

Abstract
Tuberculosis is a major cause of death in mankind and BCG vaccine protects against childhood but not adult tuberculosis. BCG avoids lysosomal fusion in macrophages decreasing peptides required for activating CD4 T cells and Th1 immunity while suppressing the expression of MHC-II by antigen presenting cells (APCs). An in vitro model of antigen presentation showed that ligands for TLR-9, 7, 4 and 1/2 increased the ability of APCs to present antigen-85B of BCG to CD4 T cells, which correlated with an increase in MHC-II expression. TLR-activation led to a down-regulation of MARCH1 ubiquitin ligase which prevents the degradation of MHC-II and decreased IL-10 also contributed to an increase in MHC-II. TLR-activation induced up-regulation of MHC-II was inhibited by the blockade of IRAK, NF-kB, and MAPKs. TLR-7 and TLR-9 ligands had the most effective adjuvant like effect on MHC-II of APCs which allowed BCG vaccine mediated activation of CD4 T cells.
Keywords: MHC-II, Macrophage, Dendritic cells, BCG vaccine, Antigen processing, Toll-like receptor (TLR), MARCH1 ubiquitin ligase, IL-10, CD86, CD80
[2] Introduction
Tuberculosis (TB) is a major public health problem with 2 million deaths per year and 8 million new cases each year. Mycobacterium bovis derived BCG is the only approved vaccine that protects children less than 5 years of age against extra-pulmonary and military disease. However, meta-analysis shows that the protective efficacy of BCG against pulmonary tuberculosis ranges from 0- 80% and it does not protect against adult disease. Several reasons have been proposed for its variability, including antigenic drift, genetic and nutritional differences among humans, exposure of humans to environmental mycobacteria, and differences in testing methods. Interestingly, Rook et al. have proposed that BCG induced Th1 responses progressively switch to Th2, which may undermine the Th1 immunity[1, 2]. Thus, the focus of BCG vaccine research has shifted to the ability of vaccine infected macrophages (MΦs) and dendritic cells (DCs) (antigen presenting cells; APCs) to activate T cells thereby amplifying Th1 immunity.
When M. tuberculosis or BCG vaccines are internalized by APCs, they are within membrane bound phagosomes, which are then sorted into lysosomes for degradation and generation of peptides [3, 4]. Peptides are bound to MHC-II complex in specialized MIIC compartments and exported to the plasma membrane for presentation to CD4 T cells. BCG secretes proteins that escape into cytosol to be processed by the proteasome and such peptides are loaded into MHC-I in the endoplasmic reticulum to be presented on the surface of APCs to CD8 T cells. CD4 and CD8 T cells together mediate the Th1 immunity to tuberculosis.
However, two types of defects in antigen processing appear to affect the ability of BCG infected APCs to prime T cells. Cell biologists have found that BCG sequesters within immature phagosomes of macrophages, which do not fuse with lysosomes [5]. Since lysosomes produce peptides from BCG through proteolytic degradation, it was apparent that activation of CD4 T cells could be less than optimal[6]. We reported the first mechanistic basis for defects during processing that sequestration of BCG within immature phagosomes reduces antigen presentation by MΦs [7]. BCG secretes a major antigen-85B (Ag85B) and others reported that limited cleaving of Ag85B and assembly into MHC-II occurs within mycobacterial phagosomes [8] [9]. Ag85B-peptide epitope loaded MHC-II complexes are exported to plasma membrane where they trigger a specific CD4 cell hybridoma to release IL-2. Using this model of in vitro antigen presentation, we demonstrated that the Cathepsin-D protease cleaved Ag85B within the BGG phagosome, but its activation was dependent upon an acidic lumen pH [7]. Paradoxically, BCG phagosome lumen had a near neutral pH due the exclusion of vacuolar proton ATPase, providing a molecular basis for the poor processing of Ag85B. Other investigators have confirmed similar processing defects by demonstrating that BCG expressing Cat-S enables better presentation of Ag85B to CD4 T cells, and that a zmp mutant of BCG which undergoes phagosome-lysosome fusion in APCs facilitates better presentation of an Ag85A derived epitope to T cells via MHC-II[10, 11]. In addition to these defects in MHC-II dependent pathways, the ability of MΦs to process peptides for MHC-I has also been found inefficient. BCG expressing the pore forming toxin listeriolysin (LLO), was reported to leak more peptides across phagosome membrane into cytosol, where they are processed and loaded into MHC-I. Recombinant BCG expressing LLO thus generated better CD8 T cell responses in mice [12]. Thus, it is evident that immunological sequestration of BCG can reduce its efficacy as a vaccine. On the other hand, genetic manipulation of BCG has been found to benefit its efficacy[13]. A major development was the over-expression of secreted Ag85B in BCG which increased its efficacy [14]. We reported that overexpressed Ag85B triggers autophagy in MΦs that sorts BCG to lysosomes, increasing antigen presentation and enhanced vaccine efficacy against tuberculosis of mice [15]. Finally, BCG expressing Th1-immunity boosting cytokines have been found marginally better vaccines [16] [17]. These observations emphasize that genetic manipulations to improve the BCG vaccine should perhaps be combined with adjuvant activation APCs to facilitate better Th1 immunity.
In this context, we hypothesized that the efficacy of BCG in humans is likely determined by two additional factors. The variable protection by BCG could be due to inefficient MHC-II dependent presentation of antigens by APCs to T cells. Indeed, variations in the ability of human HLA to present antigens are thought to exist but are probably obscured in the common variable immunodeficiency among humans (CVID) [18]. A second scenario is that BCG-derived lipids are known to suppress the expression of MHC-II in APCs of mice, and BCG could also be suppressing, comparable HLA dependent processes in humans [19, 20].
In this study, we provide a mechanistic basis for the suppression of MHC-II in BCG infected APCs and suggest an approach to avoid immune-suppression during vaccination. MHC-II molecules are synthesized in the endoplasmic reticulum and are exported to MIIC compartments for peptide loading. Defunct or membrane internalized MHC-II molecules are degraded through proteasome after they are tagged with ubiquitin by an enzyme termed MARCH1 [21]. While MHC-II molecules are constitutively synthesized, their expression can be increased through activation of MΦs either with cytokines like IFN-γ or activation of DCs via Toll-like receptors (TLRs) [22] [23]. We report that BCG derived lipids suppress the expression of MHC-II in APCs but prior activation with certain TLR-ligands can either restore or increase the expression of MHC-II, enhancing their ability to present antigen.
[3] Materials and Methods
[3.1] Macrophages and DCs
Primary bone marrow derived macrophages (MΦs) from C57Bl/6 mice (4-8 weeks old, male or female from Jackson Laboratory, USA) were grown in Iscove's medium with 10% FBS (IDM) and 10 ng/mL GM-CSF and CD11c beads (Miltenyi INc, USA; 130-052-001) were used to deplete DCs. CD11b+ CD11c- MΦs were plated in GM-CSF free medium and used for activation and infection. MΦ-derived BMA.A1 macrophage or DC2.4 dendritic cell lines (kind gift of Dr. Kenneth L. Rock, U. Massachusetts, USA) were similarly cultured in IDM and used for many experiments. MΦs or DCs were activated with TLR-ligands (1-5 μg/mL; dissolved in sterile endotoxin-free water, Invivogen, USA) and incubated overnight at 37°C. They were then infected with Mycobacterium bovis BCG (Pasteur strain, ATCC-35734; MOI=1), prepared as a single cell suspension, for 4 hr on a shaker at 37°C, washed and collected for further analysis. Viability was > 90% using the live-dead fluorescent stain (Invivogen, USA).
