

Abstract
Self-assembling peptide amphiphiles (PAs) can form hierarchically ordered membranes when brought in contact with aqueous polyelectrolytes of the opposite charge by rapidly creating a diffusion barrier composed of filamentous nanostructures parallel to the plane of the incipient membrane. Following this event, osmotic forces and charge complexation template nanofiber growth perpendicular to the plane of the membrane in a dynamic self-assembly process. In this work, we show that this hierarchical structure requires strong interactions between PA molecules and polyelectrolyte molecules, suggesting the importance of rapid diffusion barrier formation. Strong interactions are introduced here through the use of heparin-binding PAs with heparin and also with polyelectrolytes of varying charge density. Small angle x-ray scattering shows that in the case of weak PA-polyelectrolyte interaction, membranes formed display a cubic phase ordering on the nanoscale that likely results from clusters of PA nanostructures surrounded by polyelectrolyte chains.
Keywords: self-assembly, peptide amphiphiles, ordered membranes, SAXS, polyelectrolyte-supramolecular polymer complex
1. Introduction
Over the past several decades, electrostatically-driven self-assembly has been demonstrated as a powerful tool to prepare novel and complex nanostructured materials from various molecular building blocks.[1-10] Controlled self-assembly of charged nanostructures into hierarchically ordered materials similar to those found in biological systems, however, remains an interesting challenge. Recently, our laboratory reported on a unique self-assembly process at the interface of two aqueous solutions, one containing positively charged peptide amphiphiles (PAs) and the other containing negatively charged high molecular weight hyaluronic acid (HA).[9] The PAs used in the study consist of an alkyl tail covalently bound to an amino acid sequence with the propensity for β-sheet formation adjacent to a charged peptide sequence.[5, 6, 11-14] These PA molecules can self-assemble into high aspect ratio cylindrical nanofibers due to hydrophobic collapse of the alkyl tails and β-sheet formation,[13] especially in the presence of electrolytes that can screen charged residues. When a solution of the positively charged PA molecules comes in contact with a solution of negatively charged HA, a dense fibrous layer forms in millisecond time scales in the plane of the liquid-liquid interface that acts as a diffusion barrier to prevent mixing of the two solutions.[9, 15] An imbalance in ionic osmotic pressure between the PA and HA solution compartments prevents diffusion of PA supramolecular aggregates and instead promotes reptation of long HA chains through the diffusion barrier into the PA solution over time. As a result, the hierarchical structure of the HA/PA membrane has been shown to include three zones (Figure 1):[9, 15] An amorphous gel layer, a region of nanofibers parallel to the interface that make up the diffusion barrier, and a third zone that thickens in time. This latter zone contains nanofibers that consist of electrostatically-complexed PA and HA and are oriented perpendicular to the interface (Figure 1A). Because electrostatic complexation between simple surfactants and polyelectrolytes typically results in particulates and precipitates,[16, 17] formation of membrane-like materials from these components generally relies on post complexation processes such as solvent evaporation or casting to form cohesive films.[18, 19, 20] The membranes discussed here, however, exhibit ordered microstructures solely due to molecular interactions between PA nanostructures and polyelectrolytes without additional processing.
Figure 1.

(A) Schematic representation of one hemisphere in a closed sac with a hierarchically structured membrane of three zones, (1) a gel phase, (2) parallel nanofibers in the plane of the membrane, and (3) nanofibers perpendicular to the plane of the membrane.(B) Chemical structures of HBPA, HA, and heparin (B).
As PAs can be designed to display bioactive peptide sequences on the surfaces of the supramolecular nanofibers they form, the assembled membranes can also incorporate bioactivity. Examples include our recent work on membranes incorporating PA displaying an anti-cancer cytotoxic sequence[21] or the heparin binding sequence LRKKLGKA to promote angiogenesis.[14, 22] This heparin-binding PA (HBPA; Figure1B) was designed to specifically bind the sulfated polysaccharide based on the Cardin-Weintraub consensus sequence XBBBXXBX (where X is a hydrophobic amino acid and B is a basic amino acid).[14] In our previous work, we found that the incorporation of heparin into the membrane in conjunction with hyaluronic acid (Figure 1B) changes membrane microstructure and mechanical properties.[22] It was found that a critical concentration of heparin (between 0.25 wt% and 0.5 wt%) in solution with HA is required to achieve the three-zone hierarchical membrane morphology. This observation resulting from the inclusion of heparin motivated the current work, which utilizes small angle X-ray scattering (SAXS) and electron microscopy to understand the mechanistic details involved in hierarchical organization of these membranes formed through electrostatic interactions.
