

Abstract
The influenza A virus NS1 protein has a nuclear localization sequence (NLS) in the amino terminal region. This NLS overlaps sequences that are important for RNA binding as well as protein dimerization. To assess the significance of the NS1 NLS on influenza virus replication, the NLS amino acids were individually mutated to alanines and recombinant viruses encoding these mutations were rescued. Viruses containing NS1 proteins with mutations at R37, R38 and K41 displayed minimal changes in replication or NS1 protein nuclear localization. Recombinant viruses encoding NS1 R35A were not recovered but viruses containing second site mutations at position D39 in addition to the R35A mutation were isolated. The mutations at position 39 were shown to partially restore NS1 protein dimerization but had minimal effects on nuclear localization. These data indicate the amino acids in the NS1 NLS region play a more important role in protein dimerization compared to nuclear localization.
Keywords: influenza, NS1, nuclear localization, RNA binding, dimer
Introduction
The RNA genome of influenza A virus is replicated in the nucleus of infected cells (Taylor, Illmensee et al. 1977). The influenza NS1 protein has been shown to localize to the nucleus and cytoplasm of infected cells through two identified nuclear localization sequences (NLS) as well as a nuclear export sequence (NES) (Melen, Kinnunen et al. 2007; Han, Cui et al. 2010; Forbes, Selman et al. 2013).. A classic monopartite NLS, NLS1, is found between amino acids 34 and 41 and is highly conserved among influenza A virus strains. It contains four basic amino acids, R35, R37, R38, and K41 (Greenspan, Palese et al. 1988; Melen, Kinnunen et al. 2007). Of these, R35, R38, and K41 have been shown to decrease interaction with importin-α if mutated to an alanine (Melen, Kinnunen et al. 2007). The second NLS (NLS2), is bipartite, located between amino acids 219 and 232 and is virus strain specific (Greenspan, Palese et al. 1988; Melen, Kinnunen et al. 2007; Melen, Tynell et al. 2012). The NES is located between amino acids 138–147 (Li, Yamakita et al. 1998). A virus strain specific nucleolar localization (NoLS) signal has also been identified which overlaps with NLS2 (Melen, Kinnunen et al. 2007; Melen, Tynell et al. 2012; Zhu, Zheng et al. 2013).
The NS1 protein is divided into two functional domains (Qian, Alonso-Caplen et al. 1994). The first 73 amino acids form the RNA binding domain while amino acids after position 74 form the effector domain (Qian, Chien et al. 1995). The RNA binding domain interacts with at least four RNA targets: dsRNA, polyA sequence, U6snRNA, and u6atac snRNA (Wang, Riedel et al. 1999) and it is important for antagonizing the innate immune response in both the cytoplasm and the nucleus (reviewed in (Hale, Randall et al. 2008)). The arginine at position 38 is critical for binding RNA and mutating this amino acid to an alanine results in loss of RNA binding (Wang, Riedel et al. 1999; Melen, Kinnunen et al. 2007). This loss in RNA binding results in viruses that are more sensitive to exogenous interferon beta treatment and virus-infected cells produce more type I interferon and cytokines than wild type virus-infected cells (Wang, Riedel et al. 1999; Donelan, Basler et al. 2003; Min and Krug 2006; Newby, Sabin et al. 2007). NS1 interaction with RNA also requires that the protein form a dimer (Nemeroff, Qian et al. 1995; Wang, Riedel et al. 1999) and a number of amino acids in the effector domain (Wang, Basler et al. 2002) and the RNA binding domain (Wang, Riedel et al. 1999) contribute to protein dimerization. Of particular interest for dimerization is the arginine at position 35 which is also part of NLS1 (Melen, Kinnunen et al. 2007).
Consistent with having both NLS and NES signals, the NS1 protein has functions in both the nucleus and cytoplasm and can traffic between the compartments (Melen, Kinnunen et al. 2007; Han, Cui et al. 2010; Forbes, Selman et al. 2013). In the nucleus, NS1 inhibits cellular mRNA maturation through interactions with multiple host factors. Interactions with cleavage and polyadenylation specificity factor (CPSF) and polyadenine binding protein II (PABPII) prevent cleavage and polyadenylation of cellular mRNAs (as reviewed in (Hale, Randall et al. 2008)), while interactions with the mRNA splicing machinery and nuclear export factors further limits cellular mRNA maturation and export (Satterly, Tsai et al. 2007). NS1 also colocalizes with ND10 in nuclear dots and interacts with nucleolin (Sato, Yoshioka et al. 2003; Murayama, Harada et al. 2007). In the cytoplasm, NS1 interacts with a number of cellular pathways. Some of these pathways, including 2′ 5′OAS/RNase L, Rig-I/Trim25 and PKR inhibit host antiviral responses (Donelan, Basler et al. 2003; Li, Min et al. 2006; Min and Krug 2006; Kochs, Garcia-Sastre et al. 2007; Min, Li et al. 2007; Opitz, Rejaibi et al. 2007; Gack, Albrecht et al. 2009). NS1 also interacts with eIF4GI, PABPI, hnRNP-f and hStaufen to increase viral protein synthesis (Lee, Kim et al. 2009)(and reviewed in (Hale, Randall et al. 2008).
To study the role of the influenza NS1 N terminal NLS on virus replication, a series of mutations were introduced into four basic amino acids (R35, R37, R38 and K41) in the NLS1 and recombinant influenza A/WSN/33 viruses encoding these mutated proteins were rescued and characterized. Viruses encoding the R37A, R38A and K41A mutations were recovered and had minimal effects on virus replication and NS1 nuclear localization. Recombinant viruses with the R35A mutation were only rescued when spontaneous, novel second site mutations at position D39 arose. Our data indicate that the amino acids comprising the NS1 NLS1 have minimal effects on NS1 nuclear localization, but at least one, R35, plays an important role in protein dimerization. We demonstrate that second site mutation at position D39 are able to correct for a loss in dimerization caused by the R35A mutation, thereby restoring virus replication.
Results
Rescue of recombinant viruses with mutations in NS1 NLS1
The A/WSN/33 NS1 protein only encodes an amino terminal NLS (NLS1). In order to study the effects of these amino acids on influenza virus replication, the basic amino acids in NLS1 were mutated to alanine, generating plasmids encoding NS1 R35A, R37A, R38A, and K41A (Table 1, top half “NLS”). A virus with the NS1 R38A mutation (rWSN NS1 R38A), which disrupts both NLS1 and RNA binding, was previously isolated (Newby, Sabin et al. 2007). The rWSN NS1 R37A and rWSN NS1 K41A viruses were successfully rescued and plaque purified. The rWSN NS1 R35A virus was not isolated in three separate rescue attempts, but recombinant viruses with amino acid substitutions at position 39 of NS1 (D39A, D39N, or D39Y, Table 1, bottom half collectively referred to as “D39X”) in addition to the R35A mutation were isolated. The mutations were all the result of single nucleotide changes from wild type GAU (D) to GCU (A), AAU (N) or TAU (Y).
Table 1.
Influenza NS1 sequence and mutations
| Virus | Sequencea | Rescued | |
|---|---|---|---|
| NLS | rWSN |
|
Yes |
| rWSN NS1 R37A |
|
Yes | |
| rWSN NS1 R38A |
|
Yes | |
| rWSN NS1 K41A |
|
Yes | |
| rWSN NS1 R35A |
|
No | |
|
| |||
| D39X | rWSN NS1 R35A D39A |
|
Yes |
| rWSN NS1 R35A D39N |
|
Yes | |
| rWSN NS1 R35A D39Y |
|
Yes | |
Bold lowercase letters indicate mutations introduced by plasmid mutagenesis. Capital letters indicate mutations that arose during the rescue of recombinant viruses.
