

Abstract
We describe the exploration of N1-aryl-substituted benzimidazoles as ligands for the hepatitis C virus (HCV) internal ribosome entry site (IRES) RNA. The design of the compounds was guided by the co-crystal structure of a benzimidazole viral translation inhibitor in complex with the RNA target. Structure-binding activity relationships of aryl-substituted benzimidazole ligands were established that were consistent with the crystal structure of the translation inhibitor complex.
Infection with hepatitis C virus (HCV), which affects over 180 million individuals worldwide, is a major cause of hepatocellular carcinoma and the leading indication for liver transplantation.1 While two classes of direct antiviral drugs are now available, including protease and polymerase inhibitors,2, 3 the high genetic variability of HCV poses a constant threat of resistance development and requires the exploration of additional targets for antiviral therapy.4–6 Since the HCV RNA genome contains several highly conserved cis-acting elements that adopt defined structures, the repertoire for antiviral intervention may be expanded to RNA targets. The internal ribosome entry site (IRES) in the 5′ untranslated region (UTR) of the viral genome stands out for its high sequence conservation in clinical isolates and serves an essential function for ribosome recruitment and initiation of HCV protein synthesis in infected host cells. We have recently established7 the subdomain IIa RNA of the HCV IRES as the target for 2-aminobenzimidazole viral translation inhibitors8 (Figure 1). Benzimidazole scaffolds have been recognized as useful platforms for the synthesis of biologically active ligands for other RNA targets, including RNA loops,9, 10 the HIV TAR element,11 microRNAs12 and RNA repeat motifs.13, 14 The 2-aminobenzimidazoles inhibit the IRES function by capturing an extended conformation of the IIa RNA target which has been proposed to block translation initiation.15 A Förster resonance energy transfer (FRET) assay has been developed to monitor the selective conformational capture by ligands and measure their target affinity for the IIa RNA.7, 16
Figure 1.

Ligand design for the internal ribosome entry site (IRES) target of hepatitis C virus (HCV). A) The IRES element comprises the first 342 nucleotides in the 5′ untranslated region (UTR) of the HCV RNA genome. The boxed region highlights the subdomain IIa target whose secondary structure is shown in on the right. B) Structures of a 2-aminobenzimidazole translation inhibitor 1 which was previously co-crystallized in complex with the subdomain IIa RNA, and aryl-substituted 2-aminobenzimidazoles 2 which were designed using guidance by the co-crystal structure of the IIa RNA in complex with 1. C) Model of the 1-phenyl-2-aminobenzimidazole core of 2 (light blue) docked at the binding site of 1 (yellow) in the subdomain IIa RNA co-crystal structure.17 Hydrogen bonds and nucleotides participating in intramolecular contacts are indicated.
Crystal structure analysis of the IIa RNA in complex with the 2-aminobenzimidazole 1 revealed the key interactions responsible for target recognition by the translation inhibitors.17 Since previous structure-binding activity relationship studies on the 2-aminobenzimidazoles had revealed the importance of a rigid core scaffold for target affinity,8 we explored N1-aryl-substituted benzimidazoles (2) which also lack the synthetically cumbersome chroman scaffold. The compounds designed here retain the amino-imidazole core required for recognition of the IIa RNA and provide a rigid aryl linker for a dimethylamino substituent that participates in interactions with a phosphate group while avoiding an entropic penalty for pre-organization of the dimethylaminopropyl chain in ligands such as 1 (Figure 1B,C). Modeling studies suggested that the twist conformation between the imidazole and aryl ring along with an appropriate meta-substitution position for the dimethylamino group would provide a rigid vector for interaction with the phosphate group of residue A109 (Figure 1C).
Here, we describe the synthesis and target affinity testing of several chemical series of N1-aryl-substituted 2-aminobenzimidazoles (Figure 2), including the compounds 3 and the 2-amide derivatives 4. To evaluate the importance of the 2-amino substituent, we prepared 2-hydroxybenzimidazole analogs 5. A small number of dimethylaminopropyl-substituted compounds 6 which carried aryl substituents at the benzene ring were included as well.
