ensemble,172,177,178 and is best measured by experimental techniques that prevent conformational averaging (Box 2).179−182
4.3. Protein Quartet
The protein quartet model proposes that protein function can arise from four types of conformational states and the transitions between them: random coil, pre-molten globule, molten globule, and folded (Figure 9).32,34 In this model, unbound disordered regions could fall into all categories except for “folded”. Proteins in the pre-molten globule state are less compact than molten globules, but still show some residual secondary structure. In contrast, proteins in the random coil state show little or no secondary structure. The pre-molten globule state has a high propensity to participate in folding upon binding events,183 which would make this structural state suitable for disordered regions acting as effectors and scaffolds. On the basis of the notion that IDPs and IDRs possess great structural and sequence heterogeneity, proteins may also be considered as modular assemblies of foldons (independently foldable regions), inducible foldons (foldable regions that can gain structure as a result of interaction with specific partners), semifoldons (regions that are always partially folded), and nonfoldons (regions that never fold).184 The four distinct conformational states of the quartet model are a subset of the continuous spectrum of differently disordered states (see section 4.1),37 which extends from fully ordered to completely structure-less proteins, with everything in between. A single description of structure (such as the quartet states) may be suitable for the conformational average of a protein, while a structural continuum is a better description of an ensemble of different conformations (see section 4.2).
Figure 9.
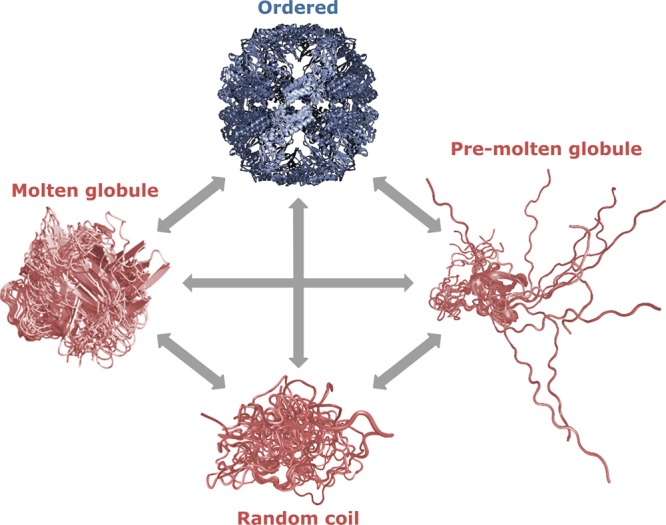
The protein quartet model of protein conformational states. In accordance with this model, protein function arises from four types of conformations of the polypeptide chain (ordered forms, molten globules, pre-molten globules, and random coils) and transitions between any of these states.
FG nucleoporins are an example of the functional significance that different disordered conformations can have. The porins make up the central part of nuclear pore complexes (NPCs) and regulate nucleocytoplasmic transport.185 Intrinsically disordered regions with multiple phenylalanine-glycine (FG) motifs make up large parts of the NPC gates. FG regions adopt various disordered conformations with specific functions.186 Some regions have the low charge characteristics of collapsed coils, while others are characterized by a high degree of charged amino acids, giving rise to relaxed and extended coil structures. Molecular dynamics simulations have shown that extended coils are more dynamic than collapsed coils, suggesting distinct functionalities for the two structural groups. Interestingly, some FG nucleoporins feature both types of disorder along their polypeptide chain. Combinations of disorder subtypes in nucleoporin domains are likely to contribute to NPC gating behavior by creating “traffic” zones with distinct physicochemical properties that influence the dynamics of substrate translocation through the nuclear envelope.186−189
4.4. Supertertiary Structure
IDRs allow for complex regulatory phenomena, as witnessed in the case of multidomain proteins in signaling and regulation.39,66,70,71,136,190 Because of the presence of structural disorder, functional domains, and short motifs, multidomain proteins are characterized by a dynamic ensemble of tertiary conformations. Some conformations are dominated by intramolecular domain–domain and domain–motif interactions and are closed and structured in nature, while other conformations are more open and disordered. This state of conformational variability within a protein lies between the tertiary structure of proteins and the quaternary structure of multiprotein assemblies, and has been termed supertertiary structure.191 Complex regulatory function stems from transitions in the ensemble of these structures, as demonstrated by several well-characterized proteins, such as the Wiskott–Aldrich syndrome protein (WASP, see section 2.4),94 the Src-family tyrosine kinase Hck,192 and the E3 ubiquitin ligase Smurf2.193
5. Sequence
The sequences of IDPs and IDRs have distinct compositional biases. They are enriched in charged and polar amino acids and depleted in bulky hydrophobic groups.31,44,194,195 These biases have led to the inference that disorder is a natural consequence of weakening the hydrophobic effects that drive folding of polypeptides into compact tertiary structures. Although disordered regions generally lack the ability to fold independently due to these biases in amino acid composition, distinct subsets of sequences that have different structural and functional characteristics can be identified within IDRs. The special sequence properties of disordered regions are the basis for many disorder prediction methods (Box 3). The following section covers sequence-based classification schemes of IDRs.
5.1. Sequence–Structural Ensemble Relationships
Systematic efforts combining experiments and computations have addressed the relationship between information encoded in amino acid sequences and the ensemble of conformations (see section 4.2) these sequences can sample in different conditions. These studies have focused on three major archetype sequences: polar tracts, polyelectrolytes, and polyampholytes.196 Polar tracts are sequence stretches enriched in polar amino acids such as glutamine, asparagine, serine, glycine, and proline, and deficient in charged as well as hydrophobic residues. These polar tracts (especially glutamine, asparagine, and glycine-rich sequences) form globules that are generally devoid of significant secondary structure preferences170,197−199 and can be as compact as well-folded domains.196 Collapse of polar tracts arises from the preference for self-solvation over solvation by the aqueous milieu. In this case, disorder derives from a lack of specificity for a single compact conformation as instead heterogeneous ensembles of conformations with similar stabilities and compactness are formed. The free energy landscape of polar tracts is weakly funneled and resembles an “egg carton”.200 Interestingly, the drive to collapse, which implies a drive to minimize the interface between the IDR and the surrounding solvent, can also give rise to the significant aggregation and solubility problems201 as is the case with several glutamine, asparagine, and glycine-rich sequences that are implicated in amyloid formation and phase separation.202
Another end of the compositional spectrum are polyelectrolytes. Their amino acid compositions are biased toward charged residues of one type such as the arginine-rich protamines166 or the Glu/Asp-rich prothymosin α.167 Experiments and simulations have shown that the tendency of polypeptide backbones to form ensembles of collapsed structures can be reversed by increasing the net charge per residue past a certain threshold (Figure 10A). The transition between globules and expanded coils is sharp, suggesting that small changes to the net charge per residue through post-translational modifications such as serine or threonine phosphorylation or lysine acetylation could cause reversible globule-to-coil transitions. These transitions might control the accessibility of SLiMs and MoRFs or even modulate the conformations of these elements.
Figure 10.

Original166 and modified204 diagram-of-states to classify predicted conformational properties of IDPs (and IDRs modeled as IDPs). (A) The original diagram predicts that sequences with a net charge per residue above 0.25 will be swollen coils. The three axes denote the fraction of positively charged residues, f+, the fraction of negatively charged residues, f–, and the hydropathy. All three parameters are calculated from the amino acid composition. Green dots correspond to 364 curated disordered sequences extracted from the DisProt database.203 These sequences have hydropathy values that designate them as being disordered; that is, they lie in the bottom portion of the pyramid by definition. Additional filters were used for chain length (more than 30 residues) and the fraction of proline residues (fpro < 0.3). 97% of sequences used in this annotation have a net charge per residue of less than 0.26 and are thus predicted to be globule formers.204 Adapted from ref (166). Copyright 2010 National Academy of Sciences of the United States of America. (B) Modified diagram-of-states from panel (A) with a focus only on the bottom portion of the pyramid (i.e., stipulating that the hydropathy is low enough to be ignored).204 The polyampholytic contribution expands the space encompassed by nonglobule-formers by subdividing the disordered globules space in panel (A) into three distinct regions of which sequences in regions 2 and 3 actually may not form globules. In these polyampholytic regions, one has to account for the total charge, in terms of the fraction of charged residues (FCR), as well as the net charge per residue (NCPR) as opposed to NCPR alone. Conformations in regions 2 and 3 are expected to be random-coil-like if oppositely charged residues are well mixed in the linear sequence. Otherwise, one can expect compact or semicompact conformations. The classification scheme uses only the amino acid sequence as input. Reprinted with permission from ref (204). Copyright 2013 National Academy of Sciences of the United States of America.
The impact of the net charge per residue on the conformational properties of IDRs can be summarized in a diagram-of-states (Figure 10A),166 which generalizes the original charge-hydropathy plot.31 The diagram classifies IDRs on the basis of their amino acid compositions. Annotation using curated disordered sequences from the DisProt database203 (Box 1) initially suggests that a vast majority (∼95%) of IDPs have amino acid compositions that predispose them to be globule formers (Figure 10A).204 However, most of these predicted globule formers are actually polyampholytes in that they are enriched in charged residues but have roughly equal numbers of positive and negative charges.204 Although such sequences are classified as globule formers on the basis of their low net charge per residue, in reality the conformational properties of polyampholytes are governed by the linear sequence distribution of oppositely charged residues. If the oppositely charged residues are segregated in the linear sequence, then electrostatic attractions between oppositely charged blocks cause chain collapse and result in hairpin or globular conformations. In sequences with well-mixed oppositely charged residues, the effects of electrostatic repulsions and attractions counterbalance. These mixed sequences adopt random-coil or globular conformations, depending on the total charge (in terms of the fraction of charged residues) (Figure 10B). Many IDPs are strong polyampholytes with well-mixed linear patterns of oppositely charged residues.204 Thus, IDPs are actually enriched in different classes of random coils that form swollen, loosely packed conformations (Figure 10B). Such random-coil sequences are likely to help improve the solubility profiles of connected structured domains (see section 9.1) and to promote the flexibility that is required for functions such as entropic tethers, which promote high local concentrations of connected protein parts, or entropic bristles, which occupy large volumes by rapid exploration of conformations. These biophysical principles of sequence–structural ensemble relationships enable the use of de novo sequence design as a tool for modulating these properties and assessing their impact on functions associated with IDPs and IDRs.
5.2. Prediction Flavors
Methods for predicting disordered regions have generally been successful (Box 3), but their prediction accuracies vary for different types of disordered regions.205 Some predictors accurately predict certain disordered regions but have lower accuracy predicting others, whereas other predictors give opposite results. Vucetic and co-workers205 classified protein disorder into three different “flavors” based on competition between disorder predictors. These V, C, and S disorder flavors (corresponding to the names of the disorder predictors that best predict them: VL-2V, VL-2C, and VL-2S) show differences in sequence composition, and combinations of flavors could be associated with different protein functions. For example, disordered regions that bind to other proteins are enriched for flavor S, while disordered ribosomal proteins predominantly belong to flavor V. Flavor C gave strong disorder predictions for sugar binding domains.
5.3. Disorder–Sequence Complexity Space
The relationship between sequence complexity and disorder propensity provides further insight into the structural and functional variations of IDRs.206 Different functional classes of proteins often show a different disorder–sequence complexity (DC) space distribution. A frequently observed DC-distribution is composed of a compact structured part and a section extending out into the low-complexity and high-disorder space before looping back into the structured region. This pattern describes a disordered linker region between structured domains. An example is the bacterial translation initiation factor, which contains a sequence that locates to the low-complexity, high-disorder region of DC space. This loop connects the N- and C-terminal domains, which are high-structure and high-complexity.206,207 Functionally related proteins have similar disorder–sequence complexity distributions, suggesting that these distributions might be useful for predicting the function of a disordered region.
5.4. Overall Degree of Disorder
Large-scale studies into IDP function often group the proteins on the basis of some measure of disorder. For example, protein sequences have been categorized on the basis of the overall degree of disorder (i.e., the fraction of residues that is shown or predicted to be disordered),68,208 resulting in groups of structured proteins (0–10% disorder), moderately disordered proteins (10–30% disorder), and highly disordered proteins (30–100% disorder). For 24% of human protein-coding genes, at least 30% of residues are predicted to be disordered (Figure 2A). Other studies classified proteins on the basis of an overall score of disorder for the whole protein,209 and the presence or absence of continuous stretches of disordered residues with a specific length.35,51,161,208 Largely structured proteins are enriched for metabolic functions, while highly disordered proteins function predominantly in regulation. Hence, classification of disordered proteins based on the level of disorder provides clues about what types of functions are likely.
5.5. Length of Disordered Regions
The length of IDRs in human follows a power law distribution: there are large numbers of short disordered regions and increasingly smaller numbers of longer ones.210 Other eukaryotic and prokaryotic proteomes show similar disorder length profiles. 44% of human protein-coding genes contain substantial disordered segments of >30 amino acids in length49 (similar data shown in Figure 2A). Short IDRs may function as linkers and contain individual linear motifs or MoRFs, whereas longer disordered regions might be entropic chains or contain combinations of motifs or domains functioning in recognition. Very long disordered regions (more than 500 residues) are typically over-represented in transcription-related functions,211 whereas proteins containing IDRs of 300–500 residues in length are enriched for kinase and phosphatase functions. Shorter IDRs (less than 50 residues) tend to be linked to metal ion binding, ion channels, and GTPase regulatory functions. Thus, the length of a disordered region can also provide a useful indication about the functional nature of the protein containing it.
5.6. Position of Disordered Regions
Almost all human proteins have some disordered residues within their terminal regions.59 For example, 97% of proteins have predicted disorder in the first or last five residues.161 Disordered N-terminal tails are common in DNA-binding proteins, and have been shown to contribute to efficient DNA scanning.212 Furthermore, proteins that are relatively rich in disordered residues at the C-terminus are often associated with transcription factor repressor and activator activities as compared to proteins rich in internal or N-terminal disorder.211 Membrane proteins, depending on their topology of insertion, also contain disordered regions in the N- or C-terminus, but their sequence composition is different as compared to disordered regions in cytosolic proteins.213 Ion channel proteins are enriched for disordered residues at the N-terminus, and the same is true to a lesser extent for C-terminal disorder.211 These terminal disordered regions are often functionally relevant, as illustrated by their role in the inactivation of voltage-gated potassium channels.214 Similarly, many G-protein-coupled receptors (GPCRs) have large disordered regions in their C-terminus, and often in the intracellular loops.215 Several of them harbor peptide motifs that link ligand binding in the transmembrane region of the receptor to intracellular effectors, or contain PTM sites or linear motifs that govern their stability.216 Finally, proteins that are relatively rich in internal disordered regions are weakly enriched for transcription regulator and DNA binding activity.211 Thus, the relative position of a disordered region in a sequence provides clues about the function of the protein containing it.
5.7. Tandem Repeats
Short tandem repeats are common in IDRs and IDPs.61,217−220 For instance, as much as 96% of polyglutamate and polyserine stretches lie within disordered regions.219 Similarly, large fractions were found for proline, glycine, glutamine, lysine, aspartate, arginine, histidine, and threonine repeats. In contrast, polyleucine stretches occur predominantly within structured regions. These observations agree with the compositional bias of disordered regions (see section 5.1); the most common tandem repeats in IDRs are made up of disorder-promoting residues44,194 and of sequence patterns that are typically associated with disorder.195 Moreover, a distinction between perfect and imperfect tandem repeats suggests that as the repeat perfection increases, so does the disorder content.219
Repeats of different composition have been linked to specific functions.218,221 Consequently, the presence of particular types of repeats is likely to contribute to IDR functioning. Descriptions and examples of different classes of disordered tandem repeats and their structural characteristics have been reviewed previously.218 For instance, polyproline and polyglutamine stretches are associated with protein and nucleic acid binding and transcription factor activity.222,223 Protein segments enriched for glutamine and asparagine often occur in disordered regions224 and are abundant in eukaryotic proteomes,225 despite their propensity to aggregate or form coiled-coil structures.226 The aggregation propensity of the Q/N-enriched segments is exploited in the formation of physiologically relevant assemblies such as P-bodies (e.g., Ccr4 and Pop2), stress granules, and processing bodies.227 However, expanded polyglutamine repeats are also associated with neurodegenerative disorders, the most well-known being Huntington’s disease.228 Moreover, several prion-like yeast proteins (e.g., Sup35p and Ure2p) contain intrinsically disordered Q/N-rich protein segments that have been implicated in the switch between a soluble and an insoluble, aggregated form.225,229 Another example of functional disordered repeats occurs in the SR protein family of splicing factors (e.g., ASF/SF2 and SRp75).230,231 SR proteins mediate the assembly of spliceosome components. They consist of an N-terminal RNA-recognition motif and a disordered C-terminus with tandem repeats of arginine and serine residues (RS domain). Phosphorylation switches the RS domain of the serine/arginine-rich splicing factor 1 (SRSF1) from a fully disordered state to a more rigid structure.232 Other disordered repeats associated with a specific function include sequences enriched in lysine, alanine, and proline in the histone H1 C-terminal domain, which are involved in the formation of 30 nm chromatin fiber by binding linker DNA between the nucleosomes.233,234 A final example is dentin sialophosphoprotein (DSPP), which contains extensively phosphorylated repeats of aspartic acid and serine involved in calcium phosphate binding (see section 9.3).235 Some repeat-containing regions are also prone to undergo phase transitions from a soluble monomeric state to an insoluble large assembly form, as demonstrated for regions rich in proline, threonine, and serine residues in mucins (see section 9.2).236
6. Protein Interactions
Disordered region-mediated molecular interactions have been proposed to work using a combination of conformational selection and induced folding.92,146,237 These mechanisms of binding are two extreme possibilities and are not mutually exclusive. Both play a role in the interaction between two proteins, the dominant mechanism depending, for example, on the concentrations of the individual proteins238 and the association rate constants.84 In conformational selection, addition of binding partners can result in a population shift in the conformational ensemble of a disordered protein (see section 4.2) toward the conformation that is most favorable for binding.119,145,173,175 This mechanism has been observed in both protein–protein and protein–nucleic acid interactions.173 Evidence for the role of conformational selection in IDP binding comes, for example, from the interaction between PDEγ and the α-subunit of transducin,239 which is important in phototransduction. The dynamic ensemble of unbound PDEγ includes a loosely folded state that resembles its structure when bound to transducin. In induced folding, a protein undergoes a disorder-to-order transition upon association with its binding partner.92,146,240 Evidence for this mechanism in IDP binding comes, for example, from a study investigating the disordered pKID region of CREB and the KIX domain of CREB-binding protein. Upon binding of pKID to the KIX domain, an ensemble of transient encounter complexes forms, which appear to be stabilized primarily by hydrophobic contacts and evolve to form the fully bound state via an intermediate state without disassociation of the two domains.91,241
6.1. Fuzzy Complexes
Although disordered protein regions frequently fold upon interacting with other proteins, complexes with IDPs often retain significant conformational freedom and can only be described as structural ensembles.242 The conformations that disordered proteins adopt in the bound state cover a continuum, similar to the structural spectrum of free, unbound IDPs,243 and range from static to dynamic, and from full to segmental disorder.242 In static disordered complexes, disordered regions can adopt multiple well-defined conformations in the complex, whereas in dynamic disorder they fluctuate between various states of an ensemble in the bound state.
Disorder in the bound state can be classified into four molecular modes of action, each of which is associated with specific molecular functions (Figure 11A–D).176,242 (i) The polymorphic model is a form of static disorder, with alternative bound conformations serving distinct functions by having different effects on the binding partner. Examples are the Tcf4 β-catenin binding domain244 and the WH2 binding domains of thymosin β4 or ciboulot,245 which have been shown to adopt several distinct conformations upon β-catenin and actin binding, respectively. Different actin–WH2 domain complexes have alternative interaction interfaces and result in actin polymers with different topologies.245 The (ii) clamp and (iii) flanking models represent forms of dynamic disorder in which complex formation either involves folding upon binding of two disordered segments that are connected by a linker that remains disordered, or the reverse situation, respectively. The cyclin-dependent kinase (Cdk) inhibitor p21, for example, acts as a clamp. It contains a dynamic helical subdomain that serves as an adaptable linker that connects two binding domains and enables these to specifically bind distinct cyclin and Cdk complex combinations.246 In both the clamp and the flanking models, disordered regions near the interacting protein segments (often short peptide motifs) contribute to binding by influencing affinity and specificity.242,247 This phenomenon relates to the importance of the sequence context in modulating disordered binding elements (see section 3). Finally, (iv) the random model is an extreme version of dynamic disorder in protein complexes, which occurs when the IDR remains largely disordered even in the bound state. In this case, interaction is achieved via linear motifs that do not get fixed upon binding. An example is the self-assembly of elastin, where solid-state NMR has provided evidence for dynamic disorder within elastin fibers, which exhibit random-coil like chemical shift values.248 Another case is the complex between the Cdk inhibitor Sic1 and the SCF ubiquitin ligase subunit Cdc4, which is formed in a phosphorylation-dependent manner.249 At any given time, only one out of nine Sic1 phosphorylation sites interact with the core Cdc4 binding site, while the others contribute to the binding energy via a secondary binding site or via long-range electrostatic interactions (Figure 12N). Hence, binding interchanges dynamically within the Sic1–Cdc4 complex to provide ultrafine tuning of the affinity.249,250
Figure 11.

