

Abstract
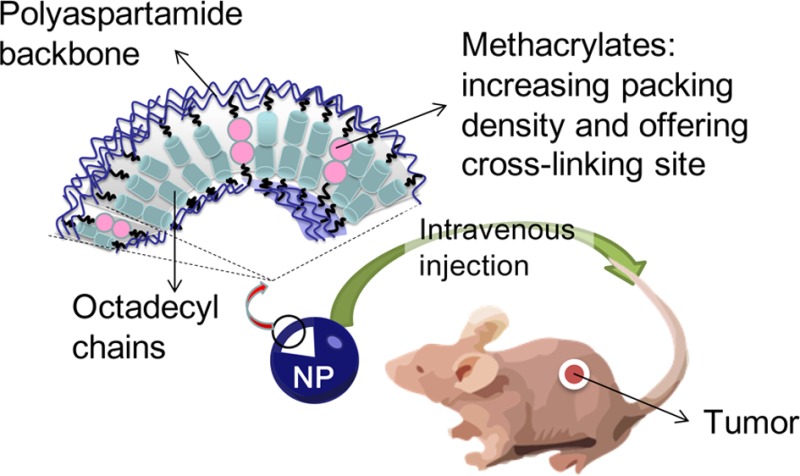
Self-assembled nanoparticles conjugated with various imaging contrast agents have been used for the detection and imaging of pathologic tissues. Inadvertently, these nanoparticles undergo fast, dilution-induced disintegration in circulation and quickly lose their capability to associate with and image the site of interest. To resolve this challenge, we hypothesize that decreasing the bilayer permeability of polymersomes can stabilize their structure, extend their lifetime in circulation, and hence improve the quality of bioimaging when the polymersome is coupled with an imaging probe. This hypothesis is examined by using poly(2-hydroxyethyl-co-octadecyl aspartamide), sequentially modified with methacrylate groups, to build model polymersomes. The bilayer permeability of the polymersome is decreased by increasing the packing density of the bilayer with methacrylate groups and is further decreased by inducing chemical cross-linking reactions between the methacrylate groups. The polymersome with decreased bilayer permeability demonstrates greater particle stability in physiological media and ultimately can better highlight tumors in mice over 2 days compared to those with higher bilayer permeability after labeling with a near-infrared (NIR) fluorescent probe. We envisage that the resulting nanoparticles will not only improve diagnosis but also further image-guided therapies.
Keywords: polyaspartamide, near-infrared (NIR) fluorescence imaging, enhanced permeability and retention (EPR) effect, polymeric vesicles (polymersomes), bilayer permeability
Introduction
Extensive efforts have been made to detect pathologic tissues (i.e., proinflammatory tissue, tumors) at an early stage, as treatment is more effective when they are found early.1,2 Common methods of diagnosis currently include biochemical screening of blood samples or whole body imaging.3 Specifically, whole body imaging offers several advantages, including the ability to locate the pathologic tissue and further assess therapeutic outcomes in a noninvasive manner. Biomedical imaging techniques that are used commonly in preclinical and clinical settings, including magnetic resonance imaging,4 optical coherence tomography,5,6 and near-infrared (NIR) fluorescence imaging,7,8 often employ molecules or nanoparticles that can provide enhanced image contrast.4−8 Furthermore, efforts are increasingly made to deliver these imaging contrast agents exclusively to the target tissue in order to pinpoint it using conventional imaging modalities.
One popular approach is to conjugate imaging contrast agents to nanoparticles that can diffuse through the leaky inflamed or tumor vasculature and accumulate in the extravascular tissue, termed as the enhanced permeability and retention (EPR) effect.9,10 Alternatively, nanoparticles conjugated with ligands can bind actively with receptors overexpressed by target pathologic cells.4−6 Integrating the EPR effect with the active targeting is also being explored in the design of advanced nanoparticles targeting tissues of interest. These efforts, however, are often plagued by the limited circulation time of the self-assembled nanoparticles in vivo.11 For example, in mice and rats, nanoparticles should continue circulating for at least 6 h to achieve the desired EPR effect.12 Accordingly, a variety of approaches have been developed to increase the circulation time of nanoparticles equipped with imaging contrast agents in the bloodstream. For instance, the nanoparticle surface is conjugated with poly(ethylene glycol) (PEG) to reduce the mononuclear phagocyte system-mediated clearance.13 Separately, the molecular weights of nanoparticle-forming molecules can be tailored to improve the structural integrity of nanoparticles: nanoparticles formed from the self-assembly of high molecular weight polymers remained more stable than small molecular weight molecules, such as lipids and surfactants.14 These approaches are often combined together to attain synergistic improvements of the particle stability.
However, self-assembled polymeric micelles were still reported to disintegrate quickly in the circulation due to dilution effects. For example, poly(caprolactone)-b-poly(ethylene oxide) and poly(d,l-lactide)-b-methoxypoly(ethylene glycol) micelles were found to dissociate within 1 h under biological conditions.15,16 Similar to the polymeric micelle, it is highly plausible that polymeric vesicles, termed polymersomes, would also quickly dissociate in circulation before achieving an effective accumulation in target tissues. To resolve this challenge, we hypothesized that reducing the bilayer permeability of polymersomes would greatly enhance the particle structural stability in circulation. Accordingly, the particles with a reduced permeability would enhance the quality of image-based diagnosis of pathologic tissues. We examined this hypothesis by introducing a controlled number of methacrylate groups into the bilayer of polymersomes formed from the self-assembly of poly(2-hydroxyethyl-co-octadecyl aspartamide) (PHEA-C18), because methacrylate groups can associate with each other and increase the packing density of the bilayer (Scheme 1). In addition, after particle assembly, the methacrylate groups were cross-linked to further decrease the bilayer permeability. Afterward, the polymer was further modified with an NIR fluorescent probe, FPR-675, and the capability of the resulting polymersome in detecting and imaging pathologic tissues was evaluated by systemically injecting them into a mouse tumor model of squamous cell carcinoma.8,17 In summary, this study should greatly serve to expand the lifetime of polymersomes under physiological conditions, contribute to the early detection of pathologic tissues, and hence take image-based diagnostics to the next level.
