

Abstract
The yeast two-hybrid method (or interaction trap) is a powerful technique for detecting protein interactions. The procedure is performed using transcriptional activation of a dual reporter system in yeast to identify interactions between a protein of interest (the bait protein) and the candidate proteins for interaction. The method can be used to screen a protein library for interactions with a bait protein or to test for association between proteins that are expected to interact based on prior evidence. Interaction mating facilitates the screening of a library with multiple bait proteins.
Keywords: protein interactions, yeast two-hybrid, interaction trap, interaction mating
INTRODUCTION
To understand the function of a particular protein, it is often useful to identify other proteins with which it associates. This can be done by a selection or screen in which novel proteins that interact specifically with a target protein of interest are isolated from a library. One particularly useful approach to detecting novel interacting proteins, the two-hybrid system or interaction trap, uses yeast as a “test tube” and transcriptional activation of a reporter system to identify associating proteins. This approach can also be used to test complex formation between two specific proteins for which there is a prior reason to expect an interaction.
In the basic version of this method (see Figs. 17.3.1 and 17.3.2), the plasmid pEG202 or a related vector is used to express the probe or “bait” protein as a fusion to the heterologous DNA-binding protein LexA. Many proteins, including transcription factors, kinases, and phosphatases, have been successfully used as bait proteins. The major requirements for the bait protein are that it should not be actively excluded from the yeast nucleus and that it should not possess the intrinsic ability to strongly activate transcription. The plasmid expressing the LexA-fused bait protein (pBait) is used to transform yeast possessing a dual reporter system responsive to transcriptional activation through the LexA operators. In the EGY48 yeast strain used here, the upstream activating sequences of the chromosomal LEU2 gene (required in the biosynthetic pathway for leucine) are replaced with LexA operators. In addition, EGY48 is transformed with the pSH18-34 plasmid, which has LexA operators upstream of the lacZ reporter gene. Basic Protocol 1 describes the transformation of EGY48 with pSH18-34 and pBait and characterization of the resulting bait strain. In the absence of a transcriptional activator (Fig. 17.3.1A), the strain should be able to express the bait protein, but should not produce viable colonies on medium that lacks leucine, and it should produce only white colonies on medium containing Xgal. A number of alternative yeast strains, plasmids, and strategies are presented that can be employed if a bait proves to have an unacceptably high level of background transcriptional activation. An additional test of the bait strain, the repression assay, is described in Alternate Protocol 1.
Figure 17.3.1.
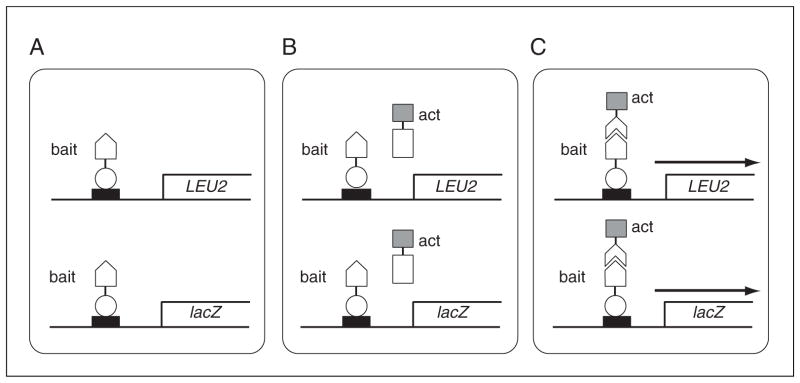
The interaction trap. (A) An EGY48 yeast cell containing two LexA operator–responsive reporters, one a chromosomally integrated copy of the LEU2 gene (required for growth on Leu medium), the second a plasmid bearing the lacZ reporter gene (causing yeast to turn blue on medium containing Xgal). The cell also contains a constitutively expressed chimeric protein, consisting of the DNA-binding domain of LexA fused to the probe or bait protein, shown as being unable to activate either of the two reporters. (B) and (C) The resulting bait strain has been additionally transformed with an activation domain (act)–fused cDNA library in pJG4-5, and the library has been induced. In (B), the encoded protein does not interact specifically with the bait protein and the two reporters are not activated. In (C), a positive interaction is shown in which the library-encoded protein interacts with bait protein, resulting in activation of the two reporters (arrow), thus causing growth on medium lacking Leu and blue color on medium containing Xgal. Symbols: black rectangle, LexA operator sequence; open circle, LexA protein; open pentagon, bait protein; open rectangle, noninteracting library protein; shaded box, activator protein (acid blob in Fig. 17.3.6); open chevron, interacting library protein.
Figure 17.3.2.

Flow chart for performing an interaction trap.
In the interaction trap (Basic Protocol 2), the bait strain is transformed with a conditionally expressed cDNA library in the vector pJG4-5. This library uses the inducible yeast GAL1 promoter to express proteins as fusions to an acidic domain (“acid blob”) that functions as a portable transcriptional activation motif (act). Expression of library-encoded proteins is induced by plating transformants on medium containing galactose. Yeast cells containing library proteins that do not interact specifically with the bait protein will fail to grow in the absence of Leu (see Fig. 17.3.1B). Yeast cells containing library proteins that do interact with the bait protein will form colonies within 2 to 5 days, and these colonies will turn blue when the cells are streaked on medium containing Xgal (see Fig. 17.3.1C). The DNA from the positive colonies is analyzed by PCR to streamline screening and detect redundant clones, and the plasmids are isolated and characterized by a series of tests to confirm specificity of the interaction (see Basic Protocol 2 and Support Protocols 2 and 3). Those found to be specific are ready for further analysis (e.g., sequencing). A flow chart for the overall procedure is shown in Figure 17.3.2.
When more than one bait will be used to screen a single library, significant time and resources can be saved by performing the screen by interaction mating (see Alternate Protocol 2). In this method, EGY48 (without the bait or lacZ plasmids) is transformed with library DNA and the transformants are collected and frozen in aliquots. For each interactor hunt, aliquots of the EGY48 library strain are thawed and mixed with aliquots of the desired bait strains. Overnight incubation of the mixture on a YPD plate results in fusion of the two strains to form diploids. The diploids are then exposed to galactose to induce expression of the library-encoded proteins, and interactors are selected in the same manner as in Basic Protocol 2. The advantage to this approach is that it requires only one high-efficiency library transformation for multiple hunts with different baits. It is also useful for bait proteins that are somewhat toxic to yeast, as yeast expressing toxic baits can be difficult to transform with the library DNA.
NOTE: All solutions and equipment coming into contact with cells must be sterile, and proper sterile technique should be used accordingly.
BASIC PROTOCOL 1: PRODUCING AND CHARACTERIZING A BAIT STRAIN
The first step in an interactor hunt is to construct a plasmid that expresses LexA fused to the protein of interest. This construct is transformed into reporter yeast strains containing LEU2 and lacZ reporter genes, and a series of control experiments is performed to establish whether the construct is suitable as is or must be modified and whether alternative yeast reporter conditions should be used. These controls establish that the bait protein is made as a stable protein in yeast and that it does not appreciably activate transcription of the LexA operator–based reporter genes. This last is the most important constraint on use of this system. The LexA-fused bait protein must not activate transcription of either reporter, i.e., the EGY48 strain that expresses the LexA fusion protein should not grow on medium lacking Leu, and the colonies should be white on medium containing Xgal. For further characterization, the bait plasmid can also be tested for its ability to bind LexA operators using the repression assay (see Alternate Protocol 1). The characterized bait protein plasmid is used for Basic Protocol 2 to screen a library for interacting proteins.
Materials
DNA encoding the protein of interest
Plasmids (see Table 17.3.1): e.g., pEG202 (Fig. 17.3.3), pSH18-34 (Fig. 17.3.4), pSH17-4, pRFHM1
Yeast strain: e.g., EGY48 (ura3 trp1 his3 3LexA-operator-LEU2; see Table 17.3.2)
-
100-mm complete minimal (CM) medium dropout plates (Treco and Lundblad, 1993) with 2% (w/v) glucose (Glu) or 2% (w/v) galactose (Gal):
Glu/CM –Ura –His
Gal/CM –Ura –His
Gal/CM –Ura –His –Leu
Glu/CM Xgal and Gal/CM Xgal plates (Treco and Lundblad, 1993)
-
CM dropout liquid medium (Treco and Lundblad, 1993) with 2% (w/v/) glucose:
Glu/CM –Ura –His
2× Laemmli sample buffer (see recipe)
Antibody to LexA or fusion domain
H2O, sterile
30°C incubator
100°C water bath
Additional reagents and equipment for subcloning (Struhl, 1987), lithium acetate transformation of yeast (Becker and Lundblad, 1993), filter lift or liquid assay for β-galactosidase (Reynolds et al., 1997), SDS-PAGE (Gallagher, 2006), and immunoblotting (Gallagher et al., 2008)
Table 17.3.1.
| Plasmid name/source | Selection
|
Comment/description | |
|---|---|---|---|
| In yeast | In E. coli | ||
| LexA fusion plasmids | |||
| pEG202c,d | HIS3 | Apr | Contains an ADH promoter that expresses LexA followed by polylinker |
| pJK202 | HIS3 | Apr | Like pEG202, but incorporates nuclear localization sequences between LexA and polylinker; used to enhance translocation of bait to nucleus |
| pNLexAd | HIS3 | Apr | Contains an ADH promoter that expresses polylinker followed by LexA for use with baits where their amino-terminal residues must remain unblocked |
| pGildad | HIS3 | Apr | Contains a GAL1 promoter that expresses same LexA and polylinker cassette as pEG202 for use with baits where their continuous presence is toxic to yeast |
| pEE202I | HIS3 | Apr | An integrating form of pEG202 that can be targeted into HIS3 following digestion with KpnI; for use where physiological screen requires lower levels of bait to be expressed |
| pRFHM1d (control) | HIS3 | Apr | Contains an ADH promoter that expresses LexA fused to the homeodomain of bicoid to produce nonactivating fusion used; as positive control for repression assay, negative control for activation and interaction assays |
| pSH17-4d (control) | HIS3 | Apr | ADH promoter expresses LexA fused to GAL4 activation domain; used as a positive control for transcriptional activation |
| pMW101e | HIS3 | Cmr | Same as pEG202, but with altered antibiotic resistance markers; basic plasmid used for cloning bait |
| pMW103e | HIS3 | Kmr | Same as pEG202, but with altered antibiotic resistance markers; basic plasmid used for cloning bait |
| pHybLex/Zeo | Zeor | Zeor | Bait cloning vector compatible with interaction trap and all other two-hybrid systems; minimal ADH promotor expresses LexA followed by extended polylinker |
| Activation domain fusion plasmids | |||
| pJG4-5c,d | TRP1 | Apr | Contains a GAL1 promoter that expresses nuclear localization domain, transcriptional activation domain, HA epitope tag, cloning sites; used to express cDNA libraries |
| pJG4-5I | TRP1 | Apr | An integrating form of pJG4-5 that can be targeted into TRP1 by digestion with Bsu36I (New England Biolabs); to be used with pEE202I to study interactions that occur physiologically at low protein concentrations |
| pYESTrp | TRP1 | Apr | Contains a GAL1 promoter that expresses nuclear localization domain, transcriptional activation domain, V5 epitope tag, multiple cloning sites; contains f1 ori and T7 promoter/flanking site used to express cDNA libraries (Invitrogen) |
| pMW102e | TRP1 | Kmr | Same as pJG4-5, but with altered antibiotic resistance markers; no libraries yet available |
| pMW104e | TRP1 | Cmr | Same as pJG4-5, but with altered antibiotic resistance markers; no libraries yet available |
| LacZ reporter plasmids | |||
| pSH18-34d | URA3 | Apr | Contains eight LexA operators that direct transcription of the lacZ gene; one of the most sensitive indicator plasmids for transcriptional activation |
| pJK103d | URA3 | Apr | Contains two LexA operators that direct transcription of the lacZ gene; an intermediate reporter plasmid for transcriptional activation |
| pRB1840d | URA3 | Apr | Contains one LexA operator that directs transcription of the lacZ gene; one of the most stringent reporters for transcriptional activation |
| pMW112e | URA3 | Kmr | Same as pSH18-34, but with altered antibiotic resistance marker |
| pMW109e | URA3 | Kmr | Same as pJK103, but with altered antibiotic resistance marker |
| pMW111e | URA3 | Kmr | Same as pRB1840, but with altered antibiotic resistance marker |
| pMW107e | URA3 | Cmr | Same as pSH18-34, but with altered antibiotic resistance marker |
| pMW108e | URA3 | Cmr | Same as pJK103, but with altered antibiotic resistance marker |
| pMW110e | URA3 | Cmr | Same as pRB1840, but with altered antibiotic resistance marker |
| pJK101d (control) | URA3 | Apr | Contains a GAL1 upstream activating sequence followed by two LexA operators followed by lacZ gene; used in repression assay to assess bait binding to operator sequences |
All plasmids contain a 2μm origin for maintenance in yeast, as well as a bacterial origin of replication, except where noted (pEE202I, pJG4-5I).
Interaction Trap reagents represent the work of many contributors: the original basic reagents were developed in the Brent laboratory (Gyuris et al., 1993). Plasmids with altered antibiotic resistance markers (all pMW plasmids) were constructed at Glaxo in Research Triangle Park, N.C. (Watson et al., 1996). Plasmids and strains for specialized applications have been developed by the following individuals: E. Golemis, Fox Chase Cancer Center, Philadelphia, Pa. (pEG202); cumulative efforts of I. York, Dana-Farber Cancer Center, Boston, Mass. and M. Sainz and S. Nottwehr, U. Oregon (pNLexA); D.A. Shaywitz, MIT Center for Cancer Research, Cambridge, Mass. (pGilda); R. Buckholz, Glaxo, Research Triangle Park, N.C. (pEE202I, pJG4-5I); J. Gyuris, Mitotix, Cambridge, Mass. (pJG4-5); R.L. Finley, Wayne State University School of Medicine, Detroit, Mich. (pSH17-4 pRFHM1); S. Hanes, Wadsworth Institute, Albany, N.Y. (pSH17-4, pSH18-34); J. Kamens, BASF, Worcester, Mass. (pJK101, pJK103, pJK202); R. Brent, The Molecular Sciences Institute, Berkeley, Calif. (pRB1840).
