

Abstract
Here, we describe the synthesis of the single amino acid chelator DOTAlaP and four of its derivatives. The corresponding gadolinium(III) complexes were investigated for their kinetic inertness, relaxometric properties at a range of fields and temperatures, water exchange rate, and interaction with human serum albumin (HSA). Derivatives with one inner-sphere water (q = 1) were determined to have a mean water residency time between 8 and 6 ns in phoshate-buffered saline at 37 °C. The corresponding europium complexes were also formed and used to obtain information on the hydration number of the corresponding coordination complexes. Two complexes capable of binding HSA were also synthesized, of which one, Gd(5b), contains no inner-sphere water, while the other derivative, Gd(4b), is a mixture of ca. 15% q =1 and 85% q = 0. In the presence of HSA, the latter displayed a very short mean water residency time (τM310 = 2.4 ns) and enhanced relaxivity at intermediate and high fields. The kinetic inertness of Gd(4b) with respect to complex dissociation was decreased compared to its DOTAla analogue but still 100-fold more inert than [Gd(BOPTA)(H2O)]2–. Magnetic resonance imaging in mice showed that Gd(4b) was able to provide 38% better vessel to muscle contrast compared to the clinically used HSA binding agent MS-325.
Short abstract
Converting one of the acetate groups in the single amino acid chelator DOTAla to methylenephosphonate (DOTAlaP) results in gadolinium(III) complexes with extremely fast water exchange kinetics and/or in equilibrium between monoaquated (q = 1) and unaquated (q = 0) states. The presence of phosphonate and the very fast water exchange kinetics result in stable complexes with high relaxivity at high magnetic fields, especially when bound to serum albumin.
Introduction
Magnetic resonance imaging (MRI) is a powerful noninvasive imaging technique, providing high-resolution anatomical images with excellent soft-tissue contrast. A large fraction of MRI studies utilize a contrast agent, e.g., more than half the clinical studies performed at our institution make use of a gadolinium(III)-based contrast agent. These contrast agents are simple, water-soluble, ternary gadolinium(III) complexes with gadolinium(III) coordinated by an octadentate poly(aminocarboxylato) ligand and an aqua coligand.1,2 Gadolinium(III) is advantageous for use in MRI contrast agents because of its large electron spin number (S = 7/2) and long electronic relaxation time, which result in efficient nuclear relaxation of nearby solvent water molecules. Contrast agents are characterized by their relaxivity, defined as the change in the solvent relaxation rate (1/T1 or 1/T2) normalized to the concentration of the contrast agent (units of mM–1 s–1). First-generation clinical contrast agents are monomeric gadolinium(III) complexes based on a [Gd(DOTA)(H2O)]− or [Gd(DTPA)(H2O)]2– (DTPA = diethylenetriaminepentaacetic acid) framework and have relaxivities in the range 3–5 mM–1 s–1.3,4 Second-generation contrast agents such as gadofosveset (MS-325, Ablavar) used reversible protein binding to increase the relaxivity.5 Protein binding slows the rotational tumbling rate of the complex, and as this rate approaches the proton Larmor frequency, nuclear relaxation is enhanced.6,7 Specific protein binding also serves to direct the contrast agent to specific pathologies, e.g., thrombosis, as well as enhance the relaxivity.8,9
However, the increase in the relaxivity due to protein binding is greatest by far at low magnetic fields (≤1.5 T) and falls off precipitously at higher fields. Clinically, 3 T MRI is becoming common, and the major equipment vendors all sell 7 T scanners. For small-animal imaging, even higher field strengths are common. At high fields (≥3 T), the proton Larmor frequency is much faster than the tumbling rate of protein-bound complexes but still much slower than the tumbling rate of discrete monomeric complexes. We10−13 and others14−19 have reported multimeric contrast agents with increased high-field relaxivity due to a tumbling rate intermediate between that of protein-bound agents and small molecules. One specific approach utilized the single amino acid chelate Gd(DOTAla).10,20 Multiple Gd(DOTAla) moieties could be incorporated into oligopeptides of defined structure and size, resulting in relaxivities tuned for high-field applications. The modular nature of Gd(DOTAla) also lends itself to the design of serum albumin-targeted contrast agents.20
Here, we report on the synthesis and properties of a modified derivative of the DOTAla ligand, DOTAlaP. We sought to improve the properties of the DOTAla ligand by replacing one of the acetate moieties by methylenephosphonate. The increased charge of the phosphonate is expected to increase the solubility of the resultant gadolinium(III) complexes. Gadolinium(III) complexes with phosphonate-containing ligands often yield higher relaxivities than complexes of the analogous carboxylate ligand due to an effect on water in the second coordination sphere, and this second-sphere effect persists at high fields.21 We also expected that inner-sphere water exchange should be faster for the phosphonate-substituted ligand. For gadolinium(III) complexes undergoing water exchange via a dissociative mechanism, substitution of a carboxylate with the bulkier phosphonate donor group results in faster water exchange as the energy difference between the 8-coordinate transition state and the 9-coordinate ground state becomes smaller. On the other hand, this steric crowding can also result in an 8-coordinate ground state with an inner-sphere water ligand eliminated, which would result in lower relaxivity.
In this study, we sought to address the following questions: (i) How does substitution of one of the acetate donors in DOTAla for methylenephosphonate impact the hydration number and water exchange kinetics and, in turn, the relaxivity? (ii) How does phosphonate substitution affect kinetic inertness with respect to gadolinium(III) dissociation? (iii) What is the effect of amine versus carboxylate functionalization of the propionamine arm of DOTAlaP on the hydration number, water exchange kinetics, relaxivity, and gadolinium(III) dissociation kinetics? (iv) Are any of these properties altered when a Gd(DOTAlaP) derivative is bound to serum albumin? (v) Does phosphonate substitution confer enhanced high-field relaxivity properties and, if so, do these result in an increased in vivo MRI signal?
To address these questions, we synthesized the DOTAlaP ligand by altering the previously established DOTAla synthesis.10 Additionally, we prepared four DOTAlaP derivatives substituted at either the amine or carboxylate of the propionamine arm. The gadolinium(III) complexes were prepared and characterized with respect to variable-field, variable-temperature relaxivity, water exchange kinetics, albumin binding studies, and gadolinium(III) dissociation kinetics. Europium(III) complexes were also synthesized, and luminescence lifetime measurements were performed to quantify the hydration number of these complexes.
Results
Synthesis
We synthesized DOTAlaP using a reaction pathway similar to that of DOTAla. Benzyl 2-[[(benzyloxy)carbonyl]amino]-3-(1,4,7,10-tetraazacyclododecan-1-yl)propanoate represents an intermediate common to both DOTAla and DOTAlaP, so we used the previously established synthetic steps10 to furnish this first intermediate (Scheme 1). In order to introduce a protected phophonate arm onto the monoderivatized cyclen macrocycle, tri-tert-butyl phosphite was synthesized according to ref (22) and appended using a Mannich-type reaction. The reaction was monitored for disappearance of the monofunctionalized macrocycle using liquid chromatography–mass spectrometry (LC–MS), followed by filtration and aqueous workup of the filtrate. The introduction of these two chelator arms at this stage is key: because of their steric hindrance, it is not possible to incorporate them efficiently at a later stage. The crude intermediate containing the difunctionalized macrocycle was then redissolved in acetonitrile and subjected to alkylation with tert-butyl bromoacetate, followed by preparative high-performance liquid chromatography (HPLC) to purify the protected intermediate compound 1. Orthogonal protection group chemistry provided us with a free and ready-to-couple carboxylate and primary amine on compound 2 after hydrogenation-mediated removal of the carboxybenzyl (Cbz) and benzyl protecting groups of 1. The overall number of steps starting from Cbz-NH-Ser-OH-OBn is 5, and compound 2 is obtained with an approximate 10% overall yield based on the protected serine. The lower yield is due to the formation of a greater number of side products compared to the DOTAla synthesis. One single phosphonate species (1,7-functionalized with respect to the aminopropanoate arm) is obtained and isolated, as indicated to us by 1H NMR. Compound 2 also represents the last common intermediate for the synthesis of compounds 3–5b, which all are furnished by activation of the carboxylate with 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) in the presence of diisopropylethylamine (DIPEA) as a base, followed by overnight amide coupling and subsequent HPLC purification. The coupled, purified products still carry tert-butyl ester protective groups on the acetate groups and the phosphonate. These esters are then removed using an acid-catalyzed deprotection reaction with trifluoroacetic acid in dichloromethane, yielding the final ligands without the formation of side products.