2. Results and Discussion
Membrane morphological differences using different compositions of HBPA, HA, and heparin were obvious at macroscopic scales in closed membranes taking the form of spherical sacs. The sac membrane is opaque in the absence of heparin and becomes more transparent with increasing heparin concentration (Figure S1). The micron-scale structural features shown in SEM micrographs (Figures 2 and S2) reveal that 0 wt%, 0.1 wt%, and 0.25 wt% heparin membranes are thicker and lack the three-zone hierarchical structure. As seen previously,[22] only at a heparin concentration above a critical value between 0.25 wt% - 0.5 wt% did the membrane organize into the three-zone hierarchical structure containing perpendicular nanofibers.
Figure 2.
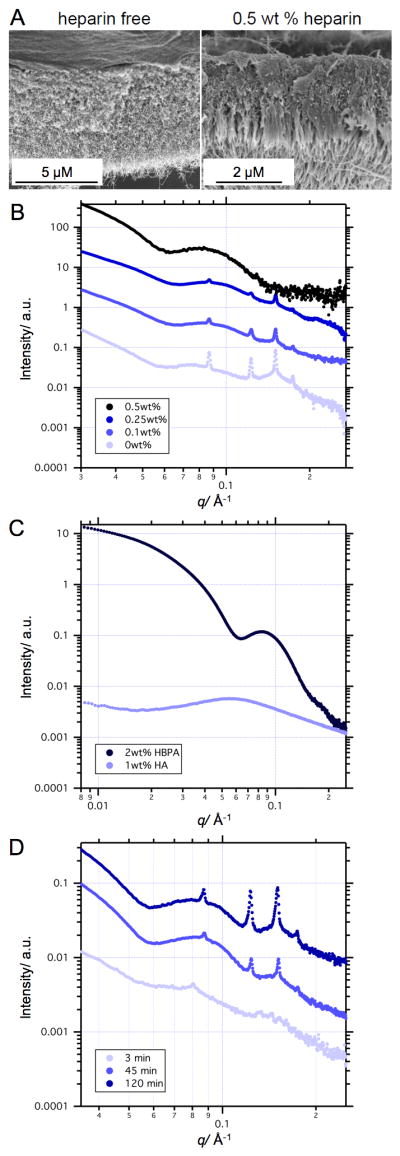
Scanning electron micrographs of membrane cross-sections of HA/HBPA sacs self-assembled with 0 wt% heparin and 0.5 wt% heparin (A). Small angle X-ray scattering (SAXS) patterns of: 1 wt %HA / 2 wt% HBPA sacs self-assembled with varying concentrations of heparin (B); solutions of 1 wt% HA and 2 wt% HBPA (C); and 1 wt% HA/ 2 wt% HBPA sacs 3 minutes, 45 minutes, and 120 minutes after formation (D). Curves were shifted vertically for visual convenience.
Using SAXS, we found that the scattering pattern as a function of the wave vector q of heparin-free membranes exhibits sharp Bragg peaks, which are characteristic of a well-ordered structure, overlaying a broad form factor peak (Figure 2B). Upon increasing heparin concentration, these Bragg peaks become smaller and eventually disappear, indicating loss of nanoscale crystalline order. At 0.5 wt% heparin, only a broad form factor (qmax = 0.08 Å-1) was observed. In order to interpret the membrane scattering patterns, we analyzed the scattering of HA and HBPA solutions alone (Figure 2C). The scattering pattern of 2 wt% HBPA was fitted to a cylindrical core-shell form factor (Figure S3) given by Equation S3, as the -1 slope in the Guinier (low q) region is indicative of a cylindrical shape. The best fit to this model, represented by the dotted blue line in Figure S3, yielded a core radius of 16.3 Å, shell thickness of 44.5 Å, and polydispersity of 0.3. These dimensions are approximately in line with the total diameter of HBPA nanofibers seen in cryo-TEM (Figure S3A) and of PA nanofibers seen previously in literature.[23] The fitted parameters are presented in the caption of Figure S3. Interestingly, the scattering pattern of this 2 wt% HBPA solution is similar to the scattering curve of the 0.5 wt% heparin sac membrane (Figure 2B). The similarity between these two scattering curves indicates that HBPA cylindrical structures are the dominant contributors in the membrane scattering profile of sacs made with HBPA above the critical concentration.