[3.2] In vitro antigen presentation assay
This has been described in detail earlier [7]. Briefly, MΦs were treated or not with either mycobacterial lipid inhibitors of TLR-signaling (19 kDa lipoprotein and lipoarabinomannan) or activated with commercial TLR-ligands, followed by infection with BCG (MOI=1). Washed monolayers were incubated with BB7 T cell hybridoma (kind gift of Dr. Clifford Harding, CWRU) (1:20 ratio) and supernatants collected either 4 or 18 hr after overlay and tested for IL-2 using sandwich ELISA.
[3.3] Inhibition of TLR-signaling
Various inhibitors of mitogen activated kinases (MAPK), AP-1/CREB were used to block signaling due to TLRs. The inhibitors were used at doses recommended by the manufacturer. The inhibitors used were: Calbiochem-EMD (SB 202190, p38 map kinase inhibitor II, #559388; FR180204 ERK Inhibitor #328006; 420119 JNK Inhibitor II; # 420119; and 217505 CBP-CREB Interaction Inhibitor, # 217505. Other reagents were NF-kB inhibitor peptide set (#2004; Imgenex, USA), IRAK1/4 inhibitor (#15409, Sigma, USA) and SR 11302 AP-1 inhibitor (Tocris, USA). MΦs were treated with inhibitors for 2 hr, activated with TLR-ligands for 2 hr and infected with BCG for 2 hr prior to fixation and assay of surface MHC-II or antigen presentation. This rapid procedure was adapted to ensure >90% viability of MΦs assayed using trypan blue.
[3.4] siRNA down-regulation of MARCH1
MΦs were treated with siRNA and scrambled siRNA directed against MARCH1 (sc-106199; sense/antisense and scrambled, Santacruz Biotechnology, USA;) as recommended by the manufacturer and rested for 14 hr. They were tested naïve, or infected with BCG for 4 hr followed by surface expression of MHC-II measured using flow cytometry. The sequences of the siRNA probes were as follows: (mix of three: Sense: GCAAGUCGAUCAAGUAACAtt; Antisense: UGUUACUUGAUCGACUUGCtt Sense: GUAGUUAGCUGUCGAAAGAtt;Antisense: UCUUUCGACAGCUAACUACtt Sense: CCAUAUCCAUGAACUUUGUtt;Antisense: ACAAAGUUCAUGGAUAUGGtt (5′ → 3′ orientation).Scrambled sequence was also obtained from the same company.
[3.5] Immunofluorescent staining of cells
MΦs or DC.2.4 cell line were stained using antibodies against MHC-II, MHC-I, CD80, CD86, IL-10 and MARCH1 (MHC-II,#12-5322; CD80,#12-0801; CD86, #12-0862;IL-10, #11-7101; MHC-I, #17-5985, all from Ebiosciences USA) (MARCH1, #D16-sc104369; Santacruz Biotechnology, USA). After incubation for 30 min on ice with antibodies, cells were washed with PBS and fixed with Cytofix/Cytoperm buffer (BD biosciences, #555028). Intracellular staining was performed using antibodies for MARCH-1 and IL-10. Cells were incubated for 30 mins and washed with PBS. Cells stained for IL-10 were incubated in brefeldin-A for 30 min but the cells stained for MARCH1 assay did not require brefeldin-A since MARCH1 is not secreted.
[3.6] Flow cytometry
The fixed cells were acquired and analyzed for surface or intracellular markers using Gallios flow cytometer (Beckman Coulter) and the data analysis was carried out using Flow-Jo software (Treestar, Ashland). To calculate Percent positive macrophages staining for MHC-II or other markers, isotype stained cells were marked M1 gate and the stained cells on X-axes as M2 (BD Biosciences manual). Median Fluorescent intensity (MFI) values from two or three separate experiments were averaged and differences analyzed using unpaired t test (Graphpad prism software).
[3.7] Immunoprecipitation and Western Blot analysis of ubiquinated MHC-II
MΦs in 6 well plates were treated with TLR-ligands at a concentration of 1 μg/mL or none overnight at 37°C. They were then infected with BCG for 4 hr (MOI=1) on a shaker at 37°C. Cells were centrifuged for 10 min (3,000 × g, 4°C) and the supernatant was removed. Cells were resuspended in 30 μL of the lysis buffer (mammalian Protein Extraction Reagent (M-PER, Pierce) with 5 μM MG132, a proteasome inhibitor, and a protease inhibitor cocktail (112 μM PMSF, 3 μM aprotinin, 112 μM leupeptin, 17 μM pepstatin). Samples were incubated with shaking at 4°C for 1 hr, centrifuged for 20 min (10,000 rpm 4°C) and nuclei free supernatants were collected and estimated for protein and normalized. One μg of anti-mouse MHC-II antibody (Ebioscience, #14-5321) was added per sample, which were gently mixed overnight at 4°C. Fifteen μL of Protein A/G PLUS-Agarose beads (Santacruz Biotechnology, USA) were added to each sample and gently mixed for 4 hr at 4°C. Supernatants were removed though centrifugation (2000 rpm, 15 min) and beads were washed three times with 1× PBS. Sample buffer was added to each sample, and each tube was boiled for 5 minutes to elute proteins from beads. Samples were loaded onto a 12% SDS-PAGE gel and electroblotted onto a PVDF membrane. Western blotting was performed using an antibody directed towards polyubiquitin (Ebioscience, #14-6078).
[3.8] Statistical analysis
The unpaired t-test was used when comparing two groups and the Dunnett's procedure was used to compare several groups within a group to a control (Graphpad Prism software).
[4] Results
[4.1] Mycobacterial lipids decrease and TLR-ligands increase in vitro antigen presentation by MΦs
It has been previously reported that BCG infection leads to suppression of MHC-II expression in vitro among MΦs and in phagocytes of mice infected with BCG [19] [24]. The 19 kDa mycobacterial lipoprotein was also found to inhibit MHC-II expression in MΦs through a TLR-1/2 dependent mechanism [19]. It is well established that MΦs infected with BCG and M. tuberculosis rapidly present an epitope from secreted Ag85B to BB7 T cells in vitro [8, 9]. To determine if reduced MHC-II levels of BCG infected MΦs affected their ability to present peptides to T cells, naïve MΦs were treated with 19 kDa lipoprotein and LAM prior to infection with BCG and BB7 T cell stimulation in vitro. Fig.1a shows that the lipids derived from BCG, which are also present in M. tuberculosis, inhibited antigen presentation. Since the TLR-ligand 19 kDa lipoprotein is secreted from BCG, it follows that during vaccination, BCG could potentially affect antigen processing in MΦs. Since MΦs express multiple TLRs, we sought to determine if prior activation with TLR-ligands could affect the antigen processing. Fig.1b illustrates that, prior activation of MΦs with ligands for TLR-9, 7, 5, 4 and 1/2 enhanced the antigen presentation. These results together indicate that, at least two lipids present in BCG and M. tuberculosis are immuno-suppressive, decreasing antigen presentation. The suppressive effect can however be bypassed with activation of TLRs including additional activation with a synthetic ligand of TLR-2 (Pam3CSK4). Fig.1c illustrates that the lipids also suppressed presentation of soluble Ag85B processed by the MΦs.