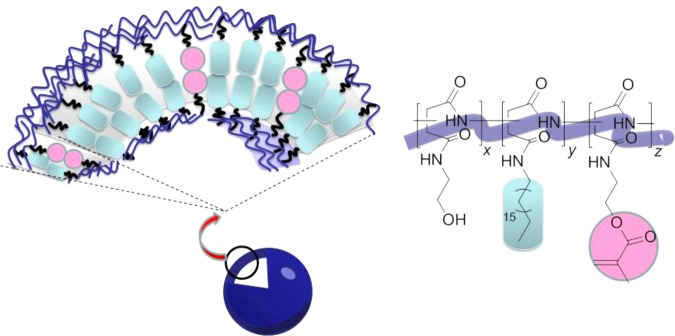
Scheme 1. Schematic Description of the Structure of a Poly(2-hydroxyethyl-co-2-methacryloxyethyl-co-octadecyl aspartamide) (PHEA-C18-MA) Polymersome, in Which the Bilayer Became Less Permeable Due to the Increased Packing Density Resulting from the Higher Degree of Substitution of Methacrylate (DSMA) and from the Cross-Linking of Methacrylate Groups.
Results
Preparation and Characterizations of PHEA-C18 and PHEA-C18-MA
Poly(2-hydroxyethyl-co-2-methacryloxyethyl-co-octadecyl aspartamide) (PHEA-C18-MA) was synthesized by modifying polysuccinimide (PSI) with octadecyl chains and methacrylate groups. PSI was prepared via the acid-catalyzed polycondensation of l-aspartic acid.18 The successive reactions of PSI with the designated amounts of octadecylamine and 2-aminoethyl methacrylate hydrochloride resulted in PSI substituted with octadecyl and methacrylate groups (Scheme 2, PSI-C18-MA). Subsequent addition of an excess of ethanolamine to the polymer solution led to the substitution of all remaining succiminide units with hydroxyl groups (Scheme 2, PHEA-C18-MA), as confirmed with 1H NMR (Supporting Information Figure S1). Separately, PHEA-C18-MA without methacrylate groups (PHEA-C18) and unalkylated poly(2-hydroxyethyl aspartamide) (PHEA) were synthesized as control polymers.
Scheme 2. Synthesis of PHEA-C18-MA, Where the Polymer Backbone Was Substituted with Octadecyl Chains and a Varying Degree of Substitution of Methacrylate.

DSMA, denoted as ‘z’ in the scheme; z = 0 for PHEA-C18. x and y in the reaction scheme represent the degree of substitution of hydroxyl groups and the degree of substitution of octadecyl chains (DSC18), respectively.
The degree of substitution of octadecyl chains (DSC18) of PHEA-C18 and PHEA-C18-MA polymers, defined as the mole percent of succinimide units substituted with octadecyl chains, was tuned to the range from 36 to 37 mol % according to the integrals of the characteristic NMR peaks at 0.85 to 0.95 ppm and 4.3 to 4.7 ppm (Supporting Information Figure S1 and Table 1). The peak at 0.85 to 0.95 ppm represents the protons of methyl groups at the ends of the substituted octadecyl chains, and the peak at 4.3 to 4.7 ppm is due to the protons on the polymer backbone. Additionally, the degree of substitution of methacrylate (DSMA) of PHEA-C18-MA, defined as the mole percent of succinimide units reacted with 2-aminoethy methacrylate hydrochloride, was tuned to 2.7 and 4.8 mol %, as quantified by the integrals of the characteristic NMR peaks at 4.3 to 4.7, 5.7, and 6.1 ppm. The peaks at 5.7 and 6.1 ppm represent the two protons on the vinyl carbon of the substituted methacrylate, and the peak at 4.3 to 4.7 ppm represents the protons on the polymer backbone (Supporting Information Figure S1). For convenience, PHEA-C18 and PHEA-C18-MA polymers at DSMA of 2.7 and 4.8 mol % are termed as MA-0.0, MA-2.7, and MA-4.8, respectively.
Table 1. Molecular Analysis of PHEA-C18 and PHEA-C18-MA with Controlled DSMA.
| hydrodynamic
radius (RH, nm)b |
||||
|---|---|---|---|---|
| sample | DSC18 [mol %]a | DSMA [mol %]a | not cross-linked | cross-linked |
| MA-0.0 | 36.7 | 0.0 | 102 ± 2 | |
| MA-2.7 | 36.4 | 2.7 | 102 ± 8 | 99 ± 3 |
| MA-4.8 | 36.9 | 4.8 | 90 ± 5 | 85 ± 5 |
Determined with the 1H NMR spectra of the polymers.
Determined by dynamic light scattering (DLS). The particles were suspended in deionized water. Data represented are the average values followed by the standard deviation from three independent experiments.
The ability of MA-0.0, MA-2.7, and MA-4.8 to self-associate in aqueous media was analyzed by the emission intensity of pyrene incorporated into the hydrophobic domain of the polymeric assembly. It is common to assess the critical aggregation concentration (CAC) of amphiphilic molecules by measuring the increase in the pyrene emission intensity ratio (I3/I1) between 385 (I3) and 373 (I1) nm.19 Pyrene mixed with unalkylated PHEA, formed by PSI substituted solely with hydroxyl groups, showed an insignificant increase in I3/I1 with the increasing polymer concentration, while the pyrene incorporated into a suspension of MA-0.0 showed an increase of I3/I1 at a CAC of 5 × 10–2 mg/mL (Figure 1a). Furthermore, the CAC value was found to be inversely related to DSMA of the studied polymer (Figure 1b).
Figure 1.

Pyrene-based fluorescence analysis of polyaspartamide polymersomes. (a) The fluorescence intensity ratio while the pyrene was incorporated into a suspension of MA-0.0, MA-2.7, MA-4.8, and PHEA in phosphate buffered saline (PBS), respectively. (b) The critical aggregation concentration (CAC), marked by the turning point of I3/I1 values in part a, of MA-0.0, MA-2.7, and MA-4.8 in PBS.
Preparation and Analysis of PHEA-C18 and PHEA-C18-MA Polymersomes
PHEA-based polymersomes were prepared by a solvent exchange process (Supporting Information Figure S2). Irgacure 2959 and polymers were first dissolved in dimethyl sulfoxide (DMSO) to provide chain mobility sufficient for intermolecular self-assembly in aqueous media. The subsequent introduction of this mixture into deionized water and the removal of DMSO by dialysis resulted in spherical polymersomes, as confirmed with transmission electron microscopy (TEM). The polymersome suspension was further exposed to ultraviolet (UV) light to activate the cross-linking reaction within the bilayer of the polymersome. No significant difference of the particle morphology was found between the polymersome before (Figure 2a) and after (Figure 2b) the exposure to UV light.