Figure 2.
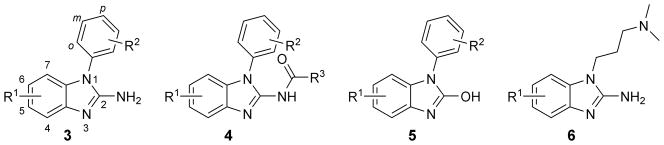
Benzimidazole derivatives for targeting the HCV IRES subdomain IIa RNA: 1-aryl-2-aminobenzimidazoles 3, 1-aryl-2-acylaminobenzimidazoles 4, 1-aryl-2-hydroxybenzimidazoles 5, N,N-dimethylaminopropyl-substituted benzimidazoles 6. Compounds made in each series are shown in Tables 1 and 2.
Synthesis of phenylether derivatives in the series 3, 4 and 5 (R1 = R4O, Scheme 1 and Table 1) was planned to progress through an N-aryl-substituted o-phenylenediamine common intermediate (12) which was cyclized to the benzimidazole by reaction with isothiocyanates or phosgene. The intermediate 12 was prepared over three steps from halogen-substituted nitrophenols 7. Alkylation of 7 yielded phenylethers 9. The parent phenol 9 (R4 = OH) was obtained as the tert-butyldimethylsilyl (TBDMS) ether derivative. Coupling with anilines 10 followed by reduction of the nitro group furnished the common intermediate 12. Cyclization of 12 with acetyl or benzoyl isothiocyanates gave the 2-amide derivatives 4 which were further hydrolyzed to the parent 2-aminobenzimidazoles 3. Treatment of 12 with phosgene yielded 2-hydroxybenzimidazole analogs 5. Two aniline derivatives in the 1-aryl-2-aminobenzimidazole series (3h and 3i, Table 1) were synthesized through isothiocyanate cyclization of an intermediate that was obtained by two consecutive Buchwald-Hartwig aminations of 2,4-dichloro-1-nitrobenzene 13 (Scheme 2). The N,N-dimethylaminopropyl-substituted benzimidazoles 6 (Table 2) were synthesized following the route outlined in Scheme 3. Dichloronitrobenzene precursors were alkylated to install the dimethylaminopropyl chain. Coupling with aromatic amines or boronic acids led to nitro-aniline (22) and -biphenyl (23) intermediates which, after reduction, were cyclized with isothiocyanate to furnish the amides 24 whose hydrolysis gave the final products 6.
Scheme 1.

Synthesis of 1-aryl-2-aminobenzimidazoles 3, 1-aryl-2-acylaminobenzimidazoles 4 and 1-aryl-2-hydroxybenzimidazoles 5 from halo-nitrophenols 6. Preparation of 5-substituted benzimidazoles commenced from 4-chloro-3-nitrophenol (X=Cl), that of 6-substituted products from 3-fluoro-4-nitrophenol (X=F). Reagents and conditions: a) (for R4=H, 8=TBDMSCl) K2CO3, ACN, reflux, 2.5–28 h, 52–94 % yield; b) Cs2CO3, Pd2(dba)3, (±)BINAP, 19–28 h, 31–91 % yield; c) K2CO3, ACN, reflux, 21–41 h, 34–36 % yield; d) H2 (1 atm), Pd/C or PtO2, MeOH, RT, 1.5–25 h; e) acylNCS (acyl=acetyl or benzoyl), DIPC, DIPEA, ACN, 22–48 h, 20–66 % yield over two steps; f) phosgene (20 % in toluene), pyridine, DCM, RT, 3–24 h, 50–64 % yield two steps; g) 1N HCl, H2O, dioxane, reflux, 18–28 h, 27–96 % yield. Abbreviations: TBDMSCl = tert-butyldimethylsilyl chloride; ACN = acetonitrile; dba = dibenzylideneacetone; BINAP = 2,2′-bis(diphenylphosphino)-1,1′-binaphthalene; RT = room temperature; DIPC = N,N′-diisopropylcarbodiimide; DIPEA = N,N-diisopropylethylamine; DCM = dichloromethane.