Figure-1. Effect of lipid antigens of Mycobacterium bovis BCG vaccine and TLR-activation on the ability of mouse macrophages to process and present a peptide antigen 85B derived epitope from BCG to T cells.


a) Lipid antigens inhibit antigen presentation. C57Bl/6 derived primary macrophages (MΦs) were treated with or without 19 kDa lipoprotein or lipoarabinomannan (LAM) TLR-agonists as indicated, followed by infection with BCG vaccine (MOI=1), washed and overlaid with Antigen-85B specific BB7 CD4 T cells (1:20). IL-2 in supernatant (pg/mL ± SD for triplicate wells; 2 similar experiments) collected 4 hr later was measured using sandwich ELISA. b) TLR-ligands for TLR-1/2, 4, 5, 7 and 9 enhance antigen presentation. MΦs were treated with TLR-agonists as indicated (1 µg/mL), followed by BCG infection and T cells. c) MΦs were treated with or without recombinant Ag85B (10 ng/mL) for 2 hr followed by addition of lipids for 2 hr (increasing doses as in Fig.1a). MΦs were washed and overlaid with T cells for 4 hr and IL-2 determined as above. Lipids suppress presentation of soluble Ag85B to T cells. MΦs infected with BCG alone, MΦs treated with ligands alone and uninfected MΦs without T cells are shown as controls (panels b and c; 3 experiments each; p values shown above bars using t test with Dunett's comparison).
[4.2] TLR-ligands increase MHC-II expression in BCG infected MΦs
Within BCG infected MΦs, Ag85B is cleaved to produce a peptide epitope, which is loaded into MHC-II, and the complex is presented to T cells on the surface [8, 9] [7]. Thus, the surface levels of peptide loaded MHC-II determines the efficacy of T cell activation, which in this model, leads to IL-2 secretion. MHC-II molecules are continuously synthesized by MΦs, and surface expressed MHC-II are internalized, ubiquitinated via MARCH1 enzyme and targeted to proteasomes for degradation [3] [25]. To determine if TLR-activation modulated these processes, MΦs were activated with TLR-ligands, followed by infection with BCG, and analyzed using flow cytometry for surface expression of MHC-II and two other costimulatory molecules (CD80, CD86) that participate in peptide presentation to CD4 T cells. Fig.2a, illustrates that TLR-activation followed by BCG infection enhanced MHC-II expression relative to BCG infected MΦs. Interestingly, a reverse pattern was observed with MARCH1 levels, the latter being reduced in TLR-activated MΦs. TLR-activation did not affect the expression of MHC-I by MΦs. Fig2b shows the averaged MFI values of receptor expression in MΦs from three experiments. Ligands for TLR-9 & 7 were the strongest activators of MHC-II followed by those for TLR-4 and TLR-1/2. During antigen presentation to CD4 T cells, MHC-II complexes with CD80 and CD86 to optimally prime T cells [3]. TLR-ligands generally did not affect the expression of co-stimulatory molecules except that of CD86 which was enhanced by TLR-9 ligand. Finally, IL-10 has been reported to down-regulate MHC-II expression by MΦs and DCs through an up-regulation of MARCH1 [25, 26]. IL-10 secreted from T-regs induced during infection can also increase in MARCH1 and down-regulate MHC-II [27]. Data show that the ligands for TLR-9 and TLR-7 decreased the intracellular levels of IL-10.
Figure-2. MΦs infected with BCG enhance the expression of surface MHC-II which correlates with elevated intracellular MARCH-1 ubiquitin ligase levels and reduced intracellular IL-10.
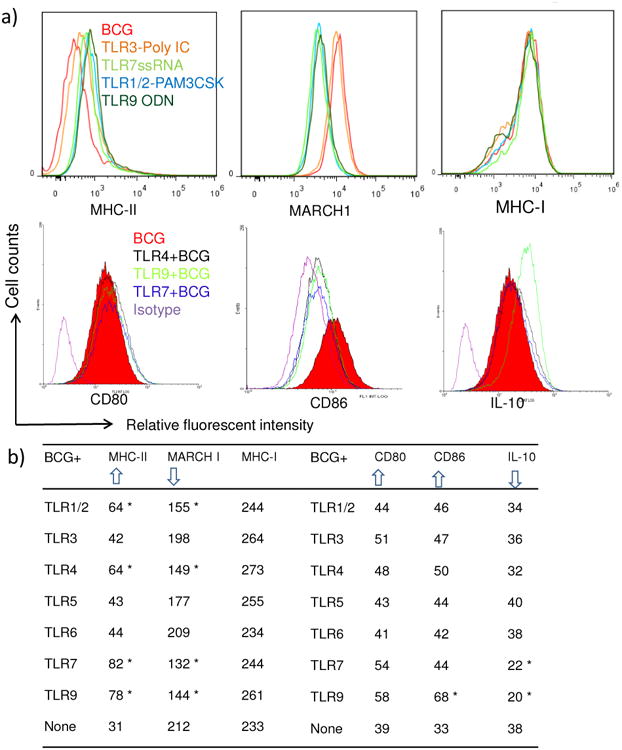
C57Bl/6 macrophage derived BMA.A4 macrophage cell line (MΦs) were activated with TLR-ligands overnight and then infected with BCG for 4 hr, washed and tested for surface expression of receptors, intracellular MARCH1 enzyme and IL-10 using specific antibodies with isotype controls followed by flow cytometric analysis. a) Representative histograms from 2 separate similar experiments illustrate that TLR-agonists enhance MHC-II expression in MΦs compared to BCG infection (top left); TLR-agonists down-regulate ubiquitin ligase MARCH-1 compared to BCG infected MΦs (top middle), and that TLR-agonists have no effect in MHC-I expression by MΦs (top right). Likewise, selected TLR-ligands are shown to affect the expression of CD80, CD86 and intracellular IL-10 in MΦs (bottom row). b) Table summarizes data of median fluorescent intensity (MFI) values averaged from three separate experiments for various groups of MΦs analyzed using flow cytometry (* p < 0.01 compared to expression of receptors in MΦs infected only with BCG, t test). Arrow heads indicate up- or down-regulation of receptors, MARCH1 and IL-10.