Scheme 1. Synthetic Scheme for Reported Chelators 3, 4a, 4b, 5a, and 5b.

Complexes were formed under standard conditions by mixing the ligand with an aqueous solution of the lanthanide trichloride salt. The pH of the solution was adjusted to 7 by the dropwise addition of a 0.1 M solution of NaOH. Complexation was followed by HPLC in order to confirm that >95% of ligand was complexed. We also used the xylenol orange test to further confirm that no free lanthanide was present.23
Water Exchange Rates in Phosphate-Buffered Saline (PBS)
According to our hypothesis, Gd(DOTAlaP) derivatives should display water exchange rates that exceed the previously reported water exchange rates of Gd(DOTAla) derivatives. We determined the water exchange rates of the gadolinium(III) complexes by variable-temperature measurement of the transverse relaxation time of H217O in the presence and absence of each gadolinium(III) complex at 11.7 T. In the case of Gd(4a), Gd(4b), and Gd(5b), we observed only minor changes in the transverse relaxation rate when the complexes were added, indicating a lack of an inner-sphere water ligand for these complexes. For instance, the maximum 17O relaxivity, r2O, was 2.5 mM–1 s–1 for these three complexes, while the maximum r2O is typically >15 mM–1 s–1 for q = 1 complexes at this field strength.24 On the other hand, Gd(3) and Gd(5a) had much higher maximum r2O values consistent with an inner-sphere water ligand (18 and 16 mM–1 s–1, respectively). Figure 1 shows the reduced transverse relaxation rate R2r as a function of the reciprocal temperature. The crossover to exchange-limited R2r occurs at a low temperature for both complexes, which indicates the predicted, very fast water exchange kinetics. We used a four-parameter model described previously25 to fit the data and obtained water residency times at 37 °C of 310τM = 8.1 ns for Gd(3) and 310τM = 6.4 ns for Gd(5a); see Table 1. While these water residency times are considerably shorter than those seen for [Gd(DOTA)(H2O)]− and [Gd(DOTAla)(H2O)]−, they are similar to the water residency time for [Gd(DO3AP)(H2O)]2–, a close structural analogue of DOTAlaP (Table 1). The coordination spheres of Gd(3) and Gd(5a) differ in that Gd(3) contains an anionic propionate oxygen donor and Gd(5a) contains a neutral propionamide oxygen donor but their water exchange kinetics are similar. The propionate to propionamide substitution does not result in different water exchange kinetics in other gadolinium(III) complexes.10,26 This is in contrast to the acetate to acetamide substitution, which is well established to reduce the water exchange rate.27 Interestingly, substitution at the propionamine [Gd(4a) and Gd(4b)] gives a ligand with the same donor set as 3, but these peripheral modifications now result in a gadolinium complex with no inner-sphere water ligand. Similarly, changing the amide substituent from Gd(5a) to Gd(5b) results in the latter complex having no inner-sphere water ligand. For complexes with exceedingly fast water exchange kinetics, these results indicate that seemingly minor modifications to the periphery of the ligand can alter the hydration number of the complex.
Figure 1.
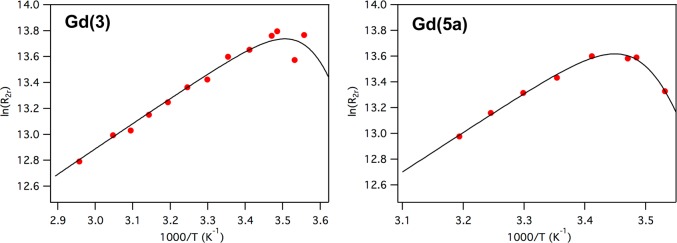
Temperature dependence of the 17O NMR (11.7 T) reduced transverse relaxation rates of Gd(3) (6.73 mM, left) and Gd(5a) in PBS (4.16 mM, right). The solid line represents a fit to the data to determine the mean water residency time τM.
Table 1. Summary of Experimentally Obtained Water Exchange Rates, Activation Enthalpies, and Mean Water Residency Time at 310 K for Gd(3) and Gd(5a) in Comparison with Previously Reported Gadolinium(III) Complexes.
Relaxivity Determination at 20 and 60 MHz in PBS
Determination of the relaxivity of the gadolinium(III) complexes also gives further indication of their hydration state. The longitudinal proton relaxivity (r1) was measured in pH 7.4 PBS at 37 °C at two Larmor frequencies, 20 and 60 MHz. For small molecules with q > 0, the relaxivity is dominated by the short rotational correlation time at these frequencies and the hydration state.20,31 Additionally, phosphonate-containing gadolinium(III) chelates have been reported to result in increased second-sphere relaxivity.30,32 If all of the complexes were q = 1, then we would expect the relaxivity to increase proportionally with the molecular weight, ranking them as follows: Gd(3) < Gd(4a) ≅ Gd(5a) < Gd(4b) ≅ Gd(5b). In fact, only Gd(3) and Gd(5a) behave as expected with a relatively high, but for q = 1 phosphonato complexes typical, relaxivity.30,31,33 On the other hand, Gd(4a), Gd(4b), and Gd(5b) show relaxivities below 4 mM–1 s–1, which is more typical for q = 0 complexes with elevated outer-sphere relaxivity (Table 2).34 These relaxivity measurements are consistent with the 17O NMR measurements, which suggests the lack of an inner-sphere water ligand for Gd(4a), Gd(4b), and Gd(5b).
Table 2. Measured Relaxivities (mM–1 s–1) in PBS at 37 °C, at 1H Larmor Frequencies of 20 and 60 MHz.
| Gd(3) | Gd(4a) | Gd(4b) | Gd(5a) | Gd(5b) | |
|---|---|---|---|---|---|
| 20 MHz | 6.4 | 3.7 | 3.9 | 5.3 | 3.2 |
| 60 MHz | 6.1 | 3.5 | 3.8 | 4.1 | 3.0 |
Determination of the Hydration Numbers Using Europium(III) Analogues
The hydration number of a gadolinium(III) complex can be estimated using a surrogate complex with the luminescent lanthanide ions Eu3+ or Tb3+. This is done by measuring the rate constants for luminescence decay in D2O and H2O solutions. An empirical expression originally reported by Horrocks and Sudnick relates these rate constants to q (Table 3).35 It is then assumed that gadolinium(III) has the same hydration number based on the similar ionic radii of these ions and their propensity for forming isostructural complexes. However, application of this method to phosphonate-containing complexes typically results in an overestimation of q by 0.3–0.6.36−38 With this consideration in mind, we find that the q values obtained mirror the relaxometric behavior of the corresponding gadolinium complexes. The hydration numbers calculated are proportional to the obtained relaxivity values in PBS at 20 and 60 MHz, with Eu(3) and Eu(5a) providing q values above 1, while the other complexes show lower q values. Figure S1 (Supporting Information) shows the correlation of the 20 MHz relaxivity values of the gadolinium(III) complex and the apparent hydration number of the corresponding europium(III) complex.