Table 1.
Activity of 1-aryl-benzimidazole derivatives 3, 4 and 5 in the FRET assay.
| Compound | R1 | R2 | R3 | EC50 [μM]a |
|---|---|---|---|---|
| 3a | 5-OH | H | H | n.a. |
| 3b/5b |
|
H | H | 250 ± 66 |
| 3c/5c |
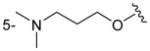
|
H | H | 210 ± 39 |
| 3d |
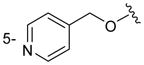
|
H | H | n.a. |
| 3e |
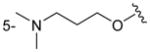
|
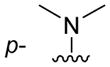
|
H | 95 ± 6 |
| 3f/5f |
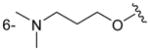
|
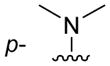
|
H | 170 ± 16 |
| 3g |
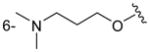
|
|
H | 120 ± 5 |
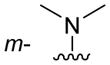
| 3h |
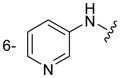
|
|
H | 74 ± 2 |
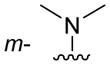
| 3i |
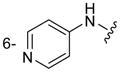
|
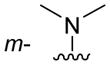
|
H | 100 ± 12 |
| 4ba |
|
H | CH3 | 100 ± 13 |
| 4ca |
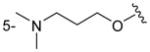
|
H | CH3 | > 500 |
| 4ab | 5-OH | H | C6H5 | n.a. |
| 4bb |
|
H | C6H5 | > 500 |
| 4cb |
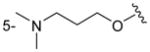
|
H | C6H5 | > 500 |
| 4eb |
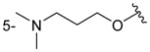
|
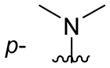
|
C6H5 | precip. |
| 4fb |
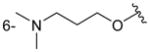
|
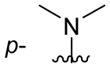
|
C6H5 | precip. |
EC50 values (± standard error from triplicate experiments) are shown for 2-aminobenzimdiazoles 3 and acyl derivates 4. All tested 2-hydroxybenzimidazoles 5 precipitated RNA at a concentration between 50–100 μM.
Scheme 2.
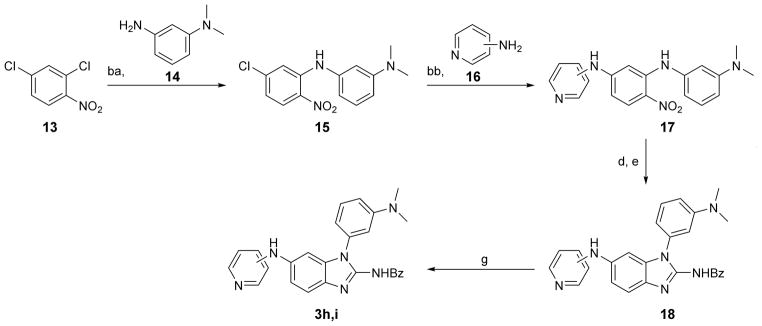
Synthesis of 1-aryl-2-aminobenzimidazoles 3h and 3i. Reagents and conditions: ba) Cs2CO3, Pd2(dba)3, (±)BINAP, toluene, reflux, 21 h, 43 % yield, two steps; 14 was synthesized from N,N-dimethyl-3-nitroaniline by reduction with H2 (1 atm), Pd/C in MeOH, 20 hr; bb) Cs2CO3, Pd2(dba)3, (±)BINAP, dioxane, reflux, 20–44 h, 34–43 % yield d) H2 (1 atm), Pd/C or PtO2, MeOH, RT, 21–23 h h; e) benzoylNCS, DIPC, DIPEA, ACN, 24–26 h, 22–32 % yield over two steps; g) 1N HCl, H2O, dioxane, reflux, 23–26 h, 27–56 % yield.