Figure 2.

Morphology characterization and bilayer permeability analysis of polyaspartamide polymersomes. (a) TEM micrographs of the self-assembled polymersomes of (I) MA-0.0, (II) MA-2.7, and (III) MA-4.8, for which all of the bilayers were not cross-linked. (b) TEM micrographs of the self-assembled polymersomes of (I) MA-2.7 and (II) MA-4.8. The bilayers of MA-2.7 and MA-4.8 polymersomes were cross-linked. All scale bars in parts a and b represent 200 nm. (c) Calcein-based fluorescence analysis of MA-2.7 and MA-4.8 polymersomes, either with cross-linked (CL) or without cross-linked (NCL) bilayers. The filled and open bars represent polymersomes incubated in deionized water and in deionized water supplemented with plasma (10%, v/v), respectively.
The average radii of MA-0.0, MA-2.7, and MA-4.8 polymersomes were also measured with dynamic light scattering (DLS) (Table 1). No significant difference in the average hydrodynamic radius (RH) was found between MA-0.0 or MA-2.7 polymersomes with or without a cross-linked bilayer. However, increasing the DSMA from 2.7 mol % to 4.8 mol % resulted in a decrease of RH from 102 to 90 nm and from 99 to 85 nm for the polymersome without cross-linked bilayers and those with, respectively.
We further analyzed how the DSMA and the subsequent cross-linking reaction influenced the bilayer permeability of polymersomes by measuring the amount of calcein released from the polymersome during the incubation in deionized water or 10% (v/v) plasma solution. Calcein is a fluorescein derivative that self-quenches as its concentration exceeds 2–3 mM.20 Therefore, encapsulated calcein at high concentrations self-quenches within polymersomes, while that released into the media generates fluorescence emission.21,22 As such, the suspension of polymersomes with the lower bilayer permeability should show a slower recovery of calcein fluorescence than those with the higher bilayer permeability. As expected, MA-2.7 polymersomes with the cross-linked bilayer showed a smaller increase of fluorescence intensity than MA-2.7 polymersomes without the cross-linked bilayers, especially when the particles were incubated in 10% (v/v) plasma aqueous solution (Figure 2c). In addition, increasing the DSMA from 2.7 to 4.8 mol % significantly limited the fluorescence recovery both in deionized water and in the 10% plasma solution, even without cross-linking the bilayer. Interestingly, at the high DSMA, chemical cross-linking of the bilayer did not make a difference in the fluorescence recovery rate (Figure 2c).
Biochemical stability of PHEA-based polymersomes was evaluated by monitoring changes of the particle radius while the polymersomes were incubated in PBS at 37 °C. MA-2.7 and MA-4.8 polymersomes displayed better stability than MA-0.0 polymersomes, even without cross-linking the bilayer. The MA-0.0 polymersomes in PBS disappeared within 48 h, following a 2-fold increase of the average RH (Figure 3a). In contrast, MA-2.7 and MA-4.8 polymersomes without cross-linked bilayers retained their original size over 12 days (open squares in Figure 3b and c). Interestingly, increasing DSMA significantly lessened the growth of RH over time. Specifically, the RH of MA-2.7 polymersomes increased from 108 to 214 nm within 1 day, followed by a gradual increase to 230 nm over the following 11 days (open squares in Figure 3b). In contrast, the RH of MA-4.8 polymersomes increased from 89 to 121 nm in the first day, followed by a minimal increase of RH over time (open squares in Figure 3c).
Figure 3.

Stability analysis of polyaspartamide polymersomes with dynamic light scattering (DLS). The changes in the hydrodynamic radius (RH) of the polymersomes of (a) MA-0.0, (b) MA-2.7, and (c) MA-4.8. The polymersomes were incubated in PBS at 37 °C. The filled and open squares represent the polymersomes with bilayers in which methacrylate groups were cross-linked and not cross-linked, respectively. *After 2 days, RH of MA-0.0 polymersomes could not be measured with DLS.
Further cross-linking of the polymersome bilayer reduced the degree of increase in RH, depending on DSMA. The RH of MA-2.7 polymersomes with a cross-linked bilayer exhibited an increase from 100 to 200 nm within the first 24 h and remained constant over the following 11 days (filled squares in Figure 3b), which showed a lessened expansion than the RH of the MA-2.7 polymersome without a cross-linked bilayer. In contrast, there was a minimal difference of the size change profile between the MA-4.8 polymersome with a cross-linked bilayer and that without a cross-linked bilayer (Figure 3c).
Incubating polymersomes at 90 °C, which is above the melting temperature of the polymer (Supporting Information Figure S3), also demonstrated different structural disintegration rates between the particles with a cross-linked bilayer and those without. The MA-0.0 polymersome disappeared within 1 day (Supporting Information Figure S4a). In contrast, both MA-2.7 and MA-4.8 polymersomes with cross-linked bilayers showed limited size changes (Supporting Information Figure S4b and S4c), while MA-4.8 polymersomes with a cross-linked bilayer had the smallest change on its size distribution before and after being incubated at the evaluated temperature. Additionally, these polymersomes demonstrated little, if any, cellular toxicity according to an analysis of cellular metabolic activity using MTT reagent. Endothelial cells incubated with 0.25 or 0.01 mg/mL of MA-2.7 or MA-4.8 polymersomes retained a level of metabolic activity similar to untreated cells (Supporting Information Figure S5).
Functionalization of Polymersomes with an NIR Fluorescent Probe and In Vivo Assessment
The polymersomes of MA-2.7 and MA-4.8 were functionalized with an NIR fluorescent probe, FPR-675, in order to use the particles as a tool to detect and image pathologic tissue innervated by abnormal, leaky vasculature. The sulfonic acid group of FPR-675 was conjugated to the hydroxyl group of PHEA-C18-MA via esterification (Supporting Information Scheme S1). Accordingly, the polymersome suspension displayed an absorbance peak at 675 nm (Figure 4a), as well as NIR fluorescence emission at 700 nm upon excitation at 675 nm (Figure 4b).