[4.3] TLR-ligands decrease the ubiquitination of MHC-II
To determine whether TLR-activation led to changes in the intracellular levels of ubiquitinated-MHC-II, MΦs were activated or not with TLR-ligands, infected with BCG and the cytosol tested for ubiquitinated-MHC-II using immune-precipitation with MHC-II antibody followed by labeling with an antibody to polyubiquitin. Fig.3a shows that TLR-ligands (7>9>5>1/2) significantly reduced the levels of ubiquitinated-MHC-II relative to those from BCG infected MΦs. To confirm the role of MARCH1 in the regulation of MHC-II, MΦs were then treated with siRNA vs. MARCH1 and surface MHC-II levels were measured using flow cytometry. Fig.3b shows that the down-regulation of MARCH1 leads to an increase in the surface expression of MHC-II among BCG infected MΦs.
Figure-3. TLR-activation decreases the ubiquitination of MHC-II in BCG infected MΦs & siRNA knock-down of MARCH1 enhance surface expression of MHC-II levels.
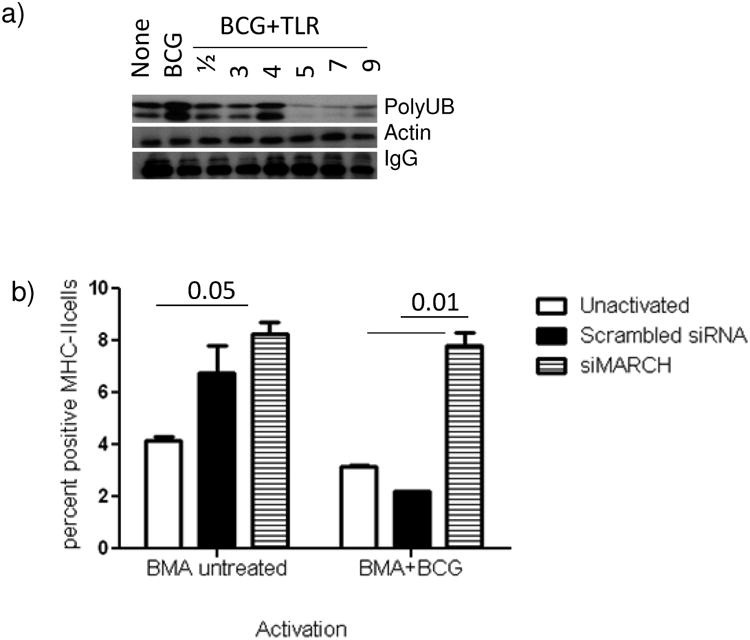
a) BMA.A4 MΦs of duplicate wells were activated with TLR-ligands (1 μg/mL for 18 hr), infected with BCG (MOI=1) for 4 hr, lysed and pooled. An antibody to MHC-II antibody was used to capture MHC-II from lysates at 4°C overnight and the immunoprecipitate was subjected to western blot using an antibody to polyubiquitin. Panel shows that MΦ-activation with TLR-agonists for 9/7/5/3 and 1/2 decrease the levels of ubiquitinated MHC-II relative to BCG infected control (one of three similar experiments shown). b) MΦs were treated with siRNA vs. MARCH1 (Santa Cruz Biotechnology) or scrambled control siRNA for 18 hr, rested for 18 hr, and infected with BCG for 4 hr. Surface expression of MHC-II was measured using flow cytometry, and data were expressed as log positive cells. Knock-down of MARCH1 increases surface expression of MHC-II, even in BCG infected macrophages. Data averaged for two separate experiments (p value indicated for groups compared, t test).
[4.4] TLR-activation mediated increase in MHC-II expression by MΦs is dependent upon MAP kinases and AP-1/CREB activation
MHC-II expression is regulated by multiple pathways in MΦs and DCs. However, most common mechanisms seem to converge at the binding of transcription factors like AP1 and CREB to the ‘Class-II transactivator’ (CIITA) prior to MHC-II synthesis (Fig.4a). There are however alternative routes for MHC-II synthesis via NF-kB activation, suggesting that TLR-ligands may differ in the mechanism of induction of MHC-II through MAPKs (p38, ERK1/2 and JNK) and NF-kB besides other down-stream effectors. To determine if TLR-signaling affected MHC-II expression, MΦs were blocked with specific inhibitors of these pathways (inhibition at steps 1-5; Fig.4a), followed by TLR-activation, BCG infection and surface staining for MHC-II. Fig.4b-g illustrates representative histograms from two similar experiments. Activation of the four TLRs (9,7,4 and 1/2) enhanced MHC-II in MΦs significantly compared to the BCG infected MΦs, while, inhibition of MAPKs led to an inhibition of MHC-II. Fig.4h shows averaged MFI values form two experiments and confirms that ligands for TLR-9 and TLR-7 followed by those for TLR-4 and TLR-1/2 are strong inducers of MHC-II in MΦs. Blockade of MAPKs and AP-1/CREB decreased the MHC-II expression.
Figure-4. TLR-activation increases MHC-II expression in macrophages and is dependent upon the activation of p38 MAPK, ERK1/2 and transcription factors AP-1 and CREB.

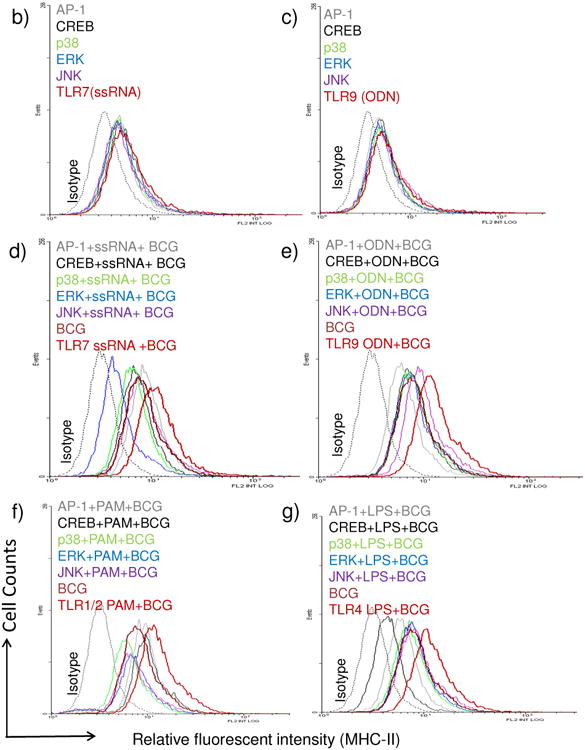

a) Cartoon outlines putative pathways of TLR-mediated activation of MHC-II expression. b-h) MΦs were incubated with inhibitors from Calbiochem, USA (Methods) for 2 hr, followed TLR-activation for 2 hr and BCG infection for 2 hrs. Trypan blue vital dye was used to ensure that macrophages were > 90% viable. MΦs were stained for surface MHC-II, fixed in 2.7% paraformaldehyde followed by flow cytometric analysis (2 separate experiments with duplicate samples in each). One histogram for each TLR-ligand illustrated. MFI values were averaged from two separate similar experiments and are shown in the Table under panel (h) (* p < 0.01 vs. BCG infected MΦs). (b-c) Inhibitors alone or TLR-ligands alone had no significant effect on MHC-II expression of MΦs. (d-e) TLR-7 (ssRNA) and TLR-9 (ODN1826) increased the expression of MHC-II, which was inhibited by blockade of p38 & ERK, AP-1 or CREB. (f-g) TLR-1/2 (Pam3CSK4) and TLR-4 (LPS) ligands induced an up-regulation of MHC-II which was differentially affected by blockade of MAPKs and AP-1 or CREB. i) MΦs were treated with a control peptide (NF-cp) or a peptide blocking NF-kB (NF-tp) or an inhibitor for IRAK1/4 (IRK) (all tested at 1 μM) for 2 hr and followed by activation with TLR-9, TLR-2 or TLR-4 ligands for 2 hr and infection with BCG for 4 hr followed by BB7 T cell overlay for 4 hr. IL-2 was determined using sandwich ELISA. Data show that both NF-kB and IRAK inhibition reduce antigen presentation among TLR-activated MΦs (**p < 0.02, t test with Dunett's comparison).