Nuclear localization mutations have minor effects on virus replication in MDCK cells
To study the effect of alanine substitutions in the NS1 NLS1 on virus replication, MDCK cells were infected at a multiplicity of infection (MOI) of 0.001 and virus production was monitored every 12 hours by TCID50 assay (Fig. 1A). There were no differences in replication between any of the rescued viruses (Fig. 1A). As another measure of virus replication, virus plaque diameter was determined on MDCK cells (Fig. 1B). While the rWSN R38A virus had smaller plaque diameter than rWSN (Newby, Sabin et al. 2007), the rWSN R37A and rWSN K41A viruses had plaque diameters similar to rWSN. There were no differences in virus replication in Vero cells after infection with an MOI of 0.001 (Fig. 1C). Taken together, these data indicate that viruses encoding NS1 protein with R37A and K41A mutations replicate in MDCK and Vero cells similarly to wild type virus. The NS1 R38A mutation slightly attenuates virus replication, as previously demonstrated (Newby, Sabin et al. 2007).
Figure 1.
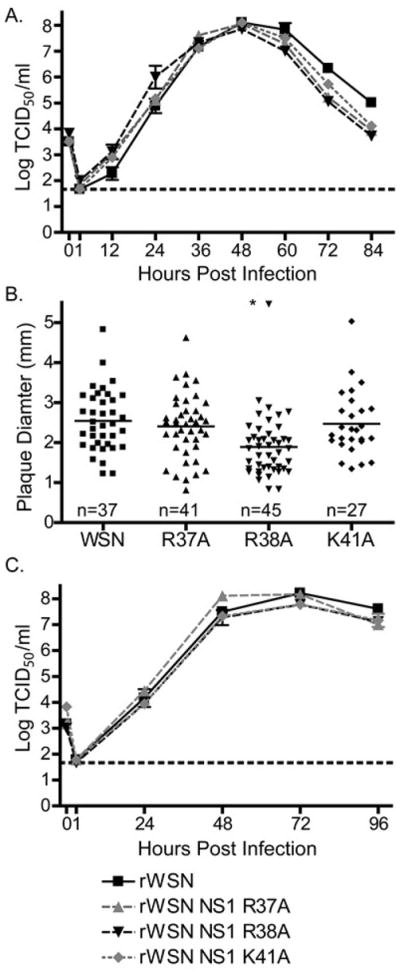
NS1 NLS viruses replicate to high titers on MDCK and Vero cells lines. (A) MDCK cells were infected with rWSN or rWSN NS1 NLS viruses at a MOI of 0.001 TCID50/cell. Supernatants were collected every 12 hours and virus titers were analyzed by TCID50 assay. The limit of detection is indicated by a dotted line at 1.67 and mean and standard error of the mean are graphed. There were no statistical differences in virus replication (two-way ANOVA). (B) MDCK cells were infected with serial dilutions of rWSN or rWSN NS1 NLS viruses and incubated under 1% agarose and media for three days then fixed. Plaque diameter was measured using ImageJ. Individual plaques are graphed and mean diameter is indicated with a solid horizontal line. Significant differences relative to rWSN are indicated with an asterisk (*), p < 0.01, one-way ANOVA and a Bonferroni multiple comparison post test. (C) Vero cells were infected with rWSN or rWSN NS1 NLS viruses at a MOI of 0.001 TCID50/cell. Supernatants were collected every 24 hours and virus titers were analyzed by TCID50 assay. The limit of detection is indicated by a dotted line at 1.67 and mean and standard error of the mean are graphed. There were no statistical differences in virus replication using a two-way ANOVA and a Bonferroni multiple comparison post test.
The levels of NS1 protein expression were not affected by the NLS mutations as determined by western blot (Fig. 2A) or mean florescent intensity (MFI) by flow cytometry (Fig. 2B) at six hours post infection with an MOI of 5. To determine whether the alanine substitutions disrupted NS1 nuclear localization, MDCK cells were infected at an MOI of 5 for 6 hours, and then examined by immunoflorescence microscopy. The NS1 protein from wild type and NLS viruses were found in both the nucleus and the cytoplasm of infected cells. As previously described, some of the cells infected with the rWSN NS1 R38A virus displayed punctate concentrations of NS1 throughout the cytoplasm (Newby, Sabin et al. 2007). The NS1 wild type protein was found in the nucleolus of some infected cells and a loss of nucleolar localization was observed for cells infected with rWSN NS1 R38A and rWSN NS1 K41A. Nucleolar localization was unaffected in rWSN NS1 R37A infected cells (Fig. 2C). Previous reports have identified little nucleolar localization of NS1 in rWSN infected cells which is consistent with our data at 3 hours post infection (data not shown) (Melen, Kinnunen et al. 2007; Newby, Sabin et al. 2007). Nucleolar localization was observed in some cells from 6 to 12 hours post infection (data not shown). Therefore, while mutations to R38 and K41 have been characterized to attenuate NS1 function and recombinant virus replication, they seem to have minimal effect on nuclear localization of the protein in virus-infected MDCK cells.
Figure 2.

The NS1 protein is express at comparable amounts and is not excluded from the nucleus by NLS mutations during infection. (A) MDCK cells were infected with rWSN or rWSN NS1 NLS viruses at a MOI of 5 TCID50/cell. At 6 hpi, the cells were lysed in 1% SDS and NS1 expression was examined by western blot. (B) MDCK cells were infected with rWSN or rWSN NS1 NLS viruses at an MOI of 5 TCID50/cell. At 6 hpi, NS1 expression was quantified by flow cytometry. The mean fluorescence intensity (MFI) of the NS1 positive cells was normalized to wt and the mean and standard error of the mean are graphed. There was no statistical difference using a one-way ANOVA and a Bonferroni multiple comparison post test. (C) MDCK cells were grown on glass coverslips and infected with rWSN or rWSN NS1 NLS viruses at a MOI of 5 TCID50/cell. The cells were fixed at 6 hpi and NS1 localization was determined by immunofluorescence microscopy using deconvolution of serial optical sections. All images were taken with a 100x objective and DAPI was used to counterstain the nucleus. Mock-infected MDCK cells were included in all experiments and showed no reactivity with the NS1 antibodies.
Viruses with NS1 R35A D39X mutations have altered NS1 subcellular localization patterns
The viruses with NS1 R35A D39X mutations were tested for virus replication, NS1 protein expression and localization (Figs. 3 and 4). The rWSN NS1 R35A D39A virus had similar replication kinetics and peak titers of infectious virus relative to rWSN, while the rWSN NS1 R35A D39N and rWSN R35A D39Y viruses reached significantly lower peak titers (Fig. 3A). When plaque diameters were measured, only the rWSN NS1 R35A D39Y virus had significantly smaller plaque diameters when compared to rWSN (Fig. 3B). All the viruses replicated with similar kinetics and to a similar extent in Vero cells (Fig. 3C).
Figure 3.
