

Abstract
A set of 5,6-fused bicyclic heteroaromatic scaffolds were investigated for their in vitro antitubercular activity versus replicating and non-replicating strains of Mycobacterium tuberculosis (Mtb) in an attempt to find an alternative scaffold to the imidazo[1,2-a]pyridine and imidazo[1,2-a]pyrimidines that were previously shown to have potent activity against replicating and drug resistant Mtb. The five new bicyclic heteroaromatic scaffolds explored in this study include a 2,6-dimethylimidazo[1,2-b]pyridazine-3-carboxamide (7), a 2,6-dimethyl-1H-indole-3-carboxamide (8), a 6-methyl-1H-indazole-3-carboxamide (9), a 7-methyl-[1,2,4]triazolo[4,3-a]pyridine-3-carboxamide (10), and a 5,7-dimethyl-[1,2,4]triazolo[1,5-a]pyrimidine-2-carboxamide (11). Additionally, imidazo[1,2-a]pyridines isomers (2 and 12) and a homologous imidazo[1,2-a]pyrimidine isomer (6) were prepared and compared. Compounds 2 and 6 were found to be the most potent against H37Rv Mtb (MIC’s of 0.1 μM and 1.3 μM) and were inactive (MIC >128 μM) against Staphylococcus aureus, Escherichia coli and Candida albicans. Against other non-tubercular mycobacteria strains, compounds 2 and 6 had activity against Mycobacterium avium (16 and 122 μM, respectively), Mycobacterium kansasii (4 and 19 μM, respectively), Mycobacterium bovis BCG (1 and 8 μM, respectively) while all the other scaffolds were inactive (>128 μM).
Keywords: Antituberculosis; Imidazo[1,2-a]pyridine; Imidazo[1,2-a]pyrimidine; 5,6-Fused bicyclic system; Scaffold hopping
Tuberculosis (TB) is a serious global health disease caused by the contagious air borne pathogen Mycobacterium tuberculosis (Mtb). In 2012, 1.3 million people died of TB and 8.6 million people fell ill of the disease.1 For the first time in 40 years, a new TB treatment (bedaquiline) was approved in 20122 giving new treatment options particularly with the estimated 450,000 patients infected with multidrug resistant Mtb.1 Our laboratories have had an interest in inhibiting Mtb with small molecule heterocyclic compounds since the serendipitous discovery of a simple synthetically attractive oxazoline scaffold3 derived from fragment screening of an iron chelating class of compounds called the mycobactins.4 Through an intensive structure activity relationship (SAR) campaign we improved the Mtb potency of the oxazolines to sub-micromolar MIC’s and expanded the chemical space to include various other heterocyclic scaffolds (oxazole, thiazole, imidazole, and pyridine).5 Further SAR effort complimented by a screening agreement with Dow AgroSciences (DAS) iteratively led to the exploration of fused heterocyclic scaffolds which resulted in the discovery of a “hit” 5,6-fused bicyclic imidazo[1,2-a]pyridine.6-8 We have since reported on the advent and advancement of various imidazo[1,2-a]pyridine-3-carboxamides (1-3), imidazo[1,2-a]pyrimidine-3-carboxamide (5, 6) and a benzyl 2,5,7-trimethylimidazo[1,2-c]pyrimidine-3-carboxylate (7) with excellent in vitro potency against replicating (H37Rv), as well as multi- (MDR) and extensively drug (XDR) resistant Mtb strains (Fig. 1).6-8 Herein, we report our efforts on “scaffold hopping”9-14 to identify new heterocyclic scaffolds that will potentially retain potency against H37Rv Mtb and subsequently have improved ADME properties while also exploring new chemical space.
Figure 1.

Previously reported imidazo[1,2-a]pyridines (1-3) and imidazo[1,2-a]pyrimidine (5) and two new analogs prepared for this SAR study the benzyl 2,6-dimethylimidazo[1,2-a]pyridine-3-carboxylate (4) and the N-benzyl-2,6-dimethylimidazo[1,2-a]pyrimidine-3-carboxamide (6). The imidazo[1,2-a]pyrimidine nitrogen is denoted in red.