The contribution of free HA chains to the scattering pattern of the sac membranes is minimal due to the complexation of HA with PA fibers. However, at the very beginning of the complexation process (i.e. short incubation times), not all of the HA has complexed with the PA and free HA thus can still contribute to the scattering. The scattering pattern of 1 wt% HA solution (Figure 2C) exhibits a correlation peak at qmax = 0.06 Å-1, which represents the distance between polymer chains and is characteristic of polyelectrolyte solutions without added salt.[24-26] A time dependence study of the heparin-free HA/HBPA membrane nanostructure (Figure 2D) shows that the first minima in the form factor moves with incubation time from smaller to larger momentum vectors. The change in minima position is likely due to the overlap of the HA correlation peak at qmax = 0.06 Å-1, which disappears with longer incubation time, with the PA form factor minima (Figure 2C).
The Bragg peaks in the scattering profile of membranes made with heparin below the critical concentration indicate that an ordered nanoscale structure emerges as early as three minutes after initiation of self-assembly among the components. This suggests that organization into a crystalline structure occurs very rapidly after the initial complexation. The well-defined ordering is clearly observed after 45 minutes and remains unchanged after four hours. Using the Bragg diffraction equation (nλ = 2dsinθ), where θ is indicated by the wave vector q, the d-spacing calculated from the position of the first Bragg peak is 7.3 nm. The relative positions of the Bragg peaks follow a spacing ratio of 1:√2:√3:√4:√5, which is the characteristic scattering pattern of a cubic phase.[27-30] This spacing ratio does not fit with Pm3n or Ia3d bicontinuous cubic phases typical for surfactant-polyelectrolyte complexes.[27, 29, 31, 32] Unfortunately, the exact ordering (e.g. simple cubic or body centered cubic) could not be determined since there are not enough peaks in the scattering scan. The size scales of ordered domains were estimated from Bragg peaks in the scattering patterns by fitting them to Gaussian profiles. The crystallite size calculated according to equation S4 was found to be 0.5-1.0 μm.
As SAXS and cryo-TEM (Figure S3) of 2 wt% HBPA solution shows cylindrical HBPA aggregates, hexagonal or lamellar packing would be expected and organization within the membrane into the observed cubic phase is therefore counterintuitive. However, a small number of spherical and shorter cylindrical nanostructures were also found to coexist with cylindrical nanofibers. Cubic phases containing spheres and short rods have been reported for several polyelectrolyte-surfactant complexes.[33-35] It has been suggested that the surfactants form small spherical or short rod-like micelles with polyelectrolyte chain loops wrapped around the clusters.[36] A membrane made with HA and a PA designed to form only spherical micelles also exhibited a cubic phase as seen by SAXS (Figure S4). The scattering pattern of this membrane shows three Bragg peaks with a spacing ratio characteristic of a face centered cubic (FCC) phase[31, 37] with a lattice constant of 7 nm (Figure S4). This result suggests that organization into the observed cubic phase is due to wrapping of HA chains around smaller HBPA aggregates.
Interestingly, SEM shows development of the hierarchical three-zone microstructure only with 0.5 wt% heparin, while SAXS data demonstrates nanoscale cubic phase ordering only with heparin concentrations of 0.25 wt% or lower. This change in the membrane structure at the nano- and micro-scales clearly points to variations in the mechanism of membrane formation above and below a critical heparin concentration. As was previously suggested, the three-zone hierarchical structure can only develop if a dense diffusion barrier is formed immediately at the interface between the PA and HA solutions.[9] The lack of this morphology in the absence of a critical heparin concentration suggests that heparin is essential for the formation of this barrier in this self-assembly system involving HPBA, heparin, and HA. HBPA molecules are designed to bind specifically to heparin;[14, 38, 39] it is therefore reasonable to assume that the HBPA-heparin association should dominate over the HBPA-HA association due to heparin specific binding. Additionally, heparin has a charge density three times larger than HA, thus at lower heparin concentrations the HBPA-heparin aggregation dominates over the HBPA-HA interactions. Such interactions with heparin can facilitate massive aggregation of HBPA molecules, which exist in aqueous solution as micelle or fiber-like nanostructures with a hydrophobic core and a highly cationic surface, upon charge complexation with HA. Assuming that HBPA-heparin interactions must be saturated for diffusion barrier formation, the HA-to-heparin charge ratio (1:3) may suggest the critical heparin concentration to be equal to 0.33 wt %, which is in good agreement with our experimental observations. We thus hypothesize that the nano-scale aggregation state of HBPA at the onset of membrane self-assembly plays a crucial role.