Figure 4.
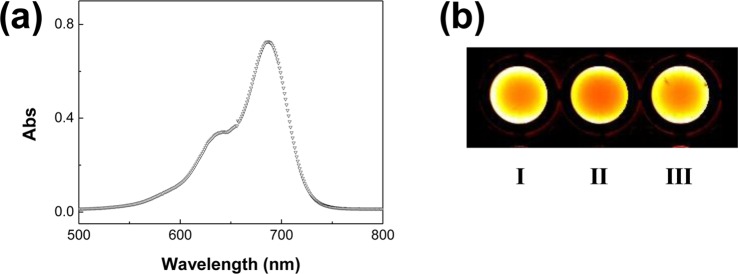
Characterization of PHEA-C18-MA polymersomes modified with the NIR probe, FPR-675. (a) UV–visible spectrum of MA-2.7 conjugated with FPR-675. (b) The NIR fluorescence image of (I) free FPR-675 solution, (II) FPR-675-labeled MA-2.7 polymersome suspension, and (III) FPR-675-labeled MA-4.8 polymersome suspension. The methacrylate groups in the bilayers of polymersomes were cross-linked.
Next, the ability of FPR-675-labeled PHEA-C18-MA polymersomes to image tumor tissue was evaluated by systemically injecting them into circulation of a tumor-bearing mouse model and imaging the whole body with a real-time NIR fluorescence imaging system. The tumor was created by subcutaneously injecting squamous cell carcinoma (SCC7) into the back of a mouse. Injection of free FPR-675 solution via tail vein yielded a low level of positive fluorescence in tumor sites throughout 48 h. The fluorescence intensity from the tumor site was comparable to the autofluorescence level of the neighboring tissue, thus making it difficult to distinguish tumor from the normal tissue (Figure 5a–i).
Figure 5.

In vivo evaluation of PHEA-C18-MA polymersomes in targeting and imaging tumors. (a) NIR fluorescence images of tumor-bearing mice after the intravenous injection of (I) free FPR-675 probes, (II) FPR-675-labeled MA-2.7 polymersomes without cross-linked bilayers, and (III) FPR-675-labeled MA-2.7 polymersomes with cross-linked bilayers. Arrows mark the tumor site. (b) NIR fluorescence intensity changes over 48 h in the tumor tissues of the corresponding mouse shown in part a. (black square) represents free FPR-675 dye, (red circle) represents FPR-675-labeled MA-2.7 polymersomes without cross-linked bilayers, and (blue triangle) represents FPR-675-labeled MA-2.7 polymersomes with cross-linked bilayers. (c) NIR fluorescence intensities of the ex vivo organ images from the tumor-bearing mice 48 h after the injection. The black, red, and blue bars represent free FPR-675 dye, FPR-675-labeled MA-2.7 polymersomes without cross-linked bilayers, and FPR-675-labeled MA-2.7 polymersomes with cross-linked bilayers, respectively.
In contrast, FPR-675-labeled MA-2.7 polymersomes without a cross-linked bilayer made a 1.4-fold increase of the fluorescence intensity compared to the free FPR-675 solution (Figure 5a and b). Although the fluorescence from the FPR-675-labeled MA-2.7 polymersome without a cross-linked bilayer in the tumor site gradually decreased throughout 48 h, it was kept at a higher level than that of a mouse injected with the free FPR-675 solution. In addition, the FRP-675-labeled MA-2.7 polymersome with a cross-linked bilayer generated 1.6 times higher fluorescence intensity at the tumor site compared to the MA-2.7 polymersome without cross-linking 1 h after injection (Figure 5b). This represented the highest intensity among the three injections over 48 h, making the tumor site clearly distinguished from the normal neighboring tissue (Figure 5a-III). However, further decreasing the permeability of the polymersome bilayer by increasing DSMA made minimal enhancements in elevating the fluorescence intensity in cancer sites over time (Supporting Information Figure S6).
Biodistribution of free FPR-675 and FPR-675-labeled polymersomes were evaluated with NIR fluorescence images of multiple organs including liver, lung, spleen, kidney, heart, and tumor, all of which were collected 48 h after the injection. Similar to the real time imaging, the highest NIR fluorescence of the ex vivo tumor was achieved with the FPR-675-labeled MA-2.7 polymersome with a cross-linked bilayer (Figure 5c). Additionally, the difference in NIR fluorescence between the tumor tissue and other organs, with the exception of the kidney, was also largest with the FPR-675-labeled, bilayer cross-linked MA-2.7 polymersome. Interestingly, absence of the cross-linked structure in the polymersome bilayer caused the particles to accumulate more in the kidney than in tumor tissue.
Discussion
In summary, this study demonstrates an advanced method to improve the imaging of pathologic, tumor tissue using PHEA-C18-MA polymersomes engineered to present less permeable bilayers. The bilayer permeability was decreased by introducing more numbers of methacrylate groups into the bilayer of the polymersome without cross-linking treatment, and also by further cross-linking the bilayer. We found that the polymersome with a larger DSMA (MA-4.8 polymersome) was more stable in physiological media than that without methacrylate groups (MA-0.0 polymersome) and that with a smaller DSMA (MA-2.7 polymersome), even without cross-linking treatment. Additionally, the cross-linking of the bilayer further decreased the permeability and enhanced the stability of the polymersome compared to its uncross-linked form, but only for the MA-2.7 polymersome. Lastly, the MA-2.7 polymersome that was modified to carry an NIR fluorescent probe and present a cross-linked bilayer significantly improved the imaging quality of tumor sites over 48 h after systemic injection.