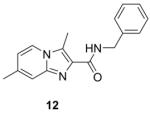
The scaffold switching that was performed in this study involved moving, incorporating or removing nitrogen(s) within the 5,6-fused bicyclic framework to give a set of analogs for screening (Fig. 2). Specifically, incorporating a nitrogen at the 5-position gave the imidazo[1,2-b]pyridazine-3-carboxamide (7), removing a nitrogen from the 4-position gave the 1H-indole-3-carboxamide (8), moving a nitrogen from the 4-position to the 2-position gave the 1H-indazole-3-carboxamide (9), incorporating a nitrogen to the 2-position gave the triazolo[4,3-a]pyridine-3-carboxamide (10), and incorporating nitrogen at the 3 and 8-positions then moving the 3-carboxamide to the 2-position gave the triazolo[1,5-a]pyrimidine-2-carboxamide (11). We also made an isomer of compound 1 wherein we switched the adjacent 3-carboxamide with the 2-methyl group to give the imidazo[1,2-a]pyridine-2-carboxamide analog (12) (Fig. 2). These various 5,6-fused bicyclic heterocyclic scaffolds (7–12) were all searched within literature but to the best of our knowledge none of these scaffolds were found to have been screened against Mtb H37Rv.
Figure 2.
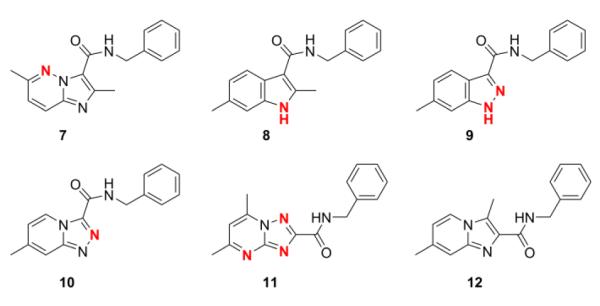
New 5,6-fused bicyclic heteroaromatic scaffold analogs (7–12) prepared to evaluate the effects of scaffold switching on anti-TB activity. The moving, incorporating or removing nitrogen(s) within the 5,6-fused bicyclic framework from that of the imidazo[1,2-a]pyridine are denoted in red.
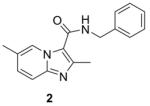
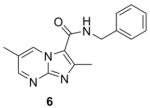
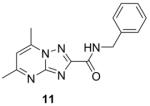
Syntheses of the imidazo[1,2-a]pyridines (1-4) and imidazo[1,2-a]pyrimidines (5 and 6) are straightforward and have been described in our previous publications5,6,8 as well as within the Supplementary data. In brief, condensation of the appropriate 2-amino-heteroaromatic (13 or 15) with either ethyl 2-bromoacetoacetate (for 13) or ethyl 2-bromoacetoacetate (for 15) followed by saponification and acidification gave the desired imidazo[1,2-a]pyridine-3-carboxylic acid (14) or imidazo[1,2-a]pyrimidine-3-carboxylic acid (15) (Scheme 1). Both carboxylic acids were coupled with benzyl amine with EDC to give compounds 2 and 6. In an efficient one step process, compounds 4 and 5 were prepared through condensation of the appropriate 2-amino-heteroaromatic precursor with benzyl 2-bromoacetoacetate.5 The N-benzyl-2,6-dimethylimidazo[1,2-b]pyridazine-3-carboxamide (7) was synthesized in an similar method by first condensation of the 3-amino-6-methylpyridazine (17) with ethyl 2-bromoacetoacetate, saponification of the ethyl ester followed by acidification then coupling to the benzyl amine with EDC. By design, all of the other 5,6-fused bicyclic heteroaromatic scaffolds investigated for “scaffold switching” were commercially available. As such, the free acids (19–22) were rapidly elaborated by EDC-mediated couplings with benzyl amine to give the desired analogs (8, 9, 11, 12) for screening (Scheme 2). However, the N-benzyl-7-methyl-[1,2,4]triazolo[4,3-a]pyridine-3-carboxamide (10) was prepared by amidation of the corresponding ethyl ester (23) with benzyl amine in a sealed tube at 90°C (Scheme 2).
Scheme 1.