Table 3. Decay Constants [s–1] for the Luminescence Lifetime of Europium Complexes in H2O and D2O Reported Here, as Well as the Calculated q Values According to the Modified Horrocks Equation39.
| Eu(3) | Eu(4a) | Eu(4b) | Eu(5a) | Eu(5b) | |
|---|---|---|---|---|---|
| k(D2O) | 0.55 | 0.51 | 0.59 | 0.52 | NDa |
| k(H2O) | 2.14 | 1.31 | 1.77 | 1.76 | NDa |
| q | 1.4 | 0.5 | 1 | 1.1 | NDa |
ND = not determined. Because of the decreased solubility of Eu(5b) compared to the other europium complexes described here, we were not able to generate a solution with high enough complex concentration required for this measurement.
Interaction of Ibuprofen-Derived Complexes with Human Serum Albumin (HSA)
We had previously conjugated ibuprofen to DOTAla and found that a 0.1 mM solution of its gadolinium(III) complex [compound Gd(6), Figure 2] was 70% bound to 4.5% (w/v) HSA, and this complex also displayed high relaxivity (at low fields) in the presence of HSA.20,25 To understand how the phosphonate modification affects protein binding and relaxivity for a slow tumbling system, we synthesized the ibuprofen conjugates Gd(4b) and Gd(5b) shown in Figure 2.
Figure 2.

Structures of ibuprofen conjugates Gd(4b) and Gd(5b) reported here, as well as previously reported compound Gd(6).
Compared to Gd(6), the affinity of Gd(4b) for HSA was increased 3-fold (Kd = 260 and 80 μM, respectively). The reason for the higher percentage of bound complex in the case of Gd(4b) is likely the double negative charge, which may provide a stronger interaction with the lysine-rich, positively charged binding pocket of HSA. On the other hand, the zwitterionic Gd(5b) has an affinity to HSA similar to that of Gd(6).
We also measured the relaxivity for these complexes in a 4.5% HSA solution (Table 4). Surprisingly, we observed very different relaxivity behaviors for these two complexes. At 20 MHz, the relaxivity of Gd(4b) increased over 5-fold when HSA was added, whereas for Gd(5b), the increase was less than 3-fold. Such a difference could not be accounted for by the difference in the fraction bound to albumin between the two complexes. In the absence of albumin, the relaxivity of Gd(4b) is about 25% higher than that of Gd(5b), but in the presence of albumin, the relaxivity of Gd(4b) is 120% and 80% higher than that of Gd(5b) at 20 and 60 MHz, respectively (Table 4). In order to better understand this phenomenon, we performed additional r1 measurements at different temperatures and additional field strengths.
Table 4. Summary of Percent Bound Probe Gd(4b), Gd(5b), and Gd(6)20 in 4.5% (w/v) HSA as Well as Corresponding Observed Relaxivity Data in PBS and 4.5% (w/v) HSA at 37 °C.
| Gd(4b)/PBS | Gd(4b)/HSA | Gd(5b)/PBS | Gd(5b)/HSA | Gd(6)/PBS | Gd(6)/HSA | |
|---|---|---|---|---|---|---|
| % bound to HSA | 88 | 70 | 70 | |||
| 20 MHz (mM–1 s–1) | 3.9 | 19 | 3.2 | 9 | 5.6 | 37 |
| 60 MHz (mM–1 s–1) | 3.8 | 14 | 3.0 | 7.8 | 4.8 | 22 |
Nuclear Magnetic Relaxation Dispersion (NMRD) of Gd(4b) and Gd(5b)
We acquired relaxivity data for Gd(4b) and Gd(5b) in PBS and 4.5% (w/v) HSA at 20, 30, 60, 100, 200, and 400 MHz and at 10, 25, 37, and 50 °C (Tables S1–S4 in the Supporting Information, SI). We also measured the fraction (fb) of Gd(4b) and Gd(5b) bound to HSA at each of these temperatures (Table S5 in the SI). Because we know the fraction bound to HSA for each complex and we measure the relaxivity of the unbound complex independently, we can calculate the relaxivity due to the HSA-bound species (r1bound) for both Gd(4b) and Gd(5b) (eq 1).
| 1 |
The magnitude and field dependences of these HSA-bound relaxivities are shown in Figure 3. The high relaxivity of Gd(4b) could not be well described by a pure second-sphere model, which assumes some adjustable number of long-lived water molecules in close proximity to the metal ion. Instead, we reasoned that the difference in the HSA-bound relaxivities between Gd(4b) and Gd(5b) was due to the presence of an inner-sphere water ligand on some fraction of the Gd(4b) complexes (eq 2). We assumed that the difference in the HSA-bound relaxivity between Gd(4b) and Gd(5b) was the contribution from this inner-sphere water (eq 3). We then calculated this difference at each temperature and field and simultaneously fit the resultant data set using Solomon–Bloembergen–Morgan theory40 (Figure 3 and eqs 2–8). While the HSA-bound relaxivity of Gd(4b) was higher than expected, it was not as high as one would anticipate for a complex with one coordinated water, and so we left the hydration number q as a temperature-dependent adjustable parameter defined by an equilibrium constant. We found that this entire data set could be reasonably well described (Figure 3) by five adjustable parameters: 310q (mole fraction of q = 1 species at 310 K), ΔH° (enthalpy change for equilibrium between q = 0 and 1 species), 310τM, ΔH⧧ (activation energy for water exchange), and Δ2 [square of the trace of zero-field-splitting (ZFS) tensor; eqs 3–8].
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
| 8 |
We found that the data were relatively insensitive to the rotational correlation time as long as this correlation time was large. This is reasonable because the very short water residency time should dominate the correlation time for relaxation. In addition, the data were insensitive to τv, the correlation time for electronic relaxation, and so we fixed this to 20 ps, in line with other studies of similar complexes.29 The fitted parameters are listed in Table 5. The quality of the fit could likely be improved by the addition of more adjustable parameters, e.g., Lipari–Szabo-type treatment,31,33,41,42 but the conclusions are unlikely to change.
Figure 3.

Relaxivity of HSA-bound Gd(5b) (filled circles) and Gd(4b) (open circles) at pH 7.4, at (A) 10 °C, (B) 25 °C, (C) 37 °C, and (D) 50 °C. The difference between the solid and open circles is the inner-sphere contribution to the Gd(4b) relaxivity, and the solid line is the fit to the data described in the text.
Table 5. Molecular Parameters for Gd(4b) Bound to HSA Derived from the Simultaneous Fitting of Variable-Temperature NMRD.
| τR (ns)a | τv (ps)a | Δ2 (s2) | 310τM (ns) | ΔH⧧ (kJ mol–1) | 310q | ΔH° (kJ mol–1) | |
|---|---|---|---|---|---|---|---|
| Gd(4b) | 30 | 20 | (4 ± 1) × 1018 | 2.4 ± 0.9 | 28 ± 5 | 0.15 ± 0.02 | 16.8 ± 2.5 |
The parameter was fixed at this value.