Classification of fuzzy complexes by topology (upper panel) and by mechanism (lower panel). Blue arrows indicate interactions between fuzzy disordered regions and structured molecules. Protein Data Bank147 identifiers for the structures are given in parentheses. Topological categories: (A) Polymorphic. The WH2 domain of ciboulot interacts with actin in alternative locations: via an 18-residue segment (3u9z) or via only three residues (2ff3). The flanking regions remain dynamically disordered. (B) Clamp. The Oct-1 transcription factor has a bipartite DNA recognition motif. The two globular binding domains are connected by a 23 residue long disordered linker (1hf0), shortening of which reduces binding affinity. (C) Flanking. The p27Kip1 cell-cycle kinase inhibitor binds to the cyclin–Cdk2 complex (1jsu). The kinase binding site is flanked by a ∼100 residue long disordered linker, which enables T187 at the C-terminus to be phosphorylated. (D) Random. UmuD2 is a dimer that is produced from UmuD by RecA-facilitated self-cleavage (1i4v). The resulting proteins exhibit a random coil signal in circular dichroism experiments at physiologically relevant concentrations. Mechanistic categories: (E) Conformational selection. The fuzzy N-terminal acidic tail of the Max transcription factor (1nkp) facilitates formation of the DNA binding helix (dark red) of the leucine zipper basic helix–loop–helix (bHLH) motif. (F) Flexibility modulation. The disordered serine/arginine-rich region of the Ets-1 transcription factor (1mdm) changes DNA binding affinity by 100–1000-fold by modulating the flexibility of the binding segment via transient interactions. (G) Competitive binding. The acidic fuzzy C-terminal tail of high-mobility group protein B1 (2gzk) competes with DNA for the positively charged binding surfaces. (H) Tethering. The binding of the virion protein 16 activation domain to the human transcriptional coactivator positive cofactor 4 (2phe) is facilitated by acidic disordered regions, which anchor the binding segments.
Figure 12.

A portrait gallery of disorder-based complexes. Illustrative examples of various interaction modes of intrinsically disordered proteins are shown. Protein Data Bank147 identifiers for the structures are given in parentheses. (A) MoRFs. Aa, α-MoRF, a complex between the botulinum neurotoxin (red helix) and its receptor (a blue cloud) (2NM1); Ab, ι-MoRF, a complex between an 18-mer cognate peptide derived from the α1 subunit of the nicotinic acetylcholine receptor from Torpedo californica (red helix) and α-cobratoxin (a blue cloud) (1LXH). (B) Wrappers. Ba, rat PP1 (blue cloud) complexed with mouse inhibitor-2 (red helices) (2O8A); Bb, a complex between the paired domain from the Drosophila paired (prd) protein and DNA (1PDN). (C) Penetrator. Ribosomal protein s12 embedded into the rRNA (1N34). (D) Huggers. Da, E. colitrp repressor dimer (1ZT9); Db, tetramerization domain of p53 (1PES); Dc, tetramerization domain of p73 (2WQI). (E) Intertwined strings. Ea, dimeric coiled coil, a basic coiled-coil protein from Eubacterium eligens ATCC 27750 (3HNW); Eb, trimeric coiled coil, salmonella trimeric autotransporter adhesin, SadA (2WPQ); Ec, tetrameric coiled coil, the virion-associated protein P3 from Caulimovirus (2O1J). (F) Long cylindrical containers. Fa, pentameric coiled coil, side and top views of the assembly domain of cartilage oligomeric matrix protein (1FBM); Fb, side and top views of the seven-helix coiled coil, engineered version of the GCN4 leucine zipper (2HY6). (G) Connectors. Ga, human heat shock factor binding protein 1 (3CI9); Gb, the bacterial cell division protein ZapA from Pseudomonas aeruginosa (1W2E). (H) Armature. Ha, side and top views of the envelope glycoprotein GP2 from Ebola virus (2EBO); Hb, side and top views of a complex between the N- and C-terminal peptides derived from the membrane fusion protein of the Visna (1JEK). (I) Tweezers or forceps. A complex between c-Jun, c-Fos, and DNA. Proteins are shown as red helices, whereas DNA is shown as a blue cloud (1FOS). (J) Grabbers. Structure of the complex between βPIX coiled coil (red helices) and Shank PDZ (blue cloud) (3L4F). (K) Tentacles. Structure of the hexameric molecular chaperone prefoldin from the archaeum Methanobacterium thermoautotrophicum (1FXK). (L) Pullers. Structure of the ClpB chaperone from Thermus thermophilus (1QVR). (M) Chameleons. The C-terminal fragment of p53 gains different types of secondary structure in complexes with four different binding partners, cyclin A (1H26), sirtuin (1MA3), CBP bromo domain (1JSP), and s100ββ (1DT7). Panels A–M reprinted with permission from ref (257). Copyright 2011 The Royal Society of Chemistry. (N) Dynamic complexes. Schematic representation of the polyelectrostatic model of the Sic1–Cdc4 interaction. An IDP (ribbon) interacts with a folded receptor (gray shape) through several distinct binding motifs and an ensemble of conformations (indicated by four representations of the interaction). The intrinsically disordered protein possesses positive and negative charges (depicted as blue and red circles, respectively) giving rise to a net charge ql, while the binding site in the receptor (light blue) has a charge qr. The effective distance ⟨r⟩ is between the binding site and the center of mass of the intrinsically disordered protein. Panel N was reprinted with permission from ref (243). Copyright 2010 John Wiley & Sons, Inc.
Bound disordered regions can impact the interaction affinity and specificity of the complex and tune interactions of folded regions176 with proteins or DNA.251 Four different mechanisms have been proposed for the formation of fuzzy complexes (Figure 11E–H). (i) The first is conformational selection, when the disordered region shifts the conformational equilibrium of the binding interface toward the bound form. The fuzzy N-terminal tail of the Max transcription factor, for example, reduces electrostatic repulsion in the basic helix–loop–helix (bHLH) domain and thereby facilitates formation of the DNA recognition helices, which increases binding affinity by 10–100-fold.252 (ii) In the second mechanism, the disordered region(s) modulate flexibility of the binding interface. The serine- and arginine-rich region of the Ets-1 transcription factor exemplifies this mechanism, which reduces DNA binding affinity by 100–1000-fold.253 (iii) The third mechanism is competitive binding of the disordered region. Here, the IDR acts as a competitive inhibitor of other regions in the same protein for binding to a partner. The acidic fuzzy C-terminal tail of high-mobility group protein B1 (HMGB1) negatively regulates interaction of the HMG DNA binding domains by occluding the basic DNA-binding surfaces.254 (iv) In the fourth mechanism, the disordered region serves to tether a weak-affinity binding region to increase its local concentration. For example, a fuzzy N-terminal domain anchors the human positive cofactor 4 (PC4) to several transactivation domains including the herpes simplex virion protein 16 (VP16).255 All mechanisms of disordered complex formation affect binding to different degrees and can be further tuned by post-translational modifications.176,251 PTMs in the disordered region may act as affinity tuners by modulating the charge available for biomolecular interactions.256
6.2. Binding Plasticity
Structural analysis of a large number of intrinsic disorder-based protein complexes resulted in another categorization of IDRs based on their binding plasticity (Figure 12).257 Examples of relatively static IDR-based complexes are (i) mono- and polyvalent complexes, which typically consist of interactions between disordered segments and one or multiple spatially distant binding sites on their binding partners, respectively, (ii) chameleons, such as p53, that have different structures when binding to different proteins, (iii) penetrators that bury significant parts of the protein inside their binding partners, and (iv) huggers, which function in protein oligomerization, for example, by coupled folding and binding of disordered monomers. In addition to these relatively static complexes involving IDRs, one can identify coiled-coil-based complexes. Regions that make up coiled coils are typically highly disordered in monomeric state and gain helical structure upon coiled-coil formation, giving rise to several distinguishable types of complexes, such as intertwined strings, connectors, armatures, and tentacles.
7. Evolution
Disordered regions typically evolve faster than structured domains.51−56,107 This behavior largely stems from a lack of constraints on maintaining packing interactions, which drives purifying selection in structured sequences.258 However, disordered residues do display a wide range of evolutionary rates (Box 2). The following section discusses the evolutionary classifications of disordered protein regions. IDRs with similar functions and properties tend to have similar evolutionary characteristics.
7.1. Sequence Conservation
While the amino acid sequence of disordered regions evolves at different rates, the property of disorder is usually conserved for functional sequences.54,159 Sequence conservation of IDRs varies according to their specific functions and provides another means for their classification.54,259,260 Three biologically distinct classes of IDRs with specific function were identified using a combination of disorder prediction and multiple sequence alignment of orthologous groups across 23 species in the yeast clade (Figure 13): (i) flexible disorder describes regions where disorder is conserved but that have quickly evolving amino acid sequences (i.e., there is a requirement to be disordered, regardless of the exact sequence), (ii) constrained disorder describes regions of conserved disorder with also highly conserved amino acid sequences, and (iii) nonconserved disorder, where not even the property of being disordered is conserved in closely related species. For flexible disorder, low sequence conservation is expected if the property of disorder itself, as opposed to disorder in combination with specific sequence, is the only requirement for function. Examples of functions that mainly require the biophysical flexibility of disordered regions are entropic springs, spacers, and flexible linkers between well-folded protein domains.37,39,57,58 The linker in RPA70 is an example where the dynamic behavior is conserved even when the sequence conservation is low.60 Flexible disorder is the most common of the three evolutionary classes with just over one-half of disordered residues in yeast. It appears to account not just for the “flexibility” functions mentioned above, but also for many of the characteristics traditionally associated with disordered regions, such as strong association with signaling and regulation processes,35,50,104,190,261,262 rapid sequence evolution,51−56,107 the presence of short linear motifs (which are themselves conserved, see below),47,72 and tight regulation (see section 8).68,263 By contrast, constrained disorder (about a third of disordered residues in yeast) is associated with different properties and functions, such as chaperone activity and RNA-binding ribosomal proteins.54 Many proteins that contain the evolutionarily constrained type of disorder can adopt a fixed conformation, suggesting that these regions might undergo folding upon binding to their targets. This structural transition might impose a high degree of local structural constraints, which results in constraints on the protein sequence alongside requirements to be flexible.54 Constrained disordered residues also occur more often in annotated protein sequence families (domains) than flexible disorder, but both types are strongly depleted in domains compared to structured regions. In human, both flexible and constrained disorder are enriched in proteins functioning in differentiation and development,264 which reflects the importance of IDPs in these processes. Finally, nonconserved disorder accounts for around 17% of disordered residues in yeast and appears to be largely nonfunctional.
Figure 13.

Classification of disordered regions according to their evolutionary conservation (constrained, flexible, and nonconserved disorder). (A) Schematic of computing disorder conservation and amino acid sequence conservation. The alignments are used to calculate the percentage of sequences in which a residue is disordered and the percentage of sequences in which the amino acid itself is conserved. A residue is considered to be conserved disordered if the property of disorder is conserved in at least one-half of the species. Similarly, the amino acid type of a residue is considered conserved if it is present in at least one-half of the species. Disordered residues in which both sequence and disorder are conserved are referred to as constrained disorder. Disordered residues in which disorder is conserved but not the amino acid sequence are referred to as flexible disorder. Residues that are disordered in S. cerevisiae but not cases of conserved disorder are referred to as nonconserved disorder. (B) Disorder splits into three distinct phenomena. Functional enrichment maps of proteins enriched in flexible disorder versus constrained disorder. The area of each rectangle is proportional to the occurrence of that type of disorder in the alignments. Related gene ontology terms are grouped based on gene overlap. Reprinted with permission from ref (54). Copyright 2011 Springer Science + Business Media.
Short linear motifs (see section 3.1)48,125 constitute a special case. Even though SLiMs almost exclusively lie within disordered regions, their own amino acid sequence tends to be conserved.48 These properties, together with the difficulty of aligning rapidly evolving disordered sequences, result in the motifs to move around when comparing their position in different sequences. In fact, not only do motifs move around (due to insertions and deletions of amino acids around the motif in the sequence67,265), they can also permute their positions with respect to other structural and functional modules. For example, SUMO modification sites in p53 are seen after and before the oligomerization domain in human and fly, respectively.266 Such behavior could emerge by convergent evolution and loss of the motif in the original site, as only a few amino acids need to mutate to make a new motif elsewhere in the sequence. As long as the position of the motif with respect to the other modules does not affect function, such permutations will not affect fitness and hence may emerge relatively easily during evolution. These are indeed confounding issues when aligning disordered regions among orthologous proteins to identify functional motifs.
In many ways, the disordered regions that contain SLiMs constitute flexible disorder as by the above classification, as their main role is to provide flexibility to enable access to the linear motif for proteins that will bind them as ligands267 or introduce post-translational modifications.47,48 Phosphorylation sites are closely related to short linear motifs that function in binding, but are often too short and weakly conserved to recognize via computational means.268 More than 90% of sites phosphorylated by the yeast Cdk1 are in predicted disordered regions,67 as consistent with previous studies highlighting the importance of IDRs as display sites for phosphorylation and other PTMs (see sections 2.2 and 3.1).45,46 Comparison of the phosphorylation sites in orthologues of the Cdk1 substrates revealed that the precise position of most phosphorylation sites is not conserved. Instead, clusters of sites move around in the alignment of rapidly evolving disordered regions.69,250,269 Another example of the role of flexible disorder in signaling and regulation is the yeast serine-arginine protein kinase Sky1, which regulates proteins involved in mRNA metabolism and cation homeostasis. The Sky1 C-terminal loop is intrinsically disordered and contains phosphosites that are important for regulating its kinase activity.270 Conservation analysis has shown that the loop is conserved for disorder but not for sequence.54
The combination of sequence conservation of IDRs and conservation of their amino acid composition between human and seven other eukaryotes (chimp, dog, rat, mouse, fly, worm, and yeast) also identifies functional preferences.260 IDRs with high residue conservation (HR) are enriched in proteins involved in transcription regulation and DNA binding. Low residue conservation in combination with high conservation of the amino acid type composition (LRHT) of the IDR (i.e., high similarity of overall amino acid composition between the human IDR and its orthologs) is often associated with ATPase and nuclease activities. Finally, IDRs that show neither conservation of sequence nor conservation of amino acid composition (LRLT) are abundant in (metal) ion binding proteins.
7.2. Lineage and Species Specificity
Increasingly complex organisms have higher abundances of disorder in their proteomes.35,271 An average of 2% of archaeal, 4% of bacterial, and 33% of eukaryotic proteins have been predicted to contain regions of disorder over 30 residues in length,35 although there is much variation within kingdoms.272,273 In human, 31% of proteins are more than 35% unstructured,68 and 44% contain stretches of disorder longer than 30 residues49,161,208 (similar data shown in Figure 2A). Human IDPs are spread relatively uniformly across the chromosomes, with percentages ranging from 38% (for genes encoding IDPs on chromosome 21) to 50% on chromosomes 12 and X.161 A computational analysis of disorder in prokaryotes has corroborated the higher abundance of disorder in Bacteria as compared to Archaea.274 Moreover, in agreement with the low abundance of disorder in prokaryotes, none of the 13 mitochondrial-encoded proteins are disordered.161 Systematic analysis of IDP occurrence in 53 archaeal species showed that disorder content is highly species-dependent.275 For example, Thermoproteales and Halobacteria proteomes have 14% and 34% disordered residues, respectively. Harsh environmental conditions seem to favor higher disorder contents, suggesting that some of the archaeal IDPs evolved to help accommodate hostile habitats.276
Structural disorder is more common in viruses than in prokaryotes.277 The characteristics of IDRs seem well suited for especially small RNA viruses with extremely compact genomes.278,279 For example, disordered regions could buffer the deleterious effects of mutations introduced by low-fidelity virus polymerases better than would structured domains.277 The flexibility of IDRs to interact with many different proteins, such as proteins of the host immune system, is another useful feature for compact viruses because it maximizes the amount of functionality they encode while minimizing the required genetic information.280 At the same time, several human innate immunity proteins have predicted disordered regions that could be important for their pathogen defense function.281 For example, the RIG-I-like receptors (RLRs) RIG-I and MDA5 recognize different types of viral double-stranded RNA (dsRNA).282 This functional divergence is partly achieved by differential flexibility of a loop that is rigid in RIG-I, but disordered in MDA5, resulting in different RNA binding preferences.283 Furthermore, the disordered linker between the RNA-binding domains and the two N-terminal CARD (caspase activation and recruitment) domains of MDA5 helps facilitate oligomerization of the CARD domains, which initiates downstream signaling.283 Activated RIG-I and MDA5 promote the formation of prion-like aggregates of the CARD domains of MAVS (mitochondrial antiviral-signaling).284 MAVS has a highly disordered central region that contains multiple phosphorylation sites and interacts with several proteins, such as TRAF2 and TRAF6 through their respective consensus binding motifs (PxQx[TS] and PxExx[FYWHDE], respectively).285 These interactions are part of a signaling pathway that activates the transcription factors IRF3/7 and NF-κB, leading to the expression of proinflammatory cytokines such as IFN-α/β and various proteins with direct antiviral activity.282 For example, to counteract viral infection, protein kinase R (PKR) phosphorylates the translation initiation factor eIF2α in the presence dsRNA, which reduces global protein synthesis in the cell.286 PKR contains a long disordered interdomain region that may become ordered upon RNA binding and could affect PKR dimerization.287,288 Interestingly, viruses counteract PKR action by mimicking eIF2α and competing for PKR binding, as has been shown in the case of the poxvirus protein K3L.289 PKR is under intense positive selection to keep recognizing eIF2α while minimizing interaction with viral antagonists.289 Many of the changing sites in PKR are in a dynamic loop near the interaction interface with both eIF2α and K3L.290 Similarly, recognition of retrovirus capsids by the restriction factor TRIM5α is mediated by disordered regions in the SPRY domain, which bear many positively selected residues that are essential for the antiviral activity.291 The SPRY domain exists as an ensemble of disordered conformations that determine the specificity and affinity of the interaction between TRIM5α and the viral capsid.292−294 In this way, the evolutionary flexibility of disordered regions (see section 7.1) provides opportunities for proteins of the host immune system to compete with rapidly changing pathogens while maintaining their functionality.
In addition to the variation in prevalence of disordered regions between species, different kingdoms of life seem to use conserved IDRs for different functions: eukaryotic and viral proteins use disorder mainly for mediating transient protein–protein interactions in signaling and regulation, while prokaryotes use disorder mainly for longer lasting interactions involved in complex formation.159 Thus, knowledge on the lineage, species, and origin of a disordered region could help in predicting its likely function.
7.3. Evolutionary History and Mechanism of Repeat Expansion
Tandem repeats are enriched for intrinsic disorder (see section 5.7), and IDRs are increasingly abundant in increasingly complex organisms (see section 7.2). The genetic instability of repetitive genomic regions in combination with the structurally permissive nature of IDRs might have driven the increase in the amount of disorder during evolution. Disordered repeat regions have been shown to fall into three categories, based on their evolutionary history and acquired functional properties (Figure 14):61 type I regions have not undergone functional diversification after repeat expansion (e.g., the titin PEVK domain), type II repeats have acquired diverse functions due to mutation or differential location within the sequence (e.g., the C-terminal domain of eukaryotic RNA polymerase II), and type III regions have gained new functions as a consequence of their expansion per se (e.g., the prion protein octarepeat region).
Figure 14.
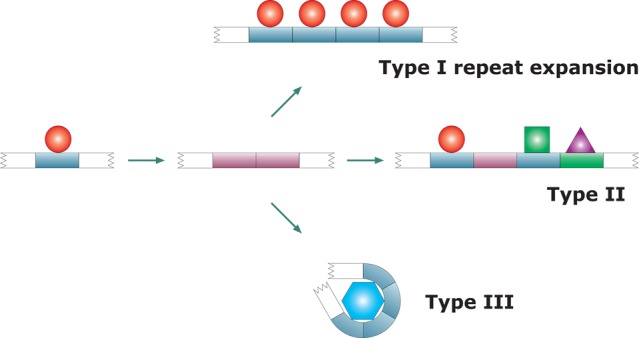
Repeat expansion creates IDRs. IDRs are abundant in repeating sequence elements, which suggests that repeat expansion is an important mechanism by which genetic material encoding for structural disorder is generated. The expanding repeats may fall into three classes (types) in terms of their functional diversification following expansion. Individual repeats may remain functionally equivalent (type I), or diversify (type II), or collectively acquire a completely new function (type III). Dark-tone red indicates structural disorder of the repeat, which may undergo full (dark-tone blue) or partial (green) induced folding upon binding to a partner. Adapted with permission from ref (61). Copyright 2003 John Wiley & Sons, Inc.
8. Regulation
Altered availability of IDPs is associated with diseases such as cancer and neurodegeneration.190,263,295−299 Indeed, genes that are harmful when overexpressed (i.e., dosage-sensitive genes) often encode proteins with disordered segments.300 Multiple mechanisms at different stages during gene expression (from transcript synthesis to protein degradation) control the availability of IDPs.68 Their tight regulation ensures that IDPs are available in appropriate levels and for the right amount of time, thereby minimizing the likelihood of ectopic interactions. Disease-causing altered availability of IDPs may result in imbalances in signaling pathways by sequestering proteins through nonfunctional interactions involving disordered segments (i.e., molecular titration263). The following section discusses possible functional roles of proteins with IDRs based on their cellular regulatory properties such as transcript abundance, alternative splicing, degradation kinetics, and post-translational processing.
8.1. Expression Patterns
Five different expression patterns were identified for transcripts encoding highly disordered proteins by investigating the mRNA levels from over 70 different human tissues and comparing the number of tissues in which IDP transcripts are expressed against the level of expression (Figure 15).208 The expression classes are associated with specific functions. (i) The first subgroup (Figure 15, light blue markers) shows constitutive high expression in all tissues and consists exclusively of large ribosomal subunit proteins, which are almost entirely disordered. (ii) The second group (blue-green) represents transcripts that show high expression levels in the majority of tissues. These often function as protease inhibitors, splicing factors, and complex assemblers. (iii) Moderately expressed transcripts (green) typically encode disordered proteins involved in DNA binding and transcription regulation. (iv) IDPs that are expressed in a tissue-specific manner (yellow) are enriched for cell organization regulators, transcription cofactors, and factors that promote complex disassembly. Finally, (v) the remaining transcripts form a group (gray) not detected to be abundant in any of the tissues studied. This low and transient expression group contains more than one-half of the IDP transcripts analyzed and has a variety of functions.
Figure 15.