Table 2.
Activity of N,N-dimethylaminopropyl-substituted benzimidazoles 6 in the FRET assay.
| Compound | R1 | EC50[μM]a |
|---|---|---|
| 6h |
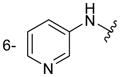
|
160 ± 15 |
| 6i |
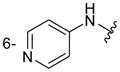
|
160 ± 29 |
| 6j |
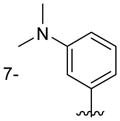
|
290 ± 100 |
EC50 values are shown ± standard error from triplicate experiments.
Scheme 3.
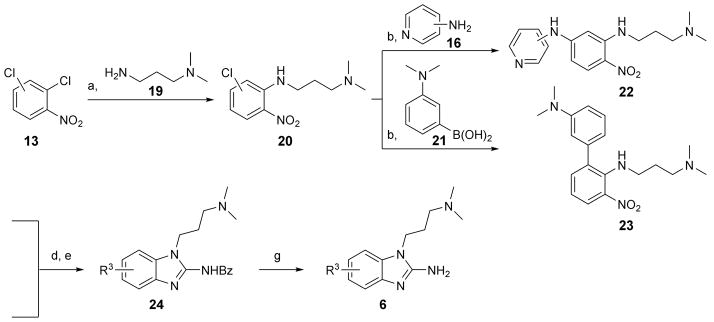
Synthesis of N,N-dimethylaminopropyl-substituted benzimidazoles 6 from dichloro-nitrobenzenes 13. Preparation of 6-substituted benzimidazoles commenced from 1,2-dichloro-4-nitrobenzene, that of 7-substituted products from 1,2-dichloro-3-nitrobenzene. Reagents and conditions: a) K2CO3, ACN, reflux, 24 h, 96–97 % yield; b) Cs2CO3, Pd2(dba)3, (±)BINAP, 19–23 h, 37–80 % yield range; d) H2 (1 atm), Pd/C or PtO2, MeOH, RT, 21–25 h; e) benzoylNCS, DIPC, DIPEA, ACN, 19–21 h, 29–33 % yield over two steps; g) 1N HCl, H2O, dioxane, reflux, 17–21 h, 22–46 % yield.
The identity of all synthesized compounds from the chemical series 3, 4, 5 and 6 was established after column chromatographic purification by mass- and NMR. Crystal structures were determined for selected derivatives, including the 2-aminobenzimidazole 3e. Compounds were tested for binding to the IRES IIa RNA by triplicate titration in the IIa FRET assay as previously described.16 Target affinity expressed as EC50 value was determined from fitting of single-site binding dose response curves to the titration data (Tables 1 and 2). See the Supporting Information for experimental procedures, spectra, crystal structure and FRET titration curves.
Of the nine 1-aryl-2-aminobenzimidazoles 3 synthesized, all but two showed binding to the IIa RNA target with affinities between 74–250 μM. Exceptions were the unsubstituted phenol 3a and the picolyl ether 3d. Inspection of the binding pocket around 1 in the co-crystal structure suggested that while the parent phenol 3a is unable to form interactions beyond the amino-imidazole scaffold, in the derivative 3d the bulky picolyl ether substituent off the 5-position might interfere with ligand docking at the G110 base of the RNA target (Figure 1C). In contrast, the more flexible aliphatic dimethylaminoalkyl ether chain was tolerated at the same position, as attested by the binding of compounds 3b and 3c (250 and 210 μM, respectively). Improvement of the affinity by 2-fold was seen in derivative 3e (95 μM) which carried a 5-dimethylaminopropyl ether substituent at the benzimidazole and an additional p-dimethyalmino group on the imidazole-linked phenyl ring. The same combination of functional groups, however, with the dimethylaminoalkyl ether chain moved to the 6-position reduced binding of compound 3f (170 μM). Relocation of the dimethylamino substituent from the para- to the meta-position at the imidazole-linked phenyl ring increased binding in derivative 3g (120 μM), yet not to the level of 3e. Finally, aromatic substituents were tolerated at the benzimidazole 6-position as indicated by the affinity of compounds 3h and 3i, which were among the most active ligands synthesized here (74 and 100 μM, respectively). In comparison, compounds 6h and 6i which carried the same aromatic pyridine substituents at the 6-position but had the m-dimethyalminophenyl ring at the imidazole replaced with a flexible dimethylaminopropyl chain showed weaker target affinity (both, 160 μM). Attachment of a m-dimethyalminophenyl group at the 7-position of the benzimidazole retained binding in compound 6j albeit only at 290 μM.