Sequence data are available for pEG202 (pLexA), accession number U89960, and pJG4-5, accession number U89961.
Plasmids and strains available from OriGene.
In pMW plasmids the ampicillin resistance gene (Apr) is replaced with the chloramphenicol resistance gene (Cmr) and the kanamycin resistance gene (Kmr) from pBC SK(+) and pBK-CMV (Stratagene), respectively. The choice between Kmr and Cmr or Apr plasmids is a matter of personal taste; use of basic Apr plasmids is described in the basic protocols. Use of the more recently developed reagents would facilitate the purification of library plasmid in later steps by eliminating the need for passage through KC8 bacteria, with substantial saving of time and effort. Apr has been maintained as marker of choice for the library plasmid because of the existence of multiple libraries already possessing this marker. These plasmids are the basic set of plasmids recommended for use.
Figure 17.3.3.

LexA fusion plasmid pEG202. The strong constitutive ADH promoter is used to express bait proteins as fusions to the DNA-binding protein LexA. Restriction sites shown in this map are based on pEG202 sequence data and include selected sites suitable for diagnostic restriction endonuclease digests. A number of restriction sites are available for insertion of coding sequences to produce protein fusions with LexA; the polylinker sequence and reading frame relative to LexA are shown below the map with unique sites marked in bold type. The sequence 5′-CGT CAG CAG AGC TTC ACC ATT G-3′ can be used to design a primer to confirm correct reading frame for LexA fusions. Plasmids contain the HIS3 selectable marker and the 2-μm origin of replication to allow propagation in yeast, and the Apr antibiotic resistance gene and the pBR origin of replication to allow propagation in E. coli. In the plasmids pMW101 and pMW103, the ampicillin resistance gene (Apr) has been replaced with the chloramphenicol resistance gene (Cmr) and the kanamycin resistance gene (Kmr), respectively (see Table 17.3.1 for details).
Figure 17.3.4.

lacZ reporter plasmid. pRB1840, pJK103, and pSH18-34 are all derivatives of LR1Δ1 (West et al., 1984) containing eight, two, or one operator for LexA (LexAop) binding inserted into the unique XhoI site located in the minimal GAL1 promoter (GAL1pro; 0.28 on map). The plasmid contains the URA3 selectable marker, the 2-μm origin to allow propagation in yeast, the ampicillin resistance gene (Apr), and the pBR322 origin (ori) to allow propagation in E. coli. Numbers indicate relative map positions. In the recently developed derivatives, the ampicillin resistance gene has been replaced with the chloramphenicol or kanamycin resistance genes (see Table 17.3.1 for details).
Table 17.3.2.
Interaction Trap Yeast Selection Strainsa
| Strain | Relevant genotype | Number of operators | Comments/description |
|---|---|---|---|
| EGY48b | MATα trp1, his3, ura3, lexAops-LEU2 | 6 | lexA operators direct transcription from the LEU2 gene; EGY48 is a basic strain used to select for interacting clones from a cDNA library |
| EGY191 | MATα trp1, his3, ura3, lexAops-LEU2 | 2 | EGY191 provides a more stringent selection than EGY48, producing lower background with baits with instrinsic ability to activate transcription |
| L40b | MATα trpl, leu2, ade2, GAL4, lexAops-HIS34, lexAops-lacZ8 | Expression driven from GAL1 promoter is constitutive in L40 (inducible in EGY strains); selection is for HIS prototrophy. Integrated lacZ reporter is considerably less sensitive than pSH18-34 maintained in EGY strains. |
Interaction Trap reagents represent the work of many contributors. The original basic reagents were developed in the Brent laboratory (Gyuris et al., 1993). Strains for specialized applications have been developed by the following individuals: E. Golemis, Fox Chase Cancer Center, Philadelphia, Pa. (EGY48, EGY191); A.B. Vojtek and S.M. Hollenberg, Fred Hutchinson Cancer Research Center, Seattle, Wash. (L40).
Strains commercially available from OriGene.
Transform yeast with the bait protein plasmid
-
1
Using standard subcloning techniques (Struhl, 1987), insert the DNA encoding the protein of interest into the polylinker of pEG202 (see Fig. 17.3.3) or other LexA fusion plasmid to make an in-frame protein fusion.
The LexA fusion protein is expressed from the strong alcohol dehydrogenase (ADH) promoter. pEG202 also contains a HIS3 selectable marker and a 2μm origin for propagation in yeast. pEG202 with the DNA encoding the protein of interest inserted is designated pBait. Uses of alternative LexA fusion plasmids are described in Background Information. -
2
Perform three separate lithium acetate transformations (Becker and Lundblad, 1993) of EGY48 using the following combinations of plasmids:
pBait + pSH18-34 (test)
pSH17-4 + pSH18-34 (positive control for activation)
-
pRFHM1 + pSH18-34 (negative control for activation).
Use of the two LexA fusions as positive and negative controls allows a rough assessment of the transcriptional activation profile of LexA bait proteins. pEG202 itself is not a good negative control because the peptide encoded by the uninterrupted polylinker sequences is itself capable of very weakly activating transcription.pSH18-34 contains a 2μm origin and a URA3 selectable marker for maintenance in yeast, as well as a bacterial origin of replication and ampicillin-resistance gene. It is the most sensitive lacZ reporter available and will detect any potential ability to activate lacZ transcription. pSH17-4 is a HIS3 2μm plasmid encoding LexA fused to the activation domain of the yeast activator protein GAL4. This fusion protein strongly activates transcription. pRFHM1 is a HIS3 2μm plasmid encoding LexA fused to the N-terminus of the Drosophila protein bicoid. This fusion protein has no ability to activate transcription.
-
3
Plate each transformation mixture on Glu/CM –Ura –His dropout plates. Incubate 2 days at 30°C to select for yeast that contain both plasmids.
-
4
Streak a Glu/CM –Ura –His master dropout plate with at least five or six independent colonies obtained from each of the three transformations (test, positive control, and negative control) and incubate overnight at 30°C.
Colonies can be tested simultaneously for protein synthesis and for activation of the lacZ and LEU2 reporters.
Assay lacZ activation by β-galactosidase assay
-
5a
For liquid or filter assay: Assay colonies on the master plate for β-galactosidase activity by liquid assay or filter lift assay (Reynolds et al., 1997).
In the filter lift assay, two or three 5-min temperature cycles (−70°C to room temperature) can be used instead of freezing in liquid nitrogen to promote better lysis; this may be worth doing if there is difficulty visualizing blue color.Acceptable results may be obtained using as little as 300 μg/ml of Xgal.It is generally useful to check the membrane after 20 min, and again after 2 to 3 hr. Strong activators will produce a blue color in 5 to 10 min, and a bait protein (LexA fusion protein) that does so is unsuitable for use in an interactor hunt using this lacZ reporter plasmid. Weak activators will produce a blue color in 1 to 6 hr (compare to negative control pRFHMI, which will itself produce a faint blue color with time) and may or may not be suitable. Weak activators should be tested using the repressor assay described in Alternate Protocol 1. -
5b
For plate assay: Streak yeast from the master plate onto Glu/CM Xgal plates and incubate at 30°C. Examine plates for color development at intervals over the next 2 to 3 days.
Strongly activating fusions should be visibly blue within 12 to 24 hr; moderate activators will be visibly blue after ~2 days.When patching from a master plate to Xgal plates (below), sufficient yeast are transferred that plasmid loss is not a major problem even in the absence of selection; this is balanced by the desire to assay sets of constructs on the same plate to eliminate batch variation in Xgal potency. Hence, master plates should be made either with complete minimal amino acid mix, or by dropping out only uracil (–Ura), to make the plates universally useful.When a bait protein appreciably activates transcription under these conditions, there are several recourses. The first and simplest is to switch to a less sensitive lacZ reporter plasmid; use of pJK103 and pRB1840 may be sufficient to reduce background to manageable levels. If this fails, it is frequently possible to generate a truncated LexA fusion that does not activate transcription.
Confirm fusion-protein synthesis by immunoblotting
-
6
From the master plates, start a 5-ml culture in Glu/CM –Ura –His liquid medium for each bait being tested and for a positive control for protein expression (i.e., RFHMI or SH17-4). Incubate overnight at 30°C.
For each construct assayed, it is a good idea to grow colonies from at least two primary transformants, as levels of bait expression are sometimes heterogeneous. -
7
From each overnight culture, start a fresh 5-ml culture in Glu/CM –Ura –His at OD600 ~0.15. Incubate again at 30°C.
-
8
When the cultures are exponentially growing (OD600 = 0.45 to 0.7, ~4 to 6 hr), transfer 1.5 ml to a microcentrifuge tube.
For some LexA fusion proteins, levels of the protein drop off rapidly in cultures approaching stationary phase, due to a combination of diminishing activity of the ADH1 promoter in late growth phases and the relative instability of particular fusion domains. Thus, it is not a good idea to let cultures become saturated in the hope of obtaining a higher yield of protein. -
9
Microcentrifuge cells 3 min at 13,000 × g, room temperature. When the pellet is visible, remove the supernatant.
Inspection of the tube should reveal a pellet of ~1 to 3 μl in volume. If the pellet is not visible, microcentrifuge another 3 min. -
10
Working rapidly, add 50 μl of 2× Laemmli sample buffer to the pellet and lyse by boiling 5 min in a 100°C water bath.
-
11
Microcentrifuge 1 min at maximum speed to pellet large cellular debris.
-
12
Perform SDS-PAGE (Gallagher, 2006) using 10 to 25 μl of lysate per lane of a minigel. To detect the fusion protein, perform immunoblotting (Gallagher et al., 2008) using an antibody to the fusion domain or LexA.
Many antibodies against LexA are available and can be found, for instance, at http://www.biocompare.com.It is generally a good idea to assay for the production of full-length LexA fusions, as some fusion proteins will be proteolytically cleaved by endogenous yeast proteases.
Test for Leu requirement
-
13
Disperse transformants from the master plate into 500 μl sterile water. Make a series of 1/10 dilutions in sterile water to cover a 1000-fold concentration range.
-
14
Plate 100 μl from each tube (undiluted, 1/10, 1/100, and 1/1000) on Gal/CM –Ura –His and Gal/CM –Ura –His –Leu dropout plates (total eight plates). Incubate overnight at 30°C.
Gal/CM –Ura –His dropout plates should show a concentration range from 10 to 10,000 colonies and Gal/CM –Ura –His –Leu dropout plates should have no colonies.Actual selection in the interactor hunt is based on the ability of the bait protein and acid-fusion pair, but not the bait protein alone, to activate transcription of LEU2 and allow growth on medium lacking Leu. Thus, the test for the Leu requirement is the most important test of whether the bait protein is likely to have an unworkably high background. The LEU2 reporter in EGY48 is more sensitive than the pSH18-34 reporter for some baits, so it is possible that a bait protein that gives little or no signal in a β-galactosidase assay would nevertheless permit some level of growth on –Leu medium. If this occurs, there are several options for proceeding, the most immediate of which is to substitute EGY191 (Table 17.3.2), a less sensitive screening strain, and repeat the assay. Nonetheless, the initial screening should use the most sensitive reporters. If activation is detected, screening can be done with increasingly less sensitive reporters (see Critical Parameters and Troubleshooting for further discussion).
ALTERNATE PROTOCOL 1: CONFIRMATION OF FUSION PROTEIN SYNTHESIS BY REPRESSION ASSAY
For LexA fusions that do not activate transcription, a repression assay (Brent and Ptashne, 1984) can be used to confirm that the LexA fusion protein is synthesized in yeast (some proteins are not), localized to the nucleus, and capable of binding LexA operator sequences (Fig. 17.3.5). The following steps can be performed concurrently with the activation assay, although the repression assay is no longer used as a standard procedure. The relative ease of screening using the interaction mating approach (Alternate Protocol 2) allows researchers to screen several baits (e.g., overlapping pieces of the same protein of interest) instead of just one, thus permitting only the minimal characterization of each bait (e.g., activation assay and immunoblotting, confirming synthesis of the fusion protein). While it is reassuring to obtain positive results in the repression assay, in the case of a negative outcome, the general recommendation would most likely be to proceed with the screen anyway.
Figure 17.3.5.

Repression assay for DNA binding. (A) The plasmid pJK101 contains the upstream activating sequence (UAS) from the GAL1 gene followed by LexA operators (ops) upstream of the lacZ coding sequence. Thus, yeast containing pJK101 will have significant β-galactosidase activity when grown on medium in which galactose is the sole carbon source because of binding of endogenous yeast GAL4 to the GALUAS. (B) LexA-fused proteins (P1-LexA) that are made, enter the nucleus, and bind the LexA ops will block activation from the GALUAS, repressing β-galactosidase activity (+) 3- to 5-fold. On glucose/Xgal medium, yeast containing pJK101 should be white because GALUAS transcription is repressed.
Additional Materials (also see Basic Protocol 1)
pBait (Basic Protocol 1)
pJK101 (Table 17.3.1)
100-mm complete minimal (CM) medium dropout plates (Treco and Lundblad, 1993) with 2% (w/v) glucose (Glu): Glu/CM –Ura
-
Transform EGY48 yeast with the following combinations of plasmids:
pBait and pJK101 (test)
pRFHM1 and pJK101 (positive control for repression)
pJK101 alone (negative control for repression).
Plate each transformation mix on Glu/CM –Ura –His dropout plates or Glu/CM –Ura dropout plates as appropriate to select cells that contain the indicated plasmids. Incubate 2 to 3 days at 30°C until colonies appear.
Streak colonies to a Glu/CM –Ura –His or Glu/CM –Ura dropout master plate and incubate overnight at 30°C.
-
Assay β-galactosidase activity of the three transformed strains by liquid assay (Reynolds et al., 1997; using Gal/CM dropout liquid medium), filter assay (Reynolds et al., 1997; using Gal/CM plates to grow overnight), or plate assay (see Basic Protocol 1; using Gal/CM –Ura Xgal plates).