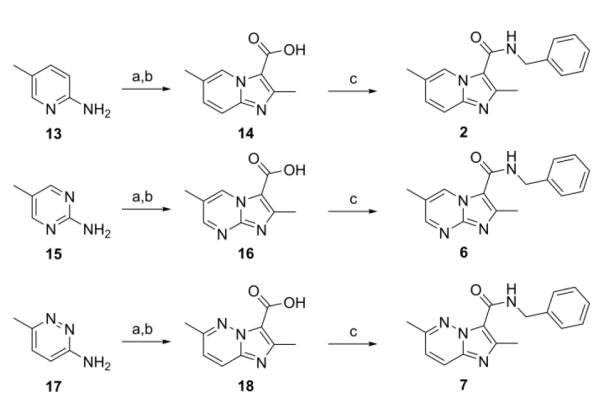
Synthesis of imidazo[1,2-a]pyridine (2), imidazo[1,2-a]pyrimidine (6), and imidazo[1,2-b]pyridazine (7). Reagents: (a) 1. Ethyl 2-chloroacetoacetate (for 13) or Ethyl 2-bromoacetoacetate (for 15 and 17), DME, reflux, 48 h.; (b) 1. LiOH, EtOH; 2. HCl, 56 h.; (c) EDC, DMAP, benzyl amine, ACN, 16 h.
Scheme 2.

Synthesis of various 5,6-fused bicyclic heterocyclic scaffold analogs (8–10) and isomeric N-benzyl-3,7-dimethylimidazo[1,2-a]pyridine-2-carboxamide (12). Reagents and conditions: (a) EDC, DMAP, benzyl amine, ACN, 16 h. (b) Benzyl amine, ACN, 90°C, 56 h.
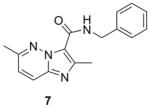
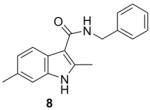
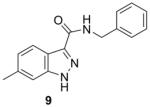
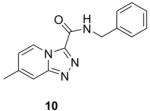
Table 1 summarizes the in vitro anti-TB activity of these six “scaffold switched” 5,6-fused bicyclic heteroaromatic analogs (6 – 12) in two different media the glycerol–alanine–salts “GAS”15 and the Middlebrook 7H1216 and their potency against non-replicating “latent” Mtb (LORA17) and an assessment of their toxicity by the VERO18 assay. The most potent 5,6-fused heteroaromatics were still the imidazo[1,2-a]pyridine (3) and imidazo[1,2-a]pyrimidine (6) scaffolds with MIC’s of <0.2 and 1.3 μM in the GAS media, respectively. However, it is not the imidazo[1,2-a]pyridine scaffold alone which imparts the majority of anti-TB activity as its isomer (12) was >700 times less active (MIC’s of 0.1 and 70 μM, respectively). N-Benzyl-2,6-dimethyl-1H-indole-3-carboxamide (8), N-benzyl-6-methyl-1H-indazole-3-carboxamide (9) and N-benzyl-2,6-dimethylimidazo[1,2-b]pyridazine-3-carboxamide (7) were found to be moderately active compounds with MIC’s of 44, 41 and 64 μM in the GAS media, respectively. N-Benzyl-7-methyl-[1,2,4]triazolo[4,3-a]pyridine-3-carboxamide (10) and N-benzyl-5,7-dimethyl-[1,2,4]triazolo[1,5-a]pyrimidine-2-carboxamide (11) had the weakest activity with MIC’s of 101 and 126 μM in the GAS media, respectively. Replacement of the 2-methyl group in compound 2 with nitrogen in compound 10 had a very negative effect (1000x) on potency (MIC of 0.1 and 101 μM, respectively). Due to the modest activity observed in most of these new scaffolds we recalculated the MIC’s in terms of μg/mL and saw MIC values which are more encouraging in the GAS media and kept the same overall trends observed above (Table 1). Part of the attraction of scaffold switching is often the enhancement of ADME properties19 which sometimes results in greater drug free fraction20 in vivo even if analogous compounds have a lower MIC. Such ADME property enhancement was demonstrated with the imidazo[1,2-a]pyridines of Ramachandran et al. at AstraZeneca.21 The SAR observed from screening in 7H12 medium again indicated that the most potent compounds were 2 and 6 while all other compounds were inactive (MIC > 128 μM). This suggests that the potency of these other 5,6-fused bicyclic heteroaromatics may be carbon source dependant, an issue that was discovered in the pyrimidine-imidazoles reported by Pethe et al.22 or due to other medium effects.23 The GAS medium uses glycerol-alanine-salts as the carbon source while the 7H12 medium uses palmitic acid. No activity was detected in the “latent” Mtb assay (LORA, MIC > 128 μM) even with compounds 2 and 6 suggesting that these compounds are only effective on replicating Mtb. The VERO toxicity assay also showed these compounds to be non-toxic with the exception of the N-benzyl-2,6-dimethylimidazo[1,2-b]pyridazine-3-carboxamide (7) which had an IC50 of 25 μM.
