

Abstract
Transmembrane channel-like (TMC) proteins 1 and 2 are necessary for hair cell mechanotransduction but their precise function is controversial. A growing body of evidence supports a direct role for TMC1 and TMC2 as components of the transduction complex. However, a number of important questions remain and alternate hypotheses have been proposed. Here we present an historical overview of the identification and cloning of Tmc genes, a discussion of mutations in TMC1 that cause deafness in mice and humans and a brief review of other members of the Tmc gene superfamily. We also examine expression of Tmc mRNAs and localization of the protein products. The review focuses on potential functions of TMC proteins and the evidence from Beethoven mice that suggests a direct role for TMC1 in hair cell mechanotransduction. Data that support alternate interpretations are also considered. The article concludes with a discussion of outstanding questions and future directions for TMC research. This article is part of a Special Issue entitled “Annual Reviews 2014”.
Keywords: auditory, vestibular, hair cell, mechanotransduction, TMC1, TMC2, sensory transduction, hearing, balance, deafness, dizziness
Historical overview
Genetic hearing loss has been categorized as either syndromic, affecting auditory function and at least one other bodily function, or non-syndromic, affecting auditory function alone. Because non-syndromic hearing loss affects the ear alone, it has been reasoned that genes that contribute to the unique mechanosensory function and cytoarchitecture of inner ear hair bundles may be identified by screens for non-syndromic hearing loss genes. Indeed, over the past twenty years this strategy has proven remarkably successful and has lead to the cloning and identification of a number of non-syndromic hearing loss genes that are critical for normal hair cell function. One example is Transmembrane channel-like 1 gene (TMC1), mutations in which cause dominant and recessive non-sydnromic hearing losses, DFNA36 and DFNB7/11, respectively (Kurima et al., 2002). Ironically, discovery of this essential hair cell gene, which may encode a component of the mechanotransduction channel, actually began over 55 years ago with one of the first descriptions of non-syndromic hearing loss in a mouse model.
Deol and Kocher (1958) characterized a mouse line that lacked auditory startle reflexes without other complications that they simply termed, deafness (dn). Although, their stimulus was crude, striking forceps against a glass bottle, the lack of an ear twitch was obvious even at the onset of hearing, postnatal day (P) 10 to P14 in control littermates. Despite the profound deafness in the dn mice, Deol and Kocher (1958) reported normal hair cell morphology until two weeks of age. Interestingly, they also reported a subtle vestibular phenotype described as “a barely perceptible abnormality”.
Further characterization of the dn mouse line would await another 22 years, until Karen Steel and Gregory Bock (1980) examined the mice electrophysiologically. Steel and Bock reported complete lack of compound action potentials in the 8th cranial nerve in response to sound stimuli that spanned the auditory frequency range for control mice (1–60 kHz) at ages between P12 and P20. In a second assay, they measured cochlear microphonics, a measure of the summed activity of hair cell receptor potentials and here again found a complete lack of response across the same frequency and age ranges, suggesting the deficit may be due to hair cell dysfunction, even as early as P12. For the third assay, they described a diminished decay rate for the endocochlear potential under anoxic conditions suggesting disruption of the normal pathway for recirculation of endolymph potassium. With these three lines of evidence and remarkable foresight Steel and Bock (1980) concluded that since “mechanical stimulation controls the opening of ion channels in the hair cell membrane…the most likely explanation for the absence of the microphonics is that the ion channels are not opening normally”. Confirmation of this notion would require another 30 years.
In the meantime, the dn mouse became a valuable animal model for human deafness as well as for investigations into the consequences of peripheral quiescence on development of central auditory pathways. Steel and Bock (1984) demonstrated normal spiral ganglion neuron density and normal responses to electrical stimulation in young animals, suggesting that although dn mice lacked sound evoked-activity, central pathways seemed to develop normally at least during early postnatal stages. At the same time the search for the defective gene in the dn mouse was well underway. Human geneticists identified that the autosomal recessive non-syndromic hearing loss locus DFNB7/11 was mapped to human chromosome 9 and may affect an orthologous gene mapped to chromosome 19 in the dn mouse (Keats et al. 1995). The search intensified as several novel genes within the DFNB7/11 interval were identified (Greinwald et al., 1997), two of which were later dismissed due to a lack of disease-causing mutations (Scott et al., 1998; 2000). In 2002 two groups, led by Friedman and Griffith and by Avraham and Steel, converged upon a common answer, but from different angles. The Friedman/Griffith group (Kurima et al., 2002) identified multiple mutations scattered throughout the coding sequence of a novel gene named Transmembrane cochlear-expressed gene 1 which was later renamed Transmembrane channel-like gene 1 (TMC1). Both dominant (DFNA36) and recessive (DFNB7/11) TMC1 mutations were identified in deaf patients. A 1.6 kb deletion in mouse Tmc1 was found to be causative disruption in the dn mouse. In a companion paper the Avraham/Steel group (Vreugde et al., 2002) identified a novel semi-dominant mutation that arose from an ENU mutagenesis screen. The mutation, termed Beethoven (Bth), resulted in a methionine to lysine substitution at amino acid position 412 in mouse TMC1. Together the two papers established a solid role for TMC1 in auditory function in mice and humans.
Tmc1 mutations in mice and men
Although the structure and function of TMC proteins are unknown, the location of pathogenic TMC1 mutations may provide some clues about this novel family of proteins. Hydropathy plots predict six to eight transmembrane domains (Kurima et al., 2002). Labay et al. (2010) used epitope tags strategically placed in several regions of the human TMC1 sequence. Using the epitope-tagged versions expressed in heterologous cells and antibodies directed against the epitopes, they predicted a TMC1 topology that includes six transmembrane domains with a long N-terminus, three extracellular loops, a large intracellular loop between transmembrane domain four and five and a short C-terminus (Figure 1). Within the S4–S5 loop there are two highly conserved domains known as the TMC domains. To date, 29 pathogenic mutations (Figure 1) have been identified in TMC1 that cause deafness in humans (Hilgert et al., 2008 and references there in; Sirmaci et al., 2009; Brownstein et al., 2011; Diaz-Horta et al., 2012; Gao et al., 2013). Twenty-three are located in coding regions and another six occur in non-coding regions and cause deletions or affect splice sites. Of the 23 mutations in the TMC1 coding sequence, three are dominant point mutations. G417R affects the human residue adjacent to the mouse Bth mutation (M412K; Yang et al., 2010) and D572 seems to be a mutational hot spot with D572N or D572H causing dominant-progressive deafness (Hilgert et al., 2009). The 26 recessive mutations are linked to DFNB7/B11 while the three dominant mutations are associated with DFNA36.