This assay should not be run for more than 1 to 2 hr for filters or 36 hr for Xgal plates, as the high basal lacZ activity will make differential activation of pJK101 impossible to see with longer incubations. It is generally most effective to use Xgal plates and inspect them at 12 to 24 hr.If the LexA protein is made (as determined by immunoblotting) but does not repress, it may be necessary to clone the sequence into a LexA fusion vector that contains a nuclear localization motif, e.g., pJK202 (see Table 17.3.1), or to modify or truncate the fusion domain to remove motifs that target it to other cellular compartments (e.g., myristoylation signals).
BASIC PROTOCOL 2: PERFORMING AN INTERACTOR HUNT
An interactor hunt involves the transformation of yeast containing LexA-fused probes and reporters (see Basic Protocol 1) with a library in pJG4-5 with a cDNA expression cassette under control of the GAL promoter. This is followed by two successive large platings of the transformants. In the first plating, yeast are plated on complete minimal (CM) medium –Ura –His –Trp dropout plates with glucose (Glu) as a sugar source to select for the library plasmid. In the second plating, which selects for yeast that contain interacting proteins, a slurry of primary transformants is plated on CM –Ura –His –Trp –Leu dropout plates with galactose/raffinose (Gal/Raff) as the sugar source. This two-step selection is encouraged for two reasons. First, a number of interesting proteins may be deleterious to the growth of yeast that bear them; these would be competed out in an initial mass plating. Second, it seems likely that immediately after simultaneous transformation and GAL induction, yeast bearing particular interacting proteins may not be able to initially express sufficient levels of these proteins to support growth on medium lacking Leu. Library plasmids from colonies identified in the second plating are screened by PCR to identify redundant clones and used to transform yeast cells for the final specificity screen.
A list of commercially available libraries for use with this system is provided in Table 17.3.3. The protocol outlined below describes the steps used to perform a single-step screen that should saturate a library derived from a mammalian cell. For screens with libraries derived from lower eukaryotes with less complex genomes, fewer plates will be required.
Table 17.3.3.
Libraries Compatible with the Interaction Trap System
| Source of RNA/DNA | Independent clones | Insert sizeb |
|---|---|---|
| Cell lines | ||
| HeLa cells (human cervical carcinoma) | 9.6 × 106 | 0.3–3.5 kb (1.5 kb) |
| WI-38 cells (human lung fibroblasts), serum-starved, cDNA | 5.7 × 106 | 0.3–3.5 kb (1.5 kb) |
| Jurkat cells (human T cell leukemia) | 5.7 × 106 | (>1.3) |
| SKOV3 human Y ovarian cancer | 5.0 × 106 | (>1.4 kb) |
| MDBK cell, bovine kidney | 5.8 × 106 | (>1.2 kb) |
| MCF7 breast cancer cells, untreated | 1.0 × 107 | (>1.5 kb) |
| MCF7 breast cancer cells, estrogen-treated | 1.0 × 107 | (>1.1 kb) |
| MCF7 breast cancer cells, serum-grown | 1.0 ×107 | 0.4–3.5 kb |
| LNCAP prostate cell line, untreated | 2.9 × 106 | (>0.8 kb) |
| LNCAP prostate cell line, androgen-treated | 4.6 × 106 | (>0.9 kb) |
| Tissues | ||
| Human liver | 1.1 × 107 | (>1 kb) |
| Human ovary | 4.6 × 106 | (>1.3 kb) |
| Human peripheral blood leukocyte | 1.0 × 107 | (>1.3 kb) |
| Human fetal kidney | 3.0 × 106 | (>1 kb) |
| Human prostate | 1.4 × 106 | (>1 kb) |
| Human prostate cancer | 1.1 × 106 | (>0.9 kb) |
| Human fetal liver | 8.6 × 106 | (>1 kb) |
| Human fetal brain | 3.5 × 106 | 0.5–1.2 kb (1.5 kb) |
| Mouse brain | 6.1 × 106 | (>1 kb) |
| Mouse breast, lactating | 1.0 × 107 | 0.4–3.1 kb |
| Mouse breast, involuting | 1.0 × 107 | 0.4–7.0 kb |
| Mouse breast, virgin | 1.0 × 107 | 0.4–5.5 kb |
| Mouse breast, 12 days pregnant | 6.3 × 106 | 0.4–5.3 kb |
| Mouse skeletal muscle | 7.2 × 106 | 0.4–3.5 kb |
| Rat adipocyte, 9-week-old Zucker rat | 1.0 × 107 | 0.4–5.0 kb |
| Rat brain | 4.5 ×106 | 0.3–3.4 kb |
| Rat testis | 8.0 × 106 | (>1.2 kb) |
| Rat thymus | 8.2 × 106 | (>1.3 kb) |
| Mouse liver | 9.5 × 106 | (>1.4 kb) |
| Mouse spleen | 1.0 × 107 | (>1 kb) |
| Mouse ovary | 4.0 × 106 | (>1.2 kb) |
| Mouse prostate | 3.7 × 106 | 0.3–0.4 kb |
| Mouse embryo, whole (19-day) | 1.0 × 105 | 0.2–2.5 kb |
| D. melanogaster, adult, cDNA | 1.8 × 106 | (>1.0 kb) |
| Miscellaneous | ||
| S. cerevisiae, S288C, genomic | 4.0 × 106 | 0.5–4.0 kb |
| C. elegans | 3.8 × 106 | (>1.2 kb) |
All libraries were constructed in conjunction with the laboratory of Roger Brent (brent@molsci.org) and are available from OriGene. All were constructed in the pJG4-5 vector, except the SKOV3 human Y ovarian cancer line, which was constructed in the pYESTrp vector.
Average insert size given in parentheses.
Occasionally, baits that seemed well-behaved during preliminary tests produce unworkably high backgrounds of “positives” during an actual screen (see Background Information and Critical Parameters and Troubleshooting). To forestall the waste of time and materials performing a screen with such a bait would entail, an alternative approach is to perform a scaled-back screen when working with a new bait (e.g., 5 rather than 30 plates of primary transformants). The results can be assessed before doing a full screen; it is then possible to switch to lower-sensitivity reporter strains and plasmids, if appropriate. Although individual baits will vary, the authors’ current default preference is to use the lacZ reporter pJK103 in conjunction with either EGY48 or EGY191.
Materials
-
Transformed yeast strains (see Basic Protocol 1), EGY48 containing:
pSH18-34 (lacZ reporter plasmid) and pBait (bait strain)
pSH18-34 and pRFHM-1 (negative control)
pSH18-34 and any nonspecific bait (nonspecific control)
-
Complete minimal (CM) dropout liquid medium (Treco and Lundblad, 1993) with 2% (w/v) glucose (Glu) or 2% (w/v) galactose (Gal)/1% (w/v) raffinose (Raff):
Glu/CM –Ura –His
Gal/Raff/CM –Ura –His –Trp
Gal/Raff/CM –Ura –His –Trp –Leu
H2O, sterile
TE buffer (pH 7.5; APPENDIX 2A), with and without 0.1 M lithium acetate
Library DNA in pJG4-5 (Table 17.3.3 and Fig. 17.3.6)
High-quality sheared salmon sperm DNA (see Support Protocol 1)
40% (w/v) polyethylene glycol 4000 (PEG 4000; filter sterilized) in 0.1 M lithium acetate/TE buffer (pH 7.5)
Dimethyl sulfoxide (DMSO)
-
Complete medium (CM) dropout plates (Treco and Lundblad, 1993; sizes indicated) with 2% (w/v) glucose (Glu) or 2% (w/v) galactose (Gal)/1% (w/v) raffinose (Raff), plus 20 μg/ml Xgal, as indicated:
Glu/CM –Ura –His –Trp, 24 × 24-cm (Nunc) and 100-mm
Gal/Raff/CM –Ura –His –Trp, 100-mm
Gal/Raff/CM –Ura –His –Trp –Leu, 100-mm
Glu/Xgal/CM –Ura –His –Trp, 100-mm
Gal/Raff/Xgal/CM –Ura –His –Trp, 100-mm
Glu/CM –Ura –His –Trp –Leu, 100-mm
Glu/CM –Ura –His, 100-mm
Glycerol solution (see recipe)
Lysis solution (see recipe)
0.7% low-melting agarose gel (see APPENDIX 1N)
HaeIII and appropriate enzyme buffer
10 μM forward primer (FP1): 5′-CGT AGT GGA GAT GCC TCC-3′
10 μM reverse primer (FP2): 5′-CTG GCA AGG TAG ACA AGC CG-3′
E. coli DH5α or other strain suitable for preparation of DNA for sequencing Restriction enzymes: EcoR1 and XhoI
pJG4-5 library vector (Fig. 17.3.6)
30°C incubator, with and without shaking
50-ml conical tubes, sterile
1.5-ml microcentrifuge tubes, sterile
42°C heating block
Glass microscope slides, sterile
Toothpicks or bacterial inoculating loop (Elbing and Brent, 2002), sterile
96-well microtiter plate
Sealing tape, e.g., wide transparent tape
150- to 212-μm glass beads, acid-washed (soak 1 hr in concentrated nitric acid, rinse thoroughly with H2O, then oven dry)
Vortexer with plate adapters
Additional reagents and equipment for performing PCR (Kramer and Coen, 2001), agarose gel electrophoresis (APPENDIX 1N), restriction endonuclease digestion (APPENDIX 1M), bacterial transformation by electroporation (Seidman et al., 1997; optional), plasmid miniprep (APPENDIX 1J; optional), and gap repair in yeast (Lundblad and Zhou, 1997)
Figure 17.3.6.

Library plasmid pJG4-5. Library plasmids express cDNAs or other coding sequences inserted into unique EcoRI and XhoI sites as a translational fusion to a cassette consisting of the SV40 nuclear localization sequence (NLS; PPKKKRKVA), the acid blob B42 domain (Ruden et al, 1991), and the hemagglutinin (HA) epitope tag (YPYDVPDYA). Expression of cassette sequences is under the control of the GAL1 galactose-inducible promoter. This map is based on the sequence data available for pJG4-5, and includes selected sites suitable for diagnostic restriction digests (shown in bold). The sequence 5′-CTG AGT GGA GAT GCC TCC-3′ can be used as a primer to identify inserts or to confirm correct reading frame. The pJG4-5 plasmid contains the TRP1 selectable marker and the 2-μm origin to allow propagation in yeast, and the Apr antibiotic resistance gene and the pUC origin to allow propagation in E. coli. In the pJG4-5 derivative plasmids pMW104 and pMW102, the ampicillin resistance gene has been replaced with the chloramphenicol resistance gene and kanamycin resistance gene, respectively (see Table 17.3.1 for details). Currently existing libraries are all made in the pJG4-5 plasmid (Gyuris et al., 1993) shown on this figure. Unique sites are marked in bold type.
Transform the library
-
1
Grow a 20-ml culture of the bait strain in Glu/CM –Ura –His liquid dropout medium overnight at 30°C.
For best results, the pBait and lacZ reporter plasmids should have been transformed into the yeast within ~7 to 10 days of commencing a screen. -
2
In the morning, dilute the culture into 300 ml Glu/CM –Ura –His liquid dropout medium to 2 × 106 cells/ml (OD600 = ~0.10). Incubate at 30°C until the culture contains ~1 × 107 cells/ml (OD600 = ~0.50).
-
3
Centrifuge 5 min at 1000 to 1500 × g, room temperature, in a low-speed centrifuge, to harvest cells. Resuspend the pellet in 30 ml sterile water and transfer to a 50-ml conical tube.
-
4
Centrifuge 5 min at 1000 to 1500 × g. Decant the supernatant and resuspend the cells in 1.5 ml TE buffer/0.1 M lithium acetate.
-
5
Add 1 μg library DNA in pJG4-5 and 50 μg high-quality sheared salmon sperm carrier DNA to each of 30 sterile 1.5-ml microcentrifuge tubes. Add 50 μl of the resuspended yeast solution from step 4 to each tube.
The total volume of library and salmon sperm DNA added should be <20 μl and preferably < 10 μl.A typical library transformation will result in 2 to 3 × 106 primary transformants. Assuming a transformation efficiency of 105/μg library DNA, this transformation requires a total of 20 to 30 μg library DNA and 1 to 2 mg carrier DNA. Doing transformations in small aliquots helps reduce the likelihood of contamination and, for reasons that are not clear, provides significantly better transformation efficiency than scaled-up versions.Do not use excess transforming library DNA per aliquot of competent yeast cells because each competent cell may take up multiple library plasmids, complicating subsequent analysis. -
6
Add 300 μl of sterile 40% PEG 4000/0.1 M lithium acetate/TE buffer, pH 7.5, and invert to mix thoroughly. Incubate 30 min at 30°C.
-
7
Add DMSO to 10% (~40 μl per tube) and invert to mix. Heat shock 10 min in a 42°C heating block.
-
8a
For 28 tubes: Plate the complete contents of one tube per 24 × 24–cm Glu/CM –Ura –His –Trp dropout plate and incubate at 30°C.
-
8b
For two remaining tubes: Plate 360 μl of each tube on a 24 × 24–cm Glu/CM –Ura –His –Trp dropout plate. Use the remaining 40 μl from each tube to make a series of 1/10 dilutions in sterile water. Plate dilutions on 100-mm Glu/CM –Ura –His – Trp dropout plates. Incubate all plates 2 to 3 days at 30°C until colonies appear.
The dilution series gives an idea of the transformation efficiency and allows an accurate estimation of the number of transformants obtained.
Collect primary transformant cells
Conventional replica plating does not work well in the selection process because so many cells are transferred to new plates that very high background levels inevitably occur. Instead, the procedure described below creates a slurry in which cells derived from >106 primary transformants are homogeneously dispersed. A precalculated number of these cells is plated for each primary transformant.
-
9
Cool all of the 24 × 24–cm plates containing transformants for several hours at 4°C to harden agar.
-
10
Wearing gloves and using a sterile glass microscope slide, gently scrape yeast cells off the plate. Pool cells from the 30 plates into one or two sterile 50-ml conical tubes.
This is the step where contamination is most likely to occur. Be careful. -
11
Wash cells by adding a volume of sterile TE buffer or water at least equal to the volume of the transferred cells. Centrifuge ~5 min at 1000 to 1500 × g, room temperature, and discard supernatant. Repeat the wash.
After the second wash, the pellet volume should be ~25 ml cells derived from 1.5 × 106 transformants. -
12
Resuspend the pellet in 1 vol glycerol solution, mix well, and store in 1-ml aliquots up to 1 year at –70°C.