[4.5] Blockade of TLR-signaling inhibits antigen presentation in DCs
Fig.4b-h illustrate that inhibition of TLR-signaling reduced MHC-II expression in MΦs. Since MHC-II is essential for antigen presentation using BB7 CD4 T cells (Fig.1), MΦs were further analyzed to determine if blockade of TLR-signaling affected antigen presentation. IRAK-4 occupies a pivotal position for ligands activating TLR-1/2, TLR-4, TLR-7 and TLR-9 (Fig.4a). NF-kB has also been found a key down-stream player during TLR-induced cytokine secretion and MHC-II synthesis [28-30]. MΦs treated with inhibitors for either IRAK1/4 or NF-kB, activated with ligands for TLR-9 TLR-2 and TLR4 followed by BCG infection showed a significant reduction in antigen presentation in vitro (Fig.4i).
[4.6] Blockade of TLR-signaling inhibits antigen presentation in DCs
DCs process vaccine derived antigens like MΦs but unlike MΦs they can more efficiently prime naïve T cells to induce a Th1 response [31]. Thus, MHC-II expression in DCs is also a critical factor inducing T cell mediated immunity. DCs were tested before and after TLR-activation as in Fig.4. Fig.5a-e illustrates that ligands for TLR-9 and TLR-7 were more potent enhancers of MHC-although they did not affect the levels of intracellular MARCH1. DCs activated with multiple TLR- ligands indicated that ligands for TLR-9>7>4>1/2 had the ability to enhance MHC-II and their effect was reduced significantly by blockade of MAPKs or AP1/CREB (Fig.5e). To confirm the effects of TLR-activation, DCs were activated and overlaid with T cells for antigen presentation. Data from two experiments show that TLR-9>7>4 and1/2 enhanced antigen presentation in vitro (Fig.5f).
Figure-5. TLR-activation increases MHC-II expression in a mouse dendritic cell line which correlates with enhanced antigen presentation.
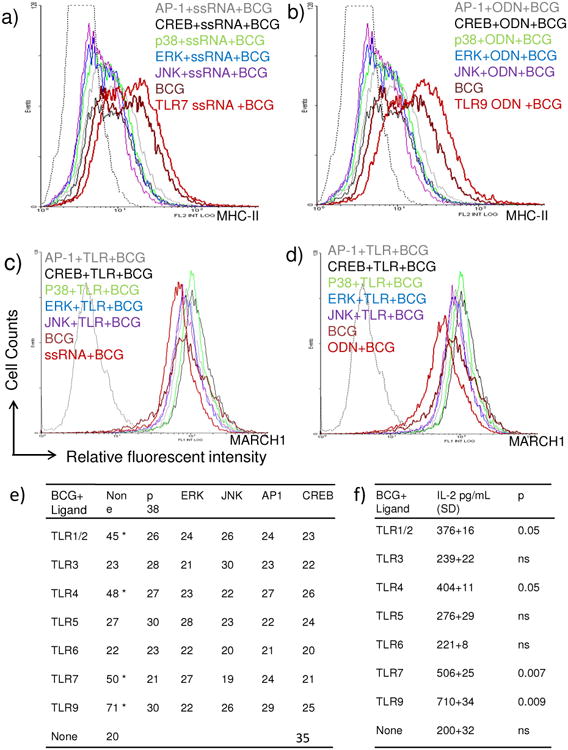
a-b) C57Bl/6 mouse bone marrow DC derived DC.2.4 cell line was activated and infected as in Fig.4. (a-b) Histograms illustrate that, TLR-7 and TLR-9 agonists enhance MHC-II and their effects are inhibited by blockade of MAPKs, AP-1 and CREB. c-d) Both agonists fail has no effect on the intracellular levels of MARCH1. e) DCs were activated with various TLR-ligands in Fig.4 and MFI values averaged from two separate experiments. Data show that ligands for TLR-9, 7, 4 and ½ enhance MHC-II. Blockade of MAPKs, AP-1 and CREB reduces expression of MHC-II or these TLR-activated DCs (* p < 0.04 vs. BCG infected DCs, t test). f) DCs were activated with various TLR-ligands in Fig.4 followed by BCG infection and IL-2 levels measured after antigen presentation. Antigen presentation by DCs is enhanced by prior activation with ligands for TLR-9, 7, 4 and 1/2 (* p values indicated for groups compared vs. BCG infected DCs, t test).
[5] Discussion
CD4 T cells play a major role in immunity against tuberculosis in animal models as well as humans. A decrease in the numbers of CD4 T cells during HIV-1 infection predisposes humans to a lethal co-infection with tuberculosis in sub-Saharan Africa and Asia, underscoring the importance of CD4 T cells. During vaccination, APCs present peptides of BCG through the MHC-II pathway and generate the CD4-dependent Th1 immunity to tuberculosis. Although BCG has been used as vaccine for over 90 years, certain inherent properties seem to prevent it from becoming a more effective vaccine. Initial studies reported immunological sequestration of BCG vaccine from human CD4 T cells [6]. In the early 90's, BCG vaccine was found sequestered in immature phagosomes of MΦs that failed to fuse with lysosomes [5]. Since mycobacterial peptides from lysosomal degradation provide epitopes which are loaded into MHC-II, we investigated the mechanisms of peptide processing and presentation in MΦs and DCs using various in vitro models, and animal models [7, 32] [33] [34]. The in vitro presentation of an Ag85B derived epitope to CD4 T cells has been used by multiple investigators for evaluation of the ability to APCs to process BCG vaccine and M. tuberculosis. [7, 10, 35]. Using this model, we found that APCs present Ag85B epitopes rather poorly from BCG [7]. Over the years we and others have activated the APCs with IFN-γ, LPS or rapamycin to enhance their ability to process BCG vaccine and increase its efficacy[36] [15]. A recombinant BCG-85B which hyper-expresses Ag85B and triggers autophagy in APCs is a stronger vaccine against tuberculosis in mice [15]. Supporting these observations, wild type BCG has been found to generate at best 1log10 decrease in the lung counts of M. tuberculosis in mice when used as vaccine while, improved recombinant vaccines like BCG-Ag85B induce nearly 2log10 decline [14] [15].