According to previous studies, polymersomes should disassemble more slowly than liposomes of a similar CAC when the concentration falls below the CAC.23−25 However, the complete disappearance of PHEA-C18 polymersomes in PBS within 48 h implies that increasing the molecular weight of self-assembling molecules does not perfectly circumvent the structural disassembly on a long-term basis. We propose that methacrylate groups linked to the PHEA-C18 polymer backbone hydrophobically associate with each other in the confined bilayer together with octadecyl chains, due to the mismatch of Hildebrand solubility parameter between ethyl methacrylate (8.61 (cal/cm3)1/2)26 and water (23.5 (cal/cm3)1/2).27 Additionally, it is unlikely that these methacrylate groups impair the vesicular structure of PHEA-C18-MA, as confirmed with TEM micrographs and supported by theoretical calculation (Supporting Information Table S1).28−30 We suggest that both the hydrophobic association and the pH-independent cross-linking bonds between methacrylate groups greatly serve to improve particle stability and subsequent targeted imaging quality, whereas other cross-linking bonds such as hydrazine and disulfide bonds may disintegrate in tissue with pH deviating from neutral. Therefore, methacrylate groups incorporated into the bilayer likely played a similar role as cholesterol in phospholipid liposome bilayers. It is well agreed that cholesterol reduces the free space in bilayers,31,32 and subsequently enhances liposome stability.33 Similarly, increasing the packing density should decrease the bilayer permeability and further decelerate the structural disintegration of polymersomes caused by dilution effects, as manifested with the slower calcein release from MA-4.8 than MA-2.7 polymersomes without cross-linking and the smallest size increase of MA-4.8 polymersomes. We therefore interpret that the stability achieved by increasing DSMA is due to the increase of packing density in the bilayer. Additionally, the decrease of RH with increasing DSMA should be attributed to an increase of the hydrophobic association between polymers and the subsequent formation of a more compact bilayer.
Additionally, cross-linking of the bilayer has further enhanced the MA-2.7 polymersome stability exclusively. The slower recovery of fluorescence from calcein in the MA-2.7 polymersome with a cross-linked bilayer can be attributed to the decreased bilayer permeability, while the limited size increase of MA-2.7 polymersomes with a cross-linked bilayer over time indicates that the particle has a greater resistance to structural disintegration. In contrast, the independence of the bilayer permeability and particle size change on the cross-linking of the MA-4.8 polymersome bilayer is likely due to the high packing density of the bilayer negating the effect of cross-linking.
Taken together, we propose that the control of bilayer permeability and particle stability serve to improve the quality of NIR fluorescence imaging of cancerous tissues. Prior studies on the EPR effect suggested that particle diameters should be smaller than 400 nm.34,35 The size of PHEA-C18-MA polymersomes used in this study fit within this particle size range. However, it is likely that lipids in blood serum accelerate the expansion or disassembly of MA-2.7 polymersomes without cross-linked bilayers,15,16,36 thus limiting the accumulation of particles in the tumor site via the EPR effect as well as accelerating the renal clearance of disassembled monomers due to the low molecular weight.37 Additionally, the accumulated particles in the tumor site can be degraded gradually and hence the fluorescence of the area of interest decreased. In contrast, the MA-2.7 polymersome with a cross-linked bilayer should remain stable in circulation over an extended time period to extravasate through leaky tumor vasculature, and the accumulated particles in the tumor site, while still degradable, were degraded slower than the polymersome without cross-linked bilayers.
Previously, some studies demonstrated that cross-linking the bilayer of a microsized gigantic polymersome results in increasing the surface elastic modulus, wall stress, and resistance to surfactant-induced disassembly than traditional liposomes.38,39 However, no efforts have been made to improve detection and imaging of cancer tissue by translating these findings into the assembly of nanosized polymersomes to date. Therefore, we envisage that our finding will be very useful to designing nanoparticles used for diagnostics and also treatment of various acute and malignant diseases, including cancer and cardiovascular diseases.40,41 Further extending the lifetime of polymersomes in circulation, as well as the time window for cancer imaging with a single dose, could potentially be achieved by conjugating PEG chains to the PHEA-C18-MA polymersome. The quality of image-guided diagnostics will be further improved by conjugating peptides or antibodies that can specifically bind with target cells to the polymersomes developed in this study. On a separate note, though PHEA-C18-MA polymersomes have been shown to demonstrate limited toxicity to the mouse endothelial cells, further investigation is needed to evaluate performance in humans.
Conclusion
In summary, this study demonstrated a nanoparticle platform engineered to present a reduced permeability that is useful for significantly improving the detection and imaging of pathologic tissue. The methacrylate groups attached to a self-assembling polyaspartamide polymer served to reduce the bilayer permeability, likely because they associated with other hydrophobic alkyl chains and increased the packing density. Cross-linking of the methacylate groups postassembly of the nanoparticles further reduced the bilayer permeability, thus leading to another increase of the particle stability in physiological media, exclusively at an intermediate degree of substitution for methacrylate groups to the polymer. Therefore, the polymersome conjugated with an NIR fluorescent probe and tailored to display reduced permeability significantly improved the imaging quality of tumor sites over 48 h after systemic injection. These results certify the importance of extending the lifetime of nanoparticles in improving their function in detecting and imaging pathologic tissues of interest. Finally, the polymersomes with a tailored permeability will be useful in future studies to modulate the release rate of various therapeutic molecules and ultimately improve the quality of diagnosis and treatment of diverse diseases.
Experimental Section
Materials were purchased from Sigma-Aldrich (U.S.A.) and used without further purification unless otherwise specified.
Synthesis of Poly(2-hydroxyethyl-co-octadecyl aspartamide) (PHEA-C18) and Poly(2-hydroxyethyl-co-2-methacryloxyethyl-co-octadecyl aspartamide) (PHEA-C18-MA)
Polysuccinimide (PSI) was synthesized by thermal condensation of L-aspartic acid (30 g) suspended in sulfolane (150 mL) at 170 °C under a nitrogen atmosphere for 14 h with phosphoric acid (0.612 mL, Fisher Scientific) as a catalyst.18 PSI was precipitated in excess methanol and successively washed with deionized water until the pH of the suspension reached neutral. The precipitate was dried by lyophilization. The molecular weight of PSI was determined by gel permeation chromatography (Breeze 2 GPC, Waters), with Styragel HT column (Waters). N,N-dimethylformamide (DMF) containing 20 mM LiBr was used as the eluent, with the elution rate of 1 mL min. Polystyrene standards were used for calibration. Mn = 13,600 g mol with PDI = 1.4.