Determine replating efficiency
-
13
Remove an aliquot of frozen transformed yeast and dilute 1/10 with Gal/Raff/CM –Ura –His –Trp dropout medium. Incubate with shaking 4 hr at 30°C to induce the GAL promoter on the library.
Raffinose (Raff) aids in growth without diminishing transcription from the GAL1 promoter. -
14
Make serial dilutions of the yeast cells using the Gal/Raff/CM –Ura –His –Trp dropout medium. Plate on 100-mm Gal/Raff/CM –Ura –His –Trp dropout plates and incubate 2 to 3 days at 30°C until colonies are visible.
-
15
Count the colonies and determine the number of colony-forming units (cfu) per aliquot of transformed yeast.
In calculating yeast concentrations, it is useful to remember that 1 OD600 unit = ~1.0 × 107 yeast cells/ml. In general, if the harvest is done carefully, viability will be >90%. Some intrepid investigators perform this step simultaneously with plating out on Leu selective medium (steps 16 and 17).
Screen for interacting proteins
-
16
Thaw the appropriate quantity of transformed yeast based on the plating efficiency, dilute, and incubate as in step 13.
-
17
Dilute cultures in Gal/Raff/CM –Ura –His –Trp –Leu medium as necessary to obtain a concentration of 107 cells/ml (OD600 = ~0.5) and plate 100 μl on each of as many 100-mm Gal/Raff/CM –Ura –His –Trp –Leu dropout plates as are necessary for full representation of transformants. Incubate 2 to 3 days at 30°C until colonies appear.
Because not all cells that contain interacting proteins plate at 100% efficiency on –Leu medium (Estojak et al., 1995), it is desirable, for actual selection, that each primary colony obtained from the transformation be represented on the selection plate by three to ten individual yeast cells. This will in some cases lead to multiple isolations of the same cDNA; however, because the slurry is not perfectly homogenous, it will increase the likelihood that all primary transformants are represented by at least one cell on the selective plate.It is easiest to visually scan for Leu+ colonies using cells plated at ~106 cfu per 100-mm plate. Plating at higher density can contribute to cross-feeding between yeast, resulting in spurious background growth. Thus, for a transformation in which 3 × 106 colonies are obtained, plate ~2 × 107 cells on a total of 20 selective plates. -
18
Using sterile toothpicks or loops, carefully pick appropriate colonies to a new Gal/Raff/CM –Ura –His –Trp –Leu master dropout plate. Incubate 2 to 7 days at 30°C until colonies appear.
A good strategy is to pick a master plate with colonies obtained on day 2, a second master plate (or set of plates) with colonies obtained on day 3, and a third with colonies obtained on day 4. Colonies from day 2 and 3 master plates should generally be characterized further. If many apparent positives are obtained, it may be worth making master plates of the much larger number of colonies likely to be obtained at day 4 (and after). See Critical Parameters and Troubleshooting and the annotation to step 20 for additional information about appropriate colony selection for the master plate.If no colonies appear within a week, those arising at later time points are likely to be artifactual. Contamination that has occurred at an earlier step (e.g., during plate scraping) is generally reflected by the growth of a very large number of colonies (>500/plate) within 24 to 48 hr after plating on selective medium.Some investigators omit use of a Gal/Raff/CM –Ura –His –Trp –Leu master plate, restreaking directly to a Glu/CM –Ura –His –Trp master plate as in step 20.
Test for Gal dependence
The following steps test for Gal dependence of the Leu+ insert and lacZ phenotypes to confirm that they are attributable to expression of the library-encoded proteins. The GAL1 promoter is turned off and –Leu selection eliminated before reinducing.
-
19
Restreak from the Gal/Raff/CM –Ura –His –Trp –Leu master dropout plate to a 100-mm Glu/CM –Ura –His –Trp master dropout plate. Incubate overnight at 30°C until colonies form.
-
20
Restreak or replica plate from this plate to the following plates:
Glu/Xgal/CM –Ura –His –Trp
Gal/Raff/Xgal/CM –Ura –His –Trp
Glu/CM –Ura –His –Trp –Leu
-
Gal/Raff/CM –Ura –His –Trp –Leu.
At this juncture, colonies and the library plasmids they contain are tentatively considered positive if they are blue on Gal/Raff/Xgal plates but not blue or only faintly blue on Glu/Xgal plates, and if they grow on Gal/Raff/CM –Leu plates but not on Glu/CM –Leu plates.The number of positives obtained will vary drastically from bait to bait. How they are processed subsequently will depend on the number initially obtained and on the preference of the individual investigator. If none are obtained using EGY48 as reporter strain, it may be worth attempting to screen a library from an additional tissue source. If a relatively small number (≤30) are obtained, proceed to step 21. However, sometimes searches will yield large numbers of colonies (>30 to 300, or more). In this case, there are several options. The first option is to warehouse the majority of the positives and work up the first 30 that arise; those growing fastest are frequently the strongest interactors. These can be checked for specificity, and restriction digests can be used to establish whether they are all independent cDNAs or represent multiple isolates of the same, or a small number, of cDNAs. If the former is true, it may be advisable to repeat the screen in a less sensitive strain background, as obtaining many different interactors can be a sign of low-affinity nonspecific background. Alternatively, if initial indications are that a few cDNAs are dominating the positives obtained, it may be useful to perform a filter hybridization with yeast (see Support Protocol 2) using these cDNAs as a probe to establish the frequency of their identification and exclude future reisolation of these plasmids. The second major option is to work up large numbers of positives to get a complete profile of isolated interactors (see Support Protocol 3). A third option is to temporarily warehouse the entire results of this first screen, and repeat the screen with a less sensitive strain such as EGY191, on the theory that it is most important to get stronger interactors first and a complete profile of interactors later. Finally, some investigators prefer to work up the entire set of positives initially obtained, even if such positives number in the hundreds.
PCR screen positive colonies
This procedure sorts a large number of positives into redundant (multiple isolates) and unique classes prior to plasmid rescue from yeast, thus greatly reducing the number of plasmid isolations that must be performed. An additional benefit is that this protocol pre-identifies positive clones containing one or multiple library plasmids; for those containing only one library plasmid, only a single colony needs to be prepared through E. coli.
-
21
Use a sterile toothpick or bacterial inoculating loop to transfer yeast from the Glu/CM –Ura –His –Trp master plate into 25 μl lysis solution in a 96-well microtiter plate. Seal the wells of the microtiter plate with sealing tape and incubate 1.5 to 3.5 hr at 37°C with shaking.
The volume of yeast transferred should not exceed ~2 to 3 μl of packed pellet; larger quantities of yeast will reduce quality of the DNA. DNA can be efficiently recovered from master plates that have been stored up to 1 week at 4°C. If yeast have been previously gridded on master plates, transfer to microtiter plates can be facilitated by using a multicolony replicator. -
22
Remove tape from the plate, add ~25 μl acid-washed glass beads to each well, and reseal with the same tape. Firmly attach the microtiter plate to a vortexer with plate adapter, and vortex 5 min at medium-high power.
-
23
Remove the tape and add ~100 μl sterile water to each well. Swirl gently to mix, then remove 0.8 to 2.0 μl sample for amplification. Press the tape back firmly to seal the microtiter plate and place in the freezer at –20°C for storage.
-
24
Amplify sample by standard PCR in a 30-μl volume using 3 μl each of the forward primer FP1 and the reverse primer FP2. Perform PCR using the following cycles:
Initial step: 2 min 94°C (denaturation) 31 cycles: 45 sec 94°C (denaturation) 45 sec 56°C (annealing) 45 sec 72°C (extension). These conditions have been used successfully to amplify fragments up to 1.8 kb in length; some modifications (e.g., increasing the extension time) are also effective. -
25
Electrophorese 20 μl PCR product on a 0.7% low-melting-temperature agarose gel (APPENDIX 1N). Based on insert sizes, group the obtained interactors into families, i.e., potential multiple independent isolates of identical cDNAs. Reserve the gel (in a gel box at room temperature or wrapped in plastic wrap at 4°C) until results of the next step are obtained.
No special precautions are needed for storing the gel. Since HaeIII digests typically yield rather small DNA fragments, running the next gel does not take a lot of time, and the delay does not exceed 45 to 60 min. -
26
While the gel is running, digest the remaining 10 μl of PCR product with HaeIII in a volume of ~20 μl (APPENDIX 1M). Based on the sizes of undigested PCR products (step 25), arrange the HaeIII digest samples so that those thought to represent a family are side by side. Resolve the digests on a 1% to 2% agarose gel (APPENDIX 1N).
Most restriction fragments will be in the 0.2- to 1.0-kb size range, so a long gel run is advisable. This analysis should produce a distinct fingerprint of insert sizes and allow definition of library cDNAs as unique isolates or related groups.A single positive yeast will sometimes contain multiple library plasmids. An advantage of this protocol is the ready detection of multiple library plasmids in PCR reactions; thus, following subsequent bacterial transformations, only a single TRP1 colony would need to be analyzed unless multiple plasmids were already known to be present. -
27
Isolate the DNA fragments from the low-melting-temperature agarose gel.
If inspection of the banding pattern on the two gels suggests that a great many reisolates of a small number of cDNAs are present, it may be worthwhile to immediately sequence PCR products representative of these clusters, but it is generally still advisable to perform the specificity tests before doing so. If the PCR products are sequenced, the FP1 (forward) primer works well in automated sequencing of PCR fragments, but the FP2 (reverse) primer is only effective in sequencing from purified plasmid.In general, priming from the AT-rich ADH terminator downstream of the polylinker/cDNA in the library plasmid is less efficient than from upstream of the cDNA, and it is hard to design effective primers in this region. -
28
Optional: Remove the microtiter plate of lysates from the freezer, thaw it, and remove 2 to 4 μl of lysed yeast for each desired positive. Electroporate DNA samples into DH5α E. coli (Seidman et al., 1997) and prepare a miniprep of plasmid DNA from the transformed bacteria (APPENDIX 1J).
KC8 E. coli should be used for electroporation when the original reagents pEG202/pJG4-5/pJK101 are used for the interaction trap.Refreeze the plate of lysates as a DNA reserve in case the bacteria fail to transform on the first pass.
Assess positive colonies with specificity tests
Much spurious background will have been removed by the previous series of controls. Other classes of false positives can be eliminated by retransforming purified plasmids into “virgin” bait strains that have not been subjected to Leu selection and verifying that interaction-dependent phenotypes are still observed. Such false positives could include mutations in the initial EGY48 yeast that favor growth on Gal medium, library-encoded cDNAs that interact with the LexA DNA-binding domain, or proteins that are sticky and interact with multiple biologically unrelated fusion domains.
-
29
In separate transformations, use the gap repair technique (Lundblad and Zhou, 1997), combining EcoRI/XhoI-digested pJG4-5 plasmid and purified PCR fragments from step 27 to transform EGY48 yeast that contain the following plasmids and are growing on Glu/CM –Ura –His plates:
pSH18-34 and pBait
pSH18-34 and pRFHM-1
pSH18-34 and a nonspecific bait (optional).
-
30
Plate each transformation mix on Glu/CM –Ura –His –Trp dropout plates and incubate 2 to 3 days at 30°C until colonies appear.
-
31
Create a Glu/CM –Ura –His –Trp master dropout plate for each library plasmid being tested. Adjacently streak five or six independent colonies derived from each of the transformation plates. Incubate overnight at 30°C.
-
32
Restreak or replica plate from this master dropout plate to the same series of test plates used for the actual screen:
Glu/Xgal/CM –Ura –His –Trp
Gal/Raff/Xgal/CM –Ura –His –Trp
Glu/CM –Ura –His –Trp –Leu
-
Gal/Raff/CM –Ura –His –Trp –Leu.
True positive cDNAs should make cells blue on Gal/Raff/Xgal but not on Glu/Xgal plates, and should make them grow on Gal/Raff/CM –Leu but not Glu/CM –Leu dropout plates, only if the cells contain LexA-bait. cDNAs that meet such criteria are ready to be sequenced (see legend to Fig. 17.3.6 for primer sequence) or otherwise characterized. Those cDNAs that encode proteins that interact with either RFHM-1 or another nonspecific bait should be discarded.It may be helpful to cross-check the isolated cDNAs with a database of cDNAs thought to be false positives. This database is available as a work in progress at http://www.fccc.edu:80/research/labs/golemis/InteractionTrapInWork.html. cDNAs reported to this database are generally those isolated only once in a screen in which obviously true interactive partners were isolated multiple times, cDNAs that may interact with more than one bait, or cDNAs for which the interaction does not appear to make biological sense in the context of the starting bait. Although some proteins in this database may ultimately turn out in fact to associate with the bait that isolated them, they are by default unlikely to possess a unique and interesting function in the context of that bait if they are well represented in the database.
-
33
If appropriate, conduct additional specificity tests. Analyze and sequence positive isolates.
The three test plasmids outlined above (pEG202, pSH18-34, and pRFHM1) represent a minimal test series. If other LexA-bait fusion proteins are available that are related to the bait protein used in the initial library screen, substantial amounts of information can be gathered in additional specificity tests. For example, if the initial bait protein was the leucine zipper of c-Fos, specificity screening of interactor-hunt positives against the leucine zippers of c-Jun or GCN4 might allow discrimination between proteins that are specific for Fos versus those that associate generically with leucine zippers.
ALTERNATE PROTOCOL 2: PERFORMING A HUNT BY INTERACTION MATING
An alternative way of conducting an interactor hunt is to mate a strain that expresses the bait protein with a second strain that has been pretransformed with the library DNA, and screen the resulting diploid cells for interactors (Bendixen et al., 1994; Finley and Brent, 1994). This “interaction mating” approach can be used for any interactor hunt and is particularly useful in three special cases. The first case is when more than one bait will be used to screen a single library. Interaction mating allows several interactor hunts with different baits to be conducted using a single high-efficiency yeast transformation with library DNA. This can be a considerable savings because the library transformation is one of the most challenging tasks in an interactor hunt. The second case is when a constitutively expressed bait interferes with yeast viability. For such baits, performing a hunt by interaction mating avoids the difficulty associated with achieving a high-efficiency library transformation of a strain expressing a toxic bait. Moreover, the actual selection for interactors will be conducted in diploid yeast, which are more vigorous than haploid yeast and can better tolerate expression of toxic proteins. The third case is when a bait cannot be used in a traditional interactor hunt using haploid yeast strains (see Basic Protocol 2) because it activates transcription of even the least sensitive reporters. In diploids the reporters are less sensitive to transcription activation than they are in haploids. Thus, the interaction mating hunt provides an additional method for reducing background from transactivating baits.