The crystal structure of compound 3e (see Supporting Information) shows a twist angle of 67° between the imidazole and the phenyl ring attached at the 1-position. As a consequence, a good fit of the 1-phenyl ring in 3e and the dimethylaminopropyl chain in 1 bound to the IIa RNA target was observed in a superposition of the two compounds (Figure 3). The meta position of the phenyl ring in 3e is pointing at the location of the dimethylamino group in 1 which forms a key hydrogen bond with the phosphate of A109 in the RNA target (Figures 1C and 3B). Comparison of the derivatives 3f and 3g suggests that higher affinity is achieved by substitution of the phenyl ring with a dimethylamino group at the meta position compared to the para position. In the superposition of 3e on the 1 target complex, the dimethylaminopropyl ether chain at the benzimidazole 5-position is directed towards a cleft lined by the sugar phosphate backbone of residues A54-U56 (Figure 3B). This region is located in a flexible internal loop of the IIa RNA target (Figure 1A) and undergoes a large conformational change upon binding of the ligand 1. Such adaptive binding might allow the accommodation of flexible substituents at the benzimidazole 5-position, for example, the dimethylaminopropyl ether chain in the 3e derivative. However, as indicated by the inactivity of compound 3d, which carries a picolyl ether substituent, bulky and more rigid groups at the 5-position may not be reconciled with target adaptation.
Figure 3.

A) Crystal structure of compound 3e (see Supporting Information, CCDC deposit number 978337). Hydrogen atoms are not shown. The dihedral twist angle between the imidazole and dimethylaniline rings is indicated. B) Model of the compound 3e crystal structure (light blue) superimposed on the HCV translation inhibitor 1 (yellow) in the subdomain IIa RNA co-crystal structure.17 Hydrogen bonds and nucleotides participating in intramolecular contacts are indicated. Residues G52 and A53, which stack below and above the ligand are shown transparent.
Acylation of the 2-amino group abolished binding as indicated by compounds in the series 4. The acetylated derivative 4ba was an exception for which a target affinity of 100 μM was measured, surpassing that of the parent compound 3b (250 μM). This observation is not readily reconciled with the crystal structure of the ligand 1 RNA complex, perhaps suggesting that compound 4ba accesses an alternative binding mode at the target. Nonspecific binding interactions might also be responsible for the strong precipitating properties of the hydroxybenzimidazoles 5. All tested compounds in this series led to rapid fluorescence quenching in the FRET assay at micromolar concentration, apparently due to precipitation of the dye labeled RNA target.
Here, we have explored N1-aryl-substituted 2-amino-benzimidazoles (2) as ligands for the IIa RNA target in the HCV IRES element. Synthesis and affinity testing of several chemical series allowed us to establish structure-binding relationships of the ligands (Figure 4). The observed pattern of compound activity in the FRET target binding assay was readily explained in the context of an earlier determined crystal structure of benzimidazole 1 bound to the IIa RNA. Ligands with the highest affinity showed binding at EC50 values around 74–100 μM which is inferior to the previously studied tricyclic benzimidazoles such as 1. Given the more facile synthetic accessibility of the chemical series 2, in combination with guidance from the clear structure-binding activity relationship established in the current work, we will explore further optimization of the N1-aryl-substituted benzimidazoles in the future.
Figure 4.