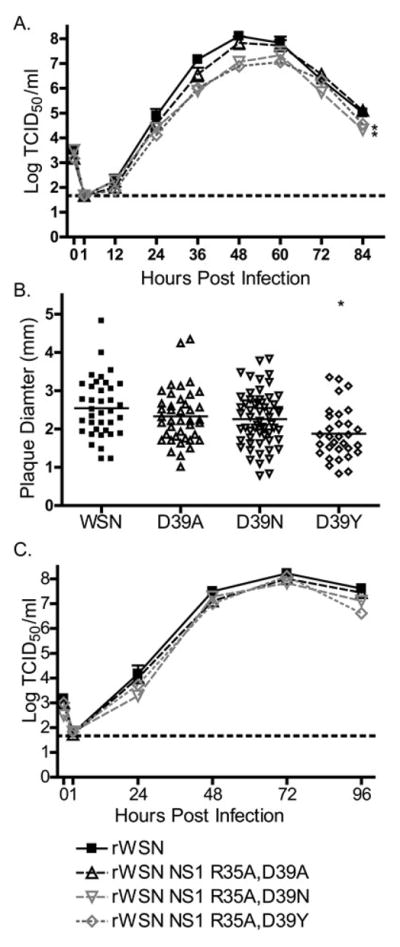
NS1 R35A D39X viruses replicate to high titers and express comparable amounts of NS1 protein. (A) MDCK cells were infected with rWSN or rWSN NS1 R35A D39X viruses at a MOI of 0.001 TCID50/cell. Supernatants were collected every 12 hours and virus titers were analyzed by TCID50 assay. The limit of detection is indicated by a dotted line at 1.67 and mean and standard error of the mean are graphed. Significant differences between the replication of NS1 R35A D39X viruses and wild type virus are indicated with an asterisk (*), p < 0.01, two-way ANOVA and a Bonferroni multiple comparison post test. (B) Plaque diameter in MDCK cells was measured using ImageJ. Individual plaques are graphed and mean diameter is indicated with a solid horizontal line. Significant differences between NS1 R35A D39X viruses and rWNV are indicated with an asterisk (*), p < 0.01, one-way ANOVA and a Bonferroni multiple comparison post test. (C) VERO cells were infected with rWSN or rWSN NS1 NLS viruses at a MOI of 0.001 TCID50/cell. Supernatants were collected every 24 hours and virus titers were analyzed by TCID50 assay. The limit of detection is indicated by a dotted line at 1.67 and mean and standard error of the mean are graphed. There were no statistical differences in virus replication using a two-way ANOVA and a Bonferroni multiple comparison post test.
Figure 4.

The NS1 protein is express at comparable amounts though intracellular localization of the NS1 protein is altered by the D39X mutations during infection. (A) MCDK cells were infected with rWSN or rWSN NS1 D39X viruses at a MOI of 5 TCID50/cell. At 6 hpi, the cultures were lysed in 1% SDS and NS1 expression was examined by western blot. (B) MCDK cells were infected with rWSN or rWSN NS1 D39X viruses at a MOI of 5 TCID50/cell. At 6 hpi, NS1 expression was quantified by flow cytometry. The MFI of the NS1 positive cells was normalized to wild type and the mean and standard error of the mean are graphed. There were no statistical differences between viruses using a one-way ANOVA and a Bonferroni multiple comparison post test (C) MDCK cells were grown on glass coverslips and infected with rWSN or rWSN NS1 D39X viruses at a MOI of 5 TCID50/cell. Infected cultures were fixed at 6 hpi and NS1 localization determined by immunofluorescence microscopy using deconvolution of serial optical sections. All images were taken with a 100x objective and DAPI was used to counterstain the nucleus. Mock-infected MDCK cells were included in all experiments and showed no reactivity with the NS1 antibodies.
To examine NS1 protein expression levels, MDCK cells were infected at an MOI of 5 and processed for western blot and flow cytometry 6 hours post infection. No significant difference was observed in the levels of NS1 protein expression (Fig. 4A and B), suggesting that altered NS1 protein levels were not responsible for the reduced virus replication of rWSN NS1 D35A D39N/Y viruses. NS1 subcellular localization was next examined in MDCK cells six hours after a MOI 5 infection. In a portion of infected cells, all three NS1 R35A D39X viruses formed fluorescent puncta in the cytoplasm not observed in wild type rWSN-infected cells (Fig. 4C). Similar to rWSN NS1 R38A- and K41A-infected cells (Fig. 2C), no nucleolar localization was seen in the D39X infected cells (Fig. 4C). These results indicate that rWSN NS1 R35A D39X viruses are able to replicate and express similar levels of NS1 protein compared to wild type virus but NS1 localization is affected. Similar to other viruses with mutations to NLS1, the NS1 protein was not excluded from the nucleus by R35A D39X mutations.
Plasmid expressed NS1 R35A and R35A D39X proteins show similar localizations
To explore how the NS1 R35A and D39X mutations were affecting NS1 expression and function, the proteins were expressed from cDNA expression vectors. Protein localization was examined by transfecting MDCK cells with wild type NS1, NS1 R35A, or one of the NS1 R35A D39X plasmids. Subcellular localization of NS1 R35A and the NS1 R35A D39X proteins at 9 hours post transfection was similar for wild type and mutant NS1 (Fig. 5), with no obvious change in nuclear localization being apparent. The distinct brightly fluorescent puncta seen in rWSN NS1 R35A D39X infected cells were not observed after cDNA expression of the proteins, indicating that the puncta form either in response to the expression of other viral proteins or are due to cellular responses to virus infection. The NS1-myc protein displayed a similar intracellular localization pattern as the wild type NS1 protein (data not shown). The expression level of the NS1 proteins in transfected cells was examined by flow cytometery (Fig. 6). Similar to virus infection, the expression levels of the NS1 R35A D39X proteins are not significantly different than wild type protein. The expression of NS1 R35A was significantly lower than any of the other NS1 proteins (Fig. 6), indicating that the R35A mutation reduces NS1 protein levels, presumably due to greater amounts of protein degradation, and that the D39X mutations are restoring protein expression levels.
Figure 5.
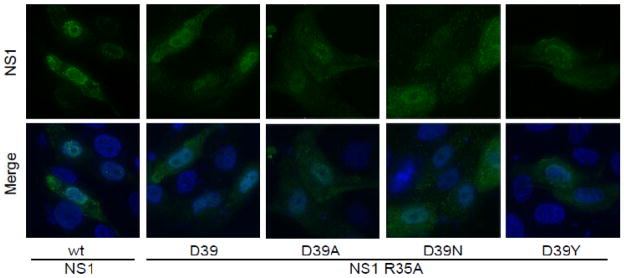
Influenza A virus NS1 protein localization is not affected by R35A and D39X mutations after cDNA transfection. MDCK cells were transfected with plasmids encoding the indicated proteins. At 9 hours post transfection, NS1 localization was determined by immunofluorescence microscopy using deconvolution of serial optical sections. All images were taken with a 100x objective and DAPI was used to counterstain the nucleus. MDCK cells transfected with control plasmids showed no reactivity with the NS1 antibody.
Figure 6.
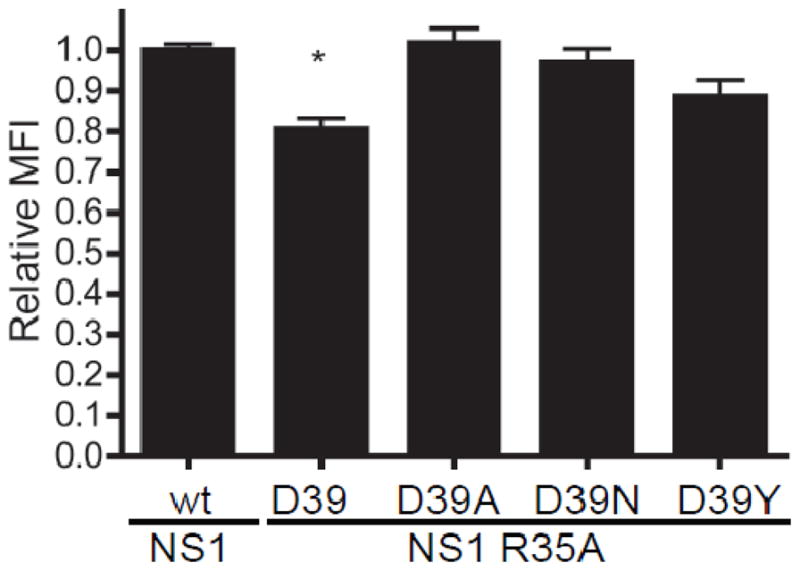
Expression levels of Influenza A virus NS1 proteins. MCDK cells were transfected with plasmids encoding the indicated proteins. At nine hours post transfection, NS1 expression was quantified by flow cytometry. The MFI of the NS1 positive cells was determined and the mean and standard error of the mean are graphed. Signficant differences between rWSN NS1 and rWSN are indicated with an asterisk (*), p < 0.01 using a one-way ANOVA and a Bonferroni multiple comparison post test. MDCK cells transfected with control plasmids showed no reactivity with the NS1 antibody.