Polyelectrolyte-surfactant complexation has been studied extensively experimentally and theoretically over the past few decades.[33, 40, 41] However, the structure of the membrane studied here develops dynamically, wherein both PA and HA components complex upon contact and further self-assembly is governed by balance between directional diffusion and aggregation.[42] The polyelectrolyte electrostatic charge (i.e. zeta potential) could significantly affect aggregation at the interface and, consequently, the structure of the membrane. To examine how the electrostatic charge of the polyelectrolyte component affects the self-assembly, we formed membranes using HBPA and several polyelectrolytes of different charge density (Figure 3) but without addition of heparin. We used λ-carrageenan that has a similar chemical composition and charge density to that of heparin and is thus expected to interact with HBPA in a similar manner. We also formed two other heparin-free HBPA sacs with alginate (twice as charged as HA) and poly(acrylic acid) (PAA). The difference in total electrostatic charge between polyelectrolytes is also evident from the zeta potential (ε) measurements of their aqueous solutions (Figure S5).[9] As expected, the highest ε was of alginate in water and the ε of PAA in water was similar to that of HA. The SAXS patterns of both the HBPA/λ-carrageenan and HBPA/alginate membranes show a similar structure to that of the 0.5 wt% heparin/HBPA/HA sac (Figure 3B). At the same time, the SAXS pattern of the HBPA/PAA sac exhibits Bragg peaks similar to that of the heparin-free HBPA/HA sac. The spacing ratio of these peaks is also characteristic of a cubic phase with a lattice parameter (calculated for a simple cubic) of 6 nm for the HBPA/PAA sac. These results suggest that formation of the nano-ordered heparin-free HA/HBPA and PAA/HBPA membranes is supported by diffusion of small HBPA aggregates into the polyelectrolyte compartment. On the other hand, addition of heparin to HBPA or use of highly anionic alginate and λ-carrageenan as the polyelectrolyte promotes the rapid assembly of HBPA into a large network of cylindrical fibers. These results suggest that the polyelectrolyte charge density can control the micro- and nanostructure of the membrane by affecting diffusion barrier formation during electrostatically-driven self-assembly of HBPA and polyelectrolyte.
Figure 3.
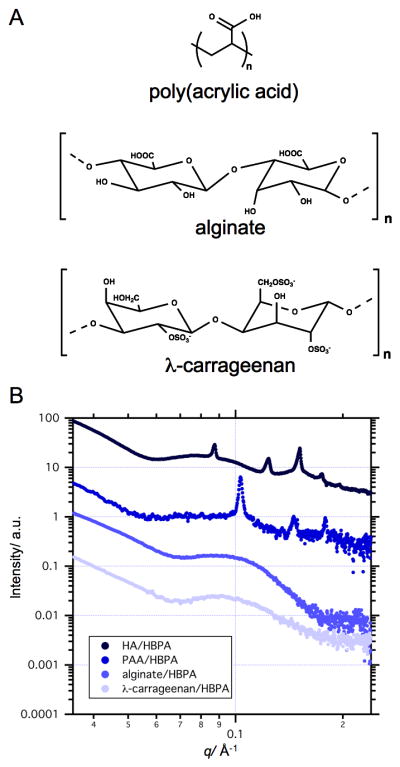
Chemical structures of λ-carrageenan, poly(acrylic acid), or alginate (A). SAXS of sacs self assembled from HBPA with λ-carrageenan, poly(acrylic acid) (PAA), or alginate (B).
3. Conclusions
We conclude that remarkable differences in the structure of membranes formed by oppositely charged polyelectrolytes and HBPA in water are dependent on the aggregation process at the interface (Figure 4). Systems that promote massive self-assembly of PA into a nanofibrous network results in hierarchically ordered membranes with orthogonally aligned nanofibers. Without such aggregation, membranes formed are non-fibrous and contain a phase with cubic symmetry and nanoscale periodicities. These differences in structured membranes could lead to profound changes in their physical properties and potential functions.
Figure 4.