Table 1.
In vitro evaluation of compounds 2, 6-12 against H37Rv Mtb in GAST media, 7H12 media and LORA as well as toxicity to VERO cells (IC50 in μM).
| Compound | Mol Wt | Calcd Clog Pa |
MIC mean +/− against replicating Mtb in medium: |
LORA (μM) |
VERO IC50 (μM) |
||
|---|---|---|---|---|---|---|---|
| GAS (μM) | GAS (μg/mL) | 7H12 (μM) | |||||
|
279.34 | 3.60 | 0.11±0.008 | 0.031±0.002 | 0.49±0.004 | >128 | >128 |
|
280.32 | 2.60 | 1.27±0.22 | 0.36±0.06 | 6.95±0.15 | >128 | >128 |
|
280.32 | 2.26 | 63.8±1.9 | 17.9±0.5 | >128 | >128 | 24.7 |
|
278.35 | 3.73 | 43.8±1.2 | 12.2±0.3 | >128 | >128 | >128 |
|
265.31 | 4.02 | 40.8±3.3 | 10.8±0.9 | >128 | >128 | >128 |
|
267.31 | 2.85 | 101±6.0 | 27±1.6 | >128 | >128 | >128 |
|
281.31 | 2.20 | 126±2.9 | 35.4±0.8 | >128 | >128 | >128 |
|
279.34 | 3.61 | 70±8.9 | 19.6±2.5 | >128 | >128 | >128 |
|
822.95 | 6.04 | 0.11 | 0.09 | 0.06 | 2.6 | 113 |
|
359.26 | 2.62 | 0.12 | 0.04 | 0.09 | 4.9 | >128 |
SD, standard deviation; MIC, minimum concentration required to inhibit growth by >90%; GAS, Glycerol-alanine-salts media; 7H12, Middlebrook7H9 broth base media with BSA, casein hydrolysate, catalase, palmitic acid; LORA, Low oxygen recovery assay using the H37Rv luxAB Mtb luminescence strain; VERO, African green monkey kidney cell line to evaluate toxicity. Values reported are the average of three individual measurements.
Calculated Clog P by ChemDraw version 12.0
Despite varying degrees of activity observed against Mtb compounds 2, 6-12 were all found to have a similar selectivity profile since they were inactive against representative Gram positive strains including Staphylococcus aureus (MIC >128 μM), Gram negative strains including Escherichia coli (MIC >128 μM), and the fungus, Candida albicans (MIC >128 μM), see Supplementary data. Additionally, when screened against various other non-tubercular mycobacteria strains only the imidazo[1,2-a]pyridine (2) and imidazo[1,2-a]pyrimidine (6) analogs had any activity and that was only against selective strains (see Supplementary data). In particular, compounds 2 and 6 inhibited Mycobacterium avium (MIC’s of 16 and 122 μM, respectively), Mycobacterium kansasii (MIC’s of 4 and 19 μM, respectively), Mycobacterium bovis BCG (MIC’s of 1 and 8 μM, respectively) while all the other analogs (7–12) were inactive (>128 μM) to Mycobacterium smegmatis, Mycobacterium chelonae, Mycobacterium marinum, Mycobacterium avium, Mycobacterium kansasii and Mycobacterium bovis BCG (see Supplementary data).