In striking contrast with the above studies where improved BCG vaccines were developed, this study addresses the issue that BCG has an inherent capacity to suppress MHC-II responses in APCs of mice. The 19 kDa lipoprotein of M. tuberculosis and BCG seems to suppress MHC-II through TLR-2 dependent processes, although other lipids could also be involved [35] [19] [20]. This issue has a major impact on the ability of APCs to activate CD4 T cells since, even if more peptide epitopes from genetically improved BCG are produced within APCs, sufficient MHC-II molecules are required on their surface to present them to CD4 T cells. Recent reports support continuation of BCG vaccination and even revaccination in developing countries[37]. Thus, the main goal of this study was to investigate whether APCs can be made to better present antigens of BCG through the MHC-II pathway.
Since BCG derived lipids affect APCs though TLR pathway, we sought to determine the effects of alternate TLR activation on their function. TLRs are single membrane-spanning, non-catalytic, receptor proteins which are similar to the protein of toll gene in Drosophila. They are present both on the membrane and in the cytosol of multiple cells in vertebrates and recognize specific microbial patterns. TLRs link the innate and adaptive immunity and allow the cell to recognize between self and non-self and while most human tissues express at least one TLR, APCs express multiple TLRs. Activation of TLRs triggers multiple pathways that lead to cytokine secretion (Fig.4a). We noted that, some TLR-ligands activated MHC-II in pig monocytes and, activation of human DCs with TLR-2, 3 and 4 ligands up-regulated HLA-DR expression coincident with the down-regulation of MARCH1[23] [38]. Infection with Toxoplasm gondii pathogen increased MHC-II levels of human DCs, through the down-regulation of MARCH1 suggesting that, ubiquitination of MHC-II is a major mechanism of regulating surface MHC-II during dendritic cell activation. It should however be noted that MHC-II molecules are continuously synthesized in response to infection, loaded with antigenic peptides in the MIIC compartment, and then exported to the plasma membrane, where, they prime CD4 T cells [3, 4]. While surface expressed MHC-II is the main determinant of CD4 T cell activation, interference with MHC-II synthesis, mechanisms of generation and loading of peptides, and sorting of MHC-II-peptide complex all seem to influence the MHC-II mediated activation of CD4 T cells. A recent study best illustrates this concept. DCs present lysozyme-derived peptides to CD4 T cells via MHC-II [39]. Interestingly, processing of lysozyme yields two types of peptide epitopes depending upon where it is cleaved. Type-I IFNs enhance presentation of early-endosome processed peptides, while, multiple TLR-ligands enhance those of late-endosome processed peptides. BCG which sequesters within immature phagosomes, but secretes the immune-dominant Ag85B, has presented a challenging model to explore antigen presentation in the context of vaccination. In this direction, we reported that BCG phagosomes are enriched with various host proteins that seem important for antigen processing but are yet to be mechanistically characterized[33, 40] [41]. Based on these studies, we proposed that sequestered BCG may not expose its repertoire of antigens to lysosome mediated degradation thereby preventing adequate expansion of T cells. However, secreted Ag85B could either be cleaved within the phagosomes [9], or they could leak to the cytosol and are processed via proteasomes or targeted to lysosomes.
The novelty of this study is that, an in vitro model was used to correlate the MHC-II levels of APCs with their ability to present BCG peptides to CD4 T cells in vitro, which in turn, is a predictor of BCG vaccine efficacy in the mouse model of tuberculosis [7, 15]. The functional assay which combined MHC-II studies with peptide presentation by MΦs revealed several interesting observations. Ligands for TLR-3 and TLR-6 were not effective in increasing peptide presentation by BCG infected MΦs (Fig. 1). The TLR-5 ligand flagellin enhanced both MHC-II and reduced MARCH1 but was excluded from further analysis since BCG lacks flagella. Of the remaining TLRs, ligands for TLR-9>7>4>1/2 enhanced MHC-II levels, in vitro antigen presentation and decreased the levels of MARCH1 in MΦs (Figs. 1-4). All four ligands also enhanced MHC-II in DCs although they failed to down-regulate MARCH1 in the DCs during the short term flow cytometry assay (Fig. 5). Although it is possible that time dependent down-regulation of MARCH1 could have become apparent in DCs after TLR-activation, alternate mechanisms regulating the degradation of MHC-II may also exist in DCs. Of the four ligands tested, TLR-9 and TLR-7 ligands were robust inducers of MHC-II via down-regulation of MARCH1 besides IL-10. Since IL-10 suppresses MHC-II through the induction of MARCH1, it follows that TLR-9 and TLR-7 ligands may mediate their effects on MHC-II in part through the down regulation of IL-10 [25, 42].
Traditionally, TLR-ligands have been thought to skew cytokine responses of APCs, although recent research is unraveling other mechanisms. Thus, we reported the unique mechanism of TLR-4 ligand LPS, which induced autophagic sorting of BCG to lysosomes facilitating their degradation[43]. Other investigators found that TLR-1/2, 3 and 7 ligands also induced autophagy in mycobacteria infected macrophages[44]. It is clear that certain TLR-ligands can sort BCG to lysosomes via autophagy and increase the levels of antigenic peptides which may in turn boost the overall synthesis of MHC-II molecules.
Another major observation that emerged from this study is that, TLR-9 and TLR-7 ligands were the most effective activators of MHC-II and also reduced the levels of anti-inflammatory cytokine IL-10 in MΦs. This observation has an important implication for adjuvant development for tuberculosis vaccines. TLR-ligands have been frequently used as adjuvants along with sub-unit protein vaccines against tuberculosis. TLR-4 ligands like monophosphoryl lipid-A (MPL) boost the efficacy of several sub-unit vaccines given either as primary vaccines or as boosters in animal models of tuberculosis [45]. MPL has also been combined with alum to enhance the efficacy of subunit vaccines [46]. In addition, some TLR-2 activating proteins of mycobacteria have been found to boost subunit vaccines [47] [48]. Our data suggest that TLR-9 and TLR-7 ligands are likely to be more potent adjuvants than MPL or TLR-2 stimulating adjuvants since they up-regulate MHC-II, down-regulate MARCH1 and IL-10, while TLR-7 ligands induce autophagy [44]. In conclusion, we state that TLR-ligands can affect multiple intracellular events regulating MHC-II dependent antigen presentation and a deeper understanding of these mechanisms appears necessary to optimally deliver BCG and other tuberculosis vaccines in their most immunogenic form.
Supplementary Material
Highlight.
BCG vaccine is the most widely used vaccine against tuberculosis in humans.
BCG inhibits MHC-II in macrophages reducing antigen presentation to T cells.
TLR-1/2 activation by lipids of BCG suppresses MHC-II in macrophages.
TLR-7/9 activation down-regulates MARCH1 enzyme preventing degradation of MHC-II.
TLR-7/9 activation enhances MHC-II dependent antigen presentation by macrophages.
Acknowledgments
This work was supported in part by AI78420 and AI49534 from NIH, NIAID. We are grateful to Dr. Cliff Harding of CWRU for BB7 T cells; Dr. Kenneth L. Rock (U. Massachusetts) for BMA and DC cell lines.
Footnotes
Authorship: PB, NS, ES, SM, AK, performed various experiments and prepared raw data. CJ planned and directed the study and CJ wrote the manuscript.