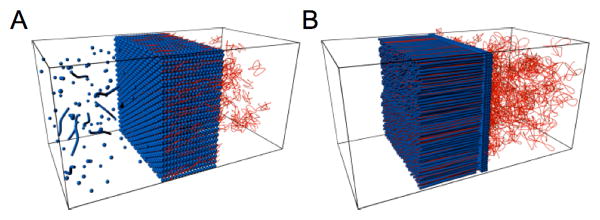
Schematic representation of the nanostructured membrane formed by peptide amphiphiles (blue) and polyelectrolytes (red) in the case of weak aggregation (A) or strong aggregation (B) at the interface. Massive interfacial aggregation of PA leads to a hierarchical membrane structure in which nanofibers grow perpendicular to the diffusion barrier, while weak interfacial aggregation creates membranes with a cubic phase of nanosphere aggregates wrapped in polyelectrolyte chains.
4. Experimental Section
HBPA Synthesis/Purification
The heparin-binding peptide amphiphile (HBPA) was synthesized using standard fluoren-9-ylmethoxycarbonyl (Fmoc) solid phase peptide synthesis as previously reported[14] and purified using reversed phase high performance liquid chromatography (HPLC) in an acetonitrile/water gradient under acidic conditions with trifluoroacetic acetate (TFA, 0.1%). To improve biocompatibility of the purified HBPA, residual TFA counter ions were exchanged by sublimation from HCl (0.01M). After lyophilization from the HCl solution, HBPA was resolubilized in deionized water and sodium hydroxide to raise the pH to 7.2. The HBPA was lyophilized again and stored at -20°C until needed.
Membrane/Sac Formation
The molecular weight of HA used in all experiments had an average molar mass of 1.76 MDa and was purchased from Lifecore Biomedical, Inc (Chaska, MN). Porcine-derived heparin sodium salt was purchased from Sigma. Four different HA/heparin biopolymer formulations containing HA (1 wt%) with heparin (0 wt%, 0.1 wt%, 0.25 wt%, or 0.5 wt%) were prepared by mixing HA and heparin as powders prior to solubilizing in nanopure water unless otherwise noted. Biopolymer solutions were prepared at least 24 hours before use, and lyophilized HBPA was solubilized in nanopure water at 2 wt% immediately prior to use. For all experimental groups, a drop of polymer solution (10 μL) was immersed into PA solution (50 μL) to form a spherical, enclosed sac as previously discussed.[15] PA solution (30 μL) was added on top to seal the sac. Sacs were allowed to incubate at room temperature for 8 hrs. Sac formation using drop shape analysis (DSA) used for optical images was described previously.[15]
SEM of Membrane Cross-Section
Sacs were removed from the PA solution and washed with water to remove excess PA. Samples were fixed in glutaraldehyde (2%) with sucrose (3%) for 1 hr at 4°C then dehydrated in an ethanol gradient from 20% to 100% ethanol in water. Critical point drying was performed with a Tousimis SAMDRI-795 critical point dryer. Dried samples were manually torn to expose the cross section then coated with 15 nm of osmium using an osmium plasma coater (Structure Probe, Inc.). Samples were imaged using a Hitachi S-4800 field emission scanning electron microscopy using an accelerating voltage of 3 kV.
CryoTEM
Cryogenic transmission electron microscopy (cryo-TEM) was performed on a JEOL 1230 microscope with an accelerating voltage of 100kV. A Vitrobot Mark IV equipped with controlled humidity and temperature was used for plunging samples. A small volume of the sample (5-7 μL) was deposited on a copper TEM grid with holey carbon support film (Electron Microscopy Sciences) and held in place with tweezers mounted to the Vitrobot. The specimen was blotted in 90-95% humidity and plunged into a liquid ethane reservoir that was cooled by liquid nitrogen. The vitrified samples were transferred in liquid nitrogen to a Gatan 626 cryo-holder through a cryo-transfer stage and imaged using a Hamamatsu ORCA CCD camera.
Small Angle X-ray Scattering
SAXS measurements were performed using beam line 5ID-D, in the DuPont-Northwestern-Dow Collaborative Access team (DND-CAT) Synchrotron Research Center at the Advanced Photon Source, Argonne National Laboratory. An energy of 15keV corresponding to a wavelength λ = 0.83 Å was selected using a double-crystal monochromator. The data were collected using a CCD detector (MAR) positioned 245 cm behind the sample. The scattering intensity was recorded in the interval 0.008< q < 0.25 Å-1. The wave vector defined as q = (4 π / λ) sin(θ / 2), where θ is the scattering angle. The exposure times were between 2-8 s depending on the sample.