A summary of expression–function trends for human transcripts encoding highly disordered proteins. The x-axis represents the log10 number of tissues in which the transcript is expressed; the y-axis represents the log10 average magnitude of expression within the tissues. From the data, five distinct functional classes of highly disordered human proteins become apparent. Adapted with permission from ref (208). Copyright 2009 Springer Science + Business Media.
8.2. Alternative Splicing
Trends in transcriptional regulation (alternative promotor and polyadenylation site usage) and post-transcriptional regulation (alternative splicing by inclusion or exclusion of exons) can also be informative of the role that specific disordered protein regions play in the cell (Figure 16). Alternatively spliced exons are overall more likely to encode intrinsically disordered rather than structured protein segments.161,301−303 This tendency is even more pronounced in alternative exons whose inclusion or exclusion is regulated in a tissue-specific manner.304 IDRs that are encoded by these tissue-specific alternative exons frequently influence the choice of protein interaction partners and can be instrumental in protein regulation304,305 by embedding binding motifs, and residues that can be post-translationally modified.304 However, simple alteration of the length of a disordered region306 can also modulate the overall protein function (Figure 16). Changes in IDR length can be an effective mechanism for modifying the affinity of interactions that a protein makes, particularly in instances where a disordered region is responsible for the positioning of protein binding motifs or domains.307,308 Among the alternative exons, those that exhibit conserved splicing patterns across different species are particularly likely to have important regulatory roles. For example, tissue-specific exons, which are alternatively spliced in multiple different mammals, remarkably often contain IDRs with embedded phosphosites.309 Disordered regions encoded by these exons are hence likely to act as modulators of protein function depending on the tissue where they are expressed.309 While tissue-specific exons that are alternatively spliced in a conserved fashion often code for phosphosites, the emergence of novel exons in a gene, although at first likely detrimental,310 is a possible template for the evolution of short interaction motifs.311 Furthermore, changes in exon regulation can also be important for the emergence of novel adaptive functions. Accordingly, protein segments encoded by exons, which are alternatively spliced either in a single species or in a whole evolutionary lineage, are enriched in short binding motifs, and alternative inclusion of disordered regions encoded by these exons is conceivably a source of evolutionary novelty.312
Figure 16.

Transcriptional and post-transcriptional gene regulation can be informative of IDR function. How inclusion of exons that code for IDRs is regulated during gene transcription and alternative splicing can give insights into the functional roles of the encoded disordered regions. For example, tissue- or developmental-specific regulation of alternative splicing or alternative promoter and polyadenylation site usage can be associated with important roles of the encoded IDRs in protein regulation and cellular interactions through, for example, the presence of binding motifs and phosphosites. Additionally, information on the conservation of patterns of exon inclusion (i.e., events shared among different evolutionary lineages versus species-specific events) can aid in better characterization of the encoded IDRs. The figure illustrates a hypothetical example where an exon (largest red box) that is included in a tissue-specific manner both in human and in mouse encodes an IDR that embeds a phosphosite (P) and is involved in protein regulation. The human gene depicted in the figure has an additional exon (smallest red box), which encodes an IDR with a short interaction motif and which is also included in a tissue-specific manner in humans. Gene structures, mature mRNAs, and corresponding protein isoforms are shown for human and mouse brain and heart tissues. On the right, possible functional roles of the IDRs encoded by the brain isoforms are illustrated. The examples illustrate how protein functional space can increase due to alternative splicing of exons that encode IDRs. Adapted with permission from ref (304). Copyright 2012 Elsevier.
In addition to the tendency of cassette alternative exons to frequently encode IDRs, exons adjacent to the alternatively spliced ones are also likely to code for disordered regions around the insertion point for the alternatively spliced segment.264,302 These disordered regions not only provide the structural flexibility that tolerates both presence and absence of the alternatively spliced segment, but they can also contain interaction motifs themselves.264 Furthermore, on the transcriptional level, diversity in protein isoforms can be created through both alternative splicing and usage of alternative promoters and polyadenylation sites. Protein segments that are encoded by the two latter mechanisms can contain disordered regions with motifs that define protein localization and stability.313 Taken together, these examples illustrate how better understanding of gene regulation and knowledge of evolutionarily conserved and novel isoforms can provide insights into possible functional roles of whole proteins and specific protein regions.
8.3. Degradation Kinetics
Another emerging functionality of disordered regions is their role in protein degradation.314−321 Protein half-life generally correlates with the fraction of disordered residues,68,317 and proteins that get ubiquitinated specifically upon heat shock stress are typically disordered.322 Although ubiquitination by E3 ligases has a dominant role in recruiting proteins to the proteasome for degradation,323,324 some IDRs of sufficient length allow for efficient initiation of degradation by the proteasome independent of the ubiquitination status. This idea is supported by in vitro experiments showing that degradation of tightly folded proteins is accelerated when a disordered region is attached to model substrates.315,321 Efficient degradation only occurs when the disordered terminal region is of a certain minimal length,321 and degradation may be initiated by IDRs either at the protein terminus or internally.314−321 Proteins that contain IDRs of sufficient length may therefore have increased turnover, although the exact length requirements will depend on the substrate. At the same time, not all IDRs influence protein half-life. For example, disordered polypeptides with specific amino acid compositions such as glycine-alanine and polyglutamine repeats can attenuate rather than accelerate degradation by the proteasome.325−327 The formation of protein complexes or transient interactions with other proteins may also protect IDPs from degradation. Thus, we can distinguish a novel functional class of IDRs: those that influence protein degradation (degradation accelerators) versus those that do not. These properties might be associated with specific protein function. For example, proteins that contain IDRs of a given length are probably more susceptible to degradation, possibly linking them to functions of IDPs with low expression.
Some highly disordered proteins (e.g., p53, p73, IκBα, BimEL) can, at least in vitro, be degraded by the 20S proteasome independent of ubiquitination.328−333 Specialized proteins termed “nannies” have been shown to bind to and protect IDPs from ubiquitin-independent 20S proteasomal degradation.334 A free IDP, such as newly synthesized p53, might be degraded by the 20S proteasome, which leads to fast degradation kinetics. After a nanny binds the IDP (Hdmx in the case of p53), slower, ubiquitin-dependent degradation by the 26S proteasome takes place. This biphasic decay has been proposed as a way to distinguish structured proteins from IDPs and the proteins that protect them from degradation.334
8.4. Post-translational Processing and Secretion
The majority of secretory proteins are targeted to the endoplasmic reticulum (ER) via an N-terminal signal peptide, which helps to initiate translocation of nascent chains into the ER.335,336 Bioinformatic analysis of proteins containing N-terminal ER signal peptides has identified only 10% of these proteins as IDPs (>70% disordered), suggesting that IDPs are under-represented in the secretome.337 The fact that secreted proteins are rarely IDPs might be partially explained by the requirement for largely disordered proteins to contain an α-helical prodomain for correct import into the ER lumen,338 as demonstrated for intrinsically disordered prohormones.337 IDPs lacking this structured, α-helical domain were subjected to ER-associated degradation (ERAD) despite the presence of a signal peptide.338
Despite the relative depletion of IDPs in the secretome, a number of important IDPs are processed within the ER, including many prohormones,337,339 components of the extracellular matrix,340 and proteins involved in biomineralization (see section 9.3).117,341,342 Pre-pro-opiomelanocortin (pre-POMC) is a disordered 285 amino acid protein whose signal peptide is removed during translation to create the 241-residue pro-opiomelanocortin (POMC). This prohormone has at least eight putative basic-rich cleavage sites and is able to yield as many as 10 biologically active peptides including adrenocorticotropic hormone (ACTH) and β-endorphin. The processing of POMC is tissue-specific and depends on the type of convertase enzyme expressed.343 Other prominent examples of disordered extracellular proteins are elastin and other components of elastic fibers,344 small integrin-binding ligand N-linked glycoproteins (SIBLINGs) (see section 9.3),340−342,345 and mucins (see section 9.2).236 Thus, although secreted proteins are not particularly enriched for structural disorder overall, some IDPs are essential for biomineralization, tissue organization, and hormonal signaling. In line with the features of intracellular IDPs, extracellular structural disorder is heavily post-translationally modified and involved in extensive interactions that organize large molecular assembles while binding multiple interaction partners.117,341,342
9. Biophysical Properties
A large range of biophysical work has been carried out on structural disorder in proteins using a variety of experimental techniques (Box 2).346 Previous sections have touched on several aspects. Disordered regions rapidly shift within a continuum of variably extended or globular conformations and are best described as dynamic ensembles (see section 4). The amino acid sequence of a disordered region determines which conformations it can sample, depending for example on the charge properties (see section 5.1). Disordered proteins frequently fold upon binding, and their binding thermodynamics allow for fast, transient, but highly specific interactions (see sections 2, 3, and 6). The following section discusses three other physical properties that are essential for the biology of some IDRs and IDPs: solubility, the ability to undergo phase transitions, and the role in biomineralization.
9.1. Solubility
The solubility of a protein depends upon the favorability of its interactions with water. Globular proteins bury hydrophobic amino acids within their solvent-excluded cores, while their surfaces are generally enriched in polar and charged amino acids that interact favorably with water, leading to aqueous solubility.347,348 The presence of hydrophobic surface residues, for example, binding sites for other proteins, and the denaturation of otherwise folded proteins lead to the exposure of hydrophobic residues to water and reduce solubility, sometimes leading to aggregation and precipitation. Disordered proteins do not spontaneously fold into globular structures because their sequences are depleted in hydrophobic amino acids that, in globular proteins, drive folding (see section 5).31,44 The accompanying enrichment in polar and charged amino acids, as a general rule, causes disordered proteins to be soluble in aqueous solutions. In addition, IDPs are generally resistant to heat-induced aggregation and precipitation, because disordered proteins, in isolation, lack extensive secondary and tertiary structure that in folded, globular proteins is subject to thermal denaturation. Heat-stability was observed for some of the earliest examples of IDPs. For example, the highly disordered cyclin-dependent kinase (Cdk) inhibitor p21 remains soluble and structurally unaltered from 5 to 90 °C.28 In fact, the related Cdk inhibitor p27 was purified by boiling, although at that time it was not known to be a disordered protein.349 In that study, boiling was used as a means to release p27 from its highly stable complexes with Cdks and cyclins, which, because they are folded proteins, underwent thermal denaturation and precipitated while heat-stable p27 remained soluble. This heat-treated preparation of p27 was subsequently demonstrated to potently inhibit Cdk2-cyclin A.349
Sequence analysis algorithms have predicted a high prevalence of IDRs and IDPs in sequenced genomes (see section 7.2).35,271 To experimentally address the issue of the disordered protein content of a proteome, Galea and co-workers209 treated the soluble extract of mouse embryo fibroblast cells with heat to precipitate folded proteins and then used large-scale liquid chromatography and mass spectrometry methods to identify ∼1300 proteins that remained soluble. Disorder predictions showed that more than two-thirds of these thermostable proteins are substantially disordered. This demonstrates that disordered proteins, as a structural class, are more heat stable and soluble than their folded counterparts, consistent with their sequence features and the principles of amino acid solubility. However, disordered proteins exhibit varying degrees of compaction, which is influenced by the presence and patterning of charged residues within the polypeptide chain (see section 5.1).166−168,196 While the influence of compaction on disordered protein solubility has not been addressed, it is reasonable to expect that the extent of compaction will influence the exposure of solubility-promoting amino acids for interactions with water and therefore aqueous protein solubility.
It is possible that solubility has influenced the evolution of disordered protein sequences, with low abundance disordered proteins involved in signaling and regulation being less dependent on high solubility than other disordered proteins that are highly abundant in certain cell types (e.g., titin in muscle cells). Several extracellular IDPs use their solubility to great effect in the sequestration of inorganic molecules in the extracellular environment (see section 9.3). Apart from evolutionary considerations, there are practical applications of the high solubility associated with some disordered protein sequences. For example, proteins with higher degrees of disorder have an increased success rate of expression in a cell-free protein synthesis system.350 Furthermore, Dunker and co-workers demonstrated that fusion of a variety of disordered polypeptide tags containing repetitive, highly negatively charged sequences (termed “entropic bristles”) enhanced the aqueous solubility of many proteins previously shown to be poorly soluble upon expression in E. coli.351 Whether the solubilizing effect of these disordered tags is simply due to an increase in the fraction of solubility-promoting amino acids or to other effects, such as a potential molecular chaperone function, has not been determined. Clearly, however, disordered regions within multidomain proteins that also contain folded domains are likely to influence overall protein solubility.
9.2. Phase Transition
The involvement of IDRs in phase transitions provides another biophysical angle to the characterization of proteins that harbor disordered regions.99 Li and co-workers137 observed that interactions between recombinant proteins that contain multiple copies of an SH3 domain and IDRs with multiple instances of the proline-rich SH3 interaction motif (see section 3.1) produced sharp liquid–liquid-demixing (phase separations) that resulted in micrometer-sized liquid protein-based droplets (Figure 17A). The concentrations needed for the phase transition depend on the valency (i.e., number of repeating units) of the interacting elements. Importantly, experiments with the natural NCK–nephrin–N-WASP (neuronal Wiskott–Aldrich syndrome protein) complex, which contains multiple copies of the same SH3 interaction partners, showed the formation of similar dynamic droplets, which lead to a significant increase in the activity of the actin nucleation factor Arp2/3.137 The formation of the droplets is controlled by the degree of phosphorylation of one of the interaction partners, which potentially explains how the phase transitions may be regulated in the cell.
Figure 17.