Figure 1.

Topology of human TMC1 showing the amino acid sequence, transmembrane domains, TMC domains (shaded in grey) and mutation sites. Sites of recessive mutations are shown in red with human mutations in large upper case letters. Mouse mutations were mapped onto the human sequence and are shown in lower case. The region deleted in the dn mouse is underlined. Dominant mutations are shown in green for mouse and human. Amino acid residues are numbered in intervals of ten and membrane spanning domains are labeled S1–S6.
In mouse, five mutations have been identified in the Tmc1 coding sequence (Figure 1), all of which produce deafness. There are four recessive mutations and one dominant. The dn mutation appears to cause congenital, recessive deafness while the Bth mutation causes an early onset semi-dominant progressive deafness (Vreugde et al., 2002).
The Tmc gene family
Tmc1 is the founding member of the Tmc superfamily and is present in genomes throughout the animal kingdom ranging from model invertebrates such as fruitflies and round worms to model vertebrates including zebrafish, frogs, chickens and mice, but is not present in plants or yeast (Keresztes et al., 2003). Within the mammalian class, the Tmc1 gene is found in species ranging from alpacas to apes including, opossums, dolphins, sloths, rodents, dogs, cats and bats (Davies et al., 2012). The gene sequence is highly conserved, the TMC domains in particular (Figure 1), suggesting strong selective pressure throughout animal evolution. Invertebrates get by with just one or two Tmc genes, while mammalian genomes include eight Tmc genes (Tmc1–8). The TMC domains are signature amino acid sequences present in all TMCs but are absent in non-TMC proteins. The structural and functional significance of the TMC domains are unclear.
Figure 2 shows a dendrogram based on the amino acid sequences of all eight mouse and human TMCs and includes the TMCs from several other model organisms. The sequences have been clustered into three subfamilies that include TMCs 1–3, TMCs 5 and 6 and TMCs 4, 7 and 8 (Keresztes et al., 2003). Gene duplication may account for some of the redundancy within the subfamilies (Kurima et al., 2003). The mouse and human orthologs share 75–95% amino acid sequence identity and there is 22–57% amino acid sequence identity among TMC proteins encoded in the human genome (Kurima et al., 2003). The highly conserved amino acid sequence – 95% for mouse and human TMC1 – suggests an important function for these proteins. Indeed, normal auditory function, for which Tmc1 is necessary, has proven advantageous over the course of animal life on earth.
Figure 2.

Dendrogram showing members of the TMC gene family from several model organisms. Nematode (Ce), fruitfly (Dm), zebrafish (Dr), xenopus (Xt), mouse (Mm) and human (Hs) TMCs are shown. TMC1–3 form one group, TMC5–6 form a second and a third includes TMC4, 7 and 8. Human TMCs are shown in blue and mouse TMCs are shown in red. The dendrogram was generated using Clustal Omega for multiple sequence alignments, Protdist from PHYLIP to calculate distances and FigTree to draw the tree.
The expression pattern and function of other Tmc genes is not clear. The exception being Tmcs 6 and 8 which may have a role in zinc transport in cutaneous tissue (reviewed by Lazarczyk and Favre, 2008).
TMC expression and localization in hair cells
Tmc1 mRNA is expressed in inner and outer hair cells at P5 (Kurima et al., 2002) and at P15 (Vreugde et al., 2002) as shown by in situ hybridization. Tmc1 mRNA is also evident in P5 vestibular epithelia (Kurima et al., 2002), while Tmc2 mRNA signals can be detected in vestibular organs at embryonic day 17 and in auditory hair cells as early as P0 (Kawashima et al., 2011). Mutai et al. (2005) showed expression of Tmc2 mRNA isolated from chicken basilar papilla hair cells and Tmc1 expression restricted to the basal region. To further characterize Tmc1 and Tmc2 expression patterns Kawashima et al. (2011) generated targeted disruption constructs of these genes by replacing exons 8 and 9 or exon 7, respectively, with an IRES LacZ construct. Expression of the LacZ reporter was evident in hair cells of auditory and vestibular epithelia at P28. Interestingly, LacZ expression was strongest in peripheral vestibular regions of Tmc1 mutants, while faint LacZ expression was evident throughout the sensory epithelium of Tmc2 mutants. Together, these studies confirmed expression of Tmc1 and Tmc2 in auditory and vestibular hair cells from birth through the end of the first month of age.
Localization of the Tmc1 and Tmc2 protein products has proven more challenging. Although Mutai et al. (2005) showed immunolocalization of TMC2 in the apical and lateral membranes of chicken hair cells, this result could not be reproduced in mouse for technical reasons. Kawashima et al. (2011) generated five antibodies that targeted the N-termini of mouse TMC1 or TMC2, but none were found to be specific. The chicken TMC2 antibody was directed against the C-terminus but its specificity was not confirmed due to a lack of a chicken model that lacked TMC2 protein. As an alternate approach to immunolocalization, TMC1::GFP and TMC2::GFP fusion constructs were generated and transfected into mouse and rat hair cells, respectively. Punctate GFP fluorescence was evident in stereocilia tips of transfected hair cells. Although less than ideal, localization of the fusion constructs in stereocilia confirmed that TMC fusion proteins can be targeted to the site of hair cell transduction in exogenous expression experiments. Whether endogeneous TMCs are localized at the site of hair cell transduction will require highly selective antibodies with minimal background staining patterns.