It is possible that a key to understanding the varying degree of potency against Mtb may lie in the subtle structural or electronic effects of these various 5,6-fused heteroaromatic analogs. As such, compounds 1, 7–10 were crystallized and X-ray structural studies undertaken. The resulting structures were subsequently compared with our initial imidazo[1,2-a]pyridine “hit” compound 1 (Fig. 3). This information will be particularly useful once the target(s) of these compounds (presumably the QcrB gene24) are ultimately crystallized and these structures docked.
Figure 3.
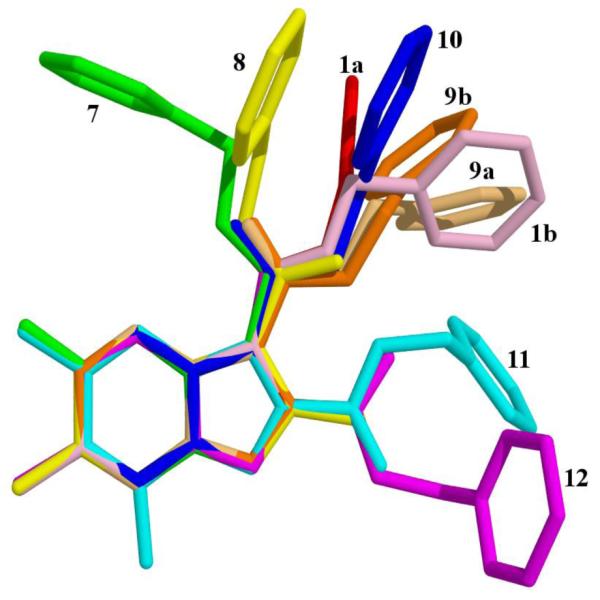
Overlay of the 5,6-fused bicyclic heteroaromatic scaffolds (7 – 12) crystal structures with potent N-benzyl-2,7-dimethyl-imidazo[1,2-a]pyridine-3-carboxamide (1). Legend. 1a – red, 1b – light pink, 7 – green, 8 – yellow, 9a – light orange, 9b – orange, 10 – blue, 11 – turquoise, 12 – magenta. Hydrogen atoms omitted for clarity. The nine atoms of 5,6-fused bicycle were used as template mapping atoms.
The overlay of the imidazo[1,2-a]pyridine-3-carboxamide (1) crystal structure with its isomeric 2-carboxamide (12) shows that these moieties lie in very different regions of space and this important structural difference is also reflected in their different MIC’s as compound 1 is 350 times more potent than 12 (MIC’s of 0.2 and 70 μM, respectively). Finally, an additional nitrogen in the 5,6-fused system did not improve activity in any scaffold other than the imidazo[1,2-a]pyrimidine (6) as the imidazo[1,2-b]pyridazine (7), triazolo[4,3-a]pyridine-3-carboxamide (10) and triazolo[1,5-a]pyrimidine-2-carboxamide (11) had the weakest activity. Curiously, in the solid state there is little correlation between the orientations of the pendant groups as can be seen in the Fig. 3 whereas in solution we expect them to dynamic and fluxional. It should be noted that in the solid state, compounds 2 and 9 have two crystallographically independent molecules present in the asymmetric unit. Though they are crystallographically independent, they are chemically identical. Compound 7 displays an unusual intra-molecular hydrogen-bond from the amide nitrogen to the imidazopyridine bridge nitrogen (position 4, Fig. 1). This hydrogen bond may be strong enough to persist in solution which may explain the decreased activity of this compound.
Intrigued that the imidazo[1,2-a]pyrimidine (6), which has an additional nitrogen at position 8, had retained good potency but the imidazo[1,2-b]pyrazine (7), where the additional nitrogen is at the 5 position, had poor potency, we prepared an additional compound set (24–26) to explore the effects of placing the nitrogen at the 6-(imidazo[1,2-c]pyrimidine) and 7-positions (imidazo[1,2-a]pyrazine) (Fig. 4 and expanded Fig. 8, Supplementary data). Interestingly, the MIC’s of the imidazo[1,2-c]pyrimidine and imidazo[1,2-a]pyrazine compounds (25 and 26) were very similar to that of the analogous imidazo[1,2-a]pyrimidine (24) in that the MIC’s all ranged from as low as 1 – 2 μM to as high as 5 to 9 μM when screened in either the GAS and 7H12 media for all three compounds. This suggests that placement of the nitrogen at either the 6, 7, or 8 positions retains potency much better than at the 5 position of the 5,6-fused bicyclic system, but the most potent compounds remain the imidazo[1,2-a]pyridines lacking any additional nitrogen as the MIC’s of imidazo[1,2-a]pyridine compounds 3 and 4 are significantly lower than that of 24, 25 or 26 (see Supplementary data).