Involvement of IDRs in phase transitions. (A) Interactions between proteins that contain multiple copies of a specific domain (an SH3 domain in the figure) and IDRs with multiple instances of its interaction motif (proline-rich SH3 motif here) can, at appropriate concentrations, produce sharp liquid–liquid-demixing phase separations. This phase transition is likely to increase local “active” protein concentrations exploitable for signaling switches. (B) High concentrations of low-complexity IDRs found in certain RNA binding domains lead to a reversible phase transition with the formation of highly dynamic hydrogels. These RNA granule-like assemblies consist of heteromeric protein aggregates and allow localization and storage of functionally related but nonidentical RNA molecules. Adapted from ref (100). Copyright 2013 the Biochemical Society.
A related phenomenon occurs with RNA-binding proteins that contain IDRs of low sequence complexity. Such regions have been associated with the regulated formation of cellular RNA granules.352 Various types of RNA granules are used to modulate the fate of specific mRNAs, but their assembly mechanism has remained unclear. Kato and co-workers353 reconstituted granule-like RNA assemblies in vitro by exploiting low complexity IDRs. They demonstrated that the low-complexity IDRs of certain RNA-binding proteins were necessary for the formation of granule-like assemblies and that high concentrations of these regions lead to a reversible phase transition with a highly dynamic hydrogel state (Figure 17B). Interestingly, hydrogels formed by the low-complexity IDR of one purified member of the granules are capable of binding IDRs of other members and thereby enable the assembly of heterogeneous macromolecular structures.353 Many IDRs that can form such functional aggregates have been shown to be under tight regulation to modulate their availability in the cell.224 Regulation of IDR abundance can shift the equilibrium between the monomeric and oligomeric/aggregate form, thereby preventing formation of undesirable aggregates and keeping functional assemblies under control.224 Together, these findings indicate that the biophysical properties of certain IDRs (such as those that contain specific low-complexity regions or linear motifs) enable phase transitions that are likely to be exploited in various macromolecular assemblies and could function to bridge the length scale of proteins with that of organelles.354
Disorder-mediated phase transitions also occur extracellularly, as exemplified by the mucin family of proteins. These proteins rely on structural disorder for the formation of gel-like networks of mucus, which function in the protection of epithelial surfaces such as those in the airway and the gut.355,356 Extensive glycosylation of very large disordered regions that are rich in proline, threonine, and serine residues contributes to the formation of these structures.357 Mucin-1 can contain up to 120 such repeats, depending on the genetic variant an individual carries.358 Regulated order-to-disorder transitions of Mucin-2 are important in the formation of colon mucus aggregates.88,236,359 Mucin-2 trimers are compact structures under the conditions of the secretory pathway, where the pH is low and calcium is present, but these structures partially unfold and greatly expand in more basic environments, such as in the colon, triggering a phase transition into a mucus polymer gel.88,236,359
9.3. Biomineralization
Most animals are able to produce hard tissues for various physiological purposes by mineralization of the extracellular matrix.360,361 Bone and teeth, for example, consist of collagen and other proteins in conjunction with inorganic calcium phosphate in the form of hydroxyapatite (HA).360,362 Proteins involved in hard tissue mineralization are predicted to have very high levels of disorder,340−342 and disordered proteins are important in mineral homeostasis in general,117 indicating an important role for IDRs in these processes. For example, unfolded phosphoproteins sequester calcium phosphate by forming stable complexes in which the phosphorylated side-chains of the proteins occupy the phosphate positions on the surfaces of calcium phosphate nanoclusters.117 The disordered nature of these proteins allows them to readily adjust their shapes to surround and solubilize clusters of calcium phosphate. In this manner, proteins such as the milk caseins achieve high concentrations of calcium and phosphate while preventing the precipitation of the corresponding salts (i.e., calcification).117 Caseins belong to the highly disordered secretory calcium-binding phosphoprotein (SCPP) gene family,341 which includes bone, tooth, milk, and salivary proteins.363
Humans encode five small integrin-binding ligand N-linked glycoproteins (SIBLINGs), which are a subset of SCPPs involved specifically in regulating bone and teeth formation by bringing together hydroxyapatite, cell-surface integrins, and collagens.345,360 These are osteopontin (OPN, or bone sialoprotein 1), bone sialoprotein 2 (IBSP), dentin matrix acidic phosphoprotein 1 (DMP1), matrix extracellular phosphoglycoprotein (MEPE), and dentin sialophosphoprotein (DSPP).235 SIBLINGs are highly disordered340−342,345 and undergo extensive phosphorylation in the Golgi before they are secreted, as demonstrated in the case of DSPP, which has approximately 200 phosphoserines.235 DSPP has a particularly extreme serine and aspartic acid content, and its maturation product dentin phosphoprotein (DPP, or phosphophoryn) is likely to be one of the most acidic natural proteins known.
10. Discussion
It is likely that many of the functionally uncharacterized proteins will be similar to already characterized ones.8−10 This notion forms the basis for computational methods that aim to improve annotation coverage by predicting the function of novel and undefined proteins based on information from better-studied proteins. Databases such as Pfam22 and SCOP24 attest to the success of these approaches. However, existing methods are focused primarily on sequences that give rise to well-folded protein structures and domains. As a result, it is much harder to gain insight into the function of intrinsically disordered regions (IDRs) and proteins (IDPs), despite the increasing evidence of their prevalence and importance for protein functionality (Figure 1).50 Many important disease proteins such as p53, Myc, α-synuclein, and BRCA1 are highly disordered, underscoring the importance of disordered regions for understanding the molecular basis of human diseases.263,295,299
In this Review, we have assembled an overview of the major approaches used to classify and categorize IDRs and IDPs (Table 1). These classification schemes help us understand how disordered protein functionality is defined and could be used to enhance function prediction for disordered protein regions in general. In these final sections, we discuss the resources that are currently available for gaining insight into IDR function (Table 2), we address potential areas for improvement of the current approaches, and we propose that combinations of multiple existing classification schemes could achieve higher-quality function prediction for IDRs. Finally, we suggest areas where increased efforts are likely to advance our understanding of the functions of structural disorder in proteins.
10.1. Current Methods for Function Prediction of IDRs and IDPs
Which methods and resources can a researcher use to gain insight into the functions of the disordered regions in a protein? Current approaches (Table 2) are mainly based on the presence of functional features such as short linear motifs (SLiMs), post-translational modification (PTM) sites, molecular recognition features (MoRFs), and intrinsically disordered domains (IDDs) (see section 3). These aspects have the potential to shed light on which interaction partners an IDR may have and how many, as well as the mode of binding.
10.1.1. Linear Motif-Based Approaches
Mapping of well-characterized linear motifs onto other protein sequences holds particular promise for discovering novel functionality. For example, proteomic characterization of the motif (RxxPDG) that recruits Tankyrase ADP-ribose polymerases has led to the identification of novel Tankyrase substrates and explains the basis for mutations causing cherubism disease.364 Similarly, proteome-wide searches for the SxIP motif have resulted in the identification of previously uncharacterized microtubule plus-end tracking proteins.365 However, these types of individual studies require considerable resources.
MiniMotif126 and ELM125 are two major efforts aimed at the annotation of known instances of linear motifs, which are primarily found in IDRs, and their binding partners. The MiniMotif and ELM databases aim to categorize linear motifs of all functions based on in-depth manual annotation of experimentally validated instances from the literature. Similar approaches have also been taken specifically for PTM site motifs (see section 10.1.2). Although these resources are excellent repositories of the functional sites that occur in IDRs, they do have certain shortcomings. For example, the annotations from MiniMotif are not publicly available. Although the ELM database is the most comprehensive database of functional features within IDRs, at present it does not have the resources to annotate all motifs in the literature; ELM contains ∼200 classes of linear motifs with over 2400 instances, but more than 250 classes await annotation with this number constantly increasing.125 This has meant ELM is limited to annotating (a fraction) of the shorter motif classes and does not explicitly consider the longer binding modules in disordered regions.
Complementary to the annotation efforts, the linear motif resources employ prediction methods that map functionality onto regions of proteins with unknown function (i.e., unannotated regions). For example, MiniMotif and ELM use regular expressions derived from experimentally validated and curated motif instances to search protein sequences. These searches bring up functional descriptions of sequence instances that match the regular expressions. A major problem in the computational detection of short motifs in particular is the high false positive rate, which means that it is very difficult for users to identify the instances that are most likely to be functional from the large total of mostly nonfunctional motif instances that result from these searches. To overcome this issue, both databases have developed additional methods to improve prediction accuracy that rely on the use of additional context information, such as accessibility (using structural models366 and predictions of intrinsic disorder72), evolutionary conservation,367,368 cell compartment (based on annotation),126,369 and protein–protein interactions.128,370,371 These efforts will need to be combined in the future with a clearer user interface so researchers can more easily identify the most relevant instances.
De novo predictors make up the final category of motif resources. These predictors computationally identify putative uncharacterized motifs in protein sequences. There are two broad types: predictors that identify clusters of amino acids that are more conserved than surrounding residues (e.g., SLiMPrints372 and phylo-HMM373) or those that find short peptide patterns that are over-represented in a set of sequences (e.g., DiliMot374 and SLiMFinder375). Although both approaches have been combined with the gene ontology terms of the identified proteins, further development is required to define potential functionality.
10.1.2. PTM Site-Based Approaches
In terms of PTM sites within disordered regions, resources such as Phospho.ELM,268 PhosphoSite,376 and PHOSIDA377 curate experimentally verified phosphorylation sites and sometimes other types of modifications from the literature and genome-scale studies. Integration of such information with data on SNPs that are seen in natural populations or in cancer genomes can provide important insights into the functionality of a PTM site.378,379 Important progress has been made in identifying and cataloging peptide motifs that direct post-translational modifications. ScanSite primarily identifies linear motifs that are likely to be phosphorylated and play key roles in signaling, such as the SH2 and 14–3–3 motifs.380 Annotation of these sequence motifs is based on results from binding experiments with peptide libraries and phage display experiments.380 NetPhorest contains consensus sequence motifs of 179 kinases and 104 phosphorylation-dependent binding domains.381 In addition, approaches such as NetworKIN370 systematically integrate experimentally derived PTM sites with evolutionary information, and define motifs around the PTM sites that may be recognized by the kinase. In this manner, site-specific interactions between 123 kinases and specific PTM sites (often in disordered regions) in 5515 phosphoproteins are predicted.382 Another resource, PhosphoNET, provides predictions of potential kinases for over 650 000 putative phosphosites.383 Extending these approaches to other post-translational modifications is an area of intense research, and a number of such PTM site prediction programs currently exist,384 although linking the PTM sites to the modifying enzymes remains to be addressed for the other types of modifications.
10.1.3. Molecular Recognition Feature-Based Approaches
Two important methods exist for identifying novel binding modules in IDRs based on the concept of molecular recognition features (MoRFs). MoRFpred predicts sequences that undergo disorder-to-order transitions of all types of MoRFs (α, β, coil, and complex) using a combination of sequence alignment and machine learning predictions based on amino acid properties, predicted disorder, B-factors, and solvent accessibility.385 ANCHOR also predicts parts of disordered regions that are likely to fold upon binding with their interactors, but does so by identifying segments that cannot form enough favorable intrachain interactions to fold on their own and are likely to gain stabilizing energy by interacting with a globular partner protein.386,387
An important shortcoming of the MoRF predictions is the difficultly in identifying which of the binding sites are relevant and what their functionality might be. This is primarily because the results are not linked to known MoRF instances with annotated functions, as is the case for linear motifs, and no clues are provided regarding the potential role of a binding site or its interacting partners. The IDEAL database388 collects verified elements in disordered regions that undergo coupled folding and binding upon interaction (Box 1). The careful annotation of well-described MoRFs in terms of their sequence propensities or interaction interfaces as well as their known binding partners, and integration of these annotations with MoRF predictions, would likely improve the use of these predictions for gaining insight into IDR functionality.
10.1.4. Intrinsically Disordered Domain-Based Approaches
Few attempts have been made to systematically annotate protein domains that are largely made up of intrinsic disorder. Pfam22 models are able to predict several intrinsically disordered domains (e.g., KID, WH2, RPEL, and BH3 domains). However, this seems to be a simple consequence of the fact that these disordered domains can be described and detected by sequence profiles, rather than an effort directed at annotating long IDRs. ELM125 has also annotated a small number of long disordered domains, such as the WH2 motif; however, the main focus of the database remains on short motifs. Finally, some of the IDRs that are present in annotated domains are in fact MoRFs or linear motifs, and linear motifs also frequently fold upon binding like MoRFs, underscoring the underlying connections between linear motifs, MoRFs, and IDDs as functional elements (see section 3.4).
10.1.5. Other Approaches
Only a few IDR classifications that are not based on linear motifs, MoRFs, or IDDs have so far been exploited for function prediction. FFPred is a correlation-based approach that uses the length and position of IDRs along a sequence (see sections 5.5 and 5.6), among other general protein features, to predict the function of the protein in terms of gene ontology categories (molecular activities and biological processes).211,389−391 The DisProt database of protein disorder203 (Box 1) lists functions of individual disordered regions, when known from experiments, the major limitation here being the small number of regions for which exact function has been characterized. The Database of Disordered Protein Prediction (D2P2)49 (Box 1) stores predictions of IDRs in whole genomes, which together with information on MoRFs, PTM sites, and domains can be used to obtain insight into the possible function of the IDR and the protein containing it.
10.2. Requirement for Annotation
Future effort in the classification of IDRs and IDPs must be directed at annotation. Substantiating classes with more examples will lead to refinement of their function descriptions and will likely reveal inaccuracies in existing classification schemes. For example, there are only a limited number of well-characterized examples of proteins that contain the evolutionarily flexible (e.g., RPA70 and Sky1) or constrained types of disorder (Rpl5 and Hsp90). The same is true for the different classes of dynamic disorder in protein complexes, although efforts are ongoing there.176 In terms of the functional features of IDRs, there is a need for annotating MoRFs and longer disordered binding regions as described in the previous section. Efforts directed at short linear motifs have been very successful, but only a small fraction of the potentially thousands of motifs392 have been annotated. Pfam contains almost 15 000 curated protein families,22 while ELM contains less than 200 motif classes,125 suggesting that significant numbers of functional features are still to be identified and further annotation is required. High-quality resources that collect all of the experimentally validated functional regions of intrinsically disordered regions will provide a strong basis to map functional features onto novel proteins of unknown function.
10.3. Integration of Methods for Finding IDR and IDP Function
The current methods for finding and classifying IDR and IDP function have been successful in the area of their focus. However, not all functional characteristics of disordered regions have been fully exploited, and neither is there a resource that brings all of these aspects together. The combination of multiple categorizations and features of IDRs is likely to provide a better understanding of the functionalities encoded in these regions.
A comprehensive IDR function resource should have several aspects. It starts with a reliable consensus disorder prediction for the protein sequence of interest (Box 3), such as available in the D2P2 database (Box 1).49 Functional features, such as SLiMs (see section 3.1), MoRFs (see section 3.2), and disordered domains (see section 3.3), can then be mapped on every disordered part of the protein. The disorder profile allows for the identification of individual IDRs in the protein, as well as the calculation of disorder properties of the whole protein, such as which disorder predictors support which IDRs (see section 5.2), the overall degree of disorder (see section 5.4), the length of the individual disordered regions (see section 5.5), or the amount of disorder at the termini (see section 5.6). These can be used to assign general function to the proteins, such as gene ontology terms that correlate with these properties. Patterns in amino acid sequence could reveal additional function. For example, the presence of tandem repeats or enrichment in certain amino acids (see sections 5.7 and 7.3) may point toward involvement in certain processes. The overall sequence composition and the distribution of charges (see section 5.1) could indicate the solubility of a polypeptide chain (see section 9.1) and conformational properties such as the degree of compaction (see section 4). The combination of sequence complexity and disorder propensity could suggest function as well (see section 5.3).
Integration of other types of information will determine what classifications can additionally be used. Addition of domain information, such as Pfam, can provide insight into the role of disordered segments that are commonly associated with specific structured domains (see section 3.3). Protein–protein interactions and structures of protein complexes could indicate interacting partners of IDR binding elements and the mode of interaction (see section 6). Information about sequence conservation (see section 7.1) is another important aspect and could provide clues about evolutionarily constrained or flexible types of disorder, which are implicated in different types of functions. Knowledge on the origin of a disordered region in evolution or the species containing the protein sequence of interest suggests possible functions as well (see section 7.2). Furthermore, data describing regulatory properties such as gene expression levels (see section 8.1), alternative splicing (see section 8.2), and degradation kinetics (see section 8.3) could implicate IDRs in regulating protein availability and may suggest or reject roles as interactions hubs, for example. Finally, biophysical properties of the protein, such as the potential of multivalent elements to undergo phase transitions (see section 9.2) and occurrence inside or outside the cell (see sections 8.4 and 9.3), may suggest involvement in the spatiotemporal organization of (extra)cellular assemblies.
The hypothetical resource might be able to suggest function for some of the following examples, although it is clear that in other cases the biology will be too complicated and the outlook of function prediction as described here will be unrealistic. Therefore, the following examples should at this point be considered as speculative. A long (more than 30 residues) IDR that shows signs of evolutionarily flexible disorder and contains no short motifs or other predicted binding regions could be a flexible linker between domains or an entropic chain. A region containing a PxxPx[KR] motif flanked by evolutionarily flexible disorder that is likely to retain an open conformation in the unbound form (based on the primary structure) probably binds a class II SH3 domain, and might be involved in transcription processes if the IDR constitutes the C-terminus of a protein with an otherwise small degree of disorder. Long IDRs that are encoded by alternatively spliced exons and have several nonoverlapping functional motifs and MoRFs might be part of signaling hubs or assemble multiprotein complexes, the type of which might be inferred from the combination of binding sites present. A constitutively expressed, largely disordered IDP with an amino acid composition promoting intrinsic coil conformations and conservation of both primary and disorder sequence is likely to be a ribosomal protein or part of another rigid multisubunit complex.
It is clear that some classifications will provide more useful and direct information about function than others. Some classifications have been proposed to contrast IDPs with structured proteins, which does not necessarily make them useful for a detailed description of disorder function per se. Others have limited use for prediction because they are conceptual only, or because of overlap in the properties they describe with other schemes. Moreover, not all approaches can realistically be incorporated in a tool. Binding functionality and sequence-based predictions will generally be possible, but predictions based on other types of data may be harder. For example, assignment of evolutionarily constrained or flexible disorder requires automatic alignment of amino acid and disorder sequences, while gene expression subtypes can be derived from the wealth of microarray and RNA sequencing data. Various types of information are already brought together in the D2P2 database,49 which contains information on disordered regions, MoRFs, PTM sites, and structured domains, and in ELM,125 which shows information on linear motifs, disorder, phosphorylation, domains, protein–protein interactions, and secondary structure. Further extension of resources like these, with information on both structured and disordered regions, holds great promise toward creating a comprehensive overview of the functional elements and properties of a protein.
10.4. Future Directions
A major area of improvement in the description of disordered protein regions pertains to their dynamic behavior.172,178 IDRs fluctuate rapidly over an ensemble of heterogeneous conformations (see section 4.2), the relative free energies and propensities of which are determined by the amino acid sequence (see section 5.1). The relationship between sequence and structural ensemble is important because it describes what part of the time the chain is in a compact state, and what part of the time it is more accessible. Knowledge about these structural subtypes and about how sequence contexts and chemical modifications of the chain (e.g., by PTMs) modulate the structural ensemble is vital for the correct description of IDR behavior and has direct implications for the functional roles such regions can have in the cell.157
Classical methods are not optimally designed to take structural dynamics into account. For example, current disorder prediction technology is successful at distinguishing sequence stretches that are likely to be disordered versus those that are likely to be part of autonomously folded domains, resulting in a binary verdict (disordered versus structured) within a certain confidence limit (Box 3). Although predicted disordered regions correlate well with experimentally determined backbone dynamics,393 detailed prediction of conformational subtypes requires a more sophisticated description of disorder. A recent method for the prediction of protein backbone dynamics, trained based on order parameters estimated from experimental chemical shifts, is not only capable of distinguishing different structural organizations with varying degrees of flexibility, such as folded domains, disordered linkers, molten globules, and MoRFs, but regions that are predicted to be dynamic also correspond well with conventional predictions of IDRs.394 Furthermore, high-throughput atomistic simulations of sequence ensembles can provide information about the degree of conformational heterogeneity,395 which can be quantified by various parameters, such as an information theory measure396 or an order parameter-like measure.397 One could imagine a multiple-component scheme describing structural and dynamic characteristics that would assign, for example, residues in a random coil small values for the fractional population of secondary structure, a large value for spatial fluctuations, a fast interconversion rate, and large values for structural heterogeneity. Conversely, molten globule residues would be assigned a relatively large value for the fractional population of secondary structure, a smaller value for spatial fluctuations and structural heterogeneity, and a slower interconversion rate. Progress in the objective description of conformational ensembles will likely require development of novel structural classifications. Such efforts will be greatly encouraged by the new pE-DB database of structural ensembles (Box 1).398 There is considerable room for growth at the interface between atomistic simulations, physical theories, machine learning methods, and experiments, to enable the unmasking of the connection between disorder dynamics and molecular and system level functions of IDRs and IDPs.
Full understanding of the cellular functions of IDPs will also require knowledge of their abundance, their interactions, and their physical state in the physiological context. Are IDPs always bound to target proteins, are they chaperoned, or are there pools of unbound IDPs? Answers to these questions will vary among different IDPs and will depend on the exact context in the cell. However, the discovery of features that can help classify and categorize IDRs in terms of their cellular status will lead to more insights into their function. For example, entropic chains may mostly be disordered even in the cell, whereas effectors and assemblers may mostly be associated with other proteins in folded conformations and exchange binding partners by competition rather than by dissociation to the free, disordered state. Scavengers likely populate both disordered and ordered states, depending on whether or not their ligand is bound. Thus, investigations of the in-cell status of IDPs399 will be crucial toward understanding their biological roles.
11. Conclusion
Finally, we would like to stress that it is not all about intrinsic disorder. This Review has focused on classifications for intrinsically disordered regions and proteins, because function annotation for these regions is lagging behind annotation of structured regions. However, proteins are modular, and their functional regions can be structured or disordered, or somewhere in between. The synergy between these fundamental building blocks of proteins leads to combinatorial diversity of function. Therefore, understanding how structure and disorder work together will be crucial for uncovering the full extent of protein function.
Box 4. Evolution of Intrinsically Disordered Regions and Proteins.
IDRs generally evolve faster than their structured counterparts.51−56,107 However, comparison of the rates of evolution of structured and disordered regions in 26 protein families has shown that this is not always the case.51 To get more insight into the evolution of disordered regions, we predicted disorder in the human proteome using MULTICOM-REFINE.415 We integrated the disorder status of the protein residues with their evolutionary rates across multiple sequence alignments of homologous proteins from 53 (mostly vertebrate) species in Ensembl Compara,1 calculated using the Rate4Site program.416 As observed previously,417 protein residues that are predicted to be disordered generally evolve more quickly (i.e., have much higher evolutionary rates) than those in structured regions (Figure Box 4, P value < 10−15, Mann−Whitney U test). However, the distributions of evolutionary rates for disordered and structured residues are wide and overlap, which confirms that some disordered residues are conserved. In line with this, it has been shown that particular residue types, such as Leu, Tyr, Trp, and Pro, are more conserved in IDRs than other residue types.53 Conserved residues and elements in IDRs are potentially important for function and might be part of protein−protein interaction interfaces or peptide motifs (see section 7.1). However, sometimes, rapid divergence of disordered regions indicates functionality, as in the case of several human antiviral proteins (see section 7.2).
Figure Box 4.
Boxplots of the distributions of evolutionary rates for predicted structured (blue) and disordered (red) residues across the human proteome. Residues with a high evolutionary rate are less conserved. Boxes represent the 50% of data points in the two quartiles above and below the median (the horizontal bar within each box). Vertical lines (whiskers) connected to the boxes represent the highest and lowest nonoutlier data points, with outliers being defined as >1.5 times the interquartile range from the median. Outliers are not shown for visual clarity.
Acknowledgments
We thank Natasha Latysheva and Guilhem Chalancon for critically reading the manuscript and providing helpful comments. We acknowledge funding from MRC (MC_U105185859); HFSP (RGY0073/2010) and the Virgo consortium, funded by the Dutch government project number FES0908, and by the Netherlands Genomics Initiative (NGI) project number 050-060-452 (R.v.d.L.); the Canadian Institutes of Health Research Postdoctoral Fellowship (R.J.W.); the American Cancer Society (RSG-07-289-01-GMC) and the National Science Foundation (MCB-0939014) (G.W.D.); the Momentum program (LP2012-41) of the Hungarian Academy of Sciences and the Hungarian Science Fund (OTKA NN 106562) (M.F.); the National Institutes of Health (R01CAO82491 and R01GM083159 to R.W.K.; and P30CA21765 to St. Jude Children’s Research Hospital) and ALSAC (R.W.K.); the Odysseus grant G.0029.12 from the Research Foundation Flanders (FWO) (P.T.); the National Cancer Institute of the National Institutes of Health (CA096865) and the Skaggs Institute for Chemical Biology (P.E.W.); and the EMBO YI Programme and HFSP (RGY0073/2010) (M.M.B.).
Glossary
Abbreviations
- ACTH
adrenocorticotropic hormone
- ACTR
activator for thyroid hormone and retinoid receptors
- AFM
atomic force microscopy
- bHLH
basic helix–loop–helix domain
- bRs
basic regions
- CARD
caspase activation and recruitment domain
- CASP
critical assessment of techniques for protein structure prediction
- CATH
class, architecture, topology, homology
- CBP
CREB-binding protein
- Cdk
cyclin-dependent kinase
- CREB
cAMP response element-binding protein
- D2P2
database of disordered protein prediction
- DBM
DNA binding motif
- DC space
disorder-sequence complexity space
- DisProt
database of protein disorder
- DMP1
dentin matrix acidic phosphoprotein 1
- DPP
dentin phosphoprotein
- DSPP
dentin sialophosphoprotein
- dsRNA
double-stranded RNA
- DUF
domain of unknown function
- ELM
eukaryotic linear motif
- ER
endoplasmic reticulum
- ERAD
endoplasmic-reticulum-associated protein degradation
- FCR
fraction of charged residues
- FG motif
phenylalanine-glycine motif
- GBD
GTPase-binding domain
- GPCR
G-protein-coupled receptor
- HA
hydroxyapatite
- HMGB1
high-mobility group protein B1
- IBSP
bone sialoprotein 2
- IDD
intrinsically disordered domain
- IDEAL
intrinsically disordered proteins with extensive annotations and literature
- IDP
intrinsically disordered protein
- IDR
intrinsically disordered region
- KID
kinase-inhibitory domain
- MAP2
microtubule-associated protein 2
- MAVS
mitochondrial antiviral-signaling
- MD
molecular dynamics
- MEPE
matrix extracellular phosphoglycoprotein
- MoRE
molecular recognition element
- MoRF
molecular recognition feature
- NCBD
nuclear coactivator binding domain
- NCPR
net charge per residue
- NLS
nuclear localization signal
- NMR
nuclear magnetic resonance
- NPC
nuclear pore complex
- OPN
osteopontin
- PC4
positive cofactor 4
- PDB
Protein Data Bank
- PKR
protein kinase R
- POMC
pro-opiomelanocortin
- PPIase
peptidylprolyl cis–trans isomerase
- pRb
retinoblastoma protein
- PreSMos
prestructured motifs
- PSE
preformed structural element
- PSI
protein structure initiative
- PTM
post-translational modification
- RLR
RIG-I-like receptor
- RPA70
70 kDa subunit of replication protein A
- RS domain
arginine-serine domain
- SAXS
small-angle X-ray scattering
- SCF
Skp, Cullin, F box
- SCOP
structural classification of proteins
- SCPP
secretory calcium-binding phosphoprotein
- SH2
Src homology 2
- SH3
Src homology 3
- SIBLING
small integrin-binding ligand N-linked glycoprotein
- SLiM
short linear motif
- smFRET
single-molecule fluorescence resonance energy transfer
- SRSF1
serine/arginine-rich splicing factor 1
- SVM
support vector machine
- VP16
virion protein 16
- WASP
Wiskott–Aldrich syndrome protein
- WH2
WASP-homology domain 2
Biographies