The analysis shows that there is a 15% fraction of Gd(4b) that exists as a q = 1 complex with a short residence time for the first sphere water molecule. For the equilibrium in eq 2, the two reactions are the formation and dissociation of the q = 1 complex. In principle, there can also be water exchange on the q = 1 complex. Because we only observe free water, we cannot distinguish between these two reactions (dissociation to a stable q = 0 complex versus water exchange at the q = 1 complex). However, the latter reaction likely must proceed via the q = 0 state. Therefore, we likely are measuring the formation and dissociation of water to the gadolinium complex; i.e., τM, the residence time in the bound state, corresponds to 1/kd.
Kinetic Inertness
The relatively high relaxivity of Gd(4b) in the presence of HSA, especially at high fields, prompted us to further explore the kinetic inertness of this complex with respect to gadolinium(III) dissociation. Acetate to methylenephosphonate substitution in lanthanide(III) polyaminocarboxylates typically results in a more thermodynamically stable complex (because of the increased basicity of the phosphonate), but at the same time, the complex can become more kinetically labile.43 We measured the half-life of Gd(4b) under different forcing conditions and compared it directly to the acetate analogue Gd(6) measured under the same conditions. In the first experiment, we challenged each complex with 1 equiv of MS-325 ligand [a DTPA derivative of higher thermodynamic stability than Gd(DOTAla) derivatives] at pH 3 and 37 °C and monitored transchelation by HPLC. Under these conditions, we found no measurable decomplexation of Gd(4b), while Gd(6) underwent complete transchelation to MS-325 with a half-life of 115 h. This result suggests that Gd(4b) is much more thermodynamically stable than MS-325. In a second experiment, we challenged each complex with a 100-fold excess of citrate at pH 3 and 37 °C. With a citrate challenge, we found the opposite behavior: transchelation proceeded to completion in both cases but over 1 order of magnitude more rapidly in the case of Gd(4b) (t1/2 = 9.5 h) than Gd(6) (t1/2 = 110 h) (Figure 4).
Figure 4.

Dechelation of Gd(4b) (filled circles) and Gd(6) (open circles) at pH 3 and 37 °C in the presence of 10 mM citrate. Solid lines represent monoexponential fits of the decomplexation reaction.
This is in agreement with the previously reported kinetic data obtained for acyclic phosphonate complexes compared to their acetate analogues.44 We hypothesize that the phosphonate arm promotes the formation of ternary complexes with citrate molecules, which facilitates decomplexation in the case of Gd(4b). Although Gd(4b) was more labile than Gd(6), we note that acyclic complexes like [Gd(DTPA)(H2O)]2– or its derivatives, such as [Gd(BOPTA)(H2O)]2– (Table S5 in the SI), are much more labile under these conditions, e.g., reaching transchelation equilibrium with MS-325 ligand in under 10 min.8,10
In Vivo MRI in Mice with Gd(4b)
Because the high field relaxivity of Gd(4b) was 50% higher than that of the approved contrast agent MS-325 at 4.7 T, we decided to assess its relaxation properties in vivo. In addition, both Gd(4b) and MS-325 showed equivalent affinity to HSA, making this comparison more direct.5 We therefore performed dynamic MRI in mice with both compounds.
A catheter was placed in the tail vein so the mouse could be injected while positioned in the scanner. We performed 3D T1-weighted imaging before and immediately after injection of each compound (0.1 mmol kg–1). Immediately after injection, the blood vessel enhancement observed with Gd(4b) was 38 ± 2% higher than that observed with MS-325 (Figure 5) consistent with the higher relaxivity of the former.
Figure 5.
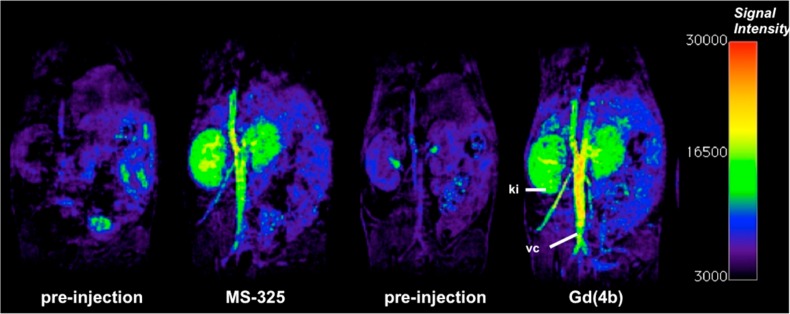
Preinjection, as well as 1 min postinjection, coronal images obtained with MS-325 (second from left) and Gd(4b) (right). Gd(4b) shows visibly better contrast in the vena cava, which can be quantified as 38 ± 2% better contrast (vs muscle). The dose of agent, time point, and image windowing was the same for both studies. Kidney (ki) and vena cava (vc) are denoted in the far right image.
Discussion
We have synthesized and evaluated the properties of five different derivatives of a new phosphonate analogue (DOTAlaP) of the single amino acid chelate DOTAla. On the basis of literature examples, we anticipated and observed an increased relaxivity due to the established second-sphere effect of the phosphonate. We also expected the phosphonate group to accelerate water exchange. However, for [Gd(DOTAla)(H2O)]− and Gd(DOTAla)-propionamide derivatives, water exchange is already very fast (310τM = 9 and 17 ns, respectively). For the Gd(DOTAlaP) complexes investigated here, we found that further acceleration of water exchange through crowding of the coordination sphere may instead lead to stabilization of the q = 0, 8-coordinate state. The five complexes investigated here comprised two that existed primarily as 9-coordinate q = 1 complexes, while the other three appear to be primarily q = 0, presumably 8-coordinate complexes. These conclusions are supported by 17O NMR and 1H relaxivity data on the gadolinium(III) complexes and by luminescence lifetime measurements on the europium(III) complexes. The energy difference between the 8- and 9-coordinate states is quite small, and peripheral changes to the macrocyclic ligand outside the inner coordination sphere can alter the coordination number in a nonpredictable manner. This delicate equilibrium between q = 0 and rapidly exchanging q = 1 underlines a challenge in the design of gadolinium(III) complexes with extremely fast water exchange rates.
Two of the investigated derivatives, Gd(4b) and Gd(5b), were designed to bind HSA. The relaxivities of these compounds in the absence of HSA were similar and low, although r1 for Gd(4b) was about 25% higher than r1 for Gd(5b). This small increase in the relaxivity could be due to the presence of a small amount of q = 1 species for Gd(4b). However, in the presence of HSA, the relaxivity difference between the complexes is magnified, and this increased difference seen when the molecular tumbling rate is decreased is indicative of a small fraction of q = 1 species. Variable-temperature NMRD allowed us to estimate that about 15% of Gd(4b) exists as a q = 1 species at 37 °C, at least while it is bound to albumin. The presence of this q = 1 complex whose coordinated water has an extremely short residency time results in remarkably high relaxivities at high field for a complex bound to HSA. For instance, the relaxivity at 4.7 T was 50% higher than that for the albumin-targeted approved agent MS-325.
The NMRD results have profound consequences for the development of slow tumbling contrast agents for high-field applications. The variable-temperature data clearly indicate that the water residency time, τM, is the dominant correlation time for modulating the relaxivity in this system. Figure 6 shows data at two temperatures and two fields to illustrate this point. At 0.47 T (20 MHz), the relaxivity increases dramatically as the temperature is lowered. At 37 °C, τM at 2.4 ns is too short for optimal relaxivity, but as the temperature is lowered, τM increases and the relaxivity goes up. On the other hand, at higher fields (>2 T), the optimal correlation time is in the 1–3 ns range.45 At 37 °C, a τM of 2.4 ns is ideal for relaxivity at 2.4 T (100 MHz). However, as the temperature is lowered and τM increases, the correlation time becomes too long for optimizing high-field relaxivity. The very short τM provides increased relaxivity at high fields and offers a strategy to increase the relaxivity of slow tumbling entities at high magnetic fields. We note that the data in Figure 6 are for a complex that is only 25% (at 10 °C) or 15% (at 37 °C) for q = 1. Thus, the relaxivity gains for a single q = 1 species are 4–6-fold higher.