Tmc function in mechanotransduction
Beginning with the initial studies of Steel and Bock (1980), it was clear that the dn mouse lacked cochlear microphonic potentials, which arise from the summed electrical activity of inner and outer hair cells. This result implicated a hair cell defect, but what? Marcotti et al. (2006) tackled this question in a large study that examined hair cell physiology in both the dn and Bth mouse models. However, for reasons that would not become clear for another five years, a role for TMC1 in hair cell transduction evaded detection. Marcotti et al. (2006) correctly reported normal mechanotransduction in homozygous dn and Bth outer hair cells excised from the cochlear apex at P6–P8. The transduction currents were similar to those of heterozygous and wild-type controls which prompted the investigators to focus their search elsewhere. They identified several physiological deficits in hair cell maturation at later stages. Beginning in the second postnatal week both inner and outer hair cells failed to acquire specific voltage-dependent currents and other mature phenotypes. They proposed that the failure to mature electrophysiologically may have been the proximal cause leading to hair cell degeneration. However, they also astutely pointed out that ion channel expression can be regulated by calcium entry, which may provide a link between Tmc1 mutations, ion channel expression and eventual hair cell degeneration.
A missing piece of the puzzle, about which Marcotti et al. (2006) had little information, was expression of the Tmc1 ortholog, Tmc2. As identified by Kawashima et al. (2011), Tmc2 expression precedes Tmc1 and appears to provide somewhat redundant functions. Kawashima et al. (2011) found that expression of Tmc2, even in the absence of functional Tmc1, is sufficient for hair cell mechanotransduction at early postnatal stages. Thus, wild-type Tmc2 was available to compensate for mutant Tmc1 in the Marcotti et al. (2006) experiments. Using a quantitative RT-PCR approach (qPCR), Kawashima et al. (2011) identified an expression switch in cochlear hair cells from Tmc2 to Tmc1 at the end of the first postnatal week. Tmc2 expression rises just prior to the developmental onset of hair cell transduction – P0–P2 in mouse (Lelli et al., 2009) – peaks during the first postnatal week and declines to very low levels around P8. In other words, robust Tmc2 expression during the first week may have accounted for the large mechanotransduction currents recorded at P6–P8 (Marcotti et al., 2006), despite the presence Tmc1 mutations. At stages beyond P8 when Tmc2 expression declines and Tmc1 expression rises, the consequences of the dn and Bth mutations are evident. Indeed, Kim and Fettiplace (2013) showed a loss in mechanotransduction during the second postnatal week in outer hair cells of dn mice. The physiological significance of the developmental switch from Tmc2 to Tmc1 in wild-type mice is unclear.
What is clear, however, is that hair cells that lack both Tmc1 and Tmc2, lack conventional mechanotransduction entirely (Kawashima et al., 2011; Pan et al., 2013; Kim et al., 2013). Hair cells from Tmc1/Tmc2 doubly deficient mice lack conventional mechanotransduction, regardless of organ, region, age or hair bundle stimulation method (Figure 3A). The data clearly show that expression of either Tmc1 or Tmc2 is required for conventional hair cell transduction.
Figure 3.
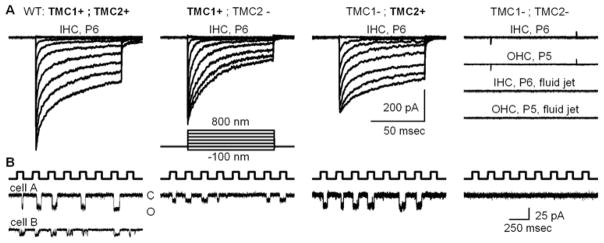
Mechanotransduction currents recorded at −84 mV from hair cells of various Tmc genotypes. Genotypes are shown at the top with the protein product expressed in each case shown in bold. A, Families of whole-cell transduction currents recorded from cochlear inner hair cells at postnatal day 5 to 6 as indicated. Each family of traces is the mean of ten datasets taken from five representative cells. The stimulus was a stiff probe driven by a piezoelectrical bimorph using the step protocol shown. At the right are four representative families recorded from hair cells deficient in both Tmc1 and Tmc2. Cochlear hair cell type and age are shown for each family. The lower two families were recorded in response to 50 Hz sine wave fluid-jet deflection of inner and outer hair bundles. The scale bar applies to all current families shown in panel A. External calcium: 1.3 mM. B, Representative concatenated traces showing single-channel currents recorded from P6 inner hair cells that expressed the protein shown above panel A. The top trace shows the concatenated 100-nm square wave protocol delivered via stiff probes designed to deflect single stereocilia. For the wild-type traces (left) two examples are shown that illustrate the range of single-channel amplitudes encountered. By convention, downward deflections represent inward currents and channel openings. The scale bar at the right applies to all traces. External calcium: 50 μM. Modified from Pan et al. (2013).
In a recent report, Kim et al. (2013) described a form of non-conventional or anomalous mechanosensitivity that differs from conventional hair-cell mechanotransduction in a number of respects. The anomalous mechanosensitivity does not require tip links or intact hair bundles and differs in its sensitivity to antagonists, calcium permeability and single-channel conductance. It also differs in its sensitivity to mechanical stimulation, being activated by fluid jet stimuli of the opposite polarity to that of conventional mechanotransduction, i.e., stimuli directed toward the tall edge of the bundle. Anomalous mechanosensitivity has also been reported in hair cells of mice that expressed mutant tip-link proteins, PCDH15 or CDH23 (Alagramam et al., 2011) and in hair cells with mutant ankle link protein, VLGR1 (Michalski et al., 2007). Interestingly, the response does not depend on expression of TMC1 or TMC2 as mutant mice that lacked wild-type Tmc1 and Tmc2, retained anomalous mechanosensitivity. Based on these observations Kim et al. (2013) proposed that TMC1 and TMC2 are not components of the conventional hair cell mechanotransduction channel. However, since conventional mechanotransduction was absent in Tmc1/2 double mutant hair cells (Kim et al., 2013) but anomalous mechanosensitivity was retained, it could be argued that TMC1/2 expression is required for the conventional but not the anomalous response. Whether there are any mechanistic or molecular similarities between conventional and anomalous mechanosensitivity has not been established.