Robin van der Lee received his B.Sc. and M.Sc. degrees in molecular life sciences Cum Laude from the Radboud University Nijmegen, The Netherlands. During his master studies, he specialized in computational biology and worked in structural bioinformatics with Prof. Gert Vriend. He then visited the group of Dr. M. Madan Babu at the MRC Laboratory of Molecular Biology in Cambridge (UK) where his fascination for intrinsically disordered proteins began. Here he started with the current Review on their classification and investigated the role of disordered segments in protein degradation (Van der Lee et al., submitted for publication). He is now a Ph.D. student in comparative genomics with Prof. Martijn Huynen at the Radboud University Medical Centre (Nijmegen, The Netherlands).

Marija Buljan’s research interest is in understanding how changes in protein sequences affect molecular networks and cellular phenotypes. She received her Ph.D. from the University of Cambridge in 2011 for work done with Dr. Alex Bateman at the Sanger Institute. She then worked as a postdoctoral fellow with Dr. M. Madan Babu at the MRC Laboratory of Molecular Biology and is now at the German Cancer Research Center, working with Prof. Michael Boutros.

Benjamin Lang studied biology in Münster, Zürich and Heidelberg before completing a Ph.D. with Dr. M. Madan Babu at the MRC Laboratory of Molecular Biology in Cambridge in 2013. His research focuses on how the complexity of post-translational regulatory systems emerges at a fundamental level, how stem cell identity is controlled in mammals, and how dysregulation of these systems can cause disease in humans. He was recently awarded an EMBL Interdisciplinary Postdoc fellowship.

Robert Weatheritt is a computational biologist who holds a joint position as a postdoctoral researcher at the MRC Laboratory of Molecular Biology in Cambridge and at the Donnelly Centre in the University of Toronto. He was awarded his Ph.D. by the European Molecular Biology Laboratory (EMBL) in Heidelberg for research undertaken in the group of Toby Gibson investigating the role of linear motifs in cell regulation.

Gary Daughdrill graduated Magna Cum Laude and Phi Beta Kappa from the University of Alabama. He received his Ph.D. from the University of Oregon working with Rick Dahlquist in the Institute of Molecular Biology. He described one of the first examples of an intrinsically disordered protein that becomes ordered when bound to another protein. Gary performed postdoctoral studies with David Lowry at Pacific Northwest National Laboratories and was an Assistant Professor at the University of Idaho. He is currently an Associate Professor in the Department of Cell Biology, Microbiology, and Molecular Biology and the Center for Drug Discovery and Innovation at the University of South Florida. His work has been funded by the National Institutes of Health, the National Science Foundation, and the American Cancer Society.

A. Keith Dunker received a broad education in chemistry (UC Berkeley), physics and biophysics (UW Madison), and structural biology (Yale University) with a 30-year focus on the structure, assembly, and molecular biology of viruses and phages. On November 15, 1995 at 12:40 pm (Chouard, T. Breaking the Protein Rules. Nature2011, 471, 151–153), Dr. Dunker switched to computational studies on intrinsically disordered proteins.

Monika Fuxreiter is an associate Professor at University of Debrecen, Hungary. She was a postdoctoral fellow with the Nobel prize winner Arieh Warshel. She worked in the Weizmann Institute, Israel and the MRC Laboratory of Molecular Biology, Cambridge, UK. She developed the concept of disordered or “fuzzy” protein complexes, currently focusing on their role in eukaryotic transcriptional regulation and on context-dependence of protein activities. She is a recipient of the L’Oréal Unesco “Women in Science” award.

Julian Gough began his research career in structural bioinformatics, but soon combined this with genomics and sequence analysis to study evolution, also producing the SUPERFAMILY database of structural domain assignments to genomes. Gough continues to examine molecular evolution across all species and applies bioinformatics at multiple levels, most recently to intrinsic protein disorder, function, and phenotype prediction, and to studying the determinants of cellular identity.

Dr. Joerg Gsponer is a biochemist and physician working in the field of computational biology. His lab develops and employs computational tools for the investigation of protein structure, folding, and interactions, with the specific focus on intrinsically disordered proteins. He received his M.D. from the University of Lausanne and his Ph.D. in computational biochemistry from the University of Zürich. After postdoctoral training at the Chemistry Department of the University of Cambridge and the MRC Laboratory of Molecular Biology, he joined the faculty of the University of British Columbia in Vancouver.

David Jones received a B.Sc. in Physics from Imperial College, M.Sc. in Biochemistry from Kings College London, and Ph.D. at University College London. After receiving his Ph.D., he was a Wellcome Trust Biomathematics Research Fellow at UCL, and later a Royal Society University Research Fellow and Reader at the University of Warwick. After taking up the first advertised UK Chair in Bioinformatics at Brunel University, he eventually returned to UCL where he is currently Professor of Bioinformatics and Director of the Bloomsbury Centre for Bioinformatics. His lab aims to develop and apply state-of-the-art mathematical and computer science techniques to problems now arising in the postgenomic era. David’s main research interests include the analysis and prediction of protein structure and function, machine learning applications in bioinformatics, and genome annotation.

Prof. Philip Kim’s main expertise is in the analysis and perturbation of protein interactions and interaction networks. He received his training at the University of Tuebingen, Germany in Biochemistry and Physics and a Ph.D. from the MIT AI Laboratory in 2003. After some business experience at McKinsey & Co. and postdoctoral work at Yale University, he set up his own laboratory at the interdisciplinary Donnelly Centre at the University of Toronto in 2009.

Richard Kriwacki received his Ph.D. from the Biophysics Division of the Department of Chemistry at Yale University in New Haven, CT, followed by postdoctoral training with Professor Peter E. Wright at the Scripps Research Institute in La Jolla, CA. In 1996 at Scripps, Drs. Kriwacki and Wright discovered that a small protein named p21Waf1/Cip1 that regulates kinases involved in controlling cell division lacked secondary and tertiary structure in isolation but folded upon binding to its kinase targets. This, together with a few following reports of functional, unstructured proteins, drew attention to what are now termed intrinsically disordered proteins in biology. In 1997, Dr. Kriwacki joined the Department of Structural Biology at St. Jude Children’s Research Hospital (St. Jude) in Memphis, TN, where he is now a Full Member. At St. Jude, Dr. Kriwacki has continued studies of disordered proteins, with focus on establishing relationships between their disordered features and biological functions, and has published more than 70 papers in the field. Dr. Kriwacki helped establish the Disordered Proteins Subgroup at the Biophysical Society, leading advocates for the disordered proteins field, and covers this topic as an Editorial Board Member at the Journal of Molecular Biology.

Christopher J. Oldfield is a graduate student in Bioinformatics in the School of Informatics and Computing at IUPUI. He is interested in the sequence determinants and functions of intrinsically disordered proteins, in particular the role of intrinsic disorder in molecular recognition and the structural implications of intrinsic disorder in biological complexes.

Rohit Pappu is a Professor of Biomedical Engineering and Director of the Center for Biological Systems Engineering at Washington University in St. Louis. He received his Ph.D. in Biological Physics from Tufts University. He joined Washington University following two postdoctoral stints at Washington University and Johns Hopkins University. Pappu’s research interests are focused on the physical principles underlying the form, function, and self-assembly of intrinsically disordered proteins.

Peter Tompa graduated in organic chemistry in Budapest in 1983, and started his research on intrinsically disordered proteins in 2000. He played an active and decisive role in the rise of the field. Currently, he is the director of VIB Department of Structural Biology, Brussels. He has published about 130 papers and the first monograph of the field “Structure and Function of Intrinsically Disordered Proteins” (2009) by Taylor and Francis, Inc. (CRC Press).

Dr. Vladimir Uversky obtained his Ph.D. in biophysics from Moscow Institute of Physics and Technology (1991) and D.Sc. in biophysics from Institute of Experimental and Theoretical Biophysics, Russian Academy of Sciences (1998). He spent his early career working on protein folding at the Institute of Protein Research and the Institute for Biological Instrumentation (Russian Academy of Sciences). In 1998, he moved to the University of California Santa Cruz to work on protein folding, misfolding, and protein intrinsic disorder. In 2004, he moved to the Center for Computational Biology and Bioinformatics at Indiana University-Purdue University Indianapolis to work on the intrinsically disordered proteins. Since 2010, he is with the Department of Molecular Biology at the University of South Florida.

Peter Wright is a Professor in the Department of Integrative Structural and Computational Biology at The Scripps Research Institute. He received his B.Sc., M.Sc., and Ph.D. degrees from the University of Auckland, New Zealand, and undertook postdoctoral studies at Oxford University, UK. His research is focused on applications of NMR to study protein folding, intrinsically disordered proteins, protein structure and dynamics, and protein–nucleic acid interactions.