The NS1 R35A mutation reduces protein dimerization and mutations at amino acid 39 restore this activity
Because a virus encoding only the NS1 R35A mutation was not rescued, it was hypothesized that the D39 mutations compensate for a loss of NS1 dimerization, as R35A was previously identified as critical for dimerization of the NS1 RNA binding domain (Wang, Riedel et al. 1999). To determine if the mutations at D39 correct for loss of dimerization, a bimolecular florescent complementation (BiFC) assay was used (Kerppola 2006). The NS1 wild type and mutant proteins were fused to either the N- (YFP1) or C-terminal (YFP2) fragments of YFP (Figure 7A). If the NS1 proteins interact, the florescent protein fragments will be brought into close proximity and refold to produce a complete, fluorescent YFP protein (Hu, Chinenov et al. 2002).
Figure 7.

NS1 protein dimerization is affected by mutations at positions R35 and D39. (A) MDCK cells were co-transfected with plasmids encoding YFP-1 and YFP-2 NS1 fusion proteins with a flexible linker (L) region separating the two domains. X indicates A, N, or Y at position 39. (B, C) At 9 hours post transfection, NS1 expression levels and YFP fluorescence were quantified by flow cytometry. The mean fluorescence intensity (MFI) of the NS1 positive cells was determined and the relative mean and standard error of the mean are graphed. Significant differences between wild type NS1 and other NS1 constructs are indicated with an asterisk (*) and significant differences between NS1 R53A and other NS1 constructs are indicated with a double cross (‡), p < 0.01, one-way analysis of variance (ANOVA) and a Bonferroni multiple comparison post test. Labels indicate the amino acid at positions 35 and 39 (35/39) with wild type NS1 sequence being R/D. Dotted line at y=0.42 represents background YFP florescence using YFP1 and YFP2-NS1. Data is normalized to NS1 wild type (R/D) constructs and averaged between three experiments.
To determine if RNA dimerization is reduced with a NS1 R35A mutation (Wang, Riedel et al. 1999), cells were transfected with plasmids encoding YFP1-NS1 and YFP2-NS1 (Fig. 7A, constructs with black NS1) or YFP1-NS1 R35A and YFP2-NS1 R35A (Fig. 7A constructs with white NS1). Transfected cells were fixed and processed for flow cytometery nine hours after transfection. NS1 expressing cells were gated on and analyzed for NS1 expression levels. Both sets of constructs expressed similar amount of NS1 protein (Fig. 7C black and white bars). YFP expression in cells expressing NS1 R35A fusion proteins was significantly lower than that observed in cells expressing NS1 wild type fusion proteins (Fig. 7B), indicating loss of NS1 dimerization in the NS1 R35A mutant protein. The YFP MFI for NS1 R35A constructs is similar to background fluorescence levels measured by transfecting cells with plasmids expressing YFP1 and YFP2-NS1 (Fig. 7B dotted line).
To determine if the loss of dimerization occurs if only one of the NS1 molecules in a dimer has the R35A mutation, cells were transfected with plasmids encoding YFP1-NS1 and YFP2-NS1 R35A or YFP1-NS1 R35A with YFP2 NS1. When NS1 wild type was expressed with NS1 R35A, YFP MFI levels were higher than that seen with NS1 R35A, but not as high as NS1 wild type (Fig. 7B grey bars). Overall levels of NS1 expression remained consistent (Fig. 7C grey bars). This indicates that each of the arginine residues contributes to the NS1 dimer stability and that having one wild type NS1 can partially stabilize the dimer.
The mutations at NS1 D39 were tested for their effects on NS1 R35A dimerization. MDCK cells were transfected with YFP1/YFP2 constructs encoding each of the NS1 R35A D39X proteins (Fig. 7A construct with diagonal lines). The YFP expression in each of the NS1 R35A D39X expressing cells was significantly higher than that observed in NS1 R35A expressing cells (Figure 7B bars with diagonal lines). The NS1 protein expression levels were similar across all constructs (Figure 7C) indicating the increased YFP was a result of increased protein dimerization and not due to increased overall protein expression levels. The cells transfected with NS1 R35A D39A constructs had similar levels of YFP florescence when compared to NS1 wild type expressing cells, while the cells expressing NS1 R35A D39N or NS1 R35A D39Y expressed YFP MFI levels that were lower than that observed in NS1 wild type expressing cells. The degree of dimerization conferred by the D39X mutation seems to correlate with the overall replication of the recombinant viruses, with the NS1 R35A D39A protein having restored NS1 dimerization and recombinant virus replication more effectively on MDCK cells than either NS1 R35A D39N or NS1 R35A D39Y (Figure 3A). These data supports the hypothesis that the D39 mutations partially restore the ability of NS1 R35A proteins to dimerize, thus allowing for recombinant virus replication.
Discussion
The NS1 N-terminal RNA binding domain has many functions important for influenza A virus replication. RNA binding requires the dimerization of NS1 which brings together the R38 and K41 residues from each monomer in a conformation optimal for interaction with the phosphate backbone of the RNA molecule (Nemeroff, Qian et al. 1995; Wang, Riedel et al. 1999). The N-terminal domain also contains a nuclear localization sequence, NLS1, which overlaps some of the same amino acids important to RNA binding and NS1 dimerization (Wang, Riedel et al. 1999). This sequence is highly conserved in NS1 among influenza A virus strains (Melen, Kinnunen et al. 2007). Changing R38 or R38/K41 to alanine causes loss of RNA binding and attenuates NS1 function (Donelan, Basler et al. 2003; Min and Krug 2006; Melen, Kinnunen et al. 2007; Newby, Sabin et al. 2007). In this study, we demonstrated that the NS1 R38A virus, which has a loss of RNA binding in addition to disrupting NLS1, has reduced virus replication and changes in NS1 protein localization, consistent with our previous results (Newby, Sabin et al. 2007). Changes at NS1 positions 37 and 41 did not significantly affect virus replication and had minimal effects on NS1 localization, including loss of nucleolar localization in the virus with the NS1 K41A mutation. Since nucleolar localization of NS1 varies widely between human influenza A virus strains and eliminating nucleolar localization of NS1 does not adversely affect the replication of influenza A/Udorn/72 (Melen, Kinnunen et al. 2007), it is unclear how significant the NS1 nucleolar localization signals are for virus replication.
Because single alanine mutations do not appear to exclude the protein from the nucleus, it may be that the dimer conformation of the NS1 protein brings enough basic amino acids together to form a functional nuclear localization signal, or that NS1 enters the nucleus by an alternate mechanism. The lack of a strong phenotype calls into question the importance of NLS1 on NS1 protein function. While the NS1 dimer is required for RNA binding, the NS1 monomer is small enough to diffuse into the nucleus unaided (Talcott and Moore 1999). The high degree of sequence conservation seen in this region of the NS1 protein may be due to other functions of those amino acids, including binding RNA or stabilization of protein dimers.