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rook GA, Seah G, Ustianowski A. M. tuberculosis: immunology and vaccination. Eur Respir J. 2001;17:537–57. doi: 10.1183/09031936.01.17305370. [DOI] [PubMed] [Google Scholar]
- 2.Rook GA, Hernandez-Pando R, Dheda K, Teng Seah G. IL-4 in tuberculosis: implications for vaccine design. Trends Immunol. 2004;25:483–8. doi: 10.1016/j.it.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 3.Bennett K, Levine T, Ellis JS, Peanasky RJ, Samloff IM, Kay J, Chain BM. Antigen processing for presentation by class II major histocompatibility complex requires cleavage by cathepsin E. Eur J Immunol. 1992;22:1519–24. doi: 10.1002/eji.1830220626. [DOI] [PubMed] [Google Scholar]
- 4.Levine TP, Chain BM. The cell biology of antigen processing. Crit Rev Biochem Mol Biol. 1991;26:439–73. doi: 10.3109/10409239109086790. [DOI] [PubMed] [Google Scholar]
- 5.Via LE, Deretic D, Ulmer RJ, Hibler NS, Huber LA, Deretic V. Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J Biol Chem. 1997;272:13326–31. doi: 10.1074/jbc.272.20.13326. [DOI] [PubMed] [Google Scholar]
- 6.Pancholi P, Mirza A, Bhardwaj N, Steinman RM. Sequestration from immune CD4+ T cells of mycobacteria growing in human macrophages. Science. 1993;260:984–6. doi: 10.1126/science.8098550. [DOI] [PubMed] [Google Scholar]
- 7.Singh CR, Moulton RA, Armitige LY, Bidani A, Snuggs M, Dhandayuthapani S, Hunter RL, Jagannath C. Processing and presentation of a mycobacterial antigen 85B epitope by murine macrophages is dependent on the phagosomal acquisition of vacuolar proton ATPase and in situ activation of cathepsin D. J Immunol. 2006;177:3250–9. doi: 10.4049/jimmunol.177.5.3250. [DOI] [PubMed] [Google Scholar]
- 8.Ramachandra L, Noss E, Boom WH, Harding CV. Phagocytic processing of antigens for presentation by class II major histocompatibility complex molecules. Cell Microbiol. 1999;1:205–14. doi: 10.1046/j.1462-5822.1999.00026.x. [DOI] [PubMed] [Google Scholar]
- 9.Ramachandra L, Noss E, Boom WH, Harding CV. Processing of Mycobacterium tuberculosis antigen 85B involves intraphagosomal formation of peptide-major histocompatibility complex II complexes and is inhibited by live bacilli that decrease phagosome maturation. J Exp Med. 2001;194:1421–32. doi: 10.1084/jem.194.10.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soualhine H, Deghmane AE, Sun J, Mak K, Talal A, Av-Gay Y, Hmama Z. Mycobacterium bovis bacillus Calmette-Guerin secreting active cathepsin S stimulates expression of mature MHC class II molecules and antigen presentation in human macrophages. J Immunol. 2007;179:5137–45. doi: 10.4049/jimmunol.179.8.5137. [DOI] [PubMed] [Google Scholar]
- 11.Johansen P, Fettelschoss A, Amstutz B, Selchow P, Waeckerle-Men Y, Keller P, Deretic V, Held L, Kundig TM, Bottger EC, Sander P. Relief from Zmp1-mediated arrest of phagosome maturation is associated with facilitated presentation and enhanced immunogenicity of mycobacterial antigens. Clin Vaccine Immunol. 18:907–13. doi: 10.1128/CVI.00015-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hess J, Miko D, Catic A, Lehmensiek V, Russell DG, Kaufmann SH. Mycobacterium bovis Bacille Calmette-Guerin strains secreting listeriolysin of Listeria monocytogenes. Proc Natl Acad Sci U S A. 1998;95:5299–304. doi: 10.1073/pnas.95.9.5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brennan MJ, Morris SL, Sizemore CF. Tuberculosis vaccine development: research, regulatory and clinical strategies. Expert Opin Biol Ther. 2004;4:1493–504. doi: 10.1517/14712598.4.9.1493. [DOI] [PubMed] [Google Scholar]
- 14.Horwitz MA. Recombinant BCG expressing Mycobacterium tuberculosis major extracellular proteins. Microbes Infect. 2005;7:947–54. doi: 10.1016/j.micinf.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 15.Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL, Jr, Eissa NT. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med. 2009;15:267–76. doi: 10.1038/nm.1928. [DOI] [PubMed] [Google Scholar]
- 16.Young SL, O'Donnell MA, Buchan GS. IL-2-secreting recombinant bacillus Calmette Guerin can overcome a Type 2 immune response and corticosteroid-induced immunosuppression to elicit a Type 1 immune response. Int Immunol. 2002;14:793–800. doi: 10.1093/intimm/dxf050. [DOI] [PubMed] [Google Scholar]
- 17.Leal IS, Smedegard B, Andersen P, Appelberg R. Interleukin-6 and interleukin-12 participate in induction of a type 1 protective T-cell response during vaccination with a tuberculosis subunit vaccine. Infect Immun. 1999;67:5747–54. doi: 10.1128/iai.67.11.5747-5754.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X, Jensen PE. MHC class II antigen presentation and immunological abnormalities due to deficiency of MHC class II and its associated genes. Exp Mol Pathol. 2008;85:40–4. doi: 10.1016/j.yexmp.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fulton SA, Reba SM, Pai RK, Pennini M, Torres M, Harding CV, Boom WH. Inhibition of major histocompatibility complex II expression and antigen processing in murine alveolar macrophages by Mycobacterium bovis BCG and the 19-kilodalton mycobacterial lipoprotein. Infect Immun. 2004;72:2101–10. doi: 10.1128/IAI.72.4.2101-2110.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gehring AJ, Dobos KM, Belisle JT, Harding CV, Boom WH. Mycobacterium tuberculosis LprG (Rv1411c): a novel TLR-2 ligand that inhibits human macrophage class II MHC antigen processing. J Immunol. 2004;173:2660–8. doi: 10.4049/jimmunol.173.4.2660. [DOI] [PubMed] [Google Scholar]
- 21.De Gassart A, Camosseto V, Thibodeau J, Ceppi M, Catalan N, Pierre P, Gatti E. MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proc Natl Acad Sci U S A. 2008;105:3491–6. doi: 10.1073/pnas.0708874105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strong BS, Unanue ER. Presentation of type B peptide-MHC complexes from hen egg white lysozyme by TLR ligands and type I IFNs independent of H2-DM regulation. J Immunol. 187:2193–201. doi: 10.4049/jimmunol.1100152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walseng E, Furuta K, Goldszmid RS, Weih KA, Sher A, Roche PA. Dendritic cell activation prevents MHC class II ubiquitination and promotes MHC class II survival regardless of the activation stimulus. J Biol Chem. 285:41749–54. doi: 10.1074/jbc.M110.157586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pecora ND, Fulton SA, Reba SM, Drage MG, Simmons DP, Urankar-Nagy NJ, Boom WH, Harding CV. Mycobacterium bovis BCG decreases MHC-II expression in vivo on murine lung macrophages and dendritic cells during aerosol infection. Cell Immunol. 