Although the interpretations of Kim et al. (2013) and Pan et al. (2013) differ, both studies showed that TMC1/2 double mutant mice lacked conventional mechanotransduction. They were also in agreement that the biophysical properties of conventional transduction differed in cells that expressed TMC1 relative to those that expressed TMC2. Pan et al. (2013) found that hair cells that expressed TMC2 had large whole-cell transduction currents, large single-channel currents (Figure 3B), high calcium permeability and slower adaptation relative to cells that expressed TMC1 ( Pan et al., 2013). Kim and Fettiplace (2013) showed that outer hair cells of dn mice, which presumably lack normal TMC1 function but expressed wild-type TMC2, had higher calcium permeability than outer hair cells of mice that expressed wild-type TMC1 in the absence of TMC2. Although Kim et al. (2013) were in agreement that single-channel conductance values differed between cells that expressed wild-type TMC1 or TMC2, the values they reported were significantly smaller than those reported by Pan et al. (2013) and smaller than those reported for conventional transduction in wild-type animals (Géléoc et al., 1997; Ricci et al., 2003; Beurg et al., 2006). Together, the data (Kawashima et al., 2011; Pan et al., 2013; Kim and Fettiplace, 2013 and Kim et al., 2013) agree that expression of TMC1 and TMC2 affect several core biophysical properties of conventional mechanotransduction in auditory and vestibular hair cells.
Beethoven provides a key
Both Vreugde et al. (2002) and Marcotti et al. (2006) reported normal transduction in hair cells of Bth mice. However, since the data were obtained from apical hair cells at P6–P8, a region and time in which wild-type TMC2 was likely available to compensate, Pan et al. (2013) chose to investigate the consequences of the Bth mutation (TMC1 p.M412K) in the absence of TMC2 and crossed Bth mice with Tmc1/Tmc2 double knockout mice. Offspring that expressed the Bth allele were profoundly deaf, had significant loss of inner hair cells but, surprisingly, the inner hair cells had large mechanotransduction currents at P8 (Figure 4A–C), suggesting that the Bth mutation was not a loss-of-function or dominant-negative mutation. Importantly, since transduction was retained, Pan et al. (2013) were able to examine the biophysical consequences of the Bth mutation in the absence of wild-type Tmc1 and Tmc2. A number of mechanotransduction properties differed from those of mice that expressed wild-type TMC1: single-channel currents were smaller; calcium selectivity was significantly reduced (Figure 4D); calcium block through open transduction channels was greater and adaptation was slower (Pan et al., 2013). Since a single point mutation altered several biophysical properties intimately associated with ion channel permeation properties, the parsimonious explanation was that TMC1 must be a component of the transduction channel itself.
Figure 4.

Data acquired from Bth mice crossed onto a Tmc1/Tmc2-deficient background. A, Auditory brainstem responses show the mice were profoundly deaf as early as P30, whereas control mice that expressed wild-type TMC1 had normal hearing thresholds. B, Counts of inner hair cell survival showed significant cell loss in Bth mice relative those that expressed wild-type TMC1. C, Inner hair cells that expressed TMC1 Bth on a Tmc1/Tmc2-deficient background had large whole-cell transduction currents suggesting that Bth is not a loss of function mutation. D, Representative mechanotransduction current–voltage relations are shown for inner hair cells that expressed either wild-type TMC1 (black) or TMC1 Bth (gray). Cells were bathed in 100 mM calcium and reversal potential was estimated from the x-intercept. The data suggest that the Bth point mutation causes a significant reduction in calcium selectivity. Modified from Pan et al. (2013).
The Bth mutation did not affect the slope of the stimulus-response curve, suggesting that the relationship between force and channel open probability was unaltered. Furthermore, this relationship did not vary among cells that expressed TMC2, TMC1 or TMC1 Bth. In other words, TMC expression does not alter the properties of channel gating and sensitivity but seems to affect permeation properties specifically. Because three of the properties that were altered in Bth hair cells are associated with ion channel permeation, the mutation may reside in the pore region or in close enough proximity to affect ion permeation.
The Bth mutation at amino acid 412 is in a predicted extracellular loop between the 3rd and 4th membrane spanning domains (Figure 1). Whether this region helps form the cation permeation pathway is not clear. One intriguing possibility that fits nicely with the Pan et al. (2013) data, is that the S3–S4 loop may form a vestibule at the mouth of the channel pore. If so, the Bth mutation which swaps a neutral methionine residue for a positively charged lysine may yield a charge screening effect. In other words, the positive charge of a single lysine or a cluster of 3 to 4 positive charges – one lysine per channel subunit – at the mouth of the pore may repel divalent cations and thus decrease the local concentration of calcium such that both the single channel currents and the effective calcium permeability are reduced. Whether this is indeed the case will require further structural information.
Although the data suggest TMC1 and TMC2 are components of the transduction channel, there are alternate explanations that could account for the constellation of observations reported thus far. TMC1 or TMC2 expression is clearly necessary for hair cell mechanotransduction and a point mutation in TMC1 affects transduction channel permeation properties. It is conceivable that TMC1 and TMC2 could serve as indispensable protein subunits and that the Bth mutation alters the permeation properties of some other pore-forming subunit. While plausible, this configuration seems less likely and is unprecedented among known ion channels. TARP proteins have been shown to modulate AMPA receptor permeation properties and Morgan and Barr-Gillespie (2013) suggested that TMCs may perform an analogous function for the hair cell transduction channel. However, TARPs are not essential subunits – AMPA receptors can function as ion channels in their absence – whereas TMCs are clearly essential for transduction channel function.
It does seem likely that other proteins may contribute the hair cell transduction channel complex. Both known and unknown hair bundle components may interact with TMC1 or TMC2. Known proteins required of normal transduction, such as PCDH15 (Kazmierczak et al., 2007), TMHS (Xiong et al., 2012) or TMIE (Gleason et al., 2009), may interact with TMCs to module hair cell transduction, yet functional interactions with TMC proteins is, thus far, hypothetical. Nonetheless, since the hair cell transduction complex is a highly specialized structure, identification of other molecular components and functional interactions will require unconventional thinking and an unbiased perspective.