Figure 6.

Inner-sphere relaxivity of Gd(4b) bound to HSA at two different temperatures (10 and 37 °C) and Larmor frequencies.
Because the relaxivity of q = 0.15 Gd(4b) was still clearly enhanced compared to q = 1 MS-325 at higher fields, we considered its evaluation for high-field in vivo imaging. For this, we first tested the kinetic inertness of Gd(4b) under forcing conditions. We found the somewhat decreased kinetic inertness, when compared with the all-acetato analogue Gd(6), is a typical reflection of the results previously reported in conjunction of phosphonato compounds.32,43 However, the complex was still quite inert compared to approved acyclic chelates.46 This encouraged the in vivo evaluation and cross-comparison with the approved HSA binding compound MS-325. The subsequent in vivo imaging experiment at 4.7 T was found to mirror the in vitro results well because we were able to obtain better angiographic contrast for Gd(4b) compared to MS-325. An increased blood signal is clearly found in the increased relaxivity for this compound at intermediate magnetic fields. We consider this outcome a promising result that encourages us to further investigate τM acceleration as a way to enhance the relaxivity at higher fields because an entirely q = 1 complex with such a fast exchange rate would have 5 times the relaxivity observed with Gd(4b). However, the somewhat unpredictable nature of the compound behavior in terms of the inner-sphere hydration number is a clear challenge.
Conclusions
In summary, we have successfully synthesized a monophosphonate analogue of the single amino acid chelator DOTAla, which we term DOTAlaP. The Gd(DOTAlaP) derivatives show enhanced second-sphere relaxivity compared to Gd(DOTAla). In two derivatives, we found increased water exchange kinetics, and this combination of fast water exchange and increased second-sphere relaxivity results in improved relaxometric behavior. We also found, however, that functionalization of this single amino acid chelate of either the nitrogen or carbon terminus with different R groups can sometimes yield gadolinium complexes with no inner-sphere water (q = 0) or a mixture of q = 0 and 1 complexes. This decrease in the hydration number appears as a side effect of accelerated water exchange and does not correlate with a specific R group or location of functionalization. For one derivative, Gd(4b), where R was chosen such that the corresponding gadolinium complex had affinity to HSA, we found that the hydration number was 0.15 in the presence of HSA, i.e., a 6:1 mixture of q = 0 to q = 1. The corresponding mean water residency time obtained for the albumin-bound complex was shorter than the rotational correlation time, which, in this case, leads to τM becoming the dominant correlation time for relaxation. Because τM is considerably shorter than τR, higher inner-sphere relaxivity was achieved for this complex at intermediate and high magnetic fields than what can be obtained with the clinically approved HSA targeting agent MS-325. We have demonstrated the effect of higher relaxivity also in vivo with higher blood vessel to muscle contrast for Gd(4b) when compared to MS-325. Future endeavors will include the identification of fast exchanging complexes that are exclusively q = 1 in order to maximize the relaxivity gains at high fields.
Experimental Procedures
General Materials and Methods
1H and 13C NMR spectra were recorded on a Varian 11.7 T NMR system equipped with a 5-mm broad-band probe. HPLC purification of intermediates was performed on a Rainin, Dynamax (Phenomenex C18 column: 250 mm × 21.2 mm, 10 μm) using method A: 0.1% trifluoroacetic acid (TFA) in water with a gradient of 5–95% (0.1% TFA in MeCN) over 20 min with a 15 mL min–1 flow rate. HPLC purity analyses (both UV and MS detection) were carried out on an Agilent 1100 system (Phenomenex Luna C18(2) column: 100 mm × 2 mm, 0.8 mL min–1 flow rate) with UV detection at 220, 254, and 280 nm and positive-mode electrospray ionization (ESI+) using the following methods. Method B: solvent A = water, solvent B = MeCN; 2–60% B over 15 min. Method C: solvent A = 10 mM NH4OAc, solvent B = 10% 10 mM NH4OAc, 90% MeCN; 5–95% B over 15 min. Method D: solvent A = 10 mM NH4OAc, solvent B = 10% 10 mM NH4OAc, 90% MeCN; 5–35% B over 15 min. The synthesis of ligands was carried out as shown in Scheme 1. Chemicals were supplied by Aldrich Chemical Co., Inc., and were used without further purification. Solvents (HPLC grade) were purchased from various commercial suppliers and used as received. The monoalkylated cyclen precursor to compound 1 was synthesized as described previously.10 Tri-tert-butyl phosphite was synthesized according to a procedure reported by Manning et al.22
General Procedure for Amide Couplings (Step To Afford Products 4a, 4b, 5a, and 5b)
Amide coupling followed by deprotection of the tert-butyl protective groups was done by activation of the carboxylate with HATU (1.2 equiv) and DIPEA (1.2 equiv) for 5 min in N,N-dimethylformamide (DMF), followed by the addition of amine (1 equiv) dissolved in DMF and stirring at room temperature for 18 h. After confirmation of the presence of the amide-coupling product by LC–MS (method B), the intermediate was isolated by preparative HPLC (method A), eluting between 11 and 13 min. The clean fractions containing the desired product were pooled and lyophilized to afford the intermediate as an off-white powder in 15–35% yield.
General Deprotection Procedure
Redissolution of intermediates described above in a 1:1 mixture of dichloromethane and TFA, followed by stirring for 18 h, afforded the final ligand with quantitative yield.
General Metal Complexation Procedure (with Lanthanides Gadolinium, Europium, and Terbium)
The ligand was dissolved in H2O (1 mL). An amount of a stock solution containing LnCl3·6H2O (0.95 equiv based on the ligand weight) is added to the ligand solution under monitoring of the pH. The pH is adjusted to 7 using a 0.1 M NaOH solution. The lightly cloudy solution is filtered and lyophilized to afford the corresponding lanthanide complex as an off-white powder. The purity of the complex was assessed using LC–MS; the presence of free lanthanides was excluded using the xylenol orange test.