Figure 4.
Positional variation of heterocyclic nitrogen.
In conclusion, various 5,6-fused heteroaromatic compounds were explored as potential surrogates to our previously reported imidazo[1,2-a]pyridine and imidazo[1,2-a]pyrimidine anti-TB scaffolds, but none of these closely related heterocycles had submicromolar potency. Additionally, there appears to be a great preference for imidazo[1,2-a]pyridine-3-carboxamide, as the imidazo[1,2-a]pyridine-2-carboxamides were found to be much less active. Therefore, while scaffold switching can often lead to great improvements in either potency or pharmacokinetic properties, in this study the first scaffold we discovered and reported (the imidazo[1,2-a]pyridine-3-carboxamides) remains the optimal scaffold for synthesis of potent 5,6-fused heteroaromatic small molecule anti-TB agents as this scaffold is the most potent and has drug-like ADME properties.8,24,25
Supplementary Material
Acknowledgments
This work was supported by Grant R01AI054193 from the National Institutes of Health (NIH) and in part by intermediates provided from Dow AgroSciences. We would like to thank the University of Notre Dame, especially the Mass Spectrometry & Proteomics Facility (Bill Boggess and Michelle Joyce), which is supported by the grant CHE-0741793 from the NIH. We thank Profs. Jennifer DuBois and Jed Fisher for meaningful scientific discussion. The excellent technical assistance of Baojie Wan and Yuehong Wang with anti-TB assays at UIC is greatly appreciated.
Footnotes
Supplementary Material Supplementary material (experimental procedures, analytical data for compounds (1–12) and X-ray crystallization data for compounds (1, 7-12) can be found as well as additional, SAR and description of the assays used) associated with this article can be found in the online version. CCDC 987940-987946 contains the supplementary crystallographic data for compounds (1, 7-12). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Global tuberculosis control WHO report 2013 WHO/HTM/TB/2013 [Google Scholar]
- 2.Mahajan R. Int. J. Appl. Basic Med. Res. 2013;3:1. doi: 10.4103/2229-516X.112228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moraski GC, Chang M, Villegas-Estrada A, Franzblau SG, Möllmann U, Miller MJ. Eur. J. Med. Chem. 2010;45:1703. doi: 10.1016/j.ejmech.2009.12.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller MJ, Walz AJ, Zhu H, Wu C, Moraski G, Möllmann U, Tristani EM, Crumbliss AL, Ferdig MT, Checkley L, Edwards RL, Boshoff HI. J. Am. Chem. Soc. 2011;133:2076. doi: 10.1021/ja109665t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moraski GC, Markley LD, Chang M, Cho S, Franzblau SG, Hwang CH, Boshoff H, Miller MJ. Bioorg. Med. Chem. 2012;7:2214. doi: 10.1016/j.bmc.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moraski GC, Markley LD, Hipskind PA, Boshoff H, Cho S, Franzblau SG, Miller MJ. ACS Med. Chem. Lett. 2011;2:466. doi: 10.1021/ml200036r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ollinger J, Bailey M-A, Moraski GC, Casey A, Florio S, Alling T, Miller MJ, Parish T. PLoS ONE. 2013;8:e60531. doi: 10.1371/journal.pone.0060531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moraski GC, Markley LD, Cramer J, Hipskind PA, Boshoff H, Bailey M-A, Alling T, Ollinger J, Parish T, Miller MJ. ACS Med. Chem. Lett. 2013;4:675. doi: 10.1021/ml400088y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sekhon SB, Bimal N. RGUHS J. Pharm. Sci. 2012;2:10. [Google Scholar]