In addition to the biophysical changes caused by the Bth mutation, there were other downstream consequences of the mutation. There was a statistically significant increase in the number of transduction channels expressed in Bth inner hair cells (Pan et al., 2013). Consistent with that observation, quantitative PCR analysis revealed an upregulation of Tmc1 mRNA levels which may explain the large transduction currents at the end of the first postnatal week. However, it is difficult to reconcile the large currents at P8 with the loss of inner hair cells and the profound deafness at the end of the first month of life. Perhaps these physiologically relevant consequences may be the result of reduced calcium permeability in hair cells that express the Bth mutation. If so, this hints at a possible mechanism for the stunted hair cell maturation and eventual hair cell death reported by Marcotti et al. (2006) and Pan et al. (2013). If hair cells employ calcium-dependent gene transcription mechanisms, those mechanisms may be disrupted in the dn and Bth mouse lines leading to loss of normal gene transcription and loss of mature physiological properties such as mature voltage-dependent currents (Marcotti et al., 2006). Indeed, this general mechanism may also contribute to hair cell loss and hence the profound deafness in humans with dominant TMC1 mutations (Yang et al., 2008).
Tmc gradients in the cochlea
Given that wild-type TMC1 and TMC2 contribute to hair cell transduction with distinct biophysical properties, developmental and tonotopic gradients in Tmc expression may contribute to frequency selectivity along the length of the cochlea. During the first postnatal week, when both Tmc1 and Tmc2 are expressed, Pan et al. (2013) found a broad range of single-channel conductances (Figure 3B). This result suggests a mechanism for variation of transduction properties along the length of the cochlea. If gradients in TMC1 and TMC2 protein expression persist into adulthood, variation in their stoichiometry may contribute to variation in single-channel properties, such as conductance and calcium permeability. Similarly, whole-cell properties such as current amplitudes and adaptation rate and extent may also vary as a function of gradients in TMC expression. Because these properties help shape the graded hair cell receptor potential, gradients in TMC expression may contribute to frequency selectivity and tuning along the cochlear tonotopic axis. Pan et al. (2013) reported four distinct single-channel conductance levels in wild-type inner hair cells. If transduction channel composition varies within a single cell and there are ~100 channels/hair cell, there could be up to 300 unique whole-cell conductance levels. Other properties such as calcium permeability and adaptation also vary along the length of the cochlea, but have not yet been measured at the single-channel level. Thus, gradients in TMC1 and TMC2 expression along the tonotopic axis of the mammalian cochlea may be sufficient to account for known variation in hair cell mechanotransduction properties. Other factors may also contribute to tonotopic variation, such as expression of other Tmcs (Tmc3–8), expression of Tmc alternative splice forms or modification by potential interacting proteins, perhaps TMHS (Xiong et al., 2012) or TMIE (Gleason et al., 2009), or gradients in other as yet, unidentified components of the transduction complex.
Future directions
A number of important questions regarding TMC function remain unanswered. The following section presents a partial list. Some of this work is currently underway, while other aspects will take considerable time and effort. Nonetheless, as TMC1 mutations cause deafness in humans and mice and since compelling evidence already exists implicating a TMC1 and TMC2 as components of the transduction channel, the following experiments should be considered high priority with considerable impact for the field of inner ear biology as well as the more extend fields of mechanotransduction and sensory biology.
Convincing localization of TMC proteins is needed. Unfortunately, immunolocalization of TMC proteins has suffered a similar fate as many other hair bundle proteins: non-specific labeling even in genetically-null mice; high background levels that prevent identification of dim signals from few proteins and so forth. To circumvent these issues, Kawashima et al. (2011) localized exogenous GFP-tagged TMC1 and TMC2. The tips of inner, outer and vestibular hair cell stereocilia were decorated with green puncta, suggesting that TMC1 and TMC2 can be trafficked to the site of hair cell transduction. Whether this was an artifact of exogenous over expression experiments in vitro or representative of the endogenous localization pattern in vivo has not been determined. To be consistent with a direct role in mechanotransduction, endogenous TMC1 and TMC2 must be localized at the site of the mechanotransduction apparatus. Localization at additional sites would not rule out a role in transduction, provided hair bundle tip localization persists. For example, if TMCs are also localized along the length or at the base of the stereocilia or in the cuticular plate region, they may be in a state not activated by conventional stimulation, perhaps in a reserve pool awaiting transport to the stereocilia tips. Of course, sub-organelle localization of TMCs within the transduction complex will also be of great interest. Are TMCs at the upper or lower end of the tip link, or both? Are they directly linked to the tip link complex or are they tethered and at some distance where they might sense tension applied through membrane stretch? Does their localization pattern change during development or regeneration of mechanotransduction as suggested for PCDH15 (Indzhykulian et al., 2013)? Answers to these questions may require reliable, specific, high-affinity TMC antibodies or endogenous tagged TMC constructs, combined with super-resolution microscopy, back scatter scanning electron microscopy or immunogold transmission electron microscopy.
Another line of evidence that could potentially support a direct role for TMCs in mechanotransduction would be reconstitution of mechanosensitivity in a heterologous cell line. This is an obvious line of investigation but thus far has proven challenging, not just for TMCs but for a number of other putative mechanosensitive channels as well. Two notable exceptions are the mechanosensitive NOMPC channel first identified in Drosophila (Walker et al., 2000), and recently shown to generate mechanosensitive currents when expressed in a heterologous cell line (Yan et al., 2013) and Piezo1 which forms mechanosensitive channels when expressed in HEK293 cells (Coste et al., 2012). Although TMCs have yet to fulfill the heterologous expression criterion, one line of evidence has suggested that C. elegans tmc-1 can be expressed in heterologous cells to yield sodium-activated currents (Chatzigeorgiou et al., 2013), suggesting that the Tmc superfamily includes genes that encode ion channels. Whether this property extends to human and mouse TMCs remains to be determined. While C. elegans tmc-1 bears some resemblance to human and mouse TMCs, they are distantly related (Figure 2). Heterologous cell lines may lack the appropriate lipid environment, appropriate binding partners or the correct cytoarchitecture that permit reconstitution of mechanosensitivity when transfected with mouse or human TMC genes. If the correct parameters can be identified that permit reconstitution of mechanosensitivity in Tmc-transfected cell lines it would not only demonstrate that TMCs form mechanosensitive ion channels, but would also provide a valuable platform for investigating TMC structure-function relationships. A principle focus would be identification of the sequence that encodes the channel pore, but other worthy investigations may focus on identification of sequences that affect calcium-dependent adaptation, channel gating and perhaps the biophysically defined gating spring. Some of these issues could be advanced by expression in a heterologous system while others may require elucidation of a TMC crystal structure.