Di-tert-butyl 2,2′-[4-[3-(benzyloxy)-2-[[(benzyloxy)carbonyl]amino]-3-oxopropyl]-10-[(di-tert-butoxyphosphoryl)methyl]-1,4,7,10-tetraazacyclododecane-1,7-diyl]diacetate (1)
2-[[(Benzyloxy)carbonyl]amino]-3-(1,4,7,10-tetraazacyclododecan-1-yl)propanoate (1.475 g, 3.05 mmol, 1 equiv) and paraformaldehyde (0.34 g, 11.45 mmol, 3.75 equiv) were dissolved in tetrahydrofuran and vigorously stirred under a dinitrogen atmosphere for 0.5 h. Tri-tert-butyl phosphite (1.145 g, 4.5 mmol, 1.5 equiv) was then added, and the reaction was stirred for 16 h. Reaction monitoring by LC–MS showed the product as [M + H]+ (m/z 690.4). The reaction mixture was then filtered, concentrated in vacuo, and redissolved in EtOAc (200 mL). The organic layer is washed with 50 mL of saturated Na2CO3 and 50 mL of brine, dried with sodium sulfate, and concentrated to afford the dialkylated intermediate (1.73 g, 2.5 mmol, 82%), which was used in the subsequent step without further characterization. The intermediate was dissolved in MeCN (75 mL). K2CO3 (0.68 g, 4.9 mmol, 2 equiv) was added, and the mixture was stirred vigorously after the addition of tert-butyl bromoacetate (0.766 g, 0.575 mL, 3.94 mmol, 1.6 equiv). After 16 h, water (70 mL) was added and most of the MeCN was removed in vacuo. The oily residue was taken up in EtOAc (80 mL) and washed with water and brine (100 mL each). The organic layer was separated, dried with sodium sulfate, and concentrated to afford the crude product, which was redissolved in MeCN (5 mL) and purified using preparative HPLC (method A). The product elutes at 12.2 min. Fractions containing the product are pooled and lyophilized to afford the product as a white powder (0.43 g, 0.46 mmol, 20% yield). 1H NMR (CDCl3, 500 MHz, ppm): 9.9 (s, br, 2H), 7.36–7.34 (m, 10H), 5.25–5.16 (m, 4H), 4.3 (m, 1H), 3.35–2.68 (m, 21H), 2.04 (s, 4H), 1.51–1.43 (m, 36H). 13C NMR (CDCl3, 125.7 MHz, ppm): 175.4, 170.6, 170.3, 135.0, 131.1, 129.4, 128.6–127.9, 81.4, 81.0, 67.7, 57.3–47.4, 30.6, 28.2, 22.1. 31P NMR (CDCl3, 202.4 MHz, ppm): 17.7. LC–ESI-MS. Calcd for C47H76N5O11P4: m/z 917.5. Found: m/z 918.6 ([M + H]+). Rt = 7.8 min (method C).
2-Amino-3-[4,10-bis[2-(tert-butoxy)-2-oxoethyl]-7-[(di-tert-butoxyphosphor ryl)methyl]-1,4,7,10-tetraazacyclododecan-1-yl]propanoic Acid (2)
Compound 1 (0.43 g, 0.47 mmol) was dissolved in EtOH (40 mL). Pd/C [10% (w/v), 0.215 g] was added, and the reaction mixture was purged first with dinitrogen and then charged with dihydrogen (1 atm). The mixture was then stirred under a dihydrogen atmosphere for 3 h, after which the reaction was found to be complete. The Pd/C was removed by filtration, and the solvent was removed in vacuo to afford the product as a colorless oil (0.32 g, 0.46 mmol, quantitative conversion). 1H NMR (CD3OD, 500 MHz, ppm): 5.15 (s, br, 1H), 4.46–2.48 (m, 24H), 1.63–1.47 (m, 36H). 13C NMR (CD3OD, 125.7 MHz, ppm): 167.9, 160.5, 86.6, 82.6, 55.2–48.5, 30.6, 29.2, 26.9. 31P NMR (CD3OD, 202.4 MHz, ppm): 16.9. LC–ESI-MS. Calcd for C32H64N5O9P: m/z 693.4. Found: m/z 694.7 ([M + H]+). Rt = 5.4 min (method B).
2,2′-[4-(2-Amino-2-carboxyethyl)-10-(phosphonomethyl)-1,4,7,10-tetraazacyclododecane-1,7-diyl]diacetic Acid (3)
1H NMR (CD3OD, 500 MHz, ppm): 5.15 (s, br, 2H), 4.26–2.46 (m, 25H). 13C NMR (CD3OD, 125.7 MHz, ppm): 175.1, 169.6, 55.4–48.9. 31P NMR (CD3OD, 202.4 MHz, ppm): 6.3. LC–ESI-MS. Calcd for C16H32N5O9P: m/z 469.2 Found: m/z 470.1 ([M + H]+). Rt = 1.1 min (method B).
2,2′-[4-[2-Carboxy-2-(2-phenylpropanamido)ethyl]-10-(phosphonomethyl)-1,4,7,10-tetraazacyclododecane-1,7-diyl]diacetic Acid (4a)
1H NMR (CD3OD, 500 MHz, ppm): 7.38–7.23 (m, 5H), 5.15 (s, br, 1H), 4.76 (m, 1H), 3.98–2.86 (m, 24H), 1.47 (t, 3H). 13C NMR (CD3OD, 125.7 MHz, ppm): 159.4, 140.9, 128.5–126.8, 116.7, 114.4, 53.3, 49.5, 46.1, 17.1. 31P NMR (CD3OD, 202.4 MHz, ppm): 0.8. LC–ESI-MS. Calcd for C25H40N5O10P: m/z 601.2. Found: m/z 602.4 ([M + H]+). Rt = 2.1 min (method B).
2,2′-[4-[2-Carboxy-2-[[1-(4-isobutylphenyl)ethyl]amino]ethyl]-10-(phosphonomethyl)-1,4,7,10-tetraazacyclododecane-1,7-diyl]diacetic Acid (4b)
1H NMR (CD3OD, 500 MHz, ppm): 7.28 (d, 2H), 7.20 (d, 2H), 4.73 (s, br, 1H), 3.91–2.87 (m, 24H), 2.45 (m, 2H), 1.84 (m, 1H), 1.48–1.29 (m, 5H), 0.89 (d, 6H). 13C NMR (CD3OD, 125.7 MHz, ppm): 172.8, 169.6, 140.5, 138.2, 129.5, 127.0, 54.7–48.2, 30.0, 21.3, 17.1. 31P NMR (CD3OD, 202.4 MHz, ppm): 3.5. LC–ESI-MS. Calcd for C28H48N5O9P: m/z 629.3. Found: m/z 630.5 ([M + H]+). Rt = 5.6 min (method B).
2,2′-[4-[2-Carboxy-2-[2-(4-isobutylphenyl)propanamido]ethyl]-10-(phosphonomethyl)-1,4,7,10-tetraazacyclododecane-1,7-diyl]diacetic Acid (5a)
1H NMR (CD3OD, 500 MHz, ppm): 7.39–7.26 (m, 5H), 5.02 (s, br, 1H), 4.15–2.46 (m, 24H), 1.64–1.46 (m, 3H). 13C NMR (CD3OD, 125.7 MHz, ppm): 169.7, 166.3, 160.4, 142.7, 128.5–125.8, 119.6, 117.3, 114.9, 112.7, 53.3–49.5, 27.2, 20.85. 31P NMR (CD3OD, 202.4 MHz, ppm): 6.1. LC–ESI-MS. Calcd for C24H41N6O8P: m/z 573.3. Found: m/z 574.6 ([M + H]+). Rt = 2.3 min (method B).
2,2′-[4-[2-Amino-3-[[2-[2-(4-isobutylphenyl)propanamido]ethyl]amino]-3-oxopropyl]-10-(phosphonomethyl)-1,4,7,10-tetraazacyclododecane-1,7-diyl]diacetic Acid (5b)
1H NMR (CD3OD, 500 MHz, ppm): 7.24 (d, 2H), 7.10 (d, 2H), 4.21–2.44 (m, 30H), 1.94–1.29 (m, 6H), 0.89 (d, 6H). 13C NMR (CD3OD, 125.7 MHz, ppm): 175.4, 169.8, 167.4, 140.2, 138.8, 128.9, 126.7, 54.6–48.1, 38.2, 30.1, 21.3, 17.1. 31P NMR (CD3OD, 202.4 MHz, ppm): 6.1. LC–ESI-MS. Calcd for C31H54N7O9P: m/z 699.3. Found: m/z 700.6 ([M + H]+). Rt = 6.8 min (method B).