When the NS1 amino acid mutation R35A was introduced into the NS segment, recombinant virus was produced, but every characterized isolate presented an additional mutation at the aspartic acid at position 39. As R35 has been identified as important to NS1 dimerization, it was hypothesized that the mutations in R39 were compensating for loss in dimerization. Previous work that investigated the NS1 protein with mutations at R35 used plasmid expressed proteins rather than infectious virus (Wang, Riedel et al. 1999; Melen, Kinnunen et al. 2007). It may be that the NS1 R35A mutation not only disrupts NS1 dimerization, but it may also adversely affect other aspects of virus replication and thus prevent recombinant virus rescue. When Li et. al. attempted to insert a six amino acid tag into the RNA binding domain specifically within the alpha helixes, they were also unable to rescue virus (Li, Lu et al. 2010). This effect is surprising as influenza A viruses with the NS1 protein deleted have been rescued in other virus strains (Garcia-Sastre, Egorov et al. 1998).
Structural studies of the NS1 RNA binding domain have described many intermolecular interactions between various amino acids at the dimer interface and between the NS1 dimer and RNA. R35 interacts with both D12 and D39 via salt bridges (Chien, Tejero et al. 1997). Direct hydrogen bonding between R35 and D12 and water-mediated hydrogen bonding between R35/D12 and R19/D39 have also been described (Liu, Lynch et al. 1997) as well as hydrogen bonding between R35 and R46 (Cheng, Wong et al. 2009). Cheng et al. examined the interactions between the NS1 dimer and RNA, and described R35 and R38 interacting directly with RNA via hydrogen bonds, while D39 and R41 interact via water-mediated hydrogen bonds (Cheng, Wong et al. 2009). A number of arginine and aspartic acid residues are found at the NS1-NS1 dimer interface near the NS1-RNA interface and protein stabilizing salt bridges commonly form between oppositely charged arginine and aspartic acid (Chien, Tejero et al. 1997; Kumar and Nussinov 2002). This is supported by previous data indicating that arginines and aspartic acids at the dimer interface are critical for NS1 dimerization (Wang, Riedel et al. 1999). When the positively charged R35 is changed to a neutral alanine, the dimerization of NS1 is disrupted (Wang, Riedel et al. 1999). Under the selective pressure in the recombinant virus rescue system, we identified compensatory mutations at D39. The location of the compensatory mutations supports a direct interaction between R35 and D39 as proposed by Chien et al. (Chien, Tejero et al. 1997). When protein dimerization was assessed with the BiFC system, the NS1 R35A proteins were not able to dimerize to the same extent as NS1 wild type. This could be due to not only loss of interaction with D39, but also weak repulsion from the unpaired negative charge of the aspartic acid at position 39. The dimerization defect could be partially corrected either by one copy of wild type NS1 in the dimer or through mutation of D39 to an amino acid with less negative charge (A, N, or Y), though in both cases the extent of dimerization was lower than NS1 wild type. When NS1 R35A interacts with wild type NS1, the interaction between R35 on the wild type protein and the D39 on the NS1 R35A mutant may be enough to stabilize the dimer even though the other 35–39 pair interaction might have a weak repulsion. Conversely, when both R35 and D39 are changed to less charged amino acids, the protein may be able to dimerize because there is no repulsion from an unpaired aspartic acid at the dimer interface and other interactions between the two monomers are enough to stabilize the dimer. These data support the idea that in addition to the previously identified, D12, R19, D29, R35, and R36 (Wang, Riedel et al. 1999), D39 should be added to the list of residues important for dimer stability.
The NS1 R35A D39X viruses were able to replicate in MDCK and Vero cells, though viruses with NS1 D39N and D39Y replicated less efficiently than wild type virus in MDCK but not Vero cells. This may indicate an incomplete recovery of NS1 function in these viruses and suggests that NS1 function is more important in MDCK cells than in the IFN-incompetent Vero cells, consistent with our previous observations (Newby, Sabin et al. 2007). While the localization of the NS1 protein was similar between the three NS1 R35A D39X viruses, it was different from wild type virus, most obviously through the appearance of cytoplasmic puncta of NS1. Similar to the viruses with NS1 R38A or K41A, no nucleolar localization of NS1 was seen in cells infected with viruses expressing the NS1 R35A D39X proteins. While the strain specific NoLS has been identified at the C terminus of the NS1 protein, interaction with nucleolin, a major nucleolar protein, has been mapped to the RNA binding domain (Melen, Kinnunen et al. 2007; Murayama, Harada et al. 2007). Changes in the RNA binding domain that disrupt the NS1-nucleolin interaction could explain the loss of nucleolar localization seen in some of the NS1 mutants. Replication of a virus encoding NS1 R38A K41A was enhanced by a mutation in NS1 (S42G) (Donelan, Basler et al. 2003). This mutation did not alter NS1 RNA binding activity but we speculate that perhaps it improved NS1 dimer formation, analogous to the D39X mutations we identified.
Dimerization of the NS1 protein is required for RNA binding, and RNA binding has an important role in NS1 function and viral pathogenesis. The BiFC assays indicate that the ability of NS1 to interact is dramatically decreased with an R35A mutation. If interfering with dimerization or the structural stability of the RNA binding domain as seen here and elsewhere (Li, Lu et al. 2010) is so detrimental that the resulting virus is not viable, then the dimerization interface of the RNA binding domain could be a viable drug target. If the R35A viruses were not viable or strongly outcompeted by low frequency D39X mutations, this indicates that blocking dimerization can have a dramatic affect on virus replication. The NS1 protein has been identified as a potential drug target (Maroto, Fernandez et al. 2008; Basu, Walkiewicz et al. 2009; Jablonski, Basu et al.; Walkiewicz, Basu et al. 2011). Previous studies on potential ligand binding sites in influenza NS1 identified 2 sites in the RNA binding domain that would interact with R35 or D39 (Darapaneni, Prabhaker et al. 2009), and at least one potential drug, baicalin has been shown to interact with R35, R38, and D39 (Nayak, Agrawal et al. 2014)
Materials and Methods
Cell culture
MDCK and 293T cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Sigma) supplemented with 100 U/ml penicillin, 100 ug/ml streptomycin (Gibco), 2mM glutamax (Gibco) and 10% fetal bovine serum (FBS, Hyclone). During infection, MDCK cells were maintained in infectious media, consisting of DMEM with penicillin/streptomycin, glutamax, 0.15% BSA (Sigma), and 4ug/ml N-acetyl trypsin (Sigma) (DMEM infectious media). During virus rescue, 293T cells were maintained in OptiMEM (Gibco) or OptiMEM with 0.1% FBS.
Plasmids
All plasmids were amplified in chemically competent DH5 alpha E. coli. Mutations to the pHH21 NS segment plasmid were introduced using four primer PCR (Table 2). Forward and reverse primers FluA6 and FluA5 were used along with mutagenesis primers NS1 R35A f and NS1 R35A r 2 for the R35A mutation, NS1 R37A F and NS1 R37A r 2 for R37A, and NS1 K41A f and NS1 K41A r for the K41A mutation. Sequences were introduced into pHH21 plasmids using restriction enzyme digest followed by DNA ligation. The NS coding region for all plasmids was confirmed by sequencing.
Table 2.