Solution samples were placed in 1.5 mm quartz capillaries. Sac samples were placed in a water-filled customized sample holder made from Aluminum and mica sheets. The 2-D SAXS images were azimuthally averaged to produce one-dimensional profiles of intensity, I vs q, using the two-dimensional data reduction program FIT2D. The scattering of sample holder (capillary or costumed cell) with and without solvent (water) were also collected and subtracted from the corresponding data. No attempt was made to convert the data to an absolute scale.
Data analysis was based on fitting the scattering curve to an appropriate model by a least-squares method using software provided by NIST (NIST SANS analysis version 7.0 on IGOR).
Supplementary Material
Figure S1. Representative images of HA/HBPA sacs self-assembled with (A) 0 wt% heparin, (B) 0.1 wt% heparin, (C) 0.25 wt% heparin, and (D) 0.5 wt% heparin.
Figure S2. Scanning electron micrographs of membrane cross-sections of HA/HBPA sacs self-assembled with (A) 0.1 wt% heparin and (B) 0.25 wt% heparin (scale bar = 5 μm).
Figure S3. (A) Cryogenic transmission electron micrograph of an aqueous solution of 2 wt% HBPA. Arrows indicate spherical micelles and smaller aggregates found with the expected cylindrical micelle morphology. Scale bar = 100 nm. (B) Fitting of polydisperse core-shell cylinder model to HBPA scattering profile indicates a core radius of 16.3 Å, shell thickness of 44.5 Å, polydispersity of 0.3, and scattering length density of 8.6e-6 Å-2 and 1.2e-5 Å-2 for the core and shell respectively. The cylinder dimensions are in line with cryo-TEM results and previously reported PA SAXS data.[23] The SLDs obtained are reasonable for a palmitic acid tail and amino acids. However, it should be noted that a cylindrical core-shell model with a gradient in the electron density of the shell should be ideally be used, but the electron density of the highly hydrated outer PA headgroup is similar to that of the solvent. Therefore, a simpler core-shell model was used, and thus the shell thickness and SLD values obtain should not be considered completely accurate.
Figure S4. (A) Schematic of a PA with the sequence C16KKK, and (B) the scanning electron micrograph of the cross-section of a membrane made with HA and this KKK PA (scale bar = 5 μm). (C) SAXS scattering of this KKK PA/HA membrane.
Figure S5. Zeta potential measurements of poly(acrylic acid), alginate, λ-carrageenan, hyaluronic acid, and heparin in milliQ water.
Acknowledgments
This work was supported by the National Institute of Biomedical Imaging and Bioengineering at the National Institutes of Health (5R01EB003806-05) and the NSF MRSEC program (DMR-0520513). R.B. acknowledges support from the Ben Gurion University of the Negev in the form of a postdoctoral fellowship. Experiments made use of the following facilities at Northwestern University: EPIC Facilities of the NUANCE Center, Biological Imaging Facility, and the Institute for BioNanotechnology in Medicine. The NUANCE Center is supported by the NSF-NSEC, NSF-MRSEC, Keck Foundation, the State of Illinois and Northwestern University. X-ray measurements were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of the Advanced Photon Source (APS). DND-CAT is supported by E.I. DuPont de Nemours & Co., The Dow Chemical Company and Northwestern University. Use of the APS, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. The authors thank Dr. Steven Weigand for X-ray assistance and fruitful discussions and Dr. Liam Palmer for the C16K3 molecules.
Footnotes
Supporting Information is available on the WWW under http://www.small-journal.com or from the author.
Contributor Information
Dr. Ronit Bitton, Institute for BioNanotechnology in Medicine, Northwestern University, Chicago, IL, 60611, United States.
Dr. Lesley W. Chow, Department of Materials Science and Engineering, Northwestern University, Evanston, IL, 60208, United States.
R. Helen Zha, Department of Materials Science and Engineering, Northwestern University, Evanston, IL, 60208, United States.
Dr. Yuri S. Velichko, Department of Materials Science and Engineering, Northwestern University, Evanston, IL, 60208, United States
Dr. E. Thomas Pashuck, Department of Materials Science and Engineering, Northwestern University, Evanston, IL, 60208, United States
Prof. Samuel I. Stupp, Email: s-stupp@northwestern.edu, Department of Materials Science and Engineering, Northwestern University, Evanston, IL 60208, United States; Department of Chemistry, Northwestern University, Evanston, IL 60208, United States; Department of Medicine, Northwestern University, Chicago, IL, 60611, United States; Institute for BioNanotechnology in Medicine, Northwestern University, Chicago, IL, 60611, United States.