Lanthanide Complexes
LC–ESI-MS. Calcd for C16H28GdN5O9P [Gd(3)]: m/z 623.1. Found: m/z 625.1 ([M + 2H]+). Calcd for C16H28EuN5O9P [Eu(3)]: m/z 618.1. Found: m/z 620.1 ([M + 2H]+). Calcd for C24H36GdN5O9P [Gd(4a)]: m/z 755.1. Found: m/z 757.2 ([M + 2H]+). Calcd for C24H36EuN5O9P [Eu(4a)]: m/z 750.1. Found: m/z 752.1 ([M + 2H]+). Calcd for C29H44GdN5O10P [Gd(4b)]: m/z 811.2. Found: m/z 813.4 ([M + 2H]+). Calcd for C29H44EuN5O10P [Eu(4b)]: m/z 806.2. Found: m/z 808.4 ([M + 2H]+). Calcd for C29H44TbN5O10P [Tb(4b)]: 812.2. Found: m/z 814.4 ([M + 2H]+). Calcd for C24H38GdN6O8P [Gd(5a)]: m/z 727.2. Found: m/z 728.2 ([M + H]+). Calcd for C24H38EuN6O8P [Eu(5a)]: m/z 722.2. Found: m/z 723.3 ([M + H]+). Calcd for C31H51GdN7O9P [Gd(5b)]: m/z 852.3. Found: m/z 853.5 ([M + H]+). Calcd for C31H51EuN7O9P [Eu(5b)]: m/z 849.3. Found: m/z 850.4 ([M + H]+).
Luminescence
Luminescence lifetime measurements of europium complexes in H2O and D2O were performed on a Hitachi f-4500 fluorescence spectrophotometer. The concentrations of the samples were 5–10 mM. For the measurements in D2O, the complexes were first dissolved in D2O (99.98% D), lyophilized, and dissolved in D2O again to reduce the amount of residual HDO. Measurements were taken with the following settings: excitation at 396 nm (Eu) emission at 616 nm (Eu) 30 replicates, 0.04 ms temporal resolution (0–20 ms), PMT voltage = 400 V. Lifetimes were obtained from monoexponential fits of the data using Igor Pro software (version 6.0, Wavemetrics, Lake Oswego, OR).
17O NMR of Gadolinium Complexes for Determination of τM
17O NMR measurements of solutions were performed at 11.7 T on 350 μL samples contained in 5 mm standard NMR tubes on a Varian spectrometer. The temperature was regulated by an air flow controlled by a Varian VT unit. The H217O transverse relaxation times of a >4 mM solution of all gadolinium complexes (pH 7.4, 10 mM PBS buffer) were measured using a CPMG sequence. The exact concentration of the sample was determined by inductively coupled plasma mass spectrometry (ICP-MS). Reduced relaxation rates, 1/T2r, were calculated from the difference of 1/T2 between the sample and water blank and then divided by the mole fraction of coordinated water, assuming q = 1. The temperature dependence of 1/T2r was fit to a four-parameter model as previously described.25 The Gd–O hyperfine coupling constant, A/ℏ, was assumed to be 3.8 × 106 rad s–1.29
Determination of the Percent Binding to HSA
Ultrafiltration measurements of protein binding solutions of a 0.1 mM gadolinium compound and 4.5% HSA (200 μL total) were added to a 5000 MW ultrafiltration unit (regenerated cellulose membrane, Millipore, Bedford, MA). The samples were incubated at a set temperature for 30 min and subsequently centrifuged to filter off the nonprotein-bound components. The filtrates were analyzed for the gadolinium concentration by ICP-MS. The concentration of bound complex in the protein samples was determined by subtracting the gadolinium concentration (unbound complex) found in the filtrate from the gadolinium concentration of the initial solution. Experiments were performed in duplicate.
Measurement of Kinetic Inertness
Stock solutions of MS-325-L (the ligand of the MS-325 complex),47 Gd(4b), and Gd(6) were prepared. For experiment 1, MS-325-L was added to solutions of the gadolinium complexes in pH 3 water and incubated at 37 °C. The final concentrations of the gadolinium complexes and MS-325-L were 0.1 mM. A 10 μL aliquot was removed for HPLC analysis and analyzed at time points 1, 20, 50, 72, 80, 96, 120,168, 192, 216, 240, 288, 312, 336, 360, and 480 h, while the remainder of the solution was incubated at 37 °C. For experiment 2, the gadolinium complexes were added to a 10 mM pH 3 citrate buffer, affording a 0.1 mM complex concentration, and incubated at 37 °C. A 10 μL aliquot was removed and analyzed at 0, 0.75, 1.5, 2, 3, 4, 5, 6, 24, 72, 96, 120, 168, 192, 216, 240, 288 312, 336, and 408 h.
Imaging Protocol for MS-325/Gd(4b)
Nu/nu mice were anesthetized with isoflurane (1–2%) and placed in a specially designed cradle with the body temperature maintained at 37 °C. The tail vein was cannulated for intravenous delivery of the contrast agent while the animal was positioned in the scanner. Imaging was performed at 4.7 T, using a custom-built volume coil to acquire coronal T1-weighted images with a field of view that covered the major organs of the thorax and abdomen (heart, stomach, liver, intestines, and kidney). The contrast agent dose was 100 μmol kg–1. T1-weighted images were acquired using a 3D Fast Low Angle Shot (FLASH) sequence: TR/TE/flip angle = 15.3 ms/1.54 ms/40°, FOV = 4.8 × 2.4 × 2.4 cm, matrix = 192 × 96 × 96, 1 average.
Acknowledgments
P.C. acknowledges the National Institute for Biomedical Imaging and Bioengineering for funding for this project (NIBIB Award R01EB009062) and the National Center for Research Resources for instrumentation grants (Grants S10OD010650, S10RR023385, and P41RR14075). E.B. thanks the Swiss National Science Foundation (advanced.postdoc mobility fellowship) for support.