Primer sequences used in plasmid development and sequencing
| Primer name | Primer sequence | Direction | Purpose |
|---|---|---|---|
| FluA5 | ATACGTCTCGTATTAGTAGAAACAAGGGTGTTTTTT | reverse | 1,2,3 |
| FluA6 | CGACGTCTCCGGGAGCAAAAGCAGGGTG | forward | 1,2,3 |
| NS1 R35A f | CCATTCCTTGATGCTCTTCGCCGAGAT | forward | 4 |
| NS1 R35A r2 | CTGATCTCGGCGAAGAGCATCAAGGAATGGG | reverse | 4 |
| NS1 R37A F | CTTGATCGGCTTGCTCGAGATCAG | forward | 4 |
| NS1 R37A r2 | CTTCTGATCTCGAGCAAGCCGATCAAGGAATGGG | reverse | 4 |
| NS1 K41A f | CGAGATCAGGCGTCCCTAAGAGGAAG | forward | 4 |
| NS1 K41A r | CTTCCTCTTAGGGACCGCTGATCTCG | reverse | 4 |
| WSN 3′ NS1 into PC | ATAATAGAGCTCACCATGGATCCAAACACTGTGTCAAGC | reverse | 5 |
| WSN 5′ NS1 into PC EcoR1 | GGCAAAGAATTCGAGCTCACCATGGATCCAAACACTGTGTCAAG | forward | 5 |
| WSN 5′ NS1 into PC | ATAAAACCCGGGTCAAACTTCTGACCTAATTGTTCCCGC | forward | 5 |
| wsnmyctag | ATAATACCCGGGTCACAAGTCTTCTTCGCTGATGAGTTTCTGCTCAACTTCTGACCTAAT | reverse | 5 |
| pCAGGS fwd for seq | GCAACGTGCTGGTTGTTGTGC | forward | 3 |
| pCAGGS rev for seq | CAAGGGGCTTCATGATGTC | reverse | 3 |
| WSN 5′ NS1 into split YFP | CGAGTCCGGAATGGATCCAAACACTGTGTCAAGC | forward | 6 |
| WSN3′NS1 into split YFP | GCATGTCTAGATCAAACTTCTGACCTAATTGTTCCCGC | reverse | 6 |
| T7 Promoter | TAATACGACTCACTATAGG | forward | 7 |
| BGH Rev | TAGAAGG CACAGTCGAGG | reverse | 7 |
1: Influenza genome RT-PCR sequencing
2: pHH21 cloning
3: sequencing
4: Introduce mutation into NS1
5: Amplifying NS1 for insertion into pCAGGs
6: Amplifying NS1 for insertion into split YFP plasmids
7: Sequencing YFP domain and NS1 in split YFP plasmids
NS1 cDNAs were cloned into the pCAGGS vector (Niwa, Yamamura et al. 1991). A myc tag with the amino acid sequence EQKLISEEDL was added to the C-terminus of NS1 using the WSN 5′ NS1 into PC and wsnmyctag primers (Table 2) and introduced into the pCAGGS plasmid using restriction enzyme digest followed by DNA ligation. NS1 R35A D39X mutations were introduced into a NS1-myc cDNA expression plasmid by amplifying NS1 sequences from virus with 5′ primers WSN 5′ NS1 into PC or WSN 5′ NS1 into PC EcoR1 (for R35A D39A) and 3′ primer WSN 3′ NS1 into PC and introduced into a pCAGGS NS1-myc plasmid by restriction enzyme digest and ligation. The NS1 coding region of all plasmids was verified by sequencing.
Plasmids used for Bimolecular Florescence Complementation (BiFC) were kindly provided by Matt Frieman, University of Maryland (Nyfeler, Michnick et al. 2005; Frieman, Yount et al. 2007). NS1 sequences were introduced downstream of either an N terminal fragment of Venus YFP (YFP1; amino acids 1–158) or the C terminal fragment of Venus YFP (YFP2; amino acids 159–239) via a 10 amino acid linker (GGGGSGGGGS) (Figure 7A). Primers WSN 5′ NS1 into split YFP and WSN 3′NS1 into split YFP (Table 2) were used to amplify the NS1 sequences that were then introduced in the YFP1 or YFP2 plasmids by restriction enzyme digest and ligation.
Viruses
Recombinant A/WSN/33 influenza viruses (rWSN) were rescued using a 12 plasmid reverse genetics system as previously described (Neumann, Watanabe et al. 1999). Mutations were introduced into the pHH21 NS plasmid as described above. Recombinant viruses were rescued by transfecting 293T cells with the 12 plasmids and 20μl TransIT-LT1 (MirusBio) in OptiMEM. Twenty four hours after transfection, media was changed to OptiMEM with 0.1% FBS. Supernatants were collected from the transfected cells every 24 hours and virus was detected by hemaggluttination assay.
Hemagglutination assay
Hemagglutination was assessed using 0.5% chicken red blood cells (CBT farms, Chestertown MD) in Alsevier’s solution. Rescue supernatants were diluted in a series of two fold dilutions in PBS. When supernatants showed hemagglutination activity, infectious virus was isolated by plaque assay.
Plaque assay
Supernatants were diluted in a serial 10 fold dilution series in DMEM infectious media and applied to monolayers of MDCK cells in 6-well plates for 1 hour with rocking at room temperature. Cells were then overlaid with 1% agarose in DMEM infectious media. Cultures were incubated for 3 days and checked for plaque formation by phase contrast microscopy. When plaques were visible, agarose plugs above individual plaques were isolated and stored in 1ml of DMEM infectious media.
Virus infections
MDCK cells were infected at the indicated multiplicity of infection (MOI). Virus was diluted to the appropriate concentration in DMEM infectious media. Cells were washed twice with phosphate buffered saline with calcium and magnesium (PBS+) (Gibco) before being infected. Cells with virus inoculum were incubated for 1 hour at room temperature with rocking and then the inoculum was removed. The cells were then washed twice in PBS+ before adding additional DMEM infection media. Seed stocks of virus were grown by infecting MDCK cells with 100μl of plaque pick solution. Working stocks were grown by infecting MDCK cells at an MOI of 0.01 TCID50/cell from seed stocks. RNA was isolated from the resulting virus stocks and the NS segment coding region was sequenced and verified.
TCID50 assay
To determine the infectious virus titer, virus supernatants were diluted in a serial 10 fold dilution series from 10^-1 to 10^-8 in DMEM infectious media. MDCK cells were grown to confluence in 96 well plates and washed twice with PBS+ before 100μl of each virus dilution was added to each of 6 wells. The cells were incubated at 37°C for 4 days. The cells were then fixed by adding an equal volume of 4% formaldehyde (Fisher Scientific), followed by staining with napthol blue-black solution. Titer was determined by calculating the number of cleared wells at each dilution using Reed & Muench calculations (Reed and Muench 1938).
Transfections
Confluent MDCK cells were trypsinized and transfected in suspension then plated at 30% density. For pCAGGS NS1-myc plasmids, 3ug of DNA was added with 6ul of Lipofectamine 2000 (Invitrogen). For Bimolecular Florescence complementation, 3 μg of each plasmid was added with 6ul of Lipofectamine 2000. DNA and Lipofectamine were first mixed separately with Optimem, incubated for 5 min, then combined and incubated for 20 minutes before being added dropwise to the MDCK cells in suspension. Nine hours post transfection, cells were process for microscopy or flow cytometry as described below.
SDS PAGE and Western blot
MDCK cells infected at an MOI of 5 TCID50/cell were washed with PBS 6 hours post infection and lysed in 1% SDS (Sigma) in PBS. Cell lysates were process by pressing through a 26 ⅝G needle (Becton Dickinson) and mixing 3:1 with a 4x SDS PAGE loading buffer before heating for 5 min at 95°C. Samples were loaded alongside Precision plus Protein all blue ladder (Biorad) onto a 15% polyacrylamide gel with a 3% polyacrylamide stacking gel for protein separation and run at 150V through the stacking gel and 100V through the separating gel. Proteins were transferred from the gel to a polyvinylidene difluoride (PVDF) Immobilon FL membrane (Millipore) at 20V overnight at 4°C. Membranes were blocked in PBS, 0.3% Tween-20, 5% non fat dry milk for 1 hour at room temperature (blocking solution). Membranes were then incubated for 1 hour in primary antibodies (mouse anti NS1 6A4 or 2H6 (kindly provided by Yee-Joo Tan (Tan, Akerstrom et al. 2010)) 1:10,000 and mouse anti beta-actin antibody (Abcam) 1:10,000 diluted in blocking solution. After incubation they were washed three times for 5 minutes each time in PBS with 0.3% Tween-20 (Sigma-Aldrich), then incubated for 1 hour in with secondary antibody goat anti mouse conjugated to Alexa Fluor 647 (Molecular Probes) 1:1000 diluted in blocking solution and again washed 3 times with PBS + 0.3% Tween-20. All membrane incubation and wash steps took place at room temperature. Protein bands were detected using a Fuji FLA 5000 phosphoimager. Mock-infected MDCK cells showed no reactivity with the NS1 antibody.