Purified PSI (79 mg) was dissolved in DMF (5 mL, Fisher Scientific), and octadecylamine (79 mg for MA-0.0, MA-2.7, and MA-4.8, respectively) was added to the reaction mixture. The reaction was stirred at 70 °C for 24 h. Next, a designated amount of 2-aminoethyl methacrylate hydrochloride (AEMA) (0 μL for MA-0.0, 195 μL for MA-2.7, and 391 μL for MA-4.8) and triethylamine (TEA, Fisher Scientific) (0 μL for MA-0.0, 10.6 μL for MA-2.7, and 21.2 μL for MA-4.8) were added to the reaction mixture, and the solution was stirred at 45 °C for 24 h. The molar ratio of AEMA to TEA was 1:3. Lastly, an excess amount of ethanolamine was added (96.8 μL for MA-0.0, 82.3 μL for MA-2.7, and 75.4 μL for MA-4.8) and the reaction was further stirred at 45 °C overnight. Following completion of the chemical reactions, the products were dialyzed (MWCO 3500 Da, Fisher Scientific) extensively in deionized water at room temperature for 2 days and then lyophilized to yield dry powders.
Measurement of the Critical Aggregation Concentration (CAC)
Pyrene molecules (Acros) were suspended at a concentration of 10−4 mg/mL in the polymer solution. The fluorescence spectra of the suspensions with varying polymer concentrations were collected using a FluoroMax-4 spectrometer (HORIBA Jobin Yvon). The excitation wavelength was set at 330 nm and the resulting emission between 350 and 450 nm was collected; the slit widths for excitation and emission were both set as 1 nm. The CAC was determined by the polymer concentration at the point where the emission intensity ratio (I3/I1) between the third vibronic peak at 385 nm (I3) and the first vibronic peak at 373 nm (I1) was significantly increased.19
Synthesis of PHEA-C18-MA Labeled with FPR-675
FPR-675 (5 μmol) (BioActs) was dissolved in 500 μL of DMSO (Merck) and added dropwise to PHEA-C18-MA dissolved in DMSO (10 mg in 2 mL). The reaction mixture was stirred overnight at room temperature in the dark. The products were dialyzed extensively in deionized water with light protection at room temperature to remove the free dye and DMSO, and then lyophilized to yield dry powders. The resulting polymers were analyzed with a UV/vis spectrophotometer Lambda 7 (PerkinElmer).
Preparations of PHEA-C18 and PHEA-C18-MA Polymersomes
PHEA-C18-MA polymers and Irgacure 2959 (Ciba Specialty Chemicals) were dissolved in DMSO at a concentration 10 and 100 mg mL, respectively. 100 μL polymer in DMSO was mixed with 20 μL initiator in DMSO, and the mixture was dropped into 1 mL deionized water. The solution was sonicated for 10–15 s and then dialyzed in deionized water extensively to remove DMSO and excess initiator overnight. During dialysis, the suspension was covered with aluminum foil to prevent exposure to light. The purified suspension was finally exposed to UV light (Model 20 CHIPhERASER, Jelight Co. Inc.) for 5 min to cross-link the bilayer of the polymersome. Separately, in preparation of PHEA-C18 or PHEA-C18-MA polymersomes without cross-linked bilayers, Irgacure 2959 was not mixed with the polymer solution; nor was the polymersome suspension exposed to UV light. In the stability analysis, 10× phosphate buffered saline (PBS) was added to adjust the ionic strength of the polymersome suspension to 1× PBS after the particle preparation.
Characterization of PHEA-C18 and PHEA-C18-MA Polymersomes
The morphology of self-assembled PHEA-C18-MA polymersomes was observed using TEM (JEOL 2100 with LaB6 emitter) operating at an accelerating voltage of 120 keV. The polymersome suspension was dropped onto a 300-mesh copper grid coated with carbon and Formvar (SPI Supplies), and the sample was stained with 2% (w/v) phosphotungstic acid solution (adjusted to pH 7.4 with NaOH). The size of PHEA-C18-MA polymersomes was measured using a Malvern Zetasizer Nano ZS (Malvern, 4 mW He–Ne laser operating at a wavelength of 633 nm) with 173° backscattering. The hydrodynamic radius was determined using cumulant analysis (International Organization for Standardization 13321:1996). All measurements were carried out in triplicate and performed at 25 °C.
Calcein Release Study
PHEA-C18-MA polymersomes were prepared by the solvent exchange process. Then, 50 mM calcein (Fisher Scientific) aqueous solution was added slowly into the polymersome suspension to a final concentration of 25 mM for diffusional loading. The suspension was incubated at room temperature with stirring for 1 day for calcein to reach equilibrium, and exposed to UV light to activate the cross-linking reaction in the bilayer of polymersomes. Certain polymersome suspensions were not exposed to UV light for a control experiment. The excess calcein that was not incorporated into the polymersomes was removed by Amicon Ultra-0.5 Centrifugal Filter Devices (MWCO 100k Da, Millipore Corporation). The polymersomes were incubated within deionized water or deionized water supplemented by 10% (v/v) human plasma (Equitech-Bio Inc.). After incubation for 30 min at room temperature, the amount of calcein released from the polymersome was measured with the emission intensity at 535 (±20) nm at the excitation of 485 (±20) nm using a microplate reader (Infinite 200Pro, Tecan Group Ltd.).
In Vivo Noninvasive NIR Fluorescence Imaging of Polymersomes in Tumor-Bearing Mice
All animal experiments were performed in compliance with the guidelines approved by the institutional ethics committee for animal care of Korea Institute of Science and Technology (KIST). To generate a tumor-bearing mouse model, squamous cell carcinoma (SCC7) tumors were induced into 5-week-old male athymic nude mice (Institute of Medical Science, Tokyo) by subcutaneous injection of 1.0 × 106 SCC7 cells. When the tumor diameter grew to approximately 10 mm, the test samples (5 mg kg) were injected into the tumor-bearing mice through the tail vein, and the whole body images were taken at 1, 3, 6, 12, 24, and 48 h after the injection, using a time-domain fluorescence imager eXplore Optix (ART Advanced Research Technologies Inc.). Laser power and count time settings were optimized at 9 μW and 0.3 s per point. Excitation and emission spots were raster-scanned in 1 mm steps over the selected region of interest to generate emission wavelength scans. A 675 nm pulsed laser diode was used to excite FPR-675 molecules. NIR fluorescence emission at 700 nm was collected and detected with a fast photomultiplier tube (Hamamatsu) and a time-correlated single photon counting system (Becker and Hickl GmbH). NIR fluorescence intensities of all samples were adjusted to the same values based on the data obtained by a 12-bit CCD camera (Kodak Imaging Station 4000 MM, New Haven) equipped with a C-mount lens and a Cy5.5 bandpass emission filter set (680 to 720 nm, Omega Optical). Furthermore, the accumulation level of samples in the tumors was evaluated by measuring the NIR fluorescence intensity at the cancer site. All data were processed using the region of interest (ROI) function of the Analysis Workstation software (ART Advanced Research Technologies Inc.).