2009;254:94–104. doi: 10.1016/j.cellimm.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thibodeau J, Bourgeois-Daigneault MC, Huppe G, Tremblay J, Aumont A, Houde M, Bartee E, Brunet A, Gauvreau ME, de Gassart A, Gatti E, Baril M, Cloutier M, Bontron S, Fruh K, Lamarre D, Steimle V. Interleukin-10-induced MARCH1 mediates intracellular sequestration of MHC class II in monocytes. Eur J Immunol. 2008;38:1225–30. doi: 10.1002/eji.200737902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galbas T, Steimle V, Lapointe R, Ishido S, Thibodeau J. MARCH1 down-regulation in IL-10-activated B cells increases MHC class II expression. Cytokine. 59:27–30. doi: 10.1016/j.cyto.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 27.Chattopadhyay G, Shevach EM. Antigen-Specific Induced T Regulatory Cells Impair Dendritic Cell Function via an IL-10/MARCH1-Dependent Mechanism. J Immunol. 2013 doi: 10.4049/jimmunol.1301693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen W, Wang J, An H, Zhou J, Zhang L, Cao X. Heat shock up-regulates TLR9 expression in human B cells through activation of ERK and NF-kappaB signal pathways. Immunol Lett. 2005;98:153–9. doi: 10.1016/j.imlet.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 29.Lee KW, Lee Y, Kim DS, Kwon HJ. Direct role of NF-kappaB activation in Toll-like receptor-triggered HLA-DRA expression. Eur J Immunol. 2006;36:1254–66. doi: 10.1002/eji.200535577. [DOI] [PubMed] [Google Scholar]
- 30.Toussi DN, Liu X, Massari P. The FomA porin from Fusobacterium nucleatum is a Toll-like receptor 2 agonist with immune adjuvant activity. Clin Vaccine Immunol. 19:1093–101. doi: 10.1128/CVI.00236-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pozzi LA, Maciaszek JW, Rock KL. Both dendritic cells and macrophages can stimulate naive CD8 T cells in vivo to proliferate, develop effector function, and differentiate into memory cells. J Immunol. 2005;175:2071–81. doi: 10.4049/jimmunol.175.4.2071. [DOI] [PubMed] [Google Scholar]
- 32.Singh CR, Bakhru P, Khan A, Li QB, Jagannath C. Cutting edge: Nicastrin and related components of gamma-secretase generate a peptide epitope facilitating immune recognition of intracellular mycobacteria, through MHC class II-dependent priming of T cells. J Immunol. 187:5495–9. doi: 10.4049/jimmunol.1100521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rao PK, Singh CR, Jagannath C, Li Q. A systems biology approach to study the phagosomal proteome modulated by mycobacterial infections. Int J Clin Exp Med. 2009;2:233–47. [PMC free article] [PubMed] [Google Scholar]
- 34.Singh CR, Roche CM, Lindsey DR, Armitige LY, Hunter RL, Jagannath C. The ΔfbpA vaccine derived from Mycobacterium tuberculosis H37Rv undergoes phagosome acidification in macrophages and dendritic cells that leads to an enhanced processing and presentation of antigens to T cells. Joural of Immunology. 2006 submitted; JI07-1120. [Google Scholar]
- 35.Noss EH, Harding CV, Boom WH. Mycobacterium tuberculosis inhibits MHC class II antigen processing in murine bone marrow macrophages. Cell Immunol. 2000;201:63–74. doi: 10.1006/cimm.2000.1633. [DOI] [PubMed] [Google Scholar]
- 36.Katti MK, Dai G, Armitige LY, Rivera Marrero C, Daniel S, Singh CR, Lindsey DR, Dhandayuthapani S, Hunter RL, Jagannath C. The Delta fbpA mutant derived from Mycobacterium tuberculosis H37Rv has an enhanced susceptibility to intracellular antimicrobial oxidative mechanisms, undergoes limited phagosome maturation and activates macrophages and dendritic cells. Cell Microbiol. 2008;10:1286–303. doi: 10.1111/j.1462-5822.2008.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dye C. Making wider use of the world's most widely used vaccine: Bacille Calmette-Guerin revaccination reconsidered. J R Soc Interface. 10:20130365. doi: 10.1098/rsif.2013.0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raymond CR, Wilkie BN. Toll-like receptor, MHC II, B7 and cytokine expression by porcine monocytes and monocyte-derived dendritic cells in response to microbial pathogen-associated molecular patterns. Vet Immunol Immunopathol. 2005;107:235–47. doi: 10.1016/j.vetimm.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 39.Strong BS, Unanue ER. Presentation of type B peptide-MHC complexes from hen egg white lysozyme by TLR ligands and type I IFNs independent of H2-DM regulation. J Immunol. 2011;187:2193–201. doi: 10.4049/jimmunol.1100152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh CR, Bakhru P, Khan A, Li QB, Jagannath C. Cutting edge: Nicastrin and related components of gamma-secretase generate a peptide epitope facilitating immune recognition of intracellular mycobacteria, through MHC class II-dependent priming of T cells. J Immunol. 2011;187:5495–9. doi: 10.4049/jimmunol.1100521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Q, S CR, M S, P ND, J C. Label-free proteomics and systems biology analysis of mycobacterial phagosomes in dendritic cells and macrophages. J Proteome Res. 2011;10:2425–2439. doi: 10.1021/pr101245u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tze LE, Horikawa K, Domaschenz H, Howard DR, Roots CM, Rigby RJ, Way DA, Ohmura-Hoshino M, Ishido S, Andoniou CE, Degli-Esposti MA, Goodnow CC. CD83 increases MHC II and CD86 on dendritic cells by opposing IL-10-driven MARCH1-mediated ubiquitination and degradation. J Exp Med. 208:149–65. doi: 10.1084/jem.20092203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–44. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. Embo J. 2008;27:1110–21. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baldwin SL, Bertholet S, Reese VA, Ching LK, Reed SG, Coler RN. The importance of adjuvant formulation in the development of a tuberculosis vaccine. J Immunol. 188:2189–97. doi: 10.4049/jimmunol.1102696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brandt L, Elhay M, Rosenkrands I, Lindblad EB, Andersen P. ESAT-6 subunit vaccination against Mycobacterium tuberculosis. Infect Immun. 2000;68:791–5. doi: 10.1128/iai.68.2.791-795.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Billeskov R, Elvang TT, Andersen PL, Dietrich J. The HyVac4 Subunit Vaccine Efficiently Boosts BCG-Primed Anti-Mycobacterial Protective Immunity. PLoS One. 7:e39909. doi: 10.1371/journal.pone.0039909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang B, Henao-Tamayo M, Harton M, Ordway D, Shanley C, Basaraba RJ, Orme IM. A Toll-like receptor-2-directed fusion protein vaccine against tuberculosis. Clin Vaccine Immunol. 2007;14:902–6. doi: 10.1128/CVI.00077-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.