M. Madan Babu heads the Regulatory Genomics and Systems Biology group at the MRC Laboratory of Molecular Biology, Cambridge, UK. He obtained his B. Tech degree in 2001 from the Centre for Biotechnology, Anna University, India. He then received a Ph.D. from the MRC Laboratory of Molecular Biology and Trinity College, Cambridge, UK. He subsequently carried out his postdoctoral research at the NCBI, U.S., and returned to the UK to become an independent Group Leader at the MRC Laboratory of Molecular Biology in 2006. He was elected as a Schlumberger Research Fellow at Darwin College, Cambridge in 2007 and was appointed as a Programme Leader in 2010. His group investigates how intrinsically disordered proteins achieve regulation at multiple levels of complexity and how this influences evolution of organisms and their genome (see http://mbgroup.mrc-lmb.cam.ac.uk/). Madan is also a Director of Studies at Trinity College, Cambridge, an Executive Editor at Nucleic Acids Research, and an Associate Editor at Molecular BioSystems.
Author Contributions
▲ These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Flicek P.; Ahmed I.; Amode M. R.; Barrell D.; Beal K.; Brent S.; Carvalho-Silva D.; Clapham P.; Coates G.; Fairley S.; Fitzgerald S.; Gil L.; Garcia-Giron C.; Gordon L.; Hourlier T.; Hunt S.; Juettemann T.; Kahari A. K.; Keenan S.; Komorowska M.; Kulesha E.; Longden I.; Maurel T.; McLaren W. M.; Muffato M.; Nag R.; Overduin B.; Pignatelli M.; Pritchard B.; Pritchard E.; Riat H. S.; Ritchie G. R.; Ruffier M.; Schuster M.; Sheppard D.; Sobral D.; Taylor K.; Thormann A.; Trevanion S.; White S.; Wilder S. P.; Aken B. L.; Birney E.; Cunningham F.; Dunham I.; Harrow J.; Herrero J.; Hubbard T. J.; Johnson N.; Kinsella R.; Parker A.; Spudich G.; Yates A.; Zadissa A.; Searle S. M. Nucleic Acids Res. 2013, 41, D48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucleic Acids Res. 2013, 41, D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodny R.; Pereyaslavets L.; Samson A. O.; Levitt M. Annu. Rev. Biophys. 2013, 42, 559. [DOI] [PubMed] [Google Scholar]
- Raes J.; Harrington E. D.; Singh A. H.; Bork P. Curr. Opin. Struct. Biol. 2007, 17, 362. [DOI] [PubMed] [Google Scholar]
- Jaroszewski L.; Li Z.; Krishna S. S.; Bakolitsa C.; Wooley J.; Deacon A. M.; Wilson I. A.; Godzik A. PLoS Biol. 2009, 7, e1000205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt M. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucleic Acids Res. 2012, 40, D71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L.; Koonin E. V. J. Mol. Biol. 1999, 287, 1023. [DOI] [PubMed] [Google Scholar]
- Gough J.; Karplus K.; Hughey R.; Chothia C. J. Mol. Biol. 2001, 313, 903. [DOI] [PubMed] [Google Scholar]
- de Lima Morais D. A.; Fang H.; Rackham O. J.; Wilson D.; Pethica R.; Chothia C.; Gough J. Nucleic Acids Res. 2011, 39, D427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L. Genome Res. 2000, 10, 1074. [DOI] [PubMed] [Google Scholar]
- Ashburner M.; Ball C. A.; Blake J. A.; Botstein D.; Butler H.; Cherry J. M.; Davis A. P.; Dolinski K.; Dwight S. S.; Eppig J. T.; Harris M. A.; Hill D. P.; Issel-Tarver L.; Kasarskis A.; Lewis S.; Matese J. C.; Richardson J. E.; Ringwald M.; Rubin G. M.; Sherlock G. Nat. Genet. 2000, 25, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D.; Marcotte E. M.; Xenarios I.; Yeates T. O. Nature 2000, 405, 823. [DOI] [PubMed] [Google Scholar]
- Thornton J. M.; Todd A. E.; Milburn D.; Borkakoti N.; Orengo C. A. Nat. Struct. Biol. 2000, 7, 991. [DOI] [PubMed] [Google Scholar]
- Whisstock J. C.; Lesk A. M. Q. Rev. Biophys. 2003, 36, 307. [DOI] [PubMed] [Google Scholar]
- Gabaldon T.; Huynen M. A. Cell. Mol. Life Sci. 2004, 61, 930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frishman D. Chem. Rev. 2007, 107, 3448. [DOI] [PubMed] [Google Scholar]
- Chothia C.; Lesk A. M. EMBO J. 1986, 5, 823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; Thornton J. M. Nat. Rev. Genet. 2008, 9, 141. [DOI] [PubMed] [Google Scholar]
- Kim S. H.; Shin D. H.; Choi I. G.; Schulze-Gahmen U.; Chen S.; Kim R. J. Struct. Funct. Genomics 2003, 4, 129. [DOI] [PubMed] [Google Scholar]
- Dessailly B. H.; Nair R.; Jaroszewski L.; Fajardo J. E.; Kouranov A.; Lee D.; Fiser A.; Godzik A.; Rost B.; Orengo C. Structure 2009, 17, 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M.; Coggill P. C.; Eberhardt R. Y.; Mistry J.; Tate J.; Boursnell C.; Pang N.; Forslund K.; Ceric G.; Clements J.; Heger A.; Holm L.; Sonnhammer E. L.; Eddy S. R.; Bateman A.; Finn R. D. Nucleic Acids Res. 2012, 40, D290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A.; Coggill P.; Finn R. D. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2010, 66, 1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin A. G.; Brenner S. E.; Hubbard T.; Chothia C. J. Mol. Biol. 1995, 247, 536. [DOI] [PubMed] [Google Scholar]
- Andreeva A.; Howorth D.; Chandonia J. M.; Brenner S. E.; Hubbard T. J.; Chothia C.; Murzin A. G. Nucleic Acids Res. 2008, 36, D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillitoe I.; Cuff A. L.; Dessailly B. H.; Dawson N. L.; Furnham N.; Lee D.; Lees J. G.; Lewis T. E.; Studer R. A.; Rentzsch R.; Yeats C.; Thornton J. M.; Orengo C. A. Nucleic Acids Res. 2013, 41, D490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis T. E.; Sillitoe I.; Andreeva A.; Blundell T. L.; Buchan D. W.; Chothia C.; Cuff A.; Dana J. M.; Filippis I.; Gough J.; Hunter S.; Jones D. T.; Kelley L. A.; Kleywegt G. J.; Minneci F.; Mitchell A.; Murzin A. G.; Ochoa-Montano B.; Rackham O. J.; Smith J.; Sternberg M. J.; Velankar S.; Yeats C.; Orengo C. Nucleic Acids Res. 2013, 41, D499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriwacki R. W.; Hengst L.; Tennant L.; Reed S. I.; Wright P. E. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daughdrill G. W.; Chadsey M. S.; Karlinsey J. E.; Hughes K. T.; Dahlquist F. W. Nat. Struct. Biol. 1997, 4, 285. [DOI] [PubMed] [Google Scholar]
- Wright P. E.; Dyson H. J. J. Mol. Biol. 1999, 293, 321. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Gillespie J. R.; Fink A. L. Proteins 2000, 41, 415. [DOI] [PubMed] [Google Scholar]
- Dunker A. K.; Lawson J. D.; Brown C. J.; Williams R. M.; Romero P.; Oh J. S.; Oldfield C. J.; Campen A. M.; Ratliff C. M.; Hipps K. W.; Ausio J.; Nissen M. S.; Reeves R.; Kang C.; Kissinger C. R.; Bailey R. W.; Griswold M. D.; Chiu W.; Garner E. C.; Obradovic Z. J. Mol. Graph. Model. 2001, 19, 26. [DOI] [PubMed] [Google Scholar]
- Tompa P. Trends Biochem. Sci. 2002, 27, 527. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. Protein Sci. 2002, 11, 739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J. J.; Sodhi J. S.; McGuffin L. J.; Buxton B. F.; Jones D. T. J. Mol. Biol. 2004, 337, 635. [DOI] [PubMed] [Google Scholar]
- Fink A. L. Curr. Opin. Struct. Biol. 2005, 15, 35. [DOI] [PubMed] [Google Scholar]
- Dyson H. J.; Wright P. E. Nat. Rev. Mol. Cell Biol. 2005, 6, 197. [DOI] [PubMed] [Google Scholar]
- Dunker A. K.; Oldfield C. J.; Meng J.; Romero P.; Yang J. Y.; Chen J. W.; Vacic V.; Obradovic Z.; Uversky V. N. BMC Genomics 2008, 9, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gsponer J.; Babu M. M. Prog. Biophys. Mol. Biol. 2009, 99, 94. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Dunker A. K. Biochim. Biophys. Acta 2010, 1804, 1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P. Trends Biochem. Sci. 2012, 37, 509. [DOI] [PubMed] [Google Scholar]
- Forman-Kay J. D.; Mittag T. Structure 2013, 21, 1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunker A. K.; Babu M. M.; Barbar E.; Blackledge M.; Bondos S. E.; Dosztányi Z.; Dyson H. J.; Forman-Kay J.; Fuxreiter M.; Gsponer J.; Han K. H.; Jones D. T.; Longhi S.; Metallo S. J.; Nishikawa K.; Nussinov R.; Obradovic Z.; Pappu R. V.; Rost B.; Selenko P.; Subramaniam V.; Sussman J. L.; Tompa P.; Uversky V. N. Intrinsically Disord. Proteins 2013, 1, e24157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero P.; Obradovic Z.; Li X.; Garner E. C.; Brown C. J.; Dunker A. K. Proteins 2001, 42, 38. [DOI] [PubMed] [Google Scholar]
- Iakoucheva L. M.; Radivojac P.; Brown C. J.; O’Connor T. R.; Sikes J. G.; Obradovic Z.; Dunker A. K. Nucleic Acids Res. 2004, 32, 1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins M. O.; Yu L.; Campuzano I.; Grant S. G.; Choudhary J. S. Mol. Cell. Proteomics 2008, 7, 1331. [DOI] [PubMed] [Google Scholar]
- Diella F.; Haslam N.; Chica C.; Budd A.; Michael S.; Brown N. P.; Trave G.; Gibson T. J. Front. Biosci. 2008, 13, 6580. [DOI] [PubMed] [Google Scholar]
- Davey N. E.; Van Roey K.; Weatheritt R. J.; Toedt G.; Uyar B.; Altenberg B.; Budd A.; Diella F.; Dinkel H.; Gibson T. J. Mol. BioSyst. 2012, 8, 268. [DOI] [PubMed] [Google Scholar]
- Oates M. E.; Romero P.; Ishida T.; Ghalwash M.; Mizianty M. J.; Xue B.; Dosztanyi Z.; Uversky V. N.; Obradovic Z.; Kurgan L.; Dunker A. K.; Gough J. Nucleic Acids Res. 2013, 41, D508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu M. M.; Kriwacki R. W.; Pappu R. V. Science 2012, 337, 1460. [DOI] [PubMed] [Google Scholar]
- Brown C. J.; Takayama S.; Campen A. M.; Vise P.; Marshall T. W.; Oldfield C. J.; Williams C. J.; Dunker A. K. J. Mol. Evol. 2002, 55, 104. [DOI] [PubMed] [Google Scholar]
- Chen J. W.; Romero P.; Uversky V. N.; Dunker A. K. J. Proteome Res. 2006, 5, 879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown C. J.; Johnson A. K.; Daughdrill G. W. Mol. Biol. Evol. 2010, 27, 609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellay J.; Han S.; Michaut M.; Kim T.; Costanzo M.; Andrews B. J.; Boone C.; Bader G. D.; Myers C. L.; Kim P. M. Genome Biol. 2011, 12, R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown C. J.; Johnson A. K.; Dunker A. K.; Daughdrill G. W. Curr. Opin. Struct. Biol. 2011, 21, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson J.; Grahn M.; Wright A. P. Genome Biol. 2011, 12, R65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunker A. K.; Brown C. J.; Lawson J. D.; Iakoucheva L. M.; Obradovic Z. Biochemistry 2002, 41, 6573. [DOI] [PubMed] [Google Scholar]
- Tompa P. FEBS Lett. 2005, 579, 3346. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. FEBS Lett. 2013, 587, 1891. [DOI] [PubMed] [Google Scholar]
- Daughdrill G. W.; Narayanaswami P.; Gilmore S. H.; Belczyk A.; Brown C. J. J. Mol. Evol. 2007, 65, 277. [DOI] [PubMed] [Google Scholar]
- Tompa P. BioEssays 2003, 25, 847. [DOI] [PubMed] [Google Scholar]
- Tskhovrebova L.; Trinick J. Nat. Rev. Mol. Cell Biol. 2003, 4, 679. [DOI] [PubMed] [Google Scholar]
- Seet B. T.; Dikic I.; Zhou M. M.; Pawson T. Nat. Rev. Mol. Cell Biol. 2006, 7, 473. [DOI] [PubMed] [Google Scholar]
- Vucetic S.; Xie H.; Iakoucheva L. M.; Oldfield C. J.; Dunker A. K.; Obradovic Z.; Uversky V. N. J. Proteome Res. 2007, 6, 1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N. Curr. Pharm. Des. 2013, 19, 4191. [DOI] [PubMed] [Google Scholar]
- Galea C. A.; Wang Y.; Sivakolundu S. G.; Kriwacki R. W. Biochemistry 2008, 47, 7598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt L. J.; Tuch B. B.; Villen J.; Johnson A. D.; Gygi S. P.; Morgan D. O. Science 2009, 325, 1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gsponer J.; Futschik M. E.; Teichmann S. A.; Babu M. M. Science 2008, 322, 1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry C. R.; Levy E. D.; Michnick S. W. Trends Genet. 2009, 25, 193. [DOI] [PubMed] [Google Scholar]
- Van Roey K.; Gibson T. J.; Davey N. E. Curr. Opin. Struct. Biol. 2012, 22, 378. [DOI] [PubMed] [Google Scholar]
- Van Roey K.; Dinkel H.; Weatheritt R. J.; Gibson T. J.; Davey N. E. Sci. Signaling 2013, 6, rs7. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M.; Tompa P.; Simon I. Bioinformatics 2007, 23, 950. [DOI] [PubMed] [Google Scholar]
- Gibson T. J. Trends Biochem. Sci. 2009, 34, 471. [DOI] [PubMed] [Google Scholar]
- Perkins J. R.; Diboun I.; Dessailly B. H.; Lees J. G.; Orengo C. Structure 2010, 18, 1233. [DOI] [PubMed] [Google Scholar]
- Bode A. M.; Dong Z. Nat. Rev. Cancer 2004, 4, 793. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Cell 2007, 128, 693. [DOI] [PubMed] [Google Scholar]
- Galea C. A.; Nourse A.; Wang Y.; Sivakolundu S. G.; Heller W. T.; Kriwacki R. W. J. Mol. Biol. 2008, 376, 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder R.; Barta A.; Semrad K. Nat. Rev. Mol. Cell Biol. 2004, 5, 908. [DOI] [PubMed] [Google Scholar]
- Young J. C.; Agashe V. R.; Siegers K.; Hartl F. U. Nat. Rev. Mol. Cell Biol. 2004, 5, 781. [DOI] [PubMed] [Google Scholar]
- Tompa P.; Csermely P. FASEB J. 2004, 18, 1169. [DOI] [PubMed] [Google Scholar]
- Ivanyi-Nagy R.; Davidovic L.; Khandjian E. W.; Darlix J. L. Cell. Mol. Life Sci. 2005, 62, 1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs D.; Tompa P. Biochem. Soc. Trans. 2012, 40, 963. [DOI] [PubMed] [Google Scholar]
- Pontius B. W. Trends Biochem. Sci. 1993, 18, 181. [DOI] [PubMed] [Google Scholar]
- Rogers J. M.; Steward A.; Clarke J. J. Am. Chem. Soc. 2013, 135, 1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorovits B. M.; Horowitz P. M. J. Biol. Chem. 1995, 270, 13057. [DOI] [PubMed] [Google Scholar]
- Lindner R. A.; Kapur A.; Mariani M.; Titmuss S. J.; Carver J. A. Eur. J. Biochem. 1998, 258, 170. [DOI] [PubMed] [Google Scholar]
- Treweek T. M.; Rekas A.; Walker M. J.; Carver J. A. Exp. Eye Res. 2010, 91, 691. [DOI] [PubMed] [Google Scholar]
- Mitrea D. M.; Kriwacki R. W. FEBS Lett. 2013, 587, 1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichmann D.; Xu Y.; Cremers C. M.; Ilbert M.; Mittelman R.; Fitzgerald M. C.; Jakob U. Cell 2012, 148, 947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell J. C.; Jakob U. Trends Biochem. Sci. 2012, 37, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugase K.; Dyson H. J.; Wright P. E. Nature 2007, 447, 1021. [DOI] [PubMed] [Google Scholar]
- Wright P. E.; Dyson H. J. Curr. Opin. Struct. Biol. 2009, 19, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucsi Z.; Hudecz F.; Hollosi M.; Tompa P.; Friedrich P. Protein Sci. 2003, 12, 2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A. S.; Kakalis L. T.; Abdul-Manan N.; Liu G. A.; Rosen M. K. Nature 2000, 404, 151. [DOI] [PubMed] [Google Scholar]
- Trudeau T.; Nassar R.; Cumberworth A.; Wong E. T.; Woollard G.; Gsponer J. Structure 2013, 21, 332. [DOI] [PubMed] [Google Scholar]
- Ferreon A. C.; Ferreon J. C.; Wright P. E.; Deniz A. A. Nature 2013, 498, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilser V. J.; Thompson E. B. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 8311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motlagh H. N.; Hilser V. J. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flock T.; Weatheritt R. J.; Latysheva N.; Babu M. M.. Curr. Opin. Struct. Biol. 2014, submitted for publication [DOI] [PubMed]
- Cumberworth A.; Lamour G.; Babu M. M.; Gsponer J. Biochem. J. 2013, 454, 361. [DOI] [PubMed] [Google Scholar]
- Wu H. Cell 2013, 153, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z.; Oldfield C. J.; Xue B.; Mizianty M. J.; Dunker A. K.; Kurgan L.; Uversky V. N.. Cell. Mol. Life Sci. 2013. [DOI] [PMC free article] [PubMed]
- Li J.; McQuade T.; Siemer A. B.; Napetschnig J.; Moriwaki K.; Hsiao Y. S.; Damko E.; Moquin D.; Walz T.; McDermott A.; Chan F. K.; Wu H. Cell 2012, 150, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxreiter M.; Tompa P.; Simon I.; Uversky V. N.; Hansen J. C.; Asturias F. J. Nat. Chem. Biol. 2008, 4, 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegyi H.; Schad E.; Tompa P. BMC Struct. Biol. 2007, 7, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes C.; Oldfield C. J.; Ji F.; Klitgord N.; Cusick M. E.; Radivojac P.; Uversky V. N.; Vidal M.; Iakoucheva L. M. PLoS Comput. Biol. 2006, 2, e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim P. M.; Sboner A.; Xia Y.; Gerstein M. Mol. Syst. Biol. 2008, 4, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekaran K.; Tsai C. J.; Kumar S.; Zanuy D.; Nussinov R. Trends Biochem. Sci. 2003, 28, 81. [DOI] [PubMed] [Google Scholar]
- Cortese M. S.; Uversky V. N.; Dunker A. K. Prog. Biophys. Mol. Biol. 2008, 98, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajani Z.; Sykes M. T.; Williamson J. R. Annu. Rev. Biochem. 2011, 80, 501. [DOI] [PubMed] [Google Scholar]
- Adilakshmi T.; Ramaswamy P.; Woodson S. A. J. Mol. Biol. 2005, 351, 508. [DOI] [PubMed] [Google Scholar]
- Timsit Y.; Acosta Z.; Allemand F.; Chiaruttini C.; Springer M. Int. J. Mol. Sci. 2009, 10, 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scripture J. B.; Huber P. W. Biochemistry 2011, 50, 3827. [DOI] [PubMed] [Google Scholar]
- Xue B.; Romero P. R.; Noutsou M.; Mauriceqq M. M.; Rudiger S. G.; William A. M. Jr.; Mizianty M. J.; Kurgan L.; Uversky V. N.; Dunker A. K. FEBS Lett. 2013, 587, 1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buday L.; Tompa P. FEBS J. 2010, 277, 4348. [DOI] [PubMed] [Google Scholar]
- Daniels A. J.; Williams R. J.; Wright P. E. Neuroscience 1978, 3, 573. [DOI] [PubMed] [Google Scholar]
- Holt C. Curr. Opin. Struct. Biol. 2013, 23, 420. [DOI] [PubMed] [Google Scholar]
- Ren S.; Uversky V. N.; Chen Z.; Dunker A. K.; Obradovic Z. BMC Genomics 2008, 9, S26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxreiter M.; Simon I.; Friedrich P.; Tompa P. J. Mol. Biol. 2004, 338, 1015. [DOI] [PubMed] [Google Scholar]
- Oldfield C. J.; Cheng Y.; Cortese M. S.; Romero P.; Uversky V. N.; Dunker A. K. Biochemistry 2005, 44, 12454. [DOI] [PubMed] [Google Scholar]
- Mohan A.; Oldfield C. J.; Radivojac P.; Vacic V.; Cortese M. S.; Dunker A. K.; Uversky V. N. J. Mol. Biol. 2006, 362, 1043. [DOI] [PubMed] [Google Scholar]
- Vacic V.; Oldfield C. J.; Mohan A.; Radivojac P.; Cortese M. S.; Uversky V. N.; Dunker A. K. J. Proteome Res. 2007, 6, 2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H.; Kim D. H.; Han J. J.; Cha E. J.; Lim J. E.; Cho Y. J.; Lee C.; Han K. H. Curr. Protein Pept. Sci. 2012, 13, 34. [DOI] [PubMed] [Google Scholar]
- Hinds M. G.; Smits C.; Fredericks-Short R.; Risk J. M.; Bailey M.; Huang D. C.; Day C. L. Cell Death Differ. 2007, 14, 128. [DOI] [PubMed] [Google Scholar]
- Dinkel H.; Van Roey K.; Michael S.; Davey N. E.; Weatheritt R. J.; Born D.; Speck T.; Kruger D.; Grebnev G.; Kuban M.; Strumillo M.; Uyar B.; Budd A.; Altenberg B.; Seiler M.; Chemes L. B.; Glavina J.; Sanchez I. E.; Diella F.; Gibson T. J. Nucleic Acids Res. 2014, 42, D259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi T.; Merlin J. C.; Deverasetty S.; Gryk M. R.; Bill T. J.; Brooks A. W.; Lee L. Y.; Rathnayake V.; Ross C. A.; Sargeant D. P.; Strong C. L.; Watts P.; Rajasekaran S.; Schiller M. R. Nucleic Acids Res. 2012, 40, D252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A.; Ceol A.; Aloy P. Nucleic Acids Res. 2011, 39, D718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatheritt R. J.; Luck K.; Petsalaki E.; Davey N. E.; Gibson T. J. Bioinformatics 2012, 28, 976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey N. E.; Trave G.; Gibson T. J. Trends Biochem. Sci. 2011, 36, 159. [DOI] [PubMed] [Google Scholar]