Ex Vivo Organ Analysis
After intravenous injection of FPR-675 labeled polymersomes, major organs and tumors were dissected from mice 48 h after the injection. NIR fluorescence images of dissected organs including liver, lung, spleen, kidney, heart, and tumors were obtained with Kodak Image Station 4000 MM. The image station was equipped with a 12-bit CCD camera, halogen lamp (150 W), and excitation/emission filter sets for FPR-675 (600–700 nm; Omega Optical). The biodistribution of polymersomes was evaluated by quantifying the NIR fluorescence intensity of the organ and tumor images processed with Kodak molecular imaging software.
Acknowledgments
This work was supported in part by the National Institutes of Health (1R01 HL109192 to H.J.K.) and the Intramural Research Program (Theragnosis) of Korea Institute of Science and Technology. M.-H. Lai appreciates the support from the Dow Chemical Company Graduate Fellowship and Study Abroad Scholarship from Ministry of Education of Republic of China (Taiwan). Transmission electron microscopy and dynamic light scattering were carried out in part in the Frederick Seitz Materials Research Laboratory Central Facilities, University of Illinois.
Supporting Information Available
Additional TEM images, 1H NMR spectra, pyrene analysis, and DSC measurement of the polymers. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Flehinger B. J.; Kimmel M.; Melamed M. R. The Effect of Surgical Treatment on Survival from Early Lung Cancer. Implications for Screening. Chest 1992, 101, 1013–1018. [DOI] [PubMed] [Google Scholar]
- Bernie T.; Les I.; Paul G.; Jan K.; David W.; Chris S. A Systematic Review of the Effects of Screening for Colorectal Cancer Using the Faecal Occult Blood Test, Hemoccult. BMJ 1998, 317, 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Optical Imaging of Cancer; Rosenthal E., Zinn K. R., Eds.; Springer: New York, 2010. [Google Scholar]
- Artemov D.; Mori N.; Okollie B.; Bhujwalla Z. M. MR Molecular Imaging of the Her-2/neu Receptor in Breast Cancer Cells Using Targeted Iron Oxide Nanoparticles. Magn. Reson. Med. 2003, 49, 403–408. [DOI] [PubMed] [Google Scholar]
- Jain P. K.; El-Sayed I. H.; El-Sayed M. A. Au Nanoparticles Target Cancer. Nano Today 2007, 2, 18–29. [Google Scholar]
- Chen J.; Saeki F.; Wiley B. J.; Cang H.; Cobb M. J.; Li Z.-Y.; Au L.; Zhang H.; Kimmey M. B.; Li; Xia Y. Gold Nanocages: Bioconjugation and Their Potential Use as Optical Imaging Contrast Agents. Nano Lett. 2005, 5, 473–477. [DOI] [PubMed] [Google Scholar]
- Ghoroghchian P. P.; Frail P. R.; Susumu K.; Blessington D.; Brannan A. K.; Bates F. S.; Chance B.; Hammer D. A.; Therien M. J. Near-Infrared-Emissive Polymersomes: Self-Assembled Soft Matter for In Vivo Optical Imaging. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 2922–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.; Kim J. H.; Park H.; Kim Y.-S.; Park K.; Nam H.; Lee S.; Park J. H.; Park R.-W.; Kim I.-S.; Choi K.; Kim S. Y.; Park K.; Kwon I. C. Tumor-Homing Multifunctional Nanoparticles for Cancer Theragnosis: Simultaneous Diagnosis, Drug Delivery, and Therapeutic Monitoring. J. Controlled Release 2010, 146, 219–227. [DOI] [PubMed] [Google Scholar]
- Matsumura Y.; Maeda H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [PubMed] [Google Scholar]
- Maeda H.; Wu J.; Sawa T.; Matsumura Y.; Hori K. Tumor Vascular Permeability and the EPR Effect in Macromolecular Therapeutics: A Review. J. Controlled Release 2000, 65, 271–284. [DOI] [PubMed] [Google Scholar]
- Gabizon A. A. Liposome Circulation Time and Tumor Targeting: Implications for Cancer Chemotherapy. Adv. Drug Delivery Rev. 1995, 16, 285–294. [Google Scholar]
- Iyer A. K.; Khaled G.; Fang J.; Maeda H. Exploiting the Enhanced Permeability and Retention Effect for Tumor Targeting. Drug Discovery Today 2006, 11, 812–818. [DOI] [PubMed] [Google Scholar]
- Harris J. M.; Martin N. E.; Modi M. Pegylation: A Novel Process for Modifying Pharmacokinetics. Clin. Pharmacokinet. 2001, 40, 539–551. [DOI] [PubMed] [Google Scholar]
- Dennis E. Discher; Eisenberg A. Polymer Vesicles. Science 2002, 297, 967–973. [DOI] [PubMed] [Google Scholar]
- Savić R.; Azzam T.; Eisenberg A.; Maysinger D. Assessment of the Integrity of Poly(caprolactone)-b-poly(ethylene oxide) Micelles under Biological Conditions: A Fluorogenic-Based Approach. Langmuir 2006, 22, 3570–3578. [DOI] [PubMed] [Google Scholar]
- Burt H. M.; Zhang X.; Toleikis P.; Embree L.; Hunter W. L. Development of Copolymers of Poly(d,l-lactide) and Methoxypolyethylene Glycol as Micellar Carriers of Paclitaxel. Colloids Surf., B 1999, 16, 161–171. [Google Scholar]
- Na J. H.; Koo H.; Lee S.; Min K. H.; Park K.; Yoo H.; Lee S. H.; Park J. H.; Kwon I. C.; Jeong S. Y.; Kim K. Real-Time and Non-invasive Optical Imaging of Tumor-Targeting Glycol Chitosan Nanoparticles in Various Tumor Models. Biomaterials 2011, 32, 5252–5261. [DOI] [PubMed] [Google Scholar]
- Tomida M.; Nakato T.; Matsunami S.; Kakuchi T. Convenient Synthesis of High Molecular Weight Poly(succinimide) by Acid-Catalysed Polycondensation of l-Aspartic Acid. Polymer 1997, 38, 4733–4736. [Google Scholar]