Immunoflorescence Microscopy
MDCK cells were grown on glass coverslips before being infected or transfected in solution and plated onto glass coverslips. At indicated times after infection or transfection, cells were washed two times with PBS+ and fixed with 2% paraformaldehyde (Sigma-Aldrich) in PBS for 15 minutes at room temperature. Cells were then washed twice with PBS and permeabilized with 0.2% Triton-X 100 (sigma X100) in PBS for 15 min at room temperature. Cells were washed after each step by dipping coverslips in PBS with 0.2% Tween-20 15 times. Cells were blocked in PBS with 3% goat serum (Sigma) and 0.5% BSA (Calbiochem) (blocking buffer) for 1 hour and then washed. Cells were then incubated in primary antibody mouse anti NS1 (1A7CL kindly provided by Robert Webster (Brown, Hinshaw et al. 1983)) at a 1:100 dilution in blocking buffer for 1 hour, washed, and then incubated with goat anti mouse antibody conjugated to Alexa Fluor 488 (molecular probes A-11029) and DAPI (Invitrogen) in blocking buffer, washed by dipping 10 times in PBS with 0.2% Tween-20 and five times in MilliQ water. Coverslips were mounted cell side down in Prolong gold antifade (Invitrogen). Cells were imaged on a Nikon 90-i microscope using Volocity software for image acquisition. Images were taken with a 100x objective in Z-stack mode and deconvoluted using Volocity software and calculated point spread functions. Mock-infected or control-transfected cells showed no reactivity with the NS1 antibody.
Flow cytometry
At 6 hours post infection or 9 hours post transfection, cells were washed two times with PBS and incubated with 0.2% trypsin-EDTA (Invitrogen) in PBS for 20 minutes. The cells were then fixed with 2% paraformaldehyde (Sigma-Aldrich) in PBS for 15 minutes at room temperature and centrifuged at 300xg for 5 minutes. Cells were then washed twice with PBS with centrifugation between each wash and permeabilized with 0.2% Triton-X 100 (Sigma) for 10 min at room temperature. Cells were washed two more times in PBS and incubated in PBS with 3% goat serum (Sigma) and 0.5% BSA (Calbiochem) (blocking buffer) for 1 hour. Cells were then incubated with mouse anti NS1 (1A7CL) at a dilution of 1:100 in blocking buffer for 1 hour at room temperature, washed three times, and then incubated with goat anti mouse antibody conjugated to Alexa Fluor 647 in blocking buffer for 1 hour at room temperature, and washed three times in PBS. All wash steps were done by centrifuging at 300xg for 5 minutes and removing the supernatant with a vacuum aspirator comb (VP Scientific BP). The NS1 antibody did not react with mock-infected MDCK cells (data not shown).
For pCAGGS NS1 myc experiments, transfected cells expressing NS1 were analyzed for Mean Florescent Intensity (MFI) using the anti-NS1 antibody (1A7CL). For BiFC experiments, transfected cells were also gated by positive NS1 florescence, and both the NS1 MFI and the YFP MFI were analyzed for this group of cells. Cells were analyzed on BD FACSCaliber and the data was processed using FlowJo 7.6.4 software.
Statistical Analysis
Low MOI growth curves were analyzed using a two-way Analysis of Variance using Graph Pad Prism 6. Growth curve treatment groups were sampled in triplicate. Plaque diameter and the MFI in flow cytometry experiments were analyzed using a one-way analysis of variance using graph pad prism 6. MFI of gated cells was normalized to wild type protein expression and represent 3 experiments combined.
Highlights.
Mutations were introduced into influenza NS1 NLS1
NS1 R37A, R38A, K41A viruses had minimal changes in replication and NS1 localization.
Viruses from NS1 R35A rescue all contained additional mutations at D39.
NS1 R35A D39X mutations recover dimerization lost in NS1 R35A mutations
These results reaffirm the importance of dimerization for NS1 protein function
Acknowledgments
We thank all the members of the Pekosz laboratory for their advice, suggestions and input on this manuscript, and especially Celeste Newby for isolating the NS1 R38A and K41A viruses. We thank Dr. Suneil Mohan for assistance in flow cytometry data processing, and Dr. Phoebe Stavride and Dr. Sabra Klein for insightful discussions. The research in this manuscript was supported by funds provided by the Marjorie Gilbert Foundation and the Eliasberg Family Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Basu D, Walkiewicz MP, et al. Novel influenza virus NS1 antagonists block replication and restore innate immune function. J Virol. 2009;83(4):1881–91. doi: 10.1128/JVI.01805-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown LE, V, Hinshaw S, et al. Antigenic variation in the influenza A virus nonstructural protein, NS1. Virology. 1983;130(1):134–43. doi: 10.1016/0042-6822(83)90123-x. [DOI] [PubMed] [Google Scholar]
- Cheng A, Wong SM, et al. Structural basis for dsRNA recognition by NS1 protein of influenza A virus. Cell Res. 2009;19(2):187–95. doi: 10.1038/cr.2008.288. [DOI] [PubMed] [Google Scholar]
- Chien CY, Tejero R, et al. A novel RNA-binding motif in influenza A virus non-structural protein 1. Nat Struct Biol. 1997;4(11):891–5. doi: 10.1038/nsb1197-891. [DOI] [PubMed] [Google Scholar]
- Darapaneni V, V, Prabhaker K, et al. Large-scale analysis of influenza A virus sequences reveals potential drug target sites of non-structural proteins. J Gen Virol. 2009;90(Pt 9):2124–33. doi: 10.1099/vir.0.011270-0. [DOI] [PubMed] [Google Scholar]
- Donelan NR, Basler CF, et al. A recombinant influenza A virus expressing an RNA-binding-defective NS1 protein induces high levels of beta interferon and is attenuated in mice. J Virol. 2003;77(24):13257–66. doi: 10.1128/JVI.77.24.13257-13266.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes N, Selman M, et al. Identification of Adaptive Mutations in the Influenza A Virus Non-Structural 1 Gene That Increase Cytoplasmic Localization and Differentially Regulate Host Gene Expression. PLoS ONE. 2013;8(12):e84673. doi: 10.1371/journal.pone.0084673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieman M, Yount B, et al. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J Virol. 2007;81(18):9812–24. doi: 10.1128/JVI.01012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Albrecht RA, et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009;5(5):439–49. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sastre A, Egorov A, et al. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology. 1998;252(2):324–30. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- Greenspan D, Palese P, et al. Two nuclear location signals in the influenza virus NS1 nonstructural protein. J Virol. 1988;62(8):3020–6. doi: 10.1128/jvi.62.8.3020-3026.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale BG, Randall RE, et al. The multifunctional NS1 protein of influenza A viruses. J Gen Virol. 2008;89(Pt 10):2359–76. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- Han H, Cui ZQ, et al. New regulatory mechanisms for the intracellular localization and trafficking of influenza A virus NS1 protein revealed by comparative analysis of A/PR/8/34 and A/Sydney/5/97. J Gen Virol. 2010;91(Pt 12):2907–17. doi: 10.1099/vir.0.024943-0. [DOI] [PubMed] [Google Scholar]