- Jurgens M. C.; Voros J.; Rautureau G. J.; Shepherd D. A.; Pye V. E.; Muldoon J.; Johnson C. M.; Ashcroft A. E.; Freund S. M.; Ferguson N. Nat. Chem. Biol. 2013, 9, 540. [DOI] [PubMed] [Google Scholar]
- Van Roey K.; Uyar B.; Weatheritt R. J.; Dinkel H.; Seiler M.; Budd A.; Gibson T. J.; Davey N. E.. Chem. Rev. 2014, in press [DOI] [PubMed]
- Pop C.; Salvesen G. S. J. Biol. Chem. 2009, 284, 21777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U.; Janicke R. U.; Schulze-Osthoff K. Cell Death Differ. 2003, 10, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines J. Biochem. J. 1995, 308, 697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X. Z.; Kops O.; Werner A.; Lu P. J.; Shen M.; Stoller G.; Kullertz G.; Stark M.; Fischer G.; Lu K. P. Mol. Cell 2000, 6, 873. [DOI] [PubMed] [Google Scholar]
- Pawson T.; Nash P. Science 2003, 300, 445. [DOI] [PubMed] [Google Scholar]
- Li P.; Banjade S.; Cheng H. C.; Kim S.; Chen B.; Guo L.; Llaguno M.; Hollingsworth J. V.; King D. S.; Banani S. F.; Russo P. S.; Jiang Q. X.; Nixon B. T.; Rosen M. K. Nature 2012, 483, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger C. M.; Kirschner M. W. Genes Dev. 2000, 14, 655. [PMC free article] [PubMed] [Google Scholar]
- He J.; Chao W. C.; Zhang Z.; Yang J.; Cronin N.; Barford D. Mol. Cell 2013, 50, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar J. R.; Pagan J. K.; Pagano M. Nat. Rev. Mol. Cell Biol. 2013, 14, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalderon D.; Roberts B. L.; Richardson W. D.; Smith A. E. Cell 1984, 39, 499. [DOI] [PubMed] [Google Scholar]
- Evans P. R.; Owen D. J. Curr. Opin. Struct. Biol. 2002, 12, 814. [DOI] [PubMed] [Google Scholar]
- Schmid E. M.; McMahon H. T. Nature 2007, 448, 883. [DOI] [PubMed] [Google Scholar]
- Balagopalan L.; Coussens N. P.; Sherman E.; Samelson L. E.; Sommers C. L. Cold Spring Harbor Perspect. Biol. 2010, 2, a005512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgen A. S.; Roberts G. C.; Feeney J. Nature 1975, 253, 753. [DOI] [PubMed] [Google Scholar]
- Espinoza-Fonseca L. M. Biochem. Biophys. Res. Commun. 2009, 382, 479. [DOI] [PubMed] [Google Scholar]
- Rose P. W.; Bi C.; Bluhm W. F.; Christie C. H.; Dimitropoulos D.; Dutta S.; Green R. K.; Goodsell D. S.; Prlic A.; Quesada M.; Quinn G. B.; Ramos A. G.; Westbrook J. D.; Young J.; Zardecki C.; Berman H. M.; Bourne P. E. Nucleic Acids Res. 2013, 41, D475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vise P. D.; Baral B.; Latos A. J.; Daughdrill G. W. Nucleic Acids Res. 2005, 33, 2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borcherds W.; Kashtanov S.; Wu H.; Daughdrill G. W. Proteins 2013, 81, 1686. [DOI] [PubMed] [Google Scholar]
- Chi S. W.; Lee S. H.; Kim D. H.; Ahn M. J.; Kim J. S.; Woo J. Y.; Torizawa T.; Kainosho M.; Han K. H. J. Biol. Chem. 2005, 280, 38795. [DOI] [PubMed] [Google Scholar]
- Bochkareva E.; Kaustov L.; Ayed A.; Yi G. S.; Lu Y.; Pineda-Lucena A.; Liao J. C.; Okorokov A. L.; Milner J.; Arrowsmith C. H.; Bochkarev A. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 15412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worrall J. A.; Gorna M.; Pei X. Y.; Spring D. R.; Nicholson R. L.; Luisi B. F. Biochem. Soc. Trans. 2007, 35, 502. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; Oldfield C. J.; Meng J.; Romero P.; Uversky V. N.; Dunker A. K. Biochemistry 2007, 46, 13468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran V.; Luisi B. F. J. Mol. Biol. 2006, 358, 8. [DOI] [PubMed] [Google Scholar]
- Nurmohamed S.; Vaidialingam B.; Callaghan A. J.; Luisi B. F. J. Mol. Biol. 2009, 389, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R. K.; Crick S. L.; Pappu R. V. J. Mol. Biol. 2012, 416, 287. [DOI] [PubMed] [Google Scholar]
- Das R. K.; Mao A. H.; Pappu R. V. Sci. Signaling 2012, 5, pe17. [DOI] [PubMed] [Google Scholar]
- Tompa P.; Fuxreiter M.; Oldfield C. J.; Simon I.; Dunker A. K.; Uversky V. N. BioEssays 2009, 31, 328. [DOI] [PubMed] [Google Scholar]
- Chen J. W.; Romero P.; Uversky V. N.; Dunker A. K. J. Proteome Res. 2006, 5, 888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buljan M.; Frankish A.; Bateman A. Genome Biol. 2010, 11, R74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentony M. M.; Jones D. T. Proteins 2010, 78, 212. [DOI] [PubMed] [Google Scholar]
- Teraguchi S.; Patil A.; Standley D. M. BMC Bioinf. 2010, 11, S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meszaros B.; Dosztanyi Z.; Simon I. PLoS One 2012, 7, e46829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarest S. J.; Martinez-Yamout M.; Chung J.; Chen H.; Xu W.; Dyson H. J.; Evans R. M.; Wright P. E. Nature 2002, 415, 549. [DOI] [PubMed] [Google Scholar]
- Dyson H. J.; Wright P. E. Nat. Struct. Biol. 1998, 5, 499. [DOI] [PubMed] [Google Scholar]
- Mao A. H.; Crick S. L.; Vitalis A.; Chicoine C. L.; Pappu R. V. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 8183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Spath S.; Soranno A.; Hirschfeld V.; Hofmann H.; Ruegger S.; Reymond L.; Nettels D.; Schuler B. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh J. A.; Forman-Kay J. D. Biophys. J. 2010, 98, 2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright P. E.; Dyson H. J.; Lerner R. A. Biochemistry 1988, 27, 7167. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S.; Krishnan R.; Lemke E. A.; Lindquist S.; Deniz A. A. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn T. R.; Radford S. E. FEBS J. 2005, 272, 5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher C. K.; Stultz C. M. Curr. Opin. Struct. Biol. 2011, 21, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr D. D.; Nussinov R.; Wright P. E. Nat. Chem. Biol. 2009, 5, 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B.; Nussinov R. Genome Biol. 2009, 10, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar G.; Keskin O.; Gursoy A.; Nussinov R. Curr. Opin. Pharmacol. 2010, 10, 715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxreiter M. Mol. BioSyst. 2012, 8, 168. [DOI] [PubMed] [Google Scholar]
- Mittag T.; Forman-Kay J. D. Curr. Opin. Struct. Biol. 2007, 17, 3. [DOI] [PubMed] [Google Scholar]
- Jensen M. R.; Ruigrok R. W.; Blackledge M. Curr. Opin. Struct. Biol. 2013, 23, 426. [DOI] [PubMed] [Google Scholar]
- Ferreon A. C.; Moran C. R.; Gambin Y.; Deniz A. A. Methods Enzymol. 2010, 472, 179. [DOI] [PubMed] [Google Scholar]
- Schuler B.; Hofmann H. Curr. Opin. Struct. Biol. 2013, 23, 36. [DOI] [PubMed] [Google Scholar]
- Banerjee P. R.; Deniz A. A. Chem. Soc. Rev. 2014, 43, 1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brucale M.; Schuler B.; Samori B.. Chem. Rev. 2014. [DOI] [PubMed]
- Receveur-Brechot V.; Bourhis J. M.; Uversky V. N.; Canard B.; Longhi S. Proteins 2006, 62, 24. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. Biochim. Biophys. Acta 2013, 1834, 932. [DOI] [PubMed] [Google Scholar]
- Alber F.; Dokudovskaya S.; Veenhoff L. M.; Zhang W.; Kipper J.; Devos D.; Suprapto A.; Karni-Schmidt O.; Williams R.; Chait B. T.; Sali A.; Rout M. P. Nature 2007, 450, 695. [DOI] [PubMed] [Google Scholar]
- Yamada J.; Phillips J. L.; Patel S.; Goldfien G.; Calestagne-Morelli A.; Huang H.; Reza R.; Acheson J.; Krishnan V. V.; Newsam S.; Gopinathan A.; Lau E. Y.; Colvin M. E.; Uversky V. N.; Rexach M. F. Mol. Cell. Proteomics 2010, 9, 2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning D. P.; Rexach M. F. Mol. Cell. Proteomics 2007, 6, 272. [DOI] [PubMed] [Google Scholar]
- Patel S. S.; Belmont B. J.; Sante J. M.; Rexach M. F. Cell 2007, 129, 83. [DOI] [PubMed] [Google Scholar]
- Krishnan V. V.; Lau E. Y.; Yamada J.; Denning D. P.; Patel S. S.; Colvin M. E.; Rexach M. F. PLoS Comput. Biol. 2008, 4, e1000145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iakoucheva L. M.; Brown C. J.; Lawson J. D.; Obradovic Z.; Dunker A. K. J. Mol. Biol. 2002, 323, 573. [DOI] [PubMed] [Google Scholar]
- Tompa P. Nat. Chem. Biol. 2012, 8, 597. [DOI] [PubMed] [Google Scholar]
- Yang S.; Blachowicz L.; Makowski L.; Roux B. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 15757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner S.; Ogunjimi A. A.; Wang H. R.; Rotin D.; Sicheri F.; Wrana J. L.; Forman-Kay J. D. Cell 2007, 130, 651. [DOI] [PubMed] [Google Scholar]
- Weathers E. A.; Paulaitis M. E.; Woolf T. B.; Hoh J. H. FEBS Lett. 2004, 576, 348. [DOI] [PubMed] [Google Scholar]
- Lise S.; Jones D. T. Proteins 2005, 58, 144. [DOI] [PubMed] [Google Scholar]
- Mao A. H.; Lyle N.; Pappu R. V. Biochem. J. 2013, 449, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick S. L.; Jayaraman M.; Frieden C.; Wetzel R.; Pappu R. V. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 16764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran H. T.; Mao A.; Pappu R. V. J. Am. Chem. Soc. 2008, 130, 7380. [DOI] [PubMed] [Google Scholar]
- Teufel D. P.; Johnson C. M.; Lum J. K.; Neuweiler H. J. Mol. Biol. 2011, 409, 250. [DOI] [PubMed] [Google Scholar]
- Papoian G. A. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 14237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappu R. V.; Wang X.; Vitalis A.; Crick S. L. Arch. Biochem. Biophys. 2008, 469, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfmann R.; Alberti S.; Krishnan R.; Lyle N.; O’Donnell C. W.; King O. D.; Berger B.; Pappu R. V.; Lindquist S. Mol. Cell 2011, 43, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmeier M.; Hamilton J. A.; LeGall T.; Vacic V.; Cortese M. S.; Tantos A.; Szabo B.; Tompa P.; Chen J.; Uversky V. N.; Obradovic Z.; Dunker A. K. Nucleic Acids Res. 2007, 35, D786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R. K.; Pappu R. V. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 13392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucetic S.; Brown C. J.; Dunker A. K.; Obradovic Z. Proteins 2003, 52, 573. [DOI] [PubMed] [Google Scholar]
- Weathers E. A.; Paulaitis M. E.; Woolf T. B.; Hoh J. H. Proteins 2007, 66, 16. [DOI] [PubMed] [Google Scholar]
- Laursen B. S.; Kjaergaard A. C.; Mortensen K. K.; Hoffman D. W.; Sperling-Petersen H. U. Protein Sci. 2004, 13, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards Y. J.; Lobley A. E.; Pentony M. M.; Jones D. T. Genome Biol. 2009, 10, R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea C. A.; High A. A.; Obenauer J. C.; Mishra A.; Park C. G.; Punta M.; Schlessinger A.; Ma J.; Rost B.; Slaughter C. A.; Kriwacki R. W. J. Proteome Res. 2009, 8, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P.; Kalmar L. J. Mol. Biol. 2010, 403, 346. [DOI] [PubMed] [Google Scholar]
- Lobley A.; Swindells M. B.; Orengo C. A.; Jones D. T. PLoS Comput. Biol. 2007, 3, e162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuzman D.; Azia A.; Levy Y. J. Mol. Biol. 2010, 396, 674. [DOI] [PubMed] [Google Scholar]
- Xue B.; Li L.; Meroueh S. O.; Uversky V. N.; Dunker A. K. Mol. BioSyst. 2009, 5, 1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magidovich E.; Fleishman S. J.; Yifrach O. Bioinformatics 2006, 22, 1546. [DOI] [PubMed] [Google Scholar]
- Jaakola V. P.; Prilusky J.; Sussman J. L.; Goldman A. Protein Eng., Des. Sel. 2005, 18, 103. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan A. J.; Deupi X.; Lebon G.; Tate C. G.; Schertler G. F.; Babu M. M. Nature 2013, 494, 185. [DOI] [PubMed] [Google Scholar]
- Simon M.; Hancock J. M. Genome Biol. 2009, 10, R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima N.; Tanaka T.; Kretsinger R. H. Protein Pept. Lett. 2009, 16, 1297. [DOI] [PubMed] [Google Scholar]
- Jorda J.; Xue B.; Uversky V. N.; Kajava A. V. FEBS J. 2010, 277, 2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanov M. Y.; Galzitskaya O. V. Mol. BioSyst. 2012, 8, 327. [DOI] [PubMed] [Google Scholar]
- Lobanov M. Y.; Sokolovskiy I. V.; Galzitskaya O. V. Nucleic Acids Res. 2014, 42, D273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber H. P.; Seipel K.; Georgiev O.; Hofferer M.; Hug M.; Rusconi S.; Schaffner W. Science 1994, 263, 808. [DOI] [PubMed] [Google Scholar]
- Lobanov M. Y.; Furletova E. I.; Bogatyreva N. S.; Roytberg M. A.; Galzitskaya O. V. PLoS Comput. Biol. 2010, 6, e1000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gsponer J.; Babu M. M. Cell Rep. 2012, 2, 1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelitsch M. D.; Weissman J. S. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiumara F.; Fioriti L.; Kandel E. R.; Hendrickson W. A. Cell 2010, 143, 1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns M. A.; Alexander R. D.; Spiller M. P.; Beggs J. D. J. Cell Sci. 2008, 121, 2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mikecz A. Trends Cell Biol. 2009, 19, 685. [DOI] [PubMed] [Google Scholar]
- DePace A. H.; Santoso A.; Hillner P.; Weissman J. S. Cell 1998, 93, 1241. [DOI] [PubMed] [Google Scholar]
- Haynes C.; Iakoucheva L. M. Nucleic Acids Res. 2006, 34, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard P. J.; Hertel K. J. Genome Biol. 2009, 10, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang S.; Gapsys V.; Kim H. Y.; Bessonov S.; Hsiao H. H.; Mohlmann S.; Klaukien V.; Ficner R.; Becker S.; Urlaub H.; Luhrmann R.; de Groot B.; Zweckstetter M. Structure 2013, 21, 2162. [DOI] [PubMed] [Google Scholar]
- Ponte I.; Vila R.; Suau P. Mol. Biol. Evol. 2003, 20, 371. [DOI] [PubMed] [Google Scholar]
- Fang H.; Clark D. J.; Hayes J. J. Nucleic Acids Res. 2012, 40, 1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci V. S.; Engel J. L.; Wen J.; Wiley S. E.; Worby C. A.; Kinch L. N.; Xiao J.; Grishin N. V.; Dixon J. E. Science 2012, 336, 1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambort D.; Johansson M. E.; Gustafsson J. K.; Nilsson H. E.; Ermund A.; Johansson B. R.; Koeck P. J.; Hebert H.; Hansson G. C. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan J.; Gianni S.; Jemth P.. Phys. Chem. Chem. Phys. 2013. [DOI] [PubMed]
- Hammes G. G.; Chang Y. C.; Oas T. G. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J.; Guo L. W.; Muradov H.; Artemyev N. O.; Ruoho A. E.; Markley J. L. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefhaber T.; Bachmann A.; Jensen K. S. Curr. Opin. Struct. Biol. 2012, 22, 21. [DOI] [PubMed] [Google Scholar]
- Eliezer D.; Palmer A. G. III. Nature 2007, 447, 920. [DOI] [PubMed] [Google Scholar]
- Tompa P.; Fuxreiter M. Trends Biochem. Sci. 2008, 33, 2. [DOI] [PubMed] [Google Scholar]
- Mittag T.; Kay L. E.; Forman-Kay J. D. J. Mol. Recognit. 2010, 23, 105. [DOI] [PubMed] [Google Scholar]
- Graham T. A.; Ferkey D. M.; Mao F.; Kimelman D.; Xu W. Nat. Struct. Biol. 2001, 8, 1048. [DOI] [PubMed] [Google Scholar]
- Renault L.; Bugyi B.; Carlier M. F. Trends Cell Biol. 2008, 18, 494. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Fisher J. C.; Mathew R.; Ou L.; Otieno S.; Sublet J.; Xiao L.; Chen J.; Roussel M. F.; Kriwacki R. W. Nat. Chem. Biol. 2011, 7, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zor T.; Mayr B. M.; Dyson H. J.; Montminy M. R.; Wright P. E. J. Biol. Chem. 2002, 277, 42241. [DOI] [PubMed] [Google Scholar]
- Pometun M. S.; Chekmenev E. Y.; Wittebort R. J. J. Biol. Chem. 2004, 279, 7982. [DOI] [PubMed] [Google Scholar]
- Mittag T.; Orlicky S.; Choy W. Y.; Tang X.; Lin H.; Sicheri F.; Kay L. E.; Tyers M.; Forman-Kay J. D. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 17772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C. S.; Jorgensen C.; Linding R. Cell Cycle 2010, 9, 1276. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M.; Simon I.; Bondos S. Trends Biochem. Sci. 2011, 36, 415. [DOI] [PubMed] [Google Scholar]
- Naud J. F.; McDuff F. O.; Sauve S.; Montagne M.; Webb B. A.; Smith S. P.; Chabot B.; Lavigne P. Biochemistry 2005, 44, 12746. [DOI] [PubMed] [Google Scholar]
- Pufall M. A.; Lee G. M.; Nelson M. L.; Kang H. S.; Velyvis A.; Kay L. E.; McIntosh L. P.; Graves B. J. Science 2005, 309, 142. [DOI] [PubMed] [Google Scholar]
- Stott K.; Watson M.; Howe F. S.; Grossmann J. G.; Thomas J. O. J. Mol. Biol. 2010, 403, 706. [DOI] [PubMed] [Google Scholar]
- Jonker H. R.; Wechselberger R. W.; Boelens R.; Kaptein R.; Folkers G. E. Biochemistry 2006, 45, 5067. [DOI] [PubMed] [Google Scholar]
- Vuzman D.; Levy Y. Mol. BioSyst. 2012, 8, 47. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. Chem. Soc. Rev. 2011, 40, 1623. [DOI] [PubMed] [Google Scholar]
- Goldman N.; Thorne J. L.; Jones D. T. Genetics 1998, 149, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellay J.; Michaut M.; Kim T.; Han S.; Colak R.; Myers C. L.; Kim P. M. Mol. BioSyst. 2012, 8, 185. [DOI] [PubMed] [Google Scholar]
- Moesa H. A.; Wakabayashi S.; Nakai K.; Patil A. Mol. BioSyst. 2012, 8, 3262. [DOI] [PubMed] [Google Scholar]
- Liu J.; Perumal N. B.; Oldfield C. J.; Su E. W.; Uversky V. N.; Dunker A. K. Biochemistry 2006, 45, 6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantos A.; Han K. H.; Tompa P. Mol. Cell. Endocrinol. 2012, 348, 457. [DOI] [PubMed] [Google Scholar]
- Babu M. M.; van der Lee R.; de Groot N. S.; Gsponer J. Curr. Opin. Struct. Biol. 2011, 21, 432. [DOI] [PubMed] [Google Scholar]
- Colak R.; Kim T.; Michaut M.; Sun M.; Irimia M.; Bellay J.; Myers C. L.; Blencowe B. J.; Kim P. M. PLoS Comput. Biol. 2013, 9, e1003030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light S.; Sagit R.; Sachenkova O.; Ekman D.; Elofsson A. Mol. Biol. Evol. 2013, 30, 2645. [DOI] [PubMed] [Google Scholar]
- Mauri F.; McNamee L. M.; Lunardi A.; Chiacchiera F.; Del Sal G.; Brodsky M. H.; Collavin L. J. Biol. Chem. 2008, 283, 20848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonikian R.; Xin X.; Toret C. P.; Gfeller D.; Landgraf C.; Panni S.; Paoluzi S.; Castagnoli L.; Currell B.; Seshagiri S.; Yu H.; Winsor B.; Vidal M.; Gerstein M. B.; Bader G. D.; Volkmer R.; Cesareni G.; Drubin D. G.; Kim P. M.; Sidhu S. S.; Boone C. PLoS Biol. 2009, 7, e1000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkel H.; Chica C.; Via A.; Gould C. M.; Jensen L. J.; Gibson T. J.; Diella F. Nucleic Acids Res. 2011, 39, D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrao P.; Trinidad J. C.; Fiedler D.; Roguev A.; Lim W. A.; Shokat K. M.; Burlingame A. L.; Krogan N. J. PLoS Biol. 2009, 7, e1000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo J. C.; Giang K.; Chakrabarti S.; Ma C. T.; Huynh N.; Hagopian J. C.; Dorrestein P. C.; Fu X. D.; Adams J. A.; Ghosh G. Mol. Cell 2008, 29, 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero P.; Obradovic Z.; Dunker A. K. Appl. Bioinf. 2004, 3, 105. [DOI] [PubMed] [Google Scholar]
- Schad E.; Tompa P.; Hegyi H. Genome Biol. 2011, 12, R120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancsa R.; Tompa P. PLoS One 2012, 7, e34687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovic-Lazetic G. M.; Mitic N. S.; Kovacevic J. J.; Obradovic Z.; Malkov S. N.; Beljanski M. V. BMC Bioinf. 2011, 12, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B.; Williams R. W.; Oldfield C. J.; Dunker A. K.; Uversky V. N. BMC Syst. Biol. 2010, 4, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burra P. V.; Kalmar L.; Tompa P. PLoS One 2010, 5, e12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuriki N.; Oldfield C. J.; Uversky V. N.; Berezovsky I. N.; Tawfik D. S. Trends Biochem. Sci. 2009, 34, 53. [DOI] [PubMed] [Google Scholar]