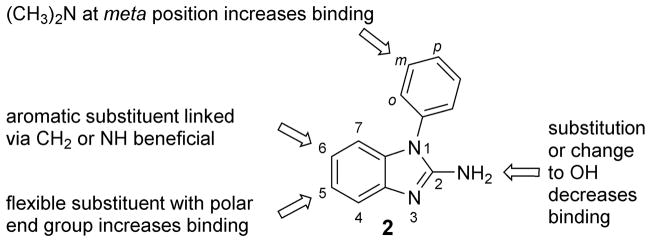
Structure-binding activity relationships discovered for 1-aryl-2-aminobenzimidazoles targeting the HCV IRES subdomain IIa RNA.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (grant No. AI72012). Support of the NMR facility by the National Science Foundation is acknowledged (CRIF grant CHE-0741968).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thomas DL. Nat Med. 2013;19:850. doi: 10.1038/nm.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pawlotsky JM. Adv Pharmacol. 2013;67:169. doi: 10.1016/B978-0-12-405880-4.00005-6. [DOI] [PubMed] [Google Scholar]
- 3.Manns MP, von Hahn T. Nat Rev Drug Discov. 2013;12:595. doi: 10.1038/nrd4050. [DOI] [PubMed] [Google Scholar]
- 4.Scheel TK, Rice CM. Nat Med. 2013;19:837. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salvatierra K, Fareleski S, Forcada A, Lopez-Labrador FX. World J Virol. 2013;2:6. doi: 10.5501/wjv.v2.i1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartenschlager R, Lohmann V, Penin F. Nat Rev Microbiol. 2013;11:482. doi: 10.1038/nrmicro3046. [DOI] [PubMed] [Google Scholar]
- 7.Parsons J, Castaldi MP, Dutta S, Dibrov SM, Wyles DL, Hermann T. Nat Chem Biol. 2009;5:823. doi: 10.1038/nchembio.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seth PP, Miyaji A, Jefferson EA, Sannes-Lowery KA, Osgood SA, Propp SS, Ranken R, Massire C, Sampath R, Ecker DJ, Swayze EE, Griffey RH. J Med Chem. 2005;48:7099. doi: 10.1021/jm050815o. [DOI] [PubMed] [Google Scholar]
- 9.Velagapudi SP, Pushechnikov A, Labuda LP, French JM, Disney MD. ACS Chem Biol. 2012;7:1902. doi: 10.1021/cb300213g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Velagapudi SP, Seedhouse SJ, French J, Disney MD. J Am Chem Soc. 2011;133:10111. doi: 10.1021/ja200212b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ranjan N, Kumar S, Watkins D, Wang D, Appella DH, Arya DP. Bioorg Med Chem Lett. 2013;23:5689. doi: 10.1016/j.bmcl.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Velagapudi SP, Gallo SM, Disney MD. Nat Chem Biol. 2014;10:291. doi: 10.1038/nchembio.1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parkesh R, Childs-Disney JL, Nakamori M, Kumar A, Wang E, Wang T, Hoskins J, Tran T, Housman D, Thornton CA, Disney MD. J Am Chem Soc. 2012;134:4731. doi: 10.1021/ja210088v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Childs-Disney JL, Hoskins J, Rzuczek SG, Thornton CA, Disney MD. ACS Chem Biol. 2012;7:856. doi: 10.1021/cb200408a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dibrov SM, Parsons J, Carnevali M, Zhou S, Rynearson KD, Ding K, Garcia Sega E, Brunn ND, Boerneke MA, Castaldi MP, Hermann T. J Med Chem. 2013 doi: 10.1021/jm401312n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou S, Rynearson KD, Ding K, Brunn ND, Hermann T. Bioorg Med Chem. 2013;21:6139. doi: 10.1016/j.bmc.2013.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dibrov SM, Ding K, Brunn ND, Parker MA, Bergdahl BM, Wyles DL, Hermann T. Proc Natl Acad Sci U S A. 2012;109:5223. doi: 10.1073/pnas.1118699109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.