- Kalyanasundaram K.; Thomas J. K. Environmental Effects on Vibronic Band Intensities in Pyrene Monomer Fluorescence and Their Application in Studies of Micellar Systems. J. Am. Chem. Soc. 1977, 99, 2039–2044. [Google Scholar]
- Hamann S.; Kiilgaard J.; Litman T.; Alvarez-Leefmans F.; Winther B.; Zeuthen T. Measurement of Cell Volume Changes by Fluorescence Self-Quenching. J. Fluoresc. 2002, 12, 139–145. [Google Scholar]
- Allen T. M.; Cleland L. G. Serum-Induced Leakage of Liposome Contents. Biochim. Biophys. Acta, Biomembr. 1980, 597, 418–426. [DOI] [PubMed] [Google Scholar]
- Cerritelli S.; Velluto D.; Hubbell J. A. PEG-SS-PPS: Reduction-Sensitive Disulfide Block Copolymer Vesicles for Intracellular Drug Delivery. Biomacromolecules 2007, 8, 1966–1972. [DOI] [PubMed] [Google Scholar]
- Kwon G. S.; Okano T. Polymeric Micelles as New Drug Carriers. Adv. Drug Delivery Rev. 1996, 21, 107–116. [Google Scholar]
- Kazunori K.; Glenn S K.; Masayuki Y.; Teruo O.; Yasuhisa S. Block Copolymer Micelles As Vehicles for Drug Delivery. J. Controlled Release 1993, 24, 119–132. [Google Scholar]
- Adams M. L.; Lavasanifar A.; Kwon G. S. Amphiphilic Block Copolymers for Drug Delivery. J. Pharm. Sci. 2003, 92, 1343–1355. [DOI] [PubMed] [Google Scholar]
- Lewin J. L.; Maerzke K. A.; Schultz N. E.; Ross R. B.; Siepmann J. I. Prediction of Hildebrand Solubility Parameters of Acrylate and Methacrylate Monomers and Their Mixtures by Molecular Simulation. J. Appl. Polym. Sci. 2010, 116, 1–9. [Google Scholar]
- Hansen C. M. The Three Dimensional Solubility Parameter—Key to Paint Component Affinities I. Solvents, Plasticizers, Polymers, and Resins. J. Paint Technol. 1967, 39, 104–117. [Google Scholar]
- Lee H. J.; Yang S. R.; An E. J.; Kim J.-D. Biodegradable Polymersomes from Poly(2-hydroxyethyl aspartamide) Grafted with Lactic Acid Oligomers in Aqueous Solution. Macromolecules 2006, 39, 4938–4940. [Google Scholar]
- Lai M.-H.; Jeong J. H.; DeVolder R. J.; Brockman C.; Schroeder C.; Kong H. Ellipsoidal Polyaspartamide Polymersomes with Enhanced Cell-Targeting Ability. Adv. Funct. Mater. 2012, 22, 3239–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J. H.; Cha C.; Kaczmarowski A.; Haan J.; Oh S.; Kong H. Polyaspartamide Vesicle Induced by Metallic Nanoparticles. Soft Matter 2012, 8, 2237–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck E.; Patra M.; Karttunen M.; Hyvönen M. T.; Vattulainen I. Lessons of Slicing Membranes: Interplay of Packing, Free Area, and Lateral Diffusion in Phospholipid/Cholesterol Bilayers. Biophys. J. 2004, 87, 1076–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alwarawrah M.; Dai J.; Huang J. A Molecular View of the Cholesterol Condensing Effect in DOPC Lipid Bilayers. J. Phys. Chem. B 2010, 114, 7516–7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder A.; Kost J.; Barenholz Y. Ultrasound, Liposomes, and Drug Delivery: Principles for Using Ultrasound to Control the Release of Drugs from Liposomes. Chem. Phys. Lipids 2009, 162, 1–16. [DOI] [PubMed] [Google Scholar]
- Soo Choi H.; Liu W.; Misra P.; Tanaka E.; Zimmer J. P.; Itty Ipe B.; Bawendi M. G.; Frangioni J. V. Renal Clearance of Quantum Dots. Nat. Biotechnol. 2007, 25, 1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae Y. H.; Park K. Targeted Drug Delivery to Tumors: Myths, Reality, and Possibility. J. Controlled Release 2011, 153, 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Kim S.; Li L.; Wang S.; Park K.; Cheng J.-X. Release of Hydrophobic Molecules from Polymer Micelles into Cell Membranes Revealed by Förster Resonance Energy Transfer Imaging. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 6596–6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour L. W.; Duncan R.; Strohalm J.; Kopeček J. Effect of Molecular Weight (Mw) of N-(2-hydroxypropyl)methacrylamide Copolymers on Body Distribution and Rate of Excretion After Subcutaneous, Intraperitoneal, and Intravenous Administration to Rats. J. Biomed. Mater. Res. 1987, 21, 1341–1358. [DOI] [PubMed] [Google Scholar]
- Discher B. M.; Bermudez H.; Hammer D. A.; Discher D. E.; Won Y.-Y.; Bates F. S. Cross-linked Polymersome Membranes: Vesicles with Broadly Adjustable Properties. J. Phys. Chem. B 2002, 106, 2848–2854. [Google Scholar]
- Ahmed F.; Hategan A.; Discher D. E.; Discher B. M. Block Copolymer Assemblies with Cross-Link Stabilization: From Single-Component Monolayers to Bilayer Blends with PEO–PLA. Langmuir 2003, 19, 6505–6511. [Google Scholar]
- Godin B.; Sakamoto J. H.; Serda R. E.; Grattoni A.; Bouamrani A.; Ferrari M. Emerging Applications of Nanomedicine for the Diagnosis and Treatment of Cardiovascular Diseases. Trends Pharmacol. Sci. 2010, 31, 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Miyoshi H.; Nakamura M. Nanomedicine for Drug Delivery and Imaging: A Promising Avenue for Cancer Therapy and Diagnosis Using Targeted Functional Nanoparticles. Int. J. Cancer 2007, 120, 2527–2537. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.