In the protocol described below, the library DNA is used to transform a strain with a LEU2 reporter (e.g., EGY48). This transformed library strain is then frozen in many aliquots, which can be thawed and used for individual interactor hunts. The bait is expressed in a strain of mating type opposite to that of the library strain, bearing the lacZ reporter. A hunt is conducted by mixing the library strain with the bait strain and allowing diploids to form on YPD medium overnight. The diploids are then induced for expression of the library-encoded proteins and screened for interactors as in Basic Protocol 2.
NOTE: Strain combinations other than those described below can also be used in an interaction-mating hunt. The key to choosing the strains is to ensure that the bait and prey strains are of opposite mating types, have auxotrophies that allow selection for the appropriate plasmids and reporter genes, and complement one another so that diploids can be selected.
Additional Materials (also see Basic Protocols 1 and 2)
Yeast strains: RFY206 (MATa ura3 trp1 his3 leu2; Finley and Brent, 1994)
YPD liquid medium and 100-mm plates (Treco and Lundblad, 1993)
Glu/CM –Trp dropout plates (Treco and Lundblad, 1993) with 2% glucose
pJG4-5 library vector (Fig. 17.3.6)
Construct the bait strain
-
1
Prepare the bait strain (see Basic Protocol 1) using the MATa yeast strain RFY206. Select transformants on Glu/CM –Ura –His plates by incubating 3 to 4 days at 30°C until colonies form. Combine three colonies for all future tests and for the mating hunt.
The bait strain can be tested by immunoblotting to ensure that the bait protein is expressed (see Basic Protocol 1), by β-galactosidase assay to test for lacZ gene activation (see Basic Protocol 1), and by repression assay to test for synthesis and nuclear localization of the bait protein (see Alternate Protocol 1).If the bait activates the lacZ reporter, a less sensitive lacZ reporter plasmid (Table 17.3.1) or an integrated version of the lacZ reporter should be tried. A bait that strongly activates the lacZ reporters usually cannot be used in a hunt based on selection of interactors with the LEU2 reporter, because the LEU2 reporters are more sensitive than the lacZ reporters. However, both reporters are less sensitive to activation by a bait in diploid cells, as compared to haploid cells. Thus, a more important test of the transactivation potential of a bait is to test the leucine requirement of diploid cells expressing it, as described below.
Prepare the library strain
-
2
Starting with EGY48 bearing no other plasmids and grown in YPD liquid medium, perform a large-scale transformation with library DNA using the lithium acetate method (see Basic Protocol 2, steps 1 to 8). Select library transformants on Glu/CM –Trp plates by incubating 3 days at 30°C.
-
3
Collect primary transformants by scraping plates, washing yeast, and resuspending in 1 pellet vol glycerol solution (see Basic Protocol 2, steps 9 to 12). Freeze 0.2- to 1.0-ml aliquots at –70°C to –80°C.
The cells will be stable for at least 1 year. Refreezing a thawed aliquot will result in loss of viability. Thus, many frozen aliquots should be made, so that each thawed aliquot can be discarded after use.
Prepare the control (library vector) strain
-
4
Transform EGY48 grown in YPD liquid medium with the empty library vector (pJG4-5) using the lithium acetate method (Becker and Lundblad, 1993). Select transformants on a single 100-mm Glu/CM –Trp plate by incubating 3 days at 30°C.
-
5
Pick and combine three transformant colonies and use them to inoculate 30 ml Glu/CM –Trp medium. Incubate 15 to 24 hr at 30° C (to OD600 3).
-
6
Centrifuge 5 min at 1000 to 1500 × g, room temperature, and remove the supernatant. Resuspend in 10 ml sterile water to wash the cells.
-
7
Centrifuge 5 min at 1000 to 1500 × g, room temperature, and remove supernatant. Resuspend in 1 pellet vol glycerol solution and freeze 100-μl aliquots at –70° to –80°C.
Determine plating efficiency of library and control strains
-
8
After freezing (at least 1 hr), thaw an aliquot of the library and control strains at room temperature. Make several serial dilutions in sterile water, including 105-fold, 106-fold, and 107-fold dilutions. Plate 100 μl of each dilution on 100-mm Glu/CM –Trp plates and incubate 2 to 3 days at 30°C.
-
9
Count the colonies and determine the number of colony-forming units (cfu) per aliquot of transformed yeast.
The plating efficiency for a typical library transformation and for the control strain will be ~1 × 109 cfu/ml.
Mate the bait strain with the library and control strains
In the following steps, an interactor hunt is conducted concurrently with testing LEU2 reporter activation by the bait itself. For most baits, this approach will be the quickest way to isolate interactors. For some baits, such as those that have a high transactivation potential or those that affect yeast mating or growth, these steps will serve as a pilot experiment to determine the optimal parameters for a subsequent hunt.
-
10
Grow a 30-ml culture of the bait strain in Glu/CM –Ura –His liquid dropout medium to mid to late log phase (OD600 = 1.0–2.0, or 2–4 × 107cells/ml).
A convenient way to grow the bait strain is to inoculate a 5-ml culture with approximately three colonies from a plate and grow it overnight at 30°C, with shaking. In the morning, measure the OD600, dilute into a 30-ml culture to a final OD600 of 0.2, and grow at 30° C, with shaking. The culture should reach mid to late log phase before the end of the day. -
11
Centrifuge the culture 5 min at 1000 to 1500 × g, room temperature, to harvest the cells. Resuspend the cell pellet in sterile water to make a final volume of 1 ml.
This should correspond to ~1 × 109 cells/ml. -
12
Set up two matings in sterile microcentrifuge tubes by mixing 200 μl of the bait strain with either 200 μl of thawed control strain or ~1 × 108 cfu (~0.1 to 1 ml) of thawed library strain.
The library mating should be set up so that it contains an ~2-fold excess of bait strain cfu over library strain cfu. Because the bait strain was harvested in log phase, most of the cells will be viable (i.e., cells/ml = ~cfu/ml), and the number of cfu can be sufficiently estimated from optical density (1 OD600 = ~1 × 107 cells/ml). Under these conditions, ~ 10% of the cfu in the library strain will mate with the bait strain. Thus, a complete screen of 107 library transformants will require a single mating with at least 108 cfu of library strain and at least 2 × 108 cfu of bait strain.To screen more library transformants, set up additional matings. The number of library transformants to screen depends on the size of the library and the number of primary transformants obtained. If the size of the library is larger than the number of transformants obtained in step 3, the goal will be to screen all of the yeast transformants. In this case, complete screening of the library will require additional transformations of EGY48 and additional interactor hunts. If the size of the library is smaller than the number of transformants obtained in step 3, the goal will be to screen at least a number of transformants equivalent to the size of the library. -
13
Centrifuge each cell mixture for 5 min at 1000 to 1500 × g, pour off the medium, and resuspend the cells in 200 μl YPD medium. Plate each suspension on a 100-mm YPD plate and incubate 12 to 15 hr at 30°C.
-
14
Add ~1 ml of Gal/Raff/CM –Ura –His –Trp dropout medium to the lawns of mated yeast on each plate. Mix the cells into the medium using a sterile applicator stick.
-
15
Transfer each slurry of mated cells to a 500-ml flask containing 100 ml of Gal/Raff/CM –Ura –His –Trp dropout medium. Incubate with shaking 6 hr at room temperature to induce the GAL1 promoter, which drives expression of the cDNA library.
-
16
Centrifuge the cell suspensions 5 min at 1000 to 1500 × g, room temperature, to harvest the cells. Wash by resuspending each pellet in 30 ml sterile water and centrifuging again. Resuspend each pellet in 5 ml sterile water. Measure OD600 and bring to a final concentration of ~1 × 108 cells/ml.
This is a mixture consisting of haploid cells that have not mated and diploid cells. Under a microscope, the two cell types can be distinguished by size (diploids are ~1.7 times bigger than haploids) and shape (diploids are slightly oblong and haploids are spherical). Because diploids grow faster than haploids, this mixture will contain ~10% to 50% diploid cells. The actual number of diploids will be determined by plating dilutions on –Ura –His –Trp medium, which will not support the growth of the parental haploids. -
17
For each mating make a series of 1/10 dilutions in sterile water, at least 200 μl each, to cover a 106-fold concentration range. Plate 100 μl from each tube (undiluted through 10–6 dilution) on 100-mm Gal/Raff/CM –Ura –His –Trp –Leu plates. Also plate 100 μl from the 10–4, 10–5, and 10–6 tubes on 100-mm Gal/Raff/CM –Ura –His –Trp plates. Incubate the plates at 30°C. Count the colonies on each plate after 2 to 5 days.
-
18
Prepare an additional 3 ml of a 10–1 dilution of the mating with the library strain. Plate 100 μl each of undiluted cells and the 10–1 dilution on each of twenty 100-mm Gal/Raff/CM –Ura –His –Trp –Leu plates (total 40 plates) and incubate at 30°C. Pick Leu+ colonies after 2 to 5 days and characterize them beginning with step 17 of Basic Protocol 2.
The number of Leu+ colonies to pick to ensure that all of the library has been screened depends on the transactivation potential of the bait protein itself. The transactivation potential is expressed as the number of Leu+ colonies that grow per cfu (Leu+/cfu) of the bait strain mated with the control strain, as determined in step 17 of this protocol. It can be calculated as the ratio of the number of colonies that grow on Gal/Raff/CM –Ura –His –Trp –Leu to the number of colonies that grow on Gal/Raff/CM –Ura –His –Trp for a given dilution of the mating between the bait strain and the control strain. A bait with essentially no transactivation potential will produce less than 10–6 Leu+/cfu. For a bait to be useful in an interactor hunt, it should not transactivate more than 10–4 Leu+/cfu.To screen all of the library, it will be necessary to pick a sufficient number of Leu+ colonies in addition to background colonies produced by the transactivation potential of the bait itself. Thus, the minimum number of Leu+ colonies that should be picked in step 18 of this protocol is given by:For example, if 107 library transformants were obtained in step 3 (and at least 108 cfu of these transformants were mated with the bait strain in step 12, since only ~10% will form diploids) and the transactivation potential of the bait is 10–4 Leu+/cfu, then at least 103 Leu+ colonies must be picked and characterized. In other words, if the rarest interactor is present in the transformed library at a frequency of 10–7, one needs to screen through at least 107 diploids from a mating of the library strain to find it. However, at least 103 of these 107 diploids would be expected to be Leu+ due to the bait background if the transactivation potential of the bait is 10–4. The true positives will be distinguished from the bait background by the galactose dependence of their Leu+ and lacZ+ phenotypes.
SUPPORT PROTOCOL 1: PREPARATION OF SHEARED SALMON SPERM CARRIER DNA
This protocol generates high-quality sheared salmon sperm DNA for use as carrier in transformation (Basic Protocol 2). This DNA is also suitable for other applications where high-quality carrier DNA is needed (e.g., hybridization). This protocol is based on Schiestl and Gietz (1989). For more details about phenol extraction or other DNA purification methods, consult APPENDIX 1G.
Materials
High-quality salmon sperm DNA (e.g., sodium salt from salmon testes, Sigma or Boehringer Mannheim), desiccated
TE buffer, pH 7.5 (APPENDIX 2A), sterile
TE-saturated buffered phenol (APPENDIX 2A)
1:1 (v/v) buffered phenol/chloroform
Chloroform
3 M sodium acetate, pH 5.2 (APPENDIX 2A)
100% and 70% (v/v) ethanol, ice cold
Magnetic stirring apparatus and stir-bar, 4°C
Sonicator with probe
50-ml conical centrifuge tube
High-speed centrifuge and appropriate tube
100°C and ice-water baths
-
Dissolve desiccated high-quality salmon sperm DNA in TE buffer, pH 7.5, at a concentration of 5 to 10 mg/ml by pipetting up and down in a 10-ml glass pipet. Place in a beaker with a stir-bar and stir overnight at 4°C to obtain a homogenous viscous solution.
It is important to use high-quality salmon sperm DNA. Sigma Type III sodium salt from salmon testes has worked well, as has a comparable grade from Boehringer Mannheim. Generally it is convenient to prepare 20- to 40-ml batches at a time. -
Shear the DNA by sonicating briefly using a large probe inserted into the beaker.
The goal of this step is to generate sheared DNA with an average size of 7 kb, but ranging from 2 to 15 kb. Oversonication (such that the average size is closer to 2 kb) drastically decreases the efficacy of carrier in enhancing transformation. The original version of this protocol (Schiestl and Gietz, 1989) called for two 30-sec pulses at three-quarter power, but optimal conditions vary between sonicators. The first time this protocol is performed, it is worthwhile to sonicate briefly, then test the size of the DNA by running out a small aliquot alongside molecular weight markers on an agarose gel containing ethidium bromide. The DNA can be sonicated further if needed. Once DNA of the appropriate size range has been obtained, extract it with an equal volume of TE-saturated buffered phenol in a 50-ml conical tube, shaking vigorously to mix.
Centrifuge 5 to 10 min at 3000 × g, room temperature, or until clear separation of phases is obtained. Transfer the upper phase containing the DNA to a clean tube.
Repeat the extraction using 1:1 (v/v) buffered phenol/chloroform, then chloroform alone. Transfer the DNA into a tube suitable for high-speed centrifugation.
Precipitate the DNA by adding 1/10 vol of 3 M sodium acetate and 2.5 vol of ice-cold 100% ethanol. Mix by inversion. Centrifuge 15 min at ~12,000 × g, room temperature.
-
Wash the pellet with 70% ethanol. Briefly dry either by air drying or by covering one end of the tube with Parafilm with a few holes poked in and placing the tube under vacuum. Resuspend the DNA in sterile TE buffer at 5 to 10 mg/ml.
Do not overdry the pellet, or it will be very difficult to resuspend. Denature the DNA by boiling 20 min in a 100°C water bath. Then immediately transfer the tube to an ice-water bath.