Lastly, there are a number of outstanding questions regarding the physiological significance of Tmc1 and Tmc2 expression patterns in the inner ear. Tmc2 is highly expressed in vestibular end organs where its expression is retained into adulthood. The physiological significance of Tmc2 expression in these organs is not clear. However, there are several aspects which seem to fit with our current understanding of vestibular function. For example, hair cells that express Tmc2 have slower adaptation rates, consistent with low frequency sensitive of vestibular organs. The larger single-channel conductance seems consistent with the lack of an endolymphatic potential in vestibular organs (Pan et al., 2013).
In the auditory organ there is a developmental switch in expression with high Tmc2 mRNA levels during the first postnatal week which drops precipitously as Tmc1 mRNA levels rise. Is Tmc2 expression in the cochlea an evolutionary hold over reflecting the possibility that auditory organs evolved as a specialization from primordial vestibular organs? Or does the high calcium permeability of TMC2 channels contribute some developmental signal, perhaps one that contributes to calcium-dependent gene transcription? The switch to TMC1 is consistent with a growing body of evidence that TMC1 may be more specialized for high frequency hearing. Kawashima et al. (2011) noted gradients in Tmc1 expression with higher levels at the basal, high frequency, end of the cochlea and Davies et al. (2012) suggested that molecular adaptations in Tmc1 may be associated with the evolution of high frequency hearing in echolocating bats and marine mammals.
Conclusions
Clarification of the precise molecular composition of the hair cell transduction channel remains an important goal worthy of further investigation. The data from Kawashima et al. (2011), Pan et al. (2013) and Kim and Fettiplace (2013) all support a role for TMC proteins that is intimately associated with hair cell mechanotransduction. Yet, many important questions remain: are TMC1 and 2 pore-forming subunits, and if so, do transduction channels consist of solely TMC proteins, or are other subunits involved? Alternatively, if they are not pore-forming subunits, are they channel components that function in some peripheral, yet essential capacity? Or could they perform other critical functions, perhaps linking mechanosensitive channels with other components of the transduction complex while still modulating channel permeation properties?
Answers to these and many other Tmc related questions will surely follow. Identification of the molecular functions of Tmc genes is not just an exercise in naming the molecules involved in hair cell mechanotransduction but is broadly significant because it may provide a handle on the fundamental mechanisms that mediate human hearing and balance. We are encouraged by recent progress and optimistic that discovery of TMC function will help accelerate the pace of auditory and vestibular research including elucidation of the precise molecular composition of the hair cell transduction complex.
Over 30 TMC1 mutations have been identified that cause deafness in mice and humans.
Eight mammalian TMC genes have been identified and a few are found in invertebrates.
Hair cells express Tmc1 and Tmc2 and the proteins can be found at stereocilia tips.
Hair cells that express TMC1 or TMC2 have distinct biophysical properties.
Hair cells that express mutant TMC1 have reduced calcium selectivity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alagramam KN, Goodyear RJ, Geng R, Furness DN, van Aken AF, Marcotti W, Kros CJ, Richardson GP. Mutations in protocadherin 15 and cadherin 23 affect tip links and mechanotransduction in mammalian sensory hair cells. PLoS One. 2011;6(4):e19183. doi: 10.1371/journal.pone.0019183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr-Gillespie PG, Nicolson T. Who needs tip links? Backwards transduction by hair cells. J Gen Physiol. 2013;142(5):481–6. doi: 10.1085/jgp.201311111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurg M, Evans MG, Hackney CM, Fettiplace R. A large-conductance calcium-selective mechanotransducer channel in mammalian cochlear hair cells. J Neurosci. 2006;26(43):10992–1000. doi: 10.1523/JNEUROSCI.2188-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownstein Z, Friedman LM, Shahin H, Oron-Karni V, Kol N, Abu Rayyan A, Parzefall T, Lev D, Shalev S, Frydman M, Davidov B, Shohat M, Rahile M, Lieberman S, Levy-Lahad E, Lee MK, Shomron N, King MC, Walsh T, Kanaan M, Avraham KB. Targeted genomic capture and massively parallel sequencing to identify genes for hereditary hearing loss in Middle Eastern families. Genome Biol. 2011;12(9):R89. doi: 10.1186/gb-2011-12-9-r89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzigeorgiou M, Bang S, Hwang SW, Schafer WR. tmc-1 encodes a sodium-sensitive channel required for salt chemosensation in C. elegans. Nature. 2013;494(7435):95–9. doi: 10.1038/nature11845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste B, Xiao B, Santos JS, Syeda R, Grandl J, Spencer KS, Kim SE, Schmidt M, Mathur J, Dubin AE, Montal M, Patapoutian A. Piezo proteins are pore-forming subunits of mechanically activated channels. Nature. 2012;483(7388):176–81. doi: 10.1038/nature10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KT, Cotton JA, Kirwan JD, Teeling EC, Rossiter SJ. Parallel signatures of sequence evolution among hearing genes in echolocating mammals: an emerging model of genetic convergence. Heredity. 2012;108(5):480–9. doi: 10.1038/hdy.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deol MS, Kocher W. A new gene for deafness in the mouse. Heredity. 1958;12:463–466. [Google Scholar]