- Hu CD, Chinenov Y, et al. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell. 2002;9(4):789–98. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- Jablonski JJ, Basu D, et al. Design, synthesis, and evaluation of novel small molecule inhibitors of the influenza virus protein NS1. Bioorg Med Chem. 2011;20(1):487–97. doi: 10.1016/j.bmc.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola TK. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat Protoc. 2006;1(3):1278–86. doi: 10.1038/nprot.2006.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochs G, Garcia-Sastre A, et al. Multiple anti-interferon actions of the influenza A virus NS1 protein. J Virol. 2007;81(13):7011–21. doi: 10.1128/JVI.02581-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Nussinov R. Close-range electrostatic interactions in proteins. Chembiochem. 2002;3(7):604–17. doi: 10.1002/1439-7633(20020703)3:7<604::AID-CBIC604>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Lee JH, Kim SH, et al. Direct interaction of cellular hnRNP-F and NS1 of influenza A virus accelerates viral replication by modulation of viral transcriptional activity and host gene expression. Virology. 2009;397(1):89–99. doi: 10.1016/j.virol.2009.10.041. [DOI] [PubMed] [Google Scholar]
- Li S, Min JY, et al. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology. 2006;349(1):13–21. doi: 10.1016/j.virol.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Li Y, Lu X, et al. Genetically engineered, biarsenically labeled influenza virus allows visualization of viral NS1 protein in living cells. J Virol. 2010;84(14):7204–13. doi: 10.1128/JVI.00203-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yamakita Y, et al. Regulation of a nuclear export signal by an adjacent inhibitory sequence: the effector domain of the influenza virus NS1 protein. Proc Natl Acad Sci U S A. 1998;95(9):4864–9. doi: 10.1073/pnas.95.9.4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lynch PA, et al. Crystal structure of the unique RNA-binding domain of the influenza virus NS1 protein. Nat Struct Biol. 1997;4(11):896–9. doi: 10.1038/nsb1197-896. [DOI] [PubMed] [Google Scholar]
- Maroto M, Fernandez Y, et al. Development of an HTS assay for the search of anti-influenza agents targeting the interaction of viral RNA with the NS1 protein. J Biomol Screen. 2008;13(7):581–90. doi: 10.1177/1087057108318754. [DOI] [PubMed] [Google Scholar]
- Melen K, Kinnunen L, et al. Nuclear and nucleolar targeting of influenza A virus NS1 protein: striking differences between different virus subtypes. J Virol. 2007;81(11):5995–6006. doi: 10.1128/JVI.01714-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melen K, Tynell J, et al. Influenza A H3N2 subtype virus NS1 protein targets into the nucleus and binds primarily via its C-terminal NLS2/NoLS to nucleolin and fibrillarin. Virology Journal. 2012;9(1):167. doi: 10.1186/1743-422X-9-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min JY, Krug RM. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′-5′ oligo (A) synthetase/RNase L pathway. Proc Natl Acad Sci U S A. 2006;103(18):7100–5. doi: 10.1073/pnas.0602184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min JY, Li S, et al. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology. 2007;363(1):236–43. doi: 10.1016/j.virol.2007.01.038. [DOI] [PubMed] [Google Scholar]
- Murayama R, Harada Y, et al. Influenza A virus non-structural protein 1 (NS1) interacts with cellular multifunctional protein nucleolin during infection. Biochem Biophys Res Commun. 2007;362(4):880–5. doi: 10.1016/j.bbrc.2007.08.091. [DOI] [PubMed] [Google Scholar]
- Nayak MK, Agrawal AS, et al. Antiviral activity of baicalin against influenza virus H1N1-pdm09 is due to modulation of NS1-mediated cellular innate immune responses. Journal of Antimicrobial Chemotherapy. 2014 doi: 10.1093/jac/dkt534. [DOI] [PubMed] [Google Scholar]
- Nemeroff ME, Qian XY, et al. The influenza virus NS1 protein forms multimers in vitro and in vivo. Virology. 1995;212(2):422–8. doi: 10.1006/viro.1995.1499. [DOI] [PubMed] [Google Scholar]
- Neumann G, Watanabe T, et al. Generation of influenza A viruses entirely from cloned cDNAs. Proc Natl Acad Sci U S A. 1999;96(16):9345–50. doi: 10.1073/pnas.96.16.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newby CM, Sabin L, et al. The RNA binding domain of influenza A virus NS1 protein affects secretion of tumor necrosis factor alpha, interleukin-6, and interferon in primary murine tracheal epithelial cells. J Virol. 2007;81(17):9469–80. doi: 10.1128/JVI.00989-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, et al. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108(2):193–9. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Nyfeler B, Michnick SW, et al. Capturing protein interactions in the secretory pathway of living cells. Proc Natl Acad Sci U S A. 2005;102(18):6350–5. doi: 10.1073/pnas.0501976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz B, Rejaibi A, et al. IFNbeta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell Microbiol. 2007;9(4):930–8. doi: 10.1111/j.1462-5822.2006.00841.x. [DOI] [PubMed] [Google Scholar]
- Qian XY, Alonso-Caplen F, et al. Two functional domains of the influenza virus NS1 protein are required for regulation of nuclear export of mRNA. J Virol. 1994;68(4):2433–41. doi: 10.1128/jvi.68.4.2433-2441.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian XY, Chien CY, et al. An amino-terminal polypeptide fragment of the influenza virus NS1 protein possesses specific RNA-binding activity and largely helical backbone structure. Rna. 1995;1(9):948–56. [PMC free article] [PubMed] [Google Scholar]
- Reed LJ, Muench H. A simple method of estimating fifty per cent endpoints. The American Journal of Hygiene. 1938;27(3):493–497. [Google Scholar]
- Sato Y, Yoshioka K, et al. Localization of influenza virus proteins to nuclear dot 10 structures in influenza virus-infected cells. Virology. 2003;310(1):29–40. doi: 10.1016/s0042-6822(03)00104-1. [DOI] [PubMed] [Google Scholar]
- Satterly N, Tsai PL, et al. Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc Natl Acad Sci U S A. 2007;104(6):1853–8. doi: 10.1073/pnas.0610977104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talcott B, Moore MS. Getting across the nuclear pore complex. Trends Cell Biol. 1999;9(8):312–8. doi: 10.1016/s0962-8924(99)01608-6. [DOI] [PubMed] [Google Scholar]
- Tan Z, Akerstrom S, et al. A new panel of NS1 antibodies for easy detection and titration of influenza A virus. J Med Virol. 2010;82(3):467–75. doi: 10.1002/jmv.21709. [DOI] [PubMed] [Google Scholar]
- Taylor JM, Illmensee R, et al. Use of specific radioactive probes to study transcription and replication of the influenza virus genome. J Virol. 1977;21(2):530–40. doi: 10.1128/jvi.21.2.530-540.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkiewicz MP, Basu D, et al. Novel inhibitor of influenza non-structural protein 1 blocks multi-cycle replication in an RNase L-dependent manner. J Gen Virol. 2011;92(Pt 1):60–70. doi: 10.1099/vir.0.025015-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Riedel K, et al. RNA binding by the novel helical domain of the influenza virus NS1 protein requires its dimer structure and a small number of specific basic amino acids. Rna. 1999;5(2):195–205. doi: 10.1017/s1355838299981621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Basler CF, et al. Functional replacement of the carboxy-terminal two-thirds of the influenza A virus NS1 protein with short heterologous dimerization domains. J Virol. 2002;76(24):12951–62. doi: 10.1128/JVI.76.24.12951-12962.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Zheng F, et al. Interaction of avian influenza virus NS1 protein and nucleolar and coiled-body phosphoprotein 1. Virus Genes. 2013;46(2):287–292. doi: 10.1007/s11262-012-0849-z. [DOI] [PMC free article] [PubMed] [Google Scholar]