-
Place aliquots of the DNA in microcentrifuge tubes and store frozen at –20°C. Thaw as needed.
DNA should be boiled again briefly (5 min) immediately before addition to transformations.Before using a new batch in a large-scale library transformation, it is a good idea to perform a small-scale transformation using suitable plasmids to determine the transformation efficiency. Optimally, use of salmon sperm DNA prepared in the manner described will yield transformation frequencies of >105 colonies/μg input plasmid DNA.
SUPPORT PROTOCOL 2: YEAST COLONY HYBRIDIZATION
This protocol is adapted from a modification of the classic protocol of Grunstein and Hogness (1975); also see Kaiser et al. (1994). It is primarily useful when a large number of putative interactors has been obtained, and initial minipreps and restriction digests have indicated that many of them derive from a small number of cDNAs. These cDNAs can then be used as probes to screen and eliminate identical cDNAs from the pool.
Materials
Glu/CM –Trp plates: CM dropout plates –Trp (Treco and Lundblad, 1993) with 2% glucose
Master dropout plate of yeast positive for Gal dependence (see Basic Protocol 2, step 19)
1 M sorbitol/20 mM EDTA/50 mM DTT (prepare fresh) 1 M sorbitol/20 mM EDTA
0.5 M NaOH
0.5 M Tris·Cl (pH 7.5)/6× SSC (APPENDIX 2A)
2 × SSC (APPENDIX 2A)
100,000 U/ml β-glucuronidase (type HP-2 crude solution from Helix pomatia; Sigma)
82-mm circular nylon membrane, sterile Whatman 3 MM paper
80 °C vacuum oven or UV cross-linker
Additional reagents and equipment for bacterial filter hybridization (Duby et al., 1993; Strauss, 1993)
-
Place a sterile nylon membrane onto a Glu/CM –Trp dropout plate. From the master dropout plate of Gal-dependent positives, gently restreak positives to be screened onto the membrane and mark the membrane to facilitate future identification of hybridizing colonies. Grow overnight (~12 hr) at 30°C.
Growth for extended periods of time (i.e., 24 hr) may result in difficulty in obtaining good lysis. It is a good idea to streak positive and negative controls for the cDNAs to be hybridized on the membrane. -
Remove the membrane from the plate. Air dry briefly. Incubate ~30 min on a sheet of Whatman 3 MM paper saturated with 1 M sorbitol/20 mM EDTA/50 mM DTT.
Optionally, before commencing chemical lysis, membranes can be placed at –70°C for 5 min, then thawed at room temperature for one or more cycles to enhance cell wall breakage. -
Cut a piece of Whatman 3 MM paper to fit inside a 100-mm petri dish. Place the paper disc in a dish and saturate it with 200 U/ml β-glucuronidase in 1 M sorbitol/20 mM EDTA (diluted 1:500 from 100,000 U/ml stock). Layer the nylon membrane on the 3 MM paper, cover the dish, and incubate up to 6 hr at 37°C until >80% of the cells lack a cell wall.
The extent of cell wall removal can be determined by removing a small quantity of cells from the filter to a drop of 1 M sorbitol/20 mM EDTA on a microscope slide and observing directly with a phase-contrast microscope at ≥60× magnification. Cells lacking a cell wall are nonrefractile. Place the membrane on Whatman 3 MM paper saturated with 0.5 M NaOH for ~8 to 10 min.
Place the membrane on Whatman 3 MM paper saturated with 0.5 M Tris·Cl (pH 7.5)/6× SSC for 5 min. Repeat with a second sheet of Whatman 3 MM paper.
Place the membrane on Whatman 3 MM paper saturated with 2× SSC for 5 min. Then place the membrane on dry Whatman paper to air dry for 10 min.
Bake the membrane 90 min at 80°C in a vacuum oven or UV cross-link.
-
Process as for bacterial filter hybridization (Duby et al., 1993; Strauss, 1993), hybridizing the membrane with probes complementary to previously isolated cDNAs.
When selecting probes, either random-primed cDNAs or oligonucleotides complementary to the cDNA sequence may be used. If the cDNA is a member of a protein family, it may be advantageous to use oligonucleotides to avoid inadvertently excluding genes related but not identical to those initially obtained.
SUPPORT PROTOCOL 3: MICROPLATE PLASMID RESCUE
In some cases, it is desirable to isolate plasmids from a large number of positive colonies (Basic Protocol 2, steps 19 and 20). The protocol described below is a batch DNA preparation protocol developed by Steve Kron (University of Chicago) as a scale-up of a basic method developed by Manuel Claros (Laboratoire de Génétique Moleculaire, Paris).
Materials
2× Glu/CM –Trp liquid medium: 2× CM –Trp liquid medium (Treco and Lundblad, 1993) with 4% glucose
Master plate of Gal-dependent yeast colonies (see Basic Protocol 2, step 18)
Rescue buffer: 50 mM Tris·Cl (pH 7.5)/10 mM EDTA/0.3% (v/v) 2-mercaptoethanol (prepare fresh)
Lysis solution: 2 to 5 mg/ml Zymolyase 100T/rescue buffer or 100,000 U/ml β-glucuronidase (type HP-2 crude solution from Helix pomatia; Sigma) diluted 1:50 in rescue buffer
10% (w/v) SDS
7.5 M ammonium acetate (APPENDIX 2A)
Isopropanol
70% (v/v) ethanol
TE buffer, pH 8.0 (APPENDIX 2A)
24-well microtiter plates
Centrifuge with rotor adapted for microtiter plates, refrigerated
Repeating micropipettor
37°C rotary shaker
Grow yeast cultures
-
1
Aliquot 2 ml of 2× Glu/CM –Trp medium into each well of a 24-well microtiter plate. Into each well, pick a putative positive colony. Grow overnight with shaking at 30°C.
The 2× minimal medium is used to maximize the yield of yeast. Four plates can generally be handled conveniently at once, based on the number that can be centrifuged simultaneously. -
2
Centrifuge 5 min at 1500 × g, 4°C. Shake off the supernatant with a snap and return the plate to upright.
-
3
Swirl or lightly vortex the plate to resuspend the cell pellets in remaining liquid. Add 1 ml water to each well and swirl lightly.
Cell pellets can most easily be resuspended in residual liquid before adding new solutions. Addition of liquid can be accomplished using a repeating pipettor. -
4
Centrifuge 5 min at 1500 × g, 4°C. Shake off the supernatant and resuspend the pellet. Add 1 ml rescue buffer.
-
5
Centrifuge 5 min at 1500 × g, 4°C. Shake off the supernatant and resuspend the pellet in the small volume of liquid remaining in the plate.
Lyse cells
-
6
To each well, add 25 μl lysis solution. Swirl or vortex to mix. Incubate (with cover on) on a rotary shaker ~1 hr at 37°C.
Lysis solution need not be completely dissolved before use. By 1 hr, lysis should be obvious as coagulation of yeast into a white precipitate.Susceptibility of yeast strains to lytic enzymes varies. If lysis occurs rapidly, then less lytic enzyme should be used. If the lysis step is allowed to go too far, too much of the partially dissolved cell wall may contaminate the final material. Lysis can be judged by examining cells with a phase-contrast microscope. Living cells are white with a dark halo and dead cells are uniformly gray. Lysis leads to release of granular cell contents into the medium. Once cells are mostly gray and many are disrupted, much of the plasmid should have been released. -
7
To each well, add 25 μl of 10% SDS. Mix gently by swirling to completely disperse the precipitates. Allow the plates to sit 1 min at room temperature.
At this point, the wells should contain a clear, somewhat viscous solution.
Purify plasmid
-
8
To each well, add 100 μl of 7.5 M ammonium acetate. Swirl gently, then incubate 15 min at –70°C or –20°C until frozen.
Addition of acetate should result in the formation of a massive white precipitate of cell debris and SDS. The freezing step appears to improve removal of inhibitors of E. coli transformation. -
9
Remove the plate from the freezer. Once it begins to thaw, centrifuge 15 min at 3000 × g, 4°C. Transfer 100 to 150 μl of the resulting clear supernatants to clean 24-well plates.
In general, some contamination of the supernatant with pelleted material cannot be avoided. However, it is better to sacrifice yield in order to maintain purity. -
10
To each well, add ~0.7 vol isopropanol. Mix by swirling and allow to precipitate 2 min at room temperature.
A cloudy fine precipitate should form immediately after the isopropanol is added. -
11
Centrifuge 15 min at 3000 × g, 4°C. Shake off the supernatant with a snap.
-
12
To each well, add ~1 ml cold 70% ethanol, mix by swirling, and centrifuge 5 min at 3000 × g, 4°C. Shake off the supernatant with a snap, invert the plates, and blot well onto a paper towel. Allow the plates to air dry.
-
13
To each well, add 100 μl TE buffer. Swirl well and allow to rest on bench several minutes, until the pellets appear fully dissolved. Transfer to microcentrifuge tubes or 96-well plates for storage at –20°C.
One to five microliters of each of the resulting preparations can be used to transform competent E. coli. Sometimes, the yield of transformants is low if E. coli carrying plasmids are not permitted time to increase the plasmid copy number above a critical threshold before the cells are placed on selective medium. Allow plenty of time for cells to express antibiotic resistance or the TRP1 gene before plating.If insufficient numbers of colonies are obtained by this approach, the final plasmid preparation can be resuspended in 20 μl instead of 100 μl TE buffer to concentrate the DNA stock.
REAGENTS AND SOLUTIONS
Use deionized, distilled water in all recipes and protocol steps. For common stock solutions, see APPENDIX 2A; for suppliers, see SUPPLIERS APPENDIX.
Glycerol solution
65% (v/v) glycerol, sterile
0.1 M MgSO4
25 mM Tris·Cl, pH 8.0 (APPENDIX 2A)
Store up to 1 year at room temperature
Laemmli sample buffer, 2×
10% (v/v) 2-mercaptoethanol (2-ME)
6% (w/v) SDS
20% (v/v) glycerol
0.2 mg/ml bromphenol blue
0.025× Laemmli stacking buffer (see recipe; optional)
-
Store up to 2 months at room temperature
This reagent can conveniently be prepared 10 ml at a time.
Laemmli stacking buffer, 2.5×
0.3 M Tris·Cl, pH 6.8 (APPENDIX 2A)
0.25% (w/v) SDS
Store up to 1 month at 4°C
Lysis solution
50 mM Tris·Cl, pH 7.5 (APPENDIX 2A)
10 mM EDTA
0.3% (v/v) 2-mercaptoethanol (2-ME), added just before use
2% (v/v) β-glucuronidase from Helix pomatia (Type HP-2; Sigma), added just before use
COMMENTARY
Background Information
Interaction-based cloning is derived from three experimental observations. In the first, Brent and Ptashne (1985) demonstrated that it was possible to assemble a novel, functional transcriptional activator by fusing the DNA-binding domain from one protein, LexA, to the activation domain from a second protein, GAL4. This allowed the use of a single reporter system containing a single DNA-binding motif, the LexA operator, to study transcriptional activation by any protein of interest. In the second, Ma and Ptashne (1988) built on this work to demonstrate that the activation domain could be brought to DNA by interaction with a DNA-binding domain. In the third, Fields and Song (1989), working independently of Ma and Ptashne, used two yeast proteins, SNF1 and SNF4, to make an SNF1 fusion to the DNA-binding domain of GAL4 and an SNF4 fusion to the GAL4 activation domain. They demonstrated that the strength of the SNF1-SNF4 interaction was sufficient to allow activation through a GAL4 DNA-binding site. This suggested the feasibility of selecting interacting proteins by performing screens of cDNA libraries made so that library-encoded proteins carried activating domains.
Several groups have developed cDNA library strategies along these lines, using a variety of DNA-binding and activation domains (see Table 17.3.4). The most frequently used DNA-binding domains, LexA and GAL4, each have different properties that should be considered when selecting a system. LexA is derived from a heterologous organism, has no known effect on the growth of yeast, possesses no residual transcriptional activity, can be used in GAL4+ yeast, and can be used with a Gal-inducible promoter. Because GAL4 is an important yeast transcriptional activator, it has the disadvantage that experiments must be performed in GAL4— yeast strains to avoid background due to activation of the reporter system by endogenous GAL4. Such GAL4— strains are frequently less healthy and more difficult to transform than wild-type strains, and either libraries must be constitutively expressed or alternate inducible systems must be used. By contrast, the GAL4 DNA-binding domain may be more efficiently localized to the nucleus and may be preferred for some proteins (for a review of GAL4-based systems, see Bartel et al., 1993). Whichever system is used, it is important to remember that the bait protein constitutes a novel fusion protein whose properties may not exactly parallel those of the original unfused protein of interest. Although systems using the two-hybrid paradigm have been developed in mammalian and bacterial cells, these have not been used extensively in library screens. It seems likely that the organism of choice for two-hybrid identification of novel partner proteins will remain yeast.
Table 17.3.4.
Two-Hybrid System Variants
| Systema | DNA-binding domain | Activation domain | Selection | Originating laboratoryb |
|---|---|---|---|---|
| Two-hybrid | GAL4 | GAL4 | lacZ, HIS3 | S. Fields |
| Improved two-hybrid | GAL4 | GAL4 | HIS3, lacZ | S. Elledge |
| ProQuest | GAL4 | GAL4 | HIS3, URA3, lacZ | Invitrogen |
| Matchmaker System 3 | GAL4 | GAL4 | ADE2, HIS3, lacZ, MEL1 | Clontech |
| Interaction trap | LexA | B42 | LEU2, lacZ | R. Brent |
| Modified two-hybrid | LexA | VP16 | HIS3, lacZ | S. Hollenberg |
| Grow‘n’Glow | LexA | B42 | LEU2, GFP | S. Cormack |
| Dual bait | LexA/cI | B42 | LEU2, lacZ / LYS2, GusA | E. Golemis |
| Two-bait interaction trap | LexA/TetR | B42 | lacZ/URA3 | R. Brent |
| Grow ‘n’ Glow ACE1 | Ace1 | Ace1 | CUP1, GFP | T. Munder |
Only key systems designed to detect protein-protein interactions are listed. A variety of specialized two-hybrid systems, including systems designed to work with self-activating and membrane proteins, are described in a recent review (Izumchenko et al., 2007).