- Diaz-Horta O, Duman D, Foster J, 2nd, Sırmacı A, Gonzalez M, Mahdieh N, Fotouhi N, Bonyadi M, Cengiz FB, Menendez I, Ulloa RH, Edwards YJ, Züchner S, Blanton S, Tekin M. Whole-exome sequencing efficiently detects rare mutations in autosomal recessive nonsyndromic hearing loss. PLoS One. 2012;7(11):e50628. doi: 10.1371/journal.pone.0050628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Su Y, Guan LP, Yuan YY, Huang SS, Lu Y, Wang GJ, Han MY, Yu F, Song YS, Zhu QY, Wu J, Dai P. Novel compound heterozygous TMC1 mutations associated with autosomal recessive hearing loss in a Chinese family. PLoS One. 2013;8(5):e63026. doi: 10.1371/journal.pone.0063026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Géléoc GS, Lennan GW, Richardson GP, Kros CJ. A quantitative comparison of mechanoelectrical transduction in vestibular and auditory hair cells of neonatal mice. Proc Biol Sci. 1997;264(1381):611–21. doi: 10.1098/rspb.1997.0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason MR, Nagiel A, Jamet S, Vologodskaia M, López-Schier H, Hudspeth AJ. The transmembrane inner ear (Tmie) protein is essential for normal hearing and balance in the zebrafish. Proc Natl Acad Sci USA. 2009;106(50):21347–52. doi: 10.1073/pnas.0911632106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greinwald JH, Jr, Scott DA, Marietta JR, Carmi R, Manaligod J, Ramesh A, Zbar RI, Kraft ML, Elbedour K, Yairi Y, Musy M, Skvorak AB, Van Camp G, Srisailapathy CR, Lovett M, Morton CC, Sheffield VC, Smith RJ. Construction of P1-derived artificial chromosome and yeast artificial chromosome contigs encompassing the DFNB7 and DFNB11 region of chromosome 9q13–21. Genome Res. 1997;7(9):879–86. doi: 10.1101/gr.7.9.879. [DOI] [PubMed] [Google Scholar]
- Hilgert N, Alasti F, Dieltjens N, Pawlik B, Wollnik B, Uyguner O, Delmaghani S, Weil D, Petit C, Danis E, Yang T, Pandelia E, Petersen MB, Goossens D, Favero JD, Sanati MH, Smith RJ, Van Camp G. Mutation analysis of TMC1 identifies four new mutations and suggests an additional deafness gene at loci DFNA36 and DFNB7/11. Clin Genet. 2008;74(3):223–32. doi: 10.1111/j.1399-0004.2008.01053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgert N, Monahan K, Kurima K, Li C, Friedman RA, Griffith AJ, Van Camp G. Amino acid 572 in TMC1: hot spot or critical functional residue for dominant mutations causing hearing impairment. J Hum Genet. 2009;54(3):188–90. doi: 10.1038/jhg.2009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indzhykulian AA, Stepanyan R, Nelina A, Spinelli KJ, Ahmed ZM, Belyantseva IA, Friedman TB, Barr-Gillespie PG, Frolenkov GI. Molecular remodeling of tip links underlies mechanosensory regeneration in auditory hair cells. PLoS Biol. 2013;11(6):e1001583. doi: 10.1371/journal.pbio.1001583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima Y, Géléoc GS, Kurima K, Labay V, Lelli A, Asai Y, Makishima T, Wu DK, Della Santina CC, Holt JR, Griffith AJ. Mechanotransduction in mouse inner ear hair cells requires transmembrane channel-like genes. J Clin Invest. 2011;121(12):4796–809. doi: 10.1172/JCI60405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak P, Sakaguchi H, Tokita J, Wilson-Kubalek EM, Milligan RA, Müller U, Kachar B. Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature. 2007;449(7158):87–91. doi: 10.1038/nature06091. [DOI] [PubMed] [Google Scholar]
- Keats BJ, Nouri N, Huang JM, Money M, Webster DB, Berlin CI. The deafness locus (dn) maps to mouse chromosome 19. Mamm Genome. 1995;6(1):8–10. doi: 10.1007/BF00350886. [DOI] [PubMed] [Google Scholar]
- Keresztes G, Mutai H, Heller S. TMC and EVER genes belong to a larger novel family, the TMC gene family encoding transmembrane proteins. BMC Genomics. 2003;4(1):24. doi: 10.1186/1471-2164-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KX, Fettiplace R. Developmental changes in the cochlear hair cell mechanotransducer channel and their regulation by transmembrane channel-like proteins. J Gen Physiol. 2013;141(1):141–8. doi: 10.1085/jgp.201210913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KX, Beurg M, Hackney CM, Furness DN, Mahendrasingam S, Fettiplace R. The role of transmembrane channel-like proteins in the operation of hair cell mechanotransducer channels. J Gen Physiol. 2013;142(5):493–505. doi: 10.1085/jgp.201311068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurima K, Yang Y, Sorber K, Griffith AJ. Characterization of the transmembrane channel-like (TMC) gene family: functional clues from hearing loss and epidermodysplasia verruciformis. Genomics. 2003;82(3):300–8. doi: 10.1016/s0888-7543(03)00154-x. [DOI] [PubMed] [Google Scholar]
- Kurima K, Peters LM, Yang Y, Riazuddin S, Ahmed ZM, Naz S, Arnaud D, Drury S, Mo J, Makishima T, Ghosh M, Menon PS, Deshmukh D, Oddoux C, Ostrer H, Khan S, Riazuddin S, Deininger PL, Hampton LL, Sullivan SL, Battey JF, Jr, Keats BJ, Wilcox ER, Friedman TB, Griffith AJ. Dominant and recessive deafness caused bymutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat Genet. 2002;30(3):277–84. doi: 10.1038/ng842. [DOI] [PubMed] [Google Scholar]