References
- 1.Tien J, Terfort A, Whitesides GM. Langmuir. 1997;13:5349. [Google Scholar]
- 2.Decher G. Science. 1997;277:1232. [Google Scholar]
- 3.Caruso F, Lichtenfeld H, Giersig M, Möhwald H. J Am Chem Soc. 1998;120:8523. [Google Scholar]
- 4.Aizenberg J, Braun PV, Wiltzius P. Phys Rev Lett. 2000;84:2997. doi: 10.1103/PhysRevLett.84.2997. [DOI] [PubMed] [Google Scholar]
- 5.Niece KL, Hartgerink JD, Donners JJJM, Stupp SI. J Am Chem Soc. 2003;125:7146. doi: 10.1021/ja028215r. [DOI] [PubMed] [Google Scholar]
- 6.Behanna HA, Donners JJJM, Gordon AC, Stupp SI. J Am Chem Soc. 2005;127:1193. doi: 10.1021/ja044863u. [DOI] [PubMed] [Google Scholar]
- 7.Leunissen ME, Christova CG, Hynninen AP, Royall CP, Campbell AI, Imhof A, Dijkstra M, van Roij R, van Blaaderen A. Nature. 2005;437:235. doi: 10.1038/nature03946. [DOI] [PubMed] [Google Scholar]
- 8.Kalsin AM, Fialkowski M, Paszewski M, Smoukov SK, Bishop KJM, Grzybowski BA. Science. 2006;312:420. doi: 10.1126/science.1125124. [DOI] [PubMed] [Google Scholar]
- 9.Capito RM, Azevedo HS, Velichko YS, Mata A, Stupp SI. Science. 2008;319:1812. doi: 10.1126/science.1154586. [DOI] [PubMed] [Google Scholar]
- 10.Cui H, Pashuck ET, Velichko YS, Weigand SJ, Cheetham AG, Newcomb CJ, Stupp SI. Science. 2010;327:555. doi: 10.1126/science.1182340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 12.Hartgerink JD, Beniash E, Stupp SI. P Natl Acad Sci USA. 2002;99:5133. doi: 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Velichko YS, Stupp SI, de la Cruz MO. J Phys Chem B. 2008;112:2326. doi: 10.1021/jp074420n. [DOI] [PubMed] [Google Scholar]
- 14.Rajangam K, Behanna HA, Hui MJ, Han X, Hulvat JF, Lomasney JW, Stupp SI. Nano Lett. 2006;6:2086. doi: 10.1021/nl0613555. [DOI] [PubMed] [Google Scholar]
- 15.Carvajal D, Bitton R, Mantei JR, Velichko YS, Stupp SI. Soft Matter. 2010;6:1816. [Google Scholar]
- 16.Langevin D. Adv Colloid Interface Sci. 2009;170:147–148. doi: 10.1016/j.cis.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 17.Thalberg K, Lindman B. J Phys Chem. 1989;93:1478. [Google Scholar]
- 18.Antonietti M, Burger C, Effing J. Adv Mater. 1995;7:751. [Google Scholar]
- 19.Thünemann AF, Lochhaas KH. Langmuir. 1999;15:4867. [Google Scholar]
- 20.Antonietti M, Conrad J, Thünemann AF. Macromolecules. 1994;27:6007. [Google Scholar]
- 21.Zha RH, Sur S, Stupp SI. Adv Healthcare Mater. 2013;2:126. doi: 10.1002/adhm.201200118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chow LW, Bitton R, Webber MJ, Carvajal D, Shull KR, Sharma AK, Stupp SI. Biomaterials. 2011;32:1574. doi: 10.1016/j.biomaterials.2010.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toft DJ, Moyer TJ, Standley SM, Ruff Y, Ugolkov A, Stupp SI, Cryns VL. ACS Nano. 2012;6:7956. doi: 10.1021/nn302503s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Popa-Nita S, Rochas C, David L, Domard A. Langmuir. 2009;25:6460. doi: 10.1021/la900061n. [DOI] [PubMed] [Google Scholar]
- 25.Wang ZY, White JW, Konno M, Saito S, Nozawa T. Biopolymers. 1995;35:227. [Google Scholar]
- 26.Josef E, Bianco-Peled H. Soft Matter. 2012;8:9156. [Google Scholar]
- 27.Leonard MJ, Strey HH. Macromolecules. 2003;36:9549. [Google Scholar]
- 28.Nilsson P, Hansson P. J Colloid Interf Sci. 2008;325:316. doi: 10.1016/j.jcis.2008.05.054. [DOI] [PubMed] [Google Scholar]