- Longhi S. Protein Pept. Lett. 2010, 17, 930. [DOI] [PubMed] [Google Scholar]
- Vidalain P. O.; Tangy F. Microbes Infect. 2010, 12, 1134. [DOI] [PubMed] [Google Scholar]
- Xue B.; Williams R. W.; Oldfield C. J.; Goh G. K.; Dunker A. K.; Uversky V. N. Protein Pept. Lett. 2010, 17, 932. [DOI] [PubMed] [Google Scholar]
- Xue B.; Uversky V. N.. J. Mol. Biol. 2013. [DOI] [PubMed]
- Goubau D.; Deddouche S.; Reis E. S. C. Immunity 2013, 38, 855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B.; Peisley A.; Richards C.; Yao H.; Zeng X.; Lin C.; Chu F.; Walz T.; Hur S. Cell 2013, 152, 276. [DOI] [PubMed] [Google Scholar]
- Hou F.; Sun L.; Zheng H.; Skaug B.; Jiang Q. X.; Chen Z. J. Cell 2011, 146, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L. G.; Wang Y. Y.; Han K. J.; Li L. Y.; Zhai Z.; Shu H. B. Mol. Cell 2005, 19, 727. [DOI] [PubMed] [Google Scholar]
- Nallagatla S. R.; Toroney R.; Bevilacqua P. C. Curr. Opin. Struct. Biol. 2011, 21, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire P. A.; Tessmer I.; Craig R.; Erie D. A.; Cole J. L. Biochemistry 2006, 45, 9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanOudenhove J.; Anderson E.; Krueger S.; Cole J. L. J. Mol. Biol. 2009, 387, 910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elde N. C.; Child S. J.; Geballe A. P.; Malik H. S. Nature 2009, 457, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar A. C.; Dever T. E.; Sicheri F. Cell 2005, 122, 887. [DOI] [PubMed] [Google Scholar]
- Sawyer S. L.; Wu L. I.; Emerman M.; Malik H. S. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biris N.; Yang Y.; Taylor A. B.; Tomashevski A.; Guo M.; Hart P. J.; Diaz-Griffero F.; Ivanov D. N. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 13278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Ji X.; Zhao G.; Ning J.; Zhao Q.; Aiken C.; Gronenborn A. M.; Zhang P.; Xiong Y. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 18372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalskyy D. B.; Ivanov D. N.. Biochemistry 2014.
- Uversky V. N.; Oldfield C. J.; Dunker A. K. Annu. Rev. Biophys. 2008, 37, 215. [DOI] [PubMed] [Google Scholar]
- Hegyi H.; Buday L.; Tompa P. PLoS Comput. Biol. 2009, 5, e1000552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N. Front. Biosci. 2009, 14, 5188. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Oldfield C. J.; Midic U.; Xie H.; Xue B.; Vucetic S.; Iakoucheva L. M.; Obradovic Z.; Dunker A. K. BMC Genomics 2009, 10, S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo S. J. Curr. Opin. Chem. Biol. 2010, 14, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vavouri T.; Semple J. I.; Garcia-Verdugo R.; Lehner B. Cell 2009, 138, 198. [DOI] [PubMed] [Google Scholar]
- Kriventseva E. V.; Koch I.; Apweiler R.; Vingron M.; Bork P.; Gelfand M. S.; Sunyaev S. Trends Genet. 2003, 19, 124. [DOI] [PubMed] [Google Scholar]
- Romero P. R.; Zaidi S.; Fang Y. Y.; Uversky V. N.; Radivojac P.; Oldfield C. J.; Cortese M. S.; Sickmeier M.; LeGall T.; Obradovic Z.; Dunker A. K. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegyi H.; Kalmar L.; Horvath T.; Tompa P. Nucleic Acids Res. 2011, 39, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buljan M.; Chalancon G.; Eustermann S.; Wagner G. P.; Fuxreiter M.; Bateman A.; Babu M. M. Mol. Cell 2012, 46, 871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis J. D.; Barrios-Rodiles M.; Colak R.; Irimia M.; Kim T.; Calarco J. A.; Wang X.; Pan Q.; O’Hanlon D.; Kim P. M.; Wrana J. L.; Blencowe B. J. Mol. Cell 2012, 46, 884. [DOI] [PubMed] [Google Scholar]
- Buljan M.; Chalancon G.; Dunker A. K.; Bateman A.; Balaji S.; Fuxreiter M.; Babu M. M. Curr. Opin. Struct. Biol. 2013, 23, 443. [DOI] [PubMed] [Google Scholar]
- Reed H. C.; Hoare T.; Thomsen S.; Weaver T. A.; White R. A.; Akam M.; Alonso C. R. Genetics 2010, 184, 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondos S. E.; Hsiao H. C. Adv. Exp. Med. Biol. 2012, 725, 86. [DOI] [PubMed] [Google Scholar]
- Merkin J.; Russell C.; Chen P.; Burge C. B. Science 2012, 338, 1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnack K.; Konig J.; Tajnik M.; Martincorena I.; Eustermann S.; Stevant I.; Reyes A.; Anders S.; Luscombe N. M.; Ule J. Cell 2013, 152, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A.; Aloy P. PLoS One 2008, 3, e2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa-Morais N. L.; Irimia M.; Pan Q.; Xiong H. Y.; Gueroussov S.; Lee L. J.; Slobodeniuc V.; Kutter C.; Watt S.; Colak R.; Kim T.; Misquitta-Ali C. M.; Wilson M. D.; Kim P. M.; Odom D. T.; Frey B. J.; Blencowe B. J. Science 2012, 338, 1587. [DOI] [PubMed] [Google Scholar]
- Weatheritt R. J.; Davey N. E.; Gibson T. J. Nucleic Acids Res. 2012, 40, 7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. W.; Corboy M. J.; DeMartino G. N.; Thomas P. J. Science 2003, 299, 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash S.; Tian L.; Ratliff K. S.; Lehotzky R. E.; Matouschek A. Nat. Struct. Mol. Biol. 2004, 11, 830. [DOI] [PubMed] [Google Scholar]
- Takeuchi J.; Chen H.; Coffino P. EMBO J. 2007, 26, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P.; Prilusky J.; Silman I.; Sussman J. L. Proteins 2008, 71, 903. [DOI] [PubMed] [Google Scholar]
- Schrader E. K.; Harstad K. G.; Matouschek A. Nat. Chem. Biol. 2009, 5, 815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov P.; Reuven N.; Prives C.; Shaul Y. J. Biol. Chem. 2009, 284, 26234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbain S.; Prakash S.; Herrig A.; Elsasser S.; Matouschek A. Nat. Commun. 2011, 2, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inobe T.; Fishbain S.; Prakash S.; Matouschek A. Nat. Chem. Biol. 2011, 7, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng A. H.; Fang N. N.; Comyn S. A.; Gsponer J.; Mayor T. Mol. Cell. Proteomics 2013, 12, 2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravid T.; Hochstrasser M. Nat. Rev. Mol. Cell Biol. 2008, 9, 679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies R. J.; Joazeiro C. A. Annu. Rev. Biochem. 2009, 78, 399. [DOI] [PubMed] [Google Scholar]
- Sharipo A.; Imreh M.; Leonchiks A.; Imreh S.; Masucci M. G. Nat. Med. 1998, 4, 939. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Coffino P. J. Biol. Chem. 2004, 279, 8635. [DOI] [PubMed] [Google Scholar]
- Tian L.; Holmgren R. A.; Matouschek A. Nat. Struct. Mol. Biol. 2005, 12, 1045. [DOI] [PubMed] [Google Scholar]
- Orlowski M.; Wilk S. Arch. Biochem. Biophys. 2003, 415, 1. [DOI] [PubMed] [Google Scholar]
- Alvarez-Castelao B.; Castano J. G. FEBS Lett. 2005, 579, 4797. [DOI] [PubMed] [Google Scholar]
- Asher G.; Tsvetkov P.; Kahana C.; Shaul Y. Genes Dev. 2005, 19, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher G.; Reuven N.; Shaul Y. BioEssays 2006, 28, 844. [DOI] [PubMed] [Google Scholar]
- Tsvetkov P.; Asher G.; Paz A.; Reuven N.; Sussman J. L.; Silman I.; Shaul Y. Proteins 2008, 70, 1357. [DOI] [PubMed] [Google Scholar]
- Wiggins C. M.; Tsvetkov P.; Johnson M.; Joyce C. L.; Lamb C. A.; Bryant N. J.; Komander D.; Shaul Y.; Cook S. J. J. Cell Sci. 2011, 124, 969. [DOI] [PubMed] [Google Scholar]
- Tsvetkov P.; Reuven N.; Shaul Y. Nat. Chem. Biol. 2009, 5, 778. [DOI] [PubMed] [Google Scholar]
- Gilmore R.; Blobel G.; Walter P. J. Cell Biol. 1982, 95, 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore R.; Walter P.; Blobel G. J. Cell Biol. 1982, 95, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirndorfer D.; Seidel R. P.; Nimrod G.; Miesbauer M.; Ben-Tal N.; Engelhard M.; Zimmermann R.; Winklhofer K. F.; Tatzelt J. J. Biol. Chem. 2013, 288, 13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesbauer M.; Pfeiffer N. V.; Rambold A. S.; Muller V.; Kiachopoulos S.; Winklhofer K. F.; Tatzelt J. J. Biol. Chem. 2009, 284, 24384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidah N. G.; Chretien M. Brain Res. 1999, 848, 45. [DOI] [PubMed] [Google Scholar]
- Peysselon F.; Xue B.; Uversky V. N.; Ricard-Blum S. Mol. BioSyst. 2011, 7, 3353. [DOI] [PubMed] [Google Scholar]
- Kalmar L.; Homola D.; Varga G.; Tompa P. Bone 2012, 51, 528. [DOI] [PubMed] [Google Scholar]
- Wojtas M.; Dobryszycki P.; Ożyhar A. In Advanced Topics in Biomineralization; Seto J., Ed.; InTech: Rijeka, Croatia, 2012; pp 1–32. [Google Scholar]
- Dores R. M.; Baron A. J. Ann. N.Y. Acad. Sci. 2011, 1220, 34. [DOI] [PubMed] [Google Scholar]
- Muiznieks L. D.; Weiss A. S.; Keeley F. W. Biochem. Cell Biol. 2010, 88, 239. [DOI] [PubMed] [Google Scholar]
- Fisher L. W.; Torchia D. A.; Fohr B.; Young M. F.; Fedarko N. S. Biochem. Biophys. Res. Commun. 2001, 280, 460. [DOI] [PubMed] [Google Scholar]
- Eliezer D. Curr. Opin. Struct. Biol. 2009, 19, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton T. E.Proteins: Structures and Molecular Properties; W.H. Freeman & Co.: New York, 1993. [Google Scholar]
- Auton M.; Bolen D. W. Methods Enzymol. 2007, 428, 397. [DOI] [PubMed] [Google Scholar]
- Hengst L.; Dulic V.; Slingerland J. M.; Lees E.; Reed S. I. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurotani A.; Takagi T.; Toyama M.; Shirouzu M.; Yokoyama S.; Fukami Y.; Tokmakov A. A. FASEB J. 2010, 24, 1095. [DOI] [PubMed] [Google Scholar]
- Santner A. A.; Croy C. H.; Vasanwala F. H.; Uversky V. N.; Van Y. Y.; Dunker A. K. Biochemistry 2012, 51, 7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N.; Ivanov P.; Anderson P. Trends Biochem. Sci. 2013, 38, 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M.; Han T. W.; Xie S.; Shi K.; Du X.; Wu L. C.; Mirzaei H.; Goldsmith E. J.; Longgood J.; Pei J.; Grishin N. V.; Frantz D. E.; Schneider J. W.; Chen S.; Li L.; Sawaya M. R.; Eisenberg D.; Tycko R.; McKnight S. L. Cell 2012, 149, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P. Intrinsically Disord. Proteins 2013, 1, e24068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd J. C.; Bresalier R. S. Cancer Metastasis Rev. 2004, 23, 77. [DOI] [PubMed] [Google Scholar]
- Thornton D. J.; Rousseau K.; McGuckin M. A. Annu. Rev. Physiol. 2008, 70, 459. [DOI] [PubMed] [Google Scholar]
- Silverman H. S.; Sutton-Smith M.; McDermott K.; Heal P.; Leir S. H.; Morris H. R.; Hollingsworth M. A.; Dell A.; Harris A. Glycobiology 2003, 13, 265. [DOI] [PubMed] [Google Scholar]
- Hanisch F. G.; Muller S. Glycobiology 2000, 10, 439. [DOI] [PubMed] [Google Scholar]
- Verdugo P. Cold Spring Harbor Perspect. Med. 2012, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George A.; Veis A. Chem. Rev. 2008, 108, 4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner S.; Addadi L. Annu. Rev. Mater. Res. 2011, 41, 21. [Google Scholar]
- Barrere F.; van Blitterswijk C. A.; de Groot K. Int. J. Nanomed. 2006, 1, 317. [PMC free article] [PubMed] [Google Scholar]
- Kawasaki K.; Lafont A. G.; Sire J. Y. Mol. Biol. Evol. 2011, 28, 2053. [DOI] [PubMed] [Google Scholar]
- Guettler S.; LaRose J.; Petsalaki E.; Gish G.; Scotter A.; Pawson T.; Rottapel R.; Sicheri F. Cell 2011, 147, 1340. [DOI] [PubMed] [Google Scholar]
- Jiang K.; Toedt G.; Montenegro Gouveia S.; Davey N. E.; Hua S.; van der Vaart B.; Grigoriev I.; Larsen J.; Pedersen L. B.; Bezstarosti K.; Lince-Faria M.; Demmers J.; Steinmetz M. O.; Gibson T. J.; Akhmanova A. Curr. Biol. 2012, 22, 1800. [DOI] [PubMed] [Google Scholar]
- Via A.; Gould C. M.; Gemund C.; Gibson T. J.; Helmer-Citterich M. BMC Bioinf. 2009, 10, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller M. R.Current Protocols in Protein Science; Wiley: New York, 2007; Chapter 2, Unit 2 12. [DOI] [PubMed] [Google Scholar]
- Chica C.; Labarga A.; Gould C. M.; Lopez R.; Gibson T. J. BMC Bioinf. 2008, 9, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould C. M.; Diella F.; Via A.; Puntervoll P.; Gemund C.; Chabanis-Davidson S.; Michael S.; Sayadi A.; Bryne J. C.; Chica C.; Seiler M.; Davey N. E.; Haslam N.; Weatheritt R. J.; Budd A.; Hughes T.; Pas J.; Rychlewski L.; Trave G.; Aasland R.; Helmer-Citterich M.; Linding R.; Gibson T. J. Nucleic Acids Res. 2010, 38, D167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linding R.; Jensen L. J.; Ostheimer G. J.; van Vugt M. A.; Jorgensen C.; Miron I. M.; Diella F.; Colwill K.; Taylor L.; Elder K.; Metalnikov P.; Nguyen V.; Pasculescu A.; Jin J.; Park J. G.; Samson L. D.; Woodgett J. R.; Russell R. B.; Bork P.; Yaffe M. B.; Pawson T. Cell 2007, 129, 1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekaran S.; Merlin J. C.; Kundeti V.; Mi T.; Oommen A.; Vyas J.; Alaniz I.; Chung K.; Chowdhury F.; Deverasatty S.; Irvey T. M.; Lacambacal D.; Lara D.; Panchangam S.; Rathnayake V.; Watts P.; Schiller M. R. Proteins 2011, 79, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey N. E.; Cowan J. L.; Shields D. C.; Gibson T. J.; Coldwell M. J.; Edwards R. J. Nucleic Acids Res. 2012, 40, 10628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Ba A. N.; Yeh B. J.; van Dyk D.; Davidson A. R.; Andrews B. J.; Weiss E. L.; Moses A. M. Sci. Signaling 2012, 5, rs1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neduva V.; Linding R.; Su-Angrand I.; Stark A.; de Masi F.; Gibson T. J.; Lewis J.; Serrano L.; Russell R. B. PLoS Biol. 2005, 3, e405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey N. E.; Haslam N. J.; Shields D. C.; Edwards R. J. Nucleic Acids Res. 2010, 38, W534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck P. V.; Chabra I.; Kornhauser J. M.; Skrzypek E.; Zhang B. Proteomics 2004, 4, 1551. [DOI] [PubMed] [Google Scholar]
- Gnad F.; Gunawardena J.; Mann M. Nucleic Acids Res. 2011, 39, D253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J.; Wagih O.; Bader G. D. Sci. Rep. 2013, 3, 2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J.; Bader G. D. Mol. Syst. Biol. 2013, 9, 637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauer J. C.; Cantley L. C.; Yaffe M. B. Nucleic Acids Res. 2003, 31, 3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. L.; Jensen L. J.; Diella F.; Jorgensen C.; Tinti M.; Li L.; Hsiung M.; Parker S. A.; Bordeaux J.; Sicheritz-Ponten T.; Olhovsky M.; Pasculescu A.; Alexander J.; Knapp S.; Blom N.; Bork P.; Li S.; Cesareni G.; Pawson T.; Turk B. E.; Yaffe M. B.; Brunak S.; Linding R. Sci. Signaling 2008, 1, ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linding R.; Jensen L. J.; Pasculescu A.; Olhovsky M.; Colwill K.; Bork P.; Yaffe M. B.; Pawson T. Nucleic Acids Res. 2008, 36, D695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safaei J.; Manuch J.; Gupta A.; Stacho L.; Pelech S. Proteome Sci. 2011, 9, S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhaber B.; Eisenhaber F. Methods Mol. Biol. 2010, 609, 365. [DOI] [PubMed] [Google Scholar]
- Disfani F. M.; Hsu W. L.; Mizianty M. J.; Oldfield C. J.; Xue B.; Dunker A. K.; Uversky V. N.; Kurgan L. Bioinformatics 2012, 28, i75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztanyi Z.; Meszaros B.; Simon I. Bioinformatics 2009, 25, 2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meszaros B.; Simon I.; Dosztanyi Z. PLoS Comput. Biol. 2009, 5, e1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuchi S.; Amemiya T.; Sakamoto S.; Nobe Y.; Hosoda K.; Kado Y.; Murakami S. D.; Koike R.; Hiroaki H.; Ota M. Nucleic Acids Res. 2014, 42, D320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobley A. E.; Nugent T.; Orengo C. A.; Jones D. T. Nucleic Acids Res. 2008, 36, W297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzetto D.; Jones D. T. Curr. Opin. Struct. Biol. 2013, 23, 467. [DOI] [PubMed] [Google Scholar]
- Minneci F.; Piovesan D.; Cozzetto D.; Jones D. T. PLoS One 2013, 8, e63754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neduva V.; Russell R. B. Curr. Opin. Biotechnol. 2006, 17, 465. [DOI] [PubMed] [Google Scholar]
- Daughdrill G. W.; Borcherds W. M.; Wu H. PLoS One 2011, 6, e29207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cilia E.; Pancsa R.; Tompa P.; Lenaerts T.; Vranken W. F. Nat. Commun. 2013, 4, 2741. [DOI] [PubMed] [Google Scholar]
- Vitalis A.; Pappu R. V. J. Comput. Chem. 2009, 30, 673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher C. K.; Stultz C. M. J. Am. Chem. Soc. 2011, 133, 10022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyle N.; Das K.; Pappu R. V. J. Chem. Phys. 2013, 139, 121907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi M.; Kosol S.; Lebrun P.; Valentini E.; Blackledge M.; Dunker A. K.; Felli I. C.; Forman-Kay J. D.; Kriwacki R. W.; Pierattelli R.; Sussman J.; Svergun D. I.; Uversky V. N.; Vendruscolo M.; Wishart D.; Wright P. E.; Tompa P. Nucleic Acids Res. 2014, 42, D326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y.; Selenko P. Curr. Opin. Struct. Biol. 2010, 20, 640. [DOI] [PubMed] [Google Scholar]
- Vucetic S.; Obradovic Z.; Vacic V.; Radivojac P.; Peng K.; Iakoucheva L. M.; Cortese M. S.; Lawson J. D.; Brown C. J.; Sikes J. G.; Newton C. D.; Dunker A. K. Bioinformatics 2005, 21, 137. [DOI] [PubMed] [Google Scholar]
- Di Domenico T.; Walsh I.; Martin A. J.; Tosatto S. C. Bioinformatics 2012, 28, 2080. [DOI] [PubMed] [Google Scholar]
- Hornbeck P. V.; Kornhauser J. M.; Tkachev S.; Zhang B.; Skrzypek E.; Murray B.; Latham V.; Sullivan M. Nucleic Acids Res. 2012, 40, D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson H. J.; Wright P. E. Chem. Rev. 2004, 104, 3607. [DOI] [PubMed] [Google Scholar]
- Sibille N.; Bernado P. Biochem. Soc. Trans. 2012, 40, 955. [DOI] [PubMed] [Google Scholar]
- Baker C. M.; Best R. B.. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2013. [DOI] [PMC free article] [PubMed]
- Bracken C.; Iakoucheva L. M.; Romero P. R.; Dunker A. K. Curr. Opin. Struct. Biol. 2004, 14, 570. [DOI] [PubMed] [Google Scholar]
- Monastyrskyy B.; Fidelis K.; Moult J.; Tramontano A.; Kryshtafovych A. Proteins 2011, 79, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monastyrskyy B.; Kryshtafovych A.; Moult J.; Tramontano A.; Fidelis K. Proteins 2014, 82, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron F.; Longhi S.; Canard B.; Karlin D. Proteins 2006, 65, 1. [DOI] [PubMed] [Google Scholar]
- Dosztanyi Z.; Meszaros B.; Simon I. Briefings Bioinf. 2010, 11, 225. [DOI] [PubMed] [Google Scholar]
- Dosztanyi Z.; Csizmok V.; Tompa P.; Simon I. Bioinformatics 2005, 21, 3433. [DOI] [PubMed] [Google Scholar]
- Prilusky J.; Felder C. E.; Zeev-Ben-Mordehai T.; Rydberg E. H.; Man O.; Beckmann J. S.; Silman I.; Sussman J. L. Bioinformatics 2005, 21, 3435. [DOI] [PubMed] [Google Scholar]
- Ishida T.; Kinoshita K. Bioinformatics 2008, 24, 1344. [DOI] [PubMed] [Google Scholar]
- Mizianty M. J.; Stach W.; Chen K.; Kedarisetti K. D.; Disfani F. M.; Kurgan L. Bioinformatics 2010, 26, i489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X.; Eickholt J.; Cheng J. BMC Bioinf. 2009, 10, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayrose I.; Graur D.; Ben-Tal N.; Pupko T. Mol. Biol. Evol. 2004, 21, 1781. [DOI] [PubMed] [Google Scholar]
- Chen S. C.; Chuang T. J.; Li W. H. Mol. Biol. Evol. 2011, 28, 2513. [DOI] [PMC free article] [PubMed] [Google Scholar]