- Labay V, Weichert RM, Makishima T, Griffith AJ. Topology of transmembrane channel-like gene 1 protein. Biochemistry. 2010;49(39):8592–8. doi: 10.1021/bi1004377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarczyk M, Favre M. Role of Zn2+ ions in host-virus interactions. J Virol. 2008;82(23):11486–94. doi: 10.1128/JVI.01314-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelli A, Asai Y, Forge A, Holt JR, Géléoc GS. Tonotopic gradient in the developmental acquisition of sensory transduction in outer hair cells of the mouse cochlea. J Neurophysiol. 2009;101(6):2961–73. doi: 10.1152/jn.00136.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, Erven A, Johnson SL, Steel KP, Kros CJ. Tmc1 is necessary for normal functional maturation and survival of inner and outer hair cells in the mouse cochlea. J Physiol. 2006;574(Pt 3):677–98. doi: 10.1113/jphysiol.2005.095661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski N, Michel V, Bahloul A, Lefèvre G, Barral J, Yagi H, Chardenoux S, Weil D, Martin P, Hardelin JP, Sato M, Petit C. Molecular characterization of the ankle-link complex in cochlear hair cells and its role in the hair bundle functioning. J Neurosci. 2007;27(24):6478–88. doi: 10.1523/JNEUROSCI.0342-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan CP, Barr-Gillespie PG. Mechanotransduction: the elusive hair cell transduction channel revealed? Curr Biol. 2013;23(19):R887–90. doi: 10.1016/j.cub.2013.08.031. [DOI] [PubMed] [Google Scholar]
- Mutai H, Mann S, Heller S. Identification of chicken transmembrane channel-like (TMC) genes: expression analysis in the cochlea. Neuroscience. 2005;132(4):1115–22. doi: 10.1016/j.neuroscience.2005.01.046. [DOI] [PubMed] [Google Scholar]
- Pan B, Géléoc GS, Asai Y, Horwitz GC, Kurima K, Ishikawa K, Kawashima Y, Griffith AJ, Holt JR. TMC1 and TMC2 are components of the mechanotransduction channel in hair cells of the mammalian inner ear. Neuron. 2013;79(3):504–15. doi: 10.1016/j.neuron.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci AJ, Crawford AC, Fettiplace R. Tonotopic variation in the conductance of the hair cell mechanotransducer channel. Neuron. 2003;40(5):983–90. doi: 10.1016/s0896-6273(03)00721-9. [DOI] [PubMed] [Google Scholar]
- Scott DA, Greinwald JH, Jr, Marietta JR, Drury S, Swiderski RE, Viñas A, DeAngelis MM, Carmi R, Ramesh A, Kraft ML, Elbedour K, Skworak AB, Friedman RA, Srikumari Srisailapathy CR, Verhoeven K, Van Gamp G, Lovett M, Deininger PL, Batzer MA, Morton CC, Keats BJ, Smith RJ, Sheffield VC. Identification and mutation analysis of a cochlear-expressed, zinc finger protein gene at the DFNB7/11 and dn hearing-loss loci on human chromosome 9q and mouse chromosome 19. Gene. 1998;215(2):461–9. doi: 10.1016/s0378-1119(98)00316-3. [DOI] [PubMed] [Google Scholar]
- Scott DA, Drury S, Sundstrom RA, Bishop J, Swiderski RE, Carmi R, Ramesh A, Elbedour K, Srikumari Srisailapathy CR, Keats BJ, Sheffield VC, Smith RJ. Refining the DFNB7–DFNB11 deafness locus using intragenic polymorphisms in a novel gene, TMEM2. Gene. 2000;246(1–2):265–74. doi: 10.1016/s0378-1119(00)00090-1. [DOI] [PubMed] [Google Scholar]
- Sirmaci A, Duman D, Oztürkmen-Akay H, Erbek S, Incesulu A, Oztürk-Hişmi B, Arici ZS, Yüksel-Konuk EB, Taşir-Yilmaz S, Tokgöz-Yilmaz S, Cengiz FB, Aslan I, Yildirim M, Hasanefendioğlu-Bayrak A, Ayçiçek A, Yilmaz I, Fitoz S, Altin F, Ozdağ H, Tekin M. Mutations in TMC1 contribute significantly to nonsyndromic autosomal recessive sensorineural hearing loss: a report of five novel mutations. Int J Pediatr Otorhinolaryngol. 2009;73(5):699–705. doi: 10.1016/j.ijporl.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Steel KP, Bock GR. Electrically-evoked responses in animals with progressive spiral ganglion degeneration. Hear Res. 1984;15(1):59–67. doi: 10.1016/0378-5955(84)90225-9. [DOI] [PubMed] [Google Scholar]
- Steel KP, Bock GR. The nature of inherited deafness in deafness mice. Nature. 1980;288(5787):159–61. doi: 10.1038/288159a0. [DOI] [PubMed] [Google Scholar]
- Vreugde S, Erven A, Kros CJ, Marcotti W, Fuchs H, Kurima K, Wilcox ER, Friedman TB, Griffith AJ, Balling R, Hrabé De Angelis M, Avraham KB, Steel KP. Beethoven, a mouse model for dominant, progressive hearing loss DFNA36. Nat Genet. 2002;30(3):257–8. doi: 10.1038/ng848. [DOI] [PubMed] [Google Scholar]
- Walker RG, Willingham AT, Zuker CS. A Drosophila mechanosensory transduction channel. Science. 2000;287(5461):2229–34. doi: 10.1126/science.287.5461.2229. [DOI] [PubMed] [Google Scholar]
- Xiong W, Grillet N, Elledge HM, Wagner TF, Zhao B, Johnson KR, Kazmierczak P, Müller U. TMHS is an integral component of the mechanotransduction machinery of cochlear hair cells. Cell. 2012;151(6):1283–95. doi: 10.1016/j.cell.2012.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Zhang W, He Y, Gorczyca D, Xiang Y, Cheng LE, Meltzer S, Jan LY, Jan YN. Drosophila NOMPC is a mechanotransduction channel subunit for gentle-touch sensation. Nature. 2013;493(7431):221–5. doi: 10.1038/nature11685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Kahrizi K, Bazazzadeghan N, Meyer N, Najmabadi H, Smith RJ. A novel mutation adjacent to the Bth mouse mutation in the TMC1 gene makes this mouse an excellent model of human deafness at the DFNA36 locus. Clin Genet. 2010;77(4):395–8. doi: 10.1111/j.1399-0004.2009.01338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]