For references see Izumchenko et al. (2007).
cDNAs that pass specificity tests are referred to as positives or “true positives.” In interactor hunts conducted to date, anywhere from zero to practically all isolated plasmids passed the final specificity test. If no positives are obtained, the tissue source for the library originally used may not be appropriate, and a different library may produce better results. However, there are some proteins for which no positives are found. Various explanations for this are provided below. Conversely, some library-encoded proteins are known to be isolated repeatedly using a series of unrelated baits, and these proteins demonstrate at least some specificity. One of these, heat shock protein 70, might be explained by positing that it assists the folding of some LexA-fused bait proteins, or that these bait proteins are not normally folded. This example illustrates the point that the physiological relevance of even quite specific interactions may sometimes be obscure.
Because the screen involves plating multiple cells to Gal/CM –Ura –His –Trp –Leu dropout medium for each primary transformant obtained, multiple reisolates of true positive cDNAs are frequently obtained. If a large number of specific positives are obtained, it is generally a good idea to attempt to sort them into classes—for example, digesting minipreps of positives with EcoRI, XhoI, and HaeIII will generate a fingerprint of sufficient resolution to determine whether multiple reisolates of a small number of clones or single isolates of many different clones have been obtained. The former situation is a good indication that the system is working well.
An important issue that arises in an inter-actor hunt is the question of how biologically relevant interacting proteins that are isolated are likely to be. This leads directly to the question of what Kd of association two molecules must have to be detected by an interactor hunt. In fact, this is not at all a simple issue. For the system described here, most fusion proteins appear to be expressed at levels ranging from 50 nM to 1 μM (Golemis and Brent, 1992). Given the strength of the GAL promoter, it is likely that many library-encoded proteins are expressed at similarly high levels (≥1 μM) in the nucleus (Golemis and Brent, 1992). At this concentration, which is in considerable excess over the nuclear concentration of operator-bound bait protein, a library-encoded protein should half-maximally occupy the DNA-bound bait protein if it possesses a Kd of 10–6 M, making it theoretically possible that very-low-affinity interactions could be detected. Such interactions have been observed in some cases. In contrast, some interactions that have been previously established using other methods, and which are predicted by known Kd to be easily detected by these means, are not detected or are detected only weakly (Finley and Brent, 1994; Estojak et al., 1995). Because of the conservation of many proteins between lower and higher eukaryotes, one explanation for this observation is that either one or both of the partners being tested is being sequestered from the desired interaction by fortuitous association with an endogenous yeast protein. A reasonably complete investigation of the degree of correlation between in vitro determinations of interaction affinity and apparent strength of interaction in the interaction trap is included in Estojak et al. (1995). The result of this investigation suggests it is important to measure the affinity of detected interactions under different conditions, using a second assay system, rather than to draw conclusions about affinity based on detection in the interaction trap.
A number of different plasmids can be used for conducting an interactor hunt. Their properties are summarized in Table 17.3.1. Because of the generous and open scientific exchange between investigators using the system, the number of available plasmids and other components has greatly expanded since the appearance of the initial two-hybrid reagents, facilitating the study of proteins inaccessible by the original system.
The original parent plasmid for generating LexA fusions, pEG202, is a derivative of 202 + PL (Ruden et al., 1991; see Fig. 17.3.3) that contains an expanded polylinker region. The available cloning sites in pEG202 include EcoRI, BamHI, SalI, NcoI, NotI, and XhoI, with the reading frame as described in the legend to Figure 17.3.3. Since the original presentation of this system, a number of groups have developed variants of this plasmid that address specialized research needs. Several currently available plasmids are listed in Table 17.3.1, along with purposes for which they are suited. pGilda, created by David A. Shaywitz, places the LexA-fusion cassette under the control of the inducible GAL1 promoter, allowing expression of the bait protein for limited times during library screening, reducing the exposure of yeast to toxic baits. pJK202, created by Joanne Kamens, adds nuclear localization sequences to pEG202, facilitating assay of the function of proteins lacking internal nuclear localization sequences. pNLexA, created by Ian York, places LexA carboxy-terminal in the fusion domain, allowing assay of interactions that require an unblocked amino-terminus on the bait protein. pEE202I, created by Mike Watson and Rich Buckholz, allows chromosomal integration of a pEG202-like bait, thus reducing expression levels so they are more physiological for bait proteins normally present at low levels intracellularly. All of these have been extensively tested by numerous researchers. pGilda, pJK202, and pEE202I work with complete reliability. pNLexA works effectively with ~50% of the fusion domains tried, but it synthesizes only very low levels of protein (relative to expression of the same fusion domain as a pEG202 fusion) with the remaining 50%. Attachment of fusion domains amino-terminal either to LexA or GAL4 has been generally problematic in the hands of many investigators; it may be that appending additional protein sequences to the amino termini of these proteins is destabilizing, although the problem has not been rigorously investigated.
A series of lacZ reporters of differing sensitivity to transcriptional activation can be used to detect interactions of varying affinity (see Table 17.3.1). These plasmids are LexA operator–containing derivatives of the plasmid LR1Δ1 (West et al., 1984). In LR1Δ1, a minimal GAL1 promoter lacking the GAL1 upstream activating sequences (GALUAS) is located upstream of the bacterial lacZ gene. In pSH18-34, eight LexA operators have been cloned into an XhoI site located 167 bp upstream of the lacZ gene (S. Hanes, unpub. observ.). pJK103 and pRB1840 contain two and one operators, respectively.
pJK101 is similar to pSH18-34, except that it contains GALUAS upstream of two LexA operator sites. A derivative of del20B (West et al., 1984), it is used in the repression assay (Brent and Ptashne, 1984; see Fig. 17.3.5) to assess LexA fusion binding to the operator.
pSH17-4 is a HIS3 2 μm plasmid that encodes LexA fused to the activation domain of the yeast activator GAL4. EGY48 cells bearing this plasmid will produce colonies in overnight growth on medium lacking Leu, and yeast that additionally contain pSH18-34 will turn deep blue on plates containing Xgal. This plasmid serves as a positive control for the activation of transcription.
pRFHM1 is a HIS3 2 μm plasmid that encodes LexA fused to the N-terminus of the Drosophila protein bicoid. The plasmid has no ability to activate transcription, so EGY48 cells that contain pRFHM1 and pSH18-34 do not grow on –Leu medium and remain white on plates containing Xgal. pRFHM1 is a good control for specificity testing, because it has been demonstrated to be sticky—that is, to associate with a number of library-encoded proteins that are clearly nonphysiological interactors (R. Finley, unpub. observ.).
This protocol uses interaction libraries (Table 17.3.3) made in pJG4-5 or its derivatives (see Fig. 17.3.6). pJG4-5 was developed to facilitate isolation and characterization of novel proteins in interactor hunts (Gyuris et al., 1993). The pJG4-5 cDNA library expression cassette is under control of the GAL1 promoter, so library proteins are expressed in the presence of galactose (Gal) but not glucose (Glu). This conditional expression has a number of advantages, the most important of which is that many false-positives obtained in screens can be easily eliminated because they do not demonstrate a Gal-dependent phenotype. The expression cassette consists of an ATG to start translation, a nuclear localization signal to extend the interaction trap’s range to include proteins that are normally predominantly localized in the cytoplasm, an activation domain (acid blob; Ma and Ptashne, 1987), the hemagglutinin epitope tag to permit rapid assessment of the size of encoded proteins, EcoRI-XhoI sites designed to receive directionally synthesized cDNAs, and the alcohol dehydrogenase (ADH) termination sequences to enhance the production of high levels of library protein. The plasmid also contains the TRP1 auxotrophy marker and 2-μm origin for propagation in yeast. A derivative plasmid, pJG4-5I, was created by Mike Watson and Richard Buckholz to facilitate chromosomal integration of the activation domain fusion expression plasmid.
A series of derivatives of pEG202, pJG4-5, and lacZ reporter plasmids (MW101 to MW112) alter the antibiotic resistance markers on these plasmids from ampicillin (Apr) to either kanamycin (Kmr) or chloramphenicol (Cmr; Watson et al., 1996). Judiciously mixing and matching these plasmids in conjunction with Apr libraries would considerably reduce work subsequent to library screening if the isolation of the actual library plasmid is required (as opposed to using PCR product).
EGY48 and EGY191 (see Table 17.3.2) are both derivatives of the strain U457 (a gift of Rodney Rothstein, Columbia University, New York) in which the endogenous LEU2 gene has been replaced by homologous recombination with LEU2 reporters carrying varying numbers of LexA operators, using a procedure detailed in Estojak et al. (1995).
A number of groups have adapted basic two-hybrid strategies to more specialized applications, and they have devised strategies to broaden their basic functionality. Interaction mating (Finley and Brent, 1994) has been used to establish extended networks of targeted protein-protein interaction. In this approach, a panel of LexA-fused proteins are transformed into a MATa haploid selective strain (such as RFY206), a panel of activation-domain-fused proteins are transformed into a suitable MATα haploid (such as EG448), and the two panels are cross-gridded against each other for mating. Selected diploids are then screened by replica plating to selective medium. This approach complements library screening in large-scale applications, such as proposed definition of interaction maps for entire genomes (Bartel et al., 1996).
Interaction mating has also provided the basis for an alternative two-hybrid hunt protocol (see Alternate Protocol 2), useful in cases when a single library will be screened with different baits. In this approach (Bendixen et al., 1994; Finley and Brent, 1994: Kolonin and Finley, 1998), a library is introduced into a single strain, like EGY48, and aliquots are stored frozen. To conduct a hunt, an aliquot is thawed and mated with a strain expressing a bait. This allows one to avoid repeated high-efficiency transformations, since a single library transformation can provide enough transformed yeast to conduct dozens of inter-actor hunts. Moreover, some yeast strains pre-transformed with libraries are becoming commercially available, which may eliminate altogether the need to conduct a high-efficiency library transformation for some researchers. In an attempt to develop a two-hybrid system variant with extended screening capacity and greater internal controls, the Dual Bait system was developed (Serebriiskii et al., 1999). It can be used to simultaneously analyze the interaction of two distinct baits with the same interactive partner (Kotova et al., 2009).
Two-hybrid approaches have been shown to be effective in identifying small peptides with biological activities on selected baits (Yang et al., 1995; Colas et al., 1996), which may prove to be useful as a guide to targeted drug design. Rapid screening protocols have been devised using custom-synthesized libraries expressing sheared plasmid DNA to facilitate rapid mapping of interaction interfaces (Stagljar et al., 1996). Osborne and coworkers have demonstrated the effectiveness of a tribrid (or tri-hybrid) approach, in which an additional plasmid expresses a tyrosine kinase to specifically modify a bait protein, allowing detection of SH2-domain-containing partner proteins that recognize specific phosphotyrosine residues (Osborne et al., 1995). A variety of more elaborate tribrid approaches, in which a DNA-binding domain fused protein is used to present an intermediate nonprotein compound for interaction with a library, have been developed and proven effective. These approaches have allowed the identification of proteins binding specific drug ligands (Chiu et al., 1994; Licitra and Liu, 1996), as well as the identification of proteins binding to RNA sequences (SenGupta et al., 1996; Wang et al., 1996). For a recent review describing a variety of specialized two-hybrid applications (including systems designed to work with self-activating and membrane proteins), see Izumchenko et al. (2007). It is expected that the range of utility of these systems will continue to expand.
Critical Parameters and Troubleshooting
To maximize chances of a successful inter-actor hunt, a number of parameters should be taken into account. Before attempting a screen, bait proteins should be carefully tested to ensure that they have little or no intrinsic ability to activate transcription. Bait proteins must be expressed at reasonably high levels and must be able to enter the yeast nucleus and bind DNA (as confirmed by the repression assay). Optimally, integrity and levels of bait proteins should be confirmed by immunoblot analysis, using an antibody to either LexA or the fused domain. In particular, bait proteins that have extensive transmembrane domains or are normally excluded from the nucleus are not likely to be productively used in a library screen. Proteins that are moderate to strong activators will need to be truncated to remove activating domains before they can be used.
If a protein neither activates nor represses, the most likely reason is that it is not being made. This can be determined by immunoblot analysis of a crude lysate protein extract of EGY48 containing the plasmid, using anti-LexA antibodies as primary antiserum (Gallagher et al., 2008; Samson et al., 1989). If the full protein is not made, it may be possible to express truncated derivatives of the protein. If the protein is made, but still does not repress, it may not enter the yeast nucleus effectively, although this appears to be a relatively rare problem. In this case, introducing the coding sequence for the fused moieity into a LexA fusion vector containing a nuclear localization motif (e.g., pJK202; Table 17.3.1) may solve the problem.
The test for the leucine (Leu) requirement is extremely important to determine whether the bait protein is likely to yield an unworkably high background. The LEU2 reporter in EGY48 is more sensitive than the pSH18-34 reporter for some baits (Estojak et al., 1995). Therefore, it is possible that a bait protein demonstrating little or no signal in a β-galactosidase assay may nevertheless permit some growth on –Leu medium. If this occurs, there are several options. First, a less sensitive strain can be used, as described in the text. Second, background can sometimes be reduced further by making the EGY strain diploid (e.g., D. Krainc, R. Finley, and R. Brent, unpub. observ.) or by performing the hunt by interaction mating as described in Alternate Protocol 2. A third option is to attempt to truncate the bait protein to remove activating function. In general, it is useful to extrapolate from the number of cells that grow on –Leu medium to the number that would be obtained in an actual library screen, and determine if this is a background level that can be tolerated. For example, if two colonies arise from 100,000 plated cells on –Leu medium, 200 to 400 would be expected in an actual screen of 106 cDNAs. Although this is a high initial number of positives, the vast majority should be eliminated immediately through easily performed controls. This is a judgment call. Finally, very rarely a bait that appears to be well behaved and negative for transcriptional activation through all characterization steps will suddenly develop a very high background of transcriptional activation following library transformation. The reason for this is currently obscure, and no means of addressing this problem has as yet been found; such baits are hence inappropriate for use in screens.