Supporting Information Available
Obtained relaxivity values, kinetic inertness challenge data, temperature-dependent fraction bound of Gd(4b) and Gd(5b) to HSA, correlation plot of europium luminescence lifetime derived q values versus relaxivity, and LC–MS traces. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Caravan P. Chem. Soc. Rev. 2006, 35, 512–523. [DOI] [PubMed] [Google Scholar]
- Chan K. W.-Y.; Wong W.-T. Coord. Chem. Rev. 2007, 251, 2428–2451. [Google Scholar]
- Helm L. Future Med. Chem. 2010, 2, 385–396. [DOI] [PubMed] [Google Scholar]
- Raymond K. N.; Pierre V. C. Bioconjugate Chem. 2005, 16, 3–8. [DOI] [PubMed] [Google Scholar]
- Caravan P.; Cloutier N. J.; Greenfield M. T.; McDermid S. A.; Dunham S. U.; Bulte J. W. M.; Amedio J. C.; Looby R. J.; Supkowski R. M.; Horrocks W. D.; McMurry T. J.; Lauffer R. B. J. Am. Chem. Soc. 2002, 124, 3152–3162. [DOI] [PubMed] [Google Scholar]
- Caravan P. Acc. Chem. Res. 2009, 42, 851–862. [DOI] [PubMed] [Google Scholar]
- Major J. L.; Meade T. J. Acc. Chem. Res. 2009, 42, 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overoye-Chan K.; Koerner S.; Looby R.; Kolodziej A.; Zech S.; Deng Q.; Chasse J.; McMurry T.; Caravan P. J. Am. Chem. Soc. 2008, 130, 6025–6039. [DOI] [PubMed] [Google Scholar]
- Uppal R.; Ay I.; Dai G.; Kim Y.; Sorensen A.; Caravan P. Stroke 2010, 41, 1271–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros E.; Polasek M.; Zhang Z.; Caravan P. J. Am. Chem. Soc. 2012, 134, 19858–19868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Kolodziej A. F.; Greenfield M. T.; Caravan P. Angew. Chem., Int. Ed. 2011, 50, 2621–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S. A.; Kolodziej A. F.; Bhole G.; Greenfield M. T.; McMurry T. J.; Caravan P. Angew. Chem., Int. Ed. 2008, 47, 4918–4921. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Greenfield M. T.; Spiller M.; McMurry T. J.; Lauffer R. B.; Caravan P. Angew. Chem., Int. Ed. 2005, 44, 6766–6769. [DOI] [PubMed] [Google Scholar]
- Livramento J. B.; Sour A.; Borel A.; Merbach A. E.; Tóth É. Chem.—Eur. J. 2006, 12, 989–1003. [DOI] [PubMed] [Google Scholar]
- Livramento J. B.; Helm L.; Sour A.; O’Neil C.; Merbach A. E.; Tóth É. Dalton Trans. 2008, 1195–1202. [DOI] [PubMed] [Google Scholar]
- Mastarone D. J.; Harrison V. S. R.; Eckermann A. L.; Parigi G.; Meade T. J. J. Am. Chem. Soc. 2011, 133, 5329–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton D. A.; Elemento E. M.; Aime S.; Chaabane L.; Botta M.; Parker D. Chem. Commun. 2006, 1064–1066. [DOI] [PubMed] [Google Scholar]
- Schühle D. T.; Schatz J.; Laurent S.; Vander Elst L.; Muller R. N.; Stuart M. C. A.; Peters J. A. Chem.—Eur. J. 2009, 15, 3290–3296. [DOI] [PubMed] [Google Scholar]
- Goswami L. N.; Ma L.; Chakravarty S.; Cai Q.; Jalisatgi S. S.; Hawthorne M. F. Inorg. Chem. 2012, 52, 1694–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros E.; Caravan P. J. Med. Chem. 2013, 56, 1782–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botta M. Eur. J. Inorg. Chem. 2000, 2000, 399–407. [Google Scholar]
- Manning H. C.; Bai M.; Anderson B. M.; Lisiak R.; Samuelson L. E.; Bornhop D. J. Tetrahedron Lett. 2005, 46, 4707–4710. [Google Scholar]
- Barge A.; Cravotto G.; Gianolio E.; Fedeli F. Contrast Media Mol. Imaging 2006, 1, 184–188. [DOI] [PubMed] [Google Scholar]
- Gale E. M.; Zhu J.; Caravan P. J. Am. Chem. Soc. 2013, 135, 18600–18608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caravan P.; Parigi G.; Chasse J. M.; Cloutier N. J.; Ellison J. J.; Lauffer R. B.; Luchinat C.; McDermid S. A.; Spiller M.; McMurry T. J. Inorg. Chem. 2007, 46, 6632–6639. [DOI] [PubMed] [Google Scholar]
- Tei L.; Gugliotta G.; Baranyai Z.; Botta M. Dalton Trans. 2009, 9712–9714. [DOI] [PubMed] [Google Scholar]
- Helm L.; Merbach A. E. Chem. Rev. 2005, 105, 1923–1960. [DOI] [PubMed] [Google Scholar]
- Ferreira M. F.; Martins A. F.; Martins J. A.; Ferreira P. M.; Toth E.; Geraldes C. F. G. C. Chem. Commun. 2009, 43, 6475–6477. [DOI] [PubMed] [Google Scholar]
- Powell D. H.; Dhubhghaill O. M. N.; Pubanz D.; Helm L.; Lebedev Y. S.; Schlaepfer W.; Merbach A. E. J. Am. Chem. Soc. 1996, 118, 9333–9346. [Google Scholar]
- Rudovský J.; Cígler P.; Kotek J.; Hermann P.; Vojtíšek P.; Lukeš I.; Peters J. A.; Vander Elst L.; Muller R. N. Chem.—Eur. J. 2005, 11, 2373–2384. [DOI] [PubMed] [Google Scholar]
- Dumas S.; Jacques V.; Sun W.-C.; Troughton J. S.; Welch J. T.; Chasse J. M.; Schmitt-Willich H.; Caravan P. Invest. Radiol. 2010, 45, 600–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polasek M.; Caravan P. Inorg. Chem. 2013, 52, 4084–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques V.; Dumas S.; Sun W.-C.; Troughton J. S.; Greenfield M. T.; Caravan P. Invest. Radiol. 2010, 45, 613–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton J. W.; Carr R.; Evans N. H.; Funk A. M.; Kenwright A. M.; Parker D.; Yufit D. S.; Botta M.; De Pinto S.; Wong K.-L. Inorg. Chem. 2012, 51, 8042–8056. [DOI] [PubMed] [Google Scholar]
- Horrocks W. D. W.; Sudnick D. R. J. Am. Chem. Soc. 1979, 101. [Google Scholar]
- Kotková Z.; Pereira G. A.; Djanashvili K.; Kotek J.; Rudovský J.; Hermann P.; Vander Elst L.; Muller R. N.; Geraldes C. F. G. C.; Lukeš I.; Peters J. A. Eur. J. Inorg. Chem. 2009, 119–136. [Google Scholar]
- Campello M. P. C.; Lacerda S.; Santos I. C.; Pereira G. A.; Geraldes C. F. G. C.; Kotek J.; Hermann P.; Vaněk J.; Lubal P.; Kubíček V.; Tóth É.; Santos I. Chem.—Eur. J. 2010, 16, 8446–8465. [DOI] [PubMed] [Google Scholar]
- Aime S.; Batsanov A. S.; Botta M.; Howard J. A. K.; Parker D.; Senanayake K.; Williams G. Inorg. Chem. 1994, 33, 4696–4706. [Google Scholar]
- Beeby A.; Clarkson I. M.; Dickins R. S.; Faulkner S.; Parker D.; Royle L.; Sousa A. S. d.; Williams J. A. G.; Woods M. J. Chem. Soc., Perkin Trans. 1999, 2, 493–503. [Google Scholar]
- Caravan P.; Ellison J. J.; McMurry T. J.; Lauffer R. B. Chem. Rev. 1999, 99, 2293–2352. [DOI] [PubMed] [Google Scholar]
- Datta A.; Hooker J. M.; Botta M.; Francis M. B.; Aime S.; Raymond K. N. J. Am. Chem. Soc. 2008, 130, 2546–2552. [DOI] [PubMed] [Google Scholar]
- Tóth É.; Helm L.; Kellar K. E.; Merbach A. E. Chem.—Eur. J. 1999, 5, 1202–1211. [Google Scholar]
- Kotek J.; Kálmán F. K.; Hermann P.; Brücher E.; Binnemans K.; Lukes I. Eur. J. Inorg. Chem. 2006, 1976–1986. [Google Scholar]
- Kotek J.; Lebduskova P.; Hermann P.; Elst L. V.; Muller R. N.; Geraldes C. F. G. C.; Maschmeyer T.; Lukes I.; Peters J. A. Chem.—Eur. J. 2003, 9. [DOI] [PubMed] [Google Scholar]
- Caravan P.; Farrar C. T.; Frullano L.; Uppal R. Contrast Media Mol. Imaging 2009, 4, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent S.; Vander Elst L.; Copoix F.; Muller R. N. Invest. Radiol. 2001, 36, 115–122. [DOI] [PubMed] [Google Scholar]
- McMurry T. J.; Parmelee D. J.; Sajiki H.; Scott D. M.; Ouellet H. S.; Walovitch R. C.; Tyeklar Z.; Dumas S.; Bernard P.; Nadler S.; Midelfort K.; Greenfield M.; Troughton J.; Lauffer R. B. J. Med. Chem. 2002, 45, 3465–3474. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.