- 10.Tresadern G, Cid JM, Macdonald GJ, Vega JA, de Lucas AI, García A, Matesanz E, Linares ML, Oehlrich D, Lavreysen H, Biesmans I, Trabanco AA. Bioorg. Med. Chem. Lett. 2010;20:175. doi: 10.1016/j.bmcl.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 11.Schneider G, Neidhart W, Giller T, Schmid G. Angew. Chem. Int. Ed. 1999;38:2894. [PubMed] [Google Scholar]
- 12.Böhm H-J, Flohr A, Stahl M. Drug Discovery Today: Technol. 2004;1:217. doi: 10.1016/j.ddtec.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 13.Brown N, Jacoby E. Mini. Rev. Med. Chem. 2006;6:1217. doi: 10.2174/138955706778742768. [DOI] [PubMed] [Google Scholar]
- 14.Langdon SR, Ertl P, Brown N. Mol. Inf. 2010;29:366. doi: 10.1002/minf.201000019. [DOI] [PubMed] [Google Scholar]
- 15.De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, Barry CE. Proc. Natl. Acad. Sci. U. S. A. 2000;97:1252. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins L, Franzblau SG. Antimicrob. Agents Chemother. 1997;41:1004. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho SH, Warit S, Wan B, Hwang CH, Pauli GF, Franzblau SG. Antimicrob. Agents Chemother. 2007;51:1380. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Falzari K, Zhou Z, Pan D, Liu H, Hongmanee P, Franzblau SG. Antimicrob. Agents Chemother. 2005;49:1447. doi: 10.1128/AAC.49.4.1447-1454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tung Y-S, Coumar MS, Wu Y-S, Shiao H-Y, Chang J-Y, Liou J-P, Shukla PS, Chang C-W, Chang C-Y, Kuo C-C, Yeh T-K, Lin C-Y, Wu J-S, Wu S-Y, Lioa C-C, Hsieh H-P. J. Med. Chem. 2011;54:3076. doi: 10.1021/jm101027s. [DOI] [PubMed] [Google Scholar]
- 20.Smith DA, Di L, Kerns EH. Nat. Rev.Drug Discov. 2010;9:929. doi: 10.1038/nrd3287. [DOI] [PubMed] [Google Scholar]
- 21.Ramachandran S, Panda M, Mukherjee K, Choudhury NR, Tantry SJ, Kedari CK, Ramachandran V, Ramya VK, Guptha S, Sambandamurthy VK. Bioorg. Med. Chem. Lett. 2013;23:4996. doi: 10.1016/j.bmcl.2013.06.043. [DOI] [PubMed] [Google Scholar]
- 22.Pethe K, Sequeira PC, Agarwalla S, Rhee K, Kuhen K, Phong WY, Patel V, Beer D, Walker JR, Duraiswamy J, Jiricek J, Keller TH, Chatterjee A, Tan MP, Ujjini M, Roa SPS, Camacho L, Bifani P, Mak PA, Ma I, Barnes SW. Nat. Commun. 2010;57:1. doi: 10.1038/ncomms1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ogawa T, Otani N. Kekkaku. 1961:138. [PubMed] [Google Scholar]
- 24.Abrahams KA, Cox JAG, Spivey VL, Loman NJ, Patten MJ, Constantinidoou C, Fernandex R, Alemparte C, Remuinan MJ, Barros D, Ballell L, Besra GS. PLoS ONE. 2012;7:e52951. doi: 10.1371/journal.pone.0052951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pethe K, Bifani P, Jang J, Kang S, Park S, Ahn S, Jiricek J, Jung J, Jeon HK, Cechetto J, Lee H, Kempf M, Jackson M, Lanaerts AJ, Pham H, Jones V, Seo MJ, Kim YM, Seo M, Seo JJ, Park D, Ko Y, Choi I, Kim R, Kim SY, Lim S-B, Yim S-A, Nam J, Kang H, Kwon H, Oh CT, Cho Y, Jang Y, Kim J, Chua A, Tan BH, Nanjundappa MB, Rao SPS, Barnes WS, Wintjens R, Walker JR, Alonso S, Lee S, Kim J, Oh S, Oh T, Nehrbass U, Han SJ, No Z, Lee J, Brodin P, Cho S-N, Nam K, Kim J. Nat. Med. 2013;19:1157. doi: 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.