The protocols described in this unit use initial screening with the most sensitive reporters followed by substitution with less sensitive reporters if activation is detected. An obvious question is, why not start out working with extremely stringent reporters and know immediately whether the system is workable? In fact, some researchers routinely use a combination of pJK103 or pRB1840 with EGY191, and obtain proteins that to date appear to be biologically relevant partners from library screens. However, extensive comparison studies using interactors of defined in vitro affinity with different combinations of lacZ and LEU2 reporters (Estojak et al., 1995) have indicated that although the most sensitive reporters (pSH18-34) may in some cases be prone to background problems, the most stringent reporters (EGY191, pRB1840) may miss some interactions that certainly are biologically relevant and occur inside cells. In the end, the choice of reporters devolves to the preference of individual investigators; the bias of the authors is to cast a broad net in the early stages of a screen, and hence to use more sensitive reporters when practicable.
It is important to move expeditiously through characterization steps and to handle yeast transformed with bait plasmids with care. In cases where yeasts have been maintained on plates for extended periods (e.g., 4 days at room temperature or >2 to 3 weeks at 4°C), unexpected problems may crop up in subsequent library screens.
The transformation protocol is a version of the lithium acetate transformation protocol described by Schiestl and Gietz (1989) and Gietz et al. (1992); also see Becker and Lundblad (1993) that maximizes transformation efficiency in Saccharomyces cerevisiae and produces up to 105 colonies/μg plasmid DNA. In contrast to Escherichia coli, the maximum efficiency of transformation for S. cerevisiae is ~104 to 105/μg input DNA. It is extremely important to optimize transformation conditions before attempting an interactor hunt. Perform small-scale pilot transformations to ensure this efficiency is attained and to avoid having to use prohibitive quantities of library DNA. In addition, as for any effort of this type, it is a good idea to obtain or construct a library from a tissue source in which the bait protein is known to be biologically relevant.
In practice, the majority of proteins isolated by interaction with a LexA fusion turn out to be specific for the fused domain; a smaller number are nonspecifically sticky, and to date there appears to have been only one isolation from a eukaryotic library of a protein specific for LexA. However, it is generally informative to retest positive clones on more than one LexA bait protein. Ideally, library-derived clones should be tested against the LexA fusion used for their isolation, several LexA fusions to proteins that are clearly unrelated to the original fusion, and, if possible, several LexA fusions to proteins related to the initial protein (e.g., if the initial probe was LexA-Fos, a good related set would include LexA-Jun and LexA-GCN4).
Colony selection for master plate production is one of the more variable parts of the procedure. For strong interactors, colonies will grow up in 2 days. However, if plates are left at 30°C, new colonies will continue to appear every day. Those that appear rapidly are most likely to reflect interactors that are biologically relevant to the bait protein. Those that appear later may or may not be relevant. However, many parameters can delay the time of colony formation of cells that contain valid interactions, including the strength of the interaction and the level of expression of the library-encoded protein.
Anticipated Results
Depending on the protein used as bait, anywhere from zero to hundreds of specific interactors will be obtained from 106 primary transformants.
Time Considerations
If all goes well, once the required constructions have been made it will take ~1 week to perform yeast transformations, obtain colonies, and determine whether bait proteins are appropriate. It will take a second week to perform library transformations, re-plate to selective medium, and obtain putative positives. A third week will be required to obtain PCR fragments from the yeast, transform fresh yeast, and confirm specificity.
Footnotes
Key Reference
Gyuris et al., 1993. See above. Initial description of interaction trap system.
Internet Resources
http://cmmg.biosci.wayne.edu/rfinley/lab.htmlSource of two-hybrid information, protocols, and links.
http://www.origene.comCommercial source for basic plasmids, strains, and libraries for interaction trap experiments.
brent@molsci.org
EA Golemis@fccc.edu
Contacts for sources of interaction trap plasmids for specialized interactions.
http://www.fccc.edu:80/research/labs/golemis/InteractionTrapInWork.htmlDatabase for false positive proteins detected in interaction trap experiments; analysis of two-hybrid usage.
Literature Cited
- Bartel PL, Chien C-T, Sternglanz R, Fields S. Using the two-hybrid system to detect protein-protein interactions. In: Hartley DA, editor. Cellular Interactions in Development: A Practical Approach. Oxford University Press; Oxford: 1993. pp. 153–179. [Google Scholar]
- Bartel PL, Roecklein JA, SenGupta D, Fields S. A protein linkage map of Escherichia coli bacteriophage T7. Nature Genet. 1996;12:72–77. doi: 10.1038/ng0196-72. [DOI] [PubMed] [Google Scholar]
- Becker DM, Lundblad V. Introduction of DNA into yeast cells. Curr Protoc Mol Biol. 1993;27:13. 7.1–13.7.10. doi: 10.1002/0471142727.mb1307s27. [DOI] [PubMed] [Google Scholar]
- Bendixen C, Gangloff S, Rothstein R. A yeast mating-selection scheme for detection of protein-protein interactions. Nucleic Acids Res. 1994;22:1778–1779. doi: 10.1093/nar/22.9.1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brent R, Ptashne M. A bacterial repressor protein or a yeast transcriptional terminator can block upstream activation of a yeast gene. Nature. 1984;312:612–615. doi: 10.1038/312612a0. [DOI] [PubMed] [Google Scholar]
- Brent R, Ptashne M. A eukaryotic transcriptional activator bearing the DNA specificity of a prokaryotic repressor. Cell. 1985;43:729–736. doi: 10.1016/0092-8674(85)90246-6. [DOI] [PubMed] [Google Scholar]
- Chiu MI, Katz H, Berlin V. RAPT1, a mammalian homolog of yeast Tor, interacts with the FKBP12/rapamycin complex. Proc Natl Acad Sci USA. 1994;91:12574–12578. doi: 10.1073/pnas.91.26.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colas P, Cohen B, Jessen T, Grishina I, McCoy J, Brent R. Genetic selection of peptide aptamers that recognize and inhibit cyclin-dependent kinase 2. Nature. 1996;380:548–550. doi: 10.1038/380548a0. [DOI] [PubMed] [Google Scholar]
- Duby A, Jacobs KA, Celeste A. Using synthetic oligonucleotides as probes. Curr Protoc Mol Biol. 1993;2:6.4.1–6.4.10. doi: 10.1002/0471142727.mb0604s09. [DOI] [PubMed] [Google Scholar]
- Estojak J, Brent R, Golemis EA. Correlation of two-hybrid affinity data with in vitro measurements. Mol Cell Biol. 1995;15:5820–5829. doi: 10.1128/mcb.15.10.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbing K, Brent R. Media preparation and bacteriological tools. Curr Protoc Mol Biol. 2002;59:1.1.1–1.1.7. doi: 10.1002/0471142727.mb0101s59. [DOI] [PubMed] [Google Scholar]
- Fields S, Song O. A novel genetic system to detect protein-protein interaction. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Finley RL, Jr, Brent R. Interaction mating reveals binary and ternary connections between Drosophila cell cycle regulators. Proc Natl Acad Sci USA. 1994;91:12980–12984. doi: 10.1073/pnas.91.26.12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher S. One-dimensional SDS gel electrophoresis of proteins. Curr Protoc Mol Biol. 2006;75:10. 2A.1–10.2A.37. doi: 10.1002/0471142727.mb1002as75. [DOI] [PubMed] [Google Scholar]
- Gallagher S, Winston SE, Fuller SA, Hurrell JGR. Immunoblotting and immunodetection. Curr Protoc Mol Biol. 2008;83:10. 8.1–10.8.28. doi: 10.1002/0471142727.mb1008s83. [DOI] [PubMed] [Google Scholar]
- Gietz D, St Jean A, Woods RA, Schiestl RH. Improved method for high-efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golemis EA, Brent R. Fused protein domains inhibit DNA binding by LexA. Mol Cell Biol. 1992;12:3006–3014. doi: 10.1128/mcb.12.7.3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunstein M, Hogness DS. Colony hybridization: A method for the isolation of cloned DNAs that contain a specific gene. Proc Natl Acad Sci USA. 1975;72:3961–3965. doi: 10.1073/pnas.72.10.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyuris J, Golemis EA, Chertkov H, Brent R. Cdi1, a human G1- and S-phase protein phosphatase that associates with Cdk2. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- Izumchenko E, Wolfson M, Golemis EA, Serebriiskii IG. Yeast hybrid approaches. In: Stansfield I, Stark M, editors. Yeast Gene Analysis. Elsevier Ltd; London: 2007. pp. 103–137. [Google Scholar]
- Kaiser C, Michaelis S, Mitchell A. Methods in Yeast Genetics, a Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 1994. pp. 135–136. [Google Scholar]
- Kolonin MG, Finley RL., Jr Targeting cycling-dependent kinases in Drosophilia with peptide aptamers. Proc Natl Acad Sci USA. 1998;95:14266–14271. doi: 10.1073/pnas.95.24.14266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotova E, Coleman T, Serebriiskii I. Two-hybrid dual bait system. Curr Protoc Mol Biol. 2009;86:20. 7.1–20.7.32. doi: 10.1002/0471142727.mb2007s86. [DOI] [PubMed] [Google Scholar]
- Kramer MF, Coen DM. Enzymatic amplification of DNA by PCR: Standard procedures and optimization. Curr Protoc Mol Biol. 2001;56:15. 1.1–15.1.14. doi: 10.1002/0471142727.mb1501s56. [DOI] [PubMed] [Google Scholar]
- Licitra EJ, Liu JO. A three-hybrid system for detecting small ligand-protein receptor interactions. Proc Natl Acad Sci USA. 1996;93:12817–12821. doi: 10.1073/pnas.93.23.12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad V, Zhou H. Manipulation of plasmids from yeast cells. Curr Protoc Mol Biol. 1997;39:13. 9.1–13.9.6. doi: 10.1002/0471142727.mb1309s39. [DOI] [PubMed] [Google Scholar]
- Ma J, Ptashne M. A new class of yeast transcriptional activators. Cell. 1987;51:113–119. doi: 10.1016/0092-8674(87)90015-8. [DOI] [PubMed] [Google Scholar]
- Ma J, Ptashne M. Converting an eukaryotic transcriptional inhibitor into an activator. Cell. 1988;55:443–446. doi: 10.1016/0092-8674(88)90030-x. [DOI] [PubMed] [Google Scholar]
- Osborne M, Dalton S, Kochan JP. The yeast tribrid system: Genetic detection of trans-phosphorylated ITAM-SH2 interactions. Bio/Technology. 1995;13:1474–1478. doi: 10.1038/nbt1295-1474. [DOI] [PubMed] [Google Scholar]
- Reynolds A, Lundblad V, Dorris D, Keaveney M. Yeast vectors and assays for expression of cloned genes. Curr Protoc Mol Biol. 1997;39:13. 6.1–13.6.6. doi: 10.1002/0471142727.mb1306s39. [DOI] [PubMed] [Google Scholar]
- Ruden DM, Ma J, Li Y, Wood K, Ptashne M. Generating yeast transcriptional activators containing no yeast protein sequences. Nature. 1991;350:426–430. doi: 10.1038/350250a0. [DOI] [PubMed] [Google Scholar]
- Samson ML, Jackson-Grusby L, Brent R. Gene activation and DNA binding by Drosophila Ubx and abd-A proteins. Cell. 1989;57:1045–1052. doi: 10.1016/0092-8674(89)90342-5. [DOI] [PubMed] [Google Scholar]
- Schiestl RH, Gietz RD. High-efficiency transformation of intact yeast cells using single-stranded nucleic acids as a carrier. Curr Genet. 1989;16:339–346. doi: 10.1007/BF00340712. [DOI] [PubMed] [Google Scholar]
- Seidman CE, Struhl K, Sheen J, Jessen T. Introduction of plasmid DNA into cells. Curr Protoc Mol Biol. 1997;37:1. 8.1–1.8.10. doi: 10.1002/0471142727.mb0108s37. [DOI] [PubMed] [Google Scholar]
- SenGupta DJ, Zhang B, Kraemer B, Pochart P, Fields S, Wickens M. A three-hybrid system to detect RNA-protein interactions in vivo. Proc Natl Acad Sci USA. 1996;93:8496–8501. doi: 10.1073/pnas.93.16.8496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serebriiskii IG, Khazak V, Golemis EA. A two- hybrid dual bait system to discriminate specificity of protein interactions. J Biol Chem. 1999;274:17080–17087. doi: 10.1074/jbc.274.24.17080. [DOI] [PubMed] [Google Scholar]
- Stagljar I, Bourquin JP, Schaffner W. Use of the two-hybrid system and random sonicated DNA to identify the interaction domain of a protein. BioTechniques. 1996;21:430–432. doi: 10.2144/96213bm20. [DOI] [PubMed] [Google Scholar]
- Strauss WM. Using DNA fragments as probes. Curr Protoc Mol Biol. 1993;24:6. 3.1–6.3.6. doi: 10.1002/0471142727.mb0603s13. [DOI] [PubMed] [Google Scholar]
- Struhl K. Subcloning of DNA fragments. Curr Protoc Mol Biol. 1987;13:3. 16.1–3.16.2. doi: 10.1002/0471142727.mb0316s13. [DOI] [PubMed] [Google Scholar]
- Treco DA, Lundblad V. Preparation of yeast media. Curr Protoc Mol Biol. 1993;23:13.1.1–13.1.7. doi: 10.1002/0471142727.mb1301s23. [DOI] [PubMed] [Google Scholar]
- Wang ZF, Whitfield ML, Ingledue TC, 3rd, Dominski A, Marzluff WF. The protein that binds the 3′ end of histone mRNA: A novel RNA-binding protein required for histone pre-mRNA processing. Genes & Dev. 1996;10:3028–3040. doi: 10.1101/gad.10.23.3028. [DOI] [PubMed] [Google Scholar]
- Watson MA, Buckholz R, Weiner MP. Vectors encoding alternative antibiotic resistance for use in the yeast two-hybrid system. BioTechniques. 1996;21:255–259. doi: 10.2144/96212st02. [DOI] [PubMed] [Google Scholar]
- West RWJ, Yocum RR, Ptashne M. Saccharomyces cerevisiae GAL1-GAL10 divergent promoter region: Location and function of the upstream activator sequence UASG. Mol Cell Biol. 1984;4:2467–2478. doi: 10.1128/mcb.4.11.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Wu Z, Fields S. Protein-peptide interactions analyzed with the yeast two-hybrid system. Nucleic Acids Res. 1995;23:1152–1156. doi: 10.1093/nar/23.7.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]