- 29.Norrman J, Lynch I, Piculell L. J Phys Chem B. 2007;111:8402. doi: 10.1021/jp067303l. [DOI] [PubMed] [Google Scholar]
- 30.Zhou S, Yeh F, Burger C, Chu B. J Phys Chem B. 1999;103:2107. [Google Scholar]
- 31.Zhou S, Yeh F, Burger C, Chu B. J Polym Sci Poly Phys. 1999;37:2165. [Google Scholar]
- 32.Nause RG, Hoagland DA, Strey HH. Macromolecules. 2008;41:4012. [Google Scholar]
- 33.Zhou SQ, Chu B. Adv Mater. 2000;12:545. [Google Scholar]
- 34.Okuzaki H, Osada Y. Macromolecules. 1995;28:380. [Google Scholar]
- 35.Trabelsi S, Guillota S, Ritacco H, Boue F, Langevin D. Eur Phys J E. 2007;23:305. doi: 10.1140/epje/i2006-10192-y. [DOI] [PubMed] [Google Scholar]
- 36.Perico A, Ciferri A. Chem-Eur J. 2009;15:6312. doi: 10.1002/chem.200900637. [DOI] [PubMed] [Google Scholar]
- 37.Zhou S, Liang DH, Burger C, Yeh F, Chu B. Biomacromolecules. 2004;5:1256. doi: 10.1021/bm034524d. [DOI] [PubMed] [Google Scholar]
- 38.Rajangam K, Arnold MS, Rocco MA, Stupp SI. Biomaterials. 2008;29:3298. doi: 10.1016/j.biomaterials.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chow LW, Wang LJ, Kaufman DB, Stupp SI. Biomaterials. 2010;31:6154. doi: 10.1016/j.biomaterials.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ober CK, Wegner G. Adv Mater. 1997;9:17. [Google Scholar]
- 41.Thünemann AF. Prog Polym Sci. 2002;27:1473. [Google Scholar]
- 42.Velichko YS, Mantei JR, Bitton R, Carvajal D, Shull KR, Stupp SI. Adv Funct Mater. 2012;22:369. doi: 10.1002/adfm.201101538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Representative images of HA/HBPA sacs self-assembled with (A) 0 wt% heparin, (B) 0.1 wt% heparin, (C) 0.25 wt% heparin, and (D) 0.5 wt% heparin.
Figure S2. Scanning electron micrographs of membrane cross-sections of HA/HBPA sacs self-assembled with (A) 0.1 wt% heparin and (B) 0.25 wt% heparin (scale bar = 5 μm).
Figure S3. (A) Cryogenic transmission electron micrograph of an aqueous solution of 2 wt% HBPA. Arrows indicate spherical micelles and smaller aggregates found with the expected cylindrical micelle morphology. Scale bar = 100 nm. (B) Fitting of polydisperse core-shell cylinder model to HBPA scattering profile indicates a core radius of 16.3 Å, shell thickness of 44.5 Å, polydispersity of 0.3, and scattering length density of 8.6e-6 Å-2 and 1.2e-5 Å-2 for the core and shell respectively. The cylinder dimensions are in line with cryo-TEM results and previously reported PA SAXS data.[23] The SLDs obtained are reasonable for a palmitic acid tail and amino acids. However, it should be noted that a cylindrical core-shell model with a gradient in the electron density of the shell should be ideally be used, but the electron density of the highly hydrated outer PA headgroup is similar to that of the solvent. Therefore, a simpler core-shell model was used, and thus the shell thickness and SLD values obtain should not be considered completely accurate.
Figure S4. (A) Schematic of a PA with the sequence C16KKK, and (B) the scanning electron micrograph of the cross-section of a membrane made with HA and this KKK PA (scale bar = 5 μm). (C) SAXS scattering of this KKK PA/HA membrane.
Figure S5. Zeta potential measurements of poly(acrylic acid), alginate, λ-carrageenan, hyaluronic acid, and heparin in milliQ water.