

Abstract
p62/sequestosome-1/A170/ZIP (hereafter referred to as p62) is a scaffold protein that has multiple functions, such as signal transduction, cell proliferation, cell survival, cell death, inflammation, tumorigenesis, and oxidative stress response. While p62 is an autophagy substrate and is degraded by autophagy, p62 serves as an autophagy receptor for selective autophagic clearance of protein aggregates and organelles. Moreover, p62 functions as a signaling hub for various signaling pathways, including NF-κB, Nrf2 and mTOR. In this review, we discuss the pathophysiological role of p62 in the liver, including formation of hepatic inclusion bodies, cholestasis, obesity, insulin resistance, liver cell death and tumorigenesis.
Keywords: p62/SQSTM1, Autophagy, Liver
Introduction
p62, also known as sequestosome-1(SQSTM1), A170, and zeta-interacting protein (ZIP), is a scaffold protein with multiple domains that functions in signal transduction, cell proliferation, cell survival, cell death, inflammation, tumorigenesis, and in the oxidative stress response 1, 2. Recent autophagy studies have revealed that p62 is an autophagy substrate that also serves as an autophagy receptor for protein aggregates and damaged or excess organelles as well as for invading microbes 3–5. At physiological conditions, p62 plays an important role in maintenance of bone and metabolic homeostasis based on studies from p62 knockout (KO) mice 6, 7. In addition, mutations of p62 have been found in human Paget’s disease of bone, which is a chronic metabolic disorder characterized by increased bone turnover, further underscoring the relevance of p62 in human diseases 8. Due to the rapid research progress on autophagy and p62, p62 has been extensively reviewed in regard to its role in selective autophagy 9, 10, metabolic homeostasis 11 and cancer 1. Therefore, our current review will discuss the role of p62 in liver physiology and pathology.
Historical Aspect of p62 and its Association with Autophagy and Liver Diseases
When searching for Src homology 2 (SH2) domain binding proteins, p62 was first identified as an unknown 62 kDa protein that bound to the SH2 domain of p56lck in 1995 by Shin 12. Shin first cloned human p62 cDNA in 1996 13. He also found that p62 preferentially bound to ubiquitinated protein aggregates, which led him to coin the name “sequestosome” (SQSTM1) for p62 14. Using differential screening, Ishii et al. cloned cDNA encoding a novel oxidative stress protein, which they called A170, from mouse peritoneal macrophages 15. A170 is approximately 90% identical to human p62. Later, using yeast two-hybrid screening, p62 was discovered to interact with the atypical protein kinase C (aPKC) 16, 17. Puls et al. then successfully cloned the rat orthologue of p62, cytoplasmic ZIP, which functions as a scaffold protein responsible for regulating PKCζ signal transduction 17. The endogenous and ectopically expressed p62 protein was originally found to display a punctate pattern when anchored together with aPKCs in the endosomal-lysosomal compartment, which was the earliest implication that p62 was associated with the autophagy-lysosomal pathway 16. Subsequently, p62 was found to co-localize with the autophagy marker, the microtubule light chain 3 (LC3), and is degraded by autophagy 18. Johansen’s group and Komatsu’s group independently showed that p62 directly binds to LC3 via a conserved LIR (LC3 interacting region) sequence, which is a motif required for autophagic degradation of p62 19, 20. Moreover, p62 levels were also highly increased in autophagy-deficient mouse livers, which further confirmed that p62 was an autophagy substrate 21.
When Denk and Zatloukal’s group tried to characterize the nature of intracytoplasmic hyaline bodies (IHBs), which are common inclusions in hepatocellular carcinoma cells, they found that one of the major components of the IHBs had an approximate molecular weight of 62 kDa by using two-dimensional gel electrophoresis 22. Sequence analysis revealed that this 62 kDa protein was identical to p62, which was reported by Shin’s group 13. This study was the first to link the accumulation of p62 to human pathological disease. The same group subsequently discovered that p62 was also a major component in Mallory-Denk bodies (MDBs), which are often associated with alcoholic and non-alcoholic steatohepatitis 23, 24. In addition, they discovered that p62 was a component of α1-antitrypsin deficiency aggregates in hepatocytes, as well as in non-liver tissues, such as Lewy bodies in Parkinson’s disease and neurofibrillary tangles in Alzheimer’s disease 24. Whether p62 is required for the formation of the inclusion bodies in the liver is currently not clear. It is possible that p62 is in these inclusions to serve as an autophagy receptor for their autophagic removal. Moreover, it has been shown that impaired clearance of p62 positive inclusions may promote liver tumorigenesis (see below discussion).
Structure and Multiple Functions of p62
Ongoing studies continue to discover functional roles of p62 as novel interaction domains are defined (Figure 1). The protein weight of 62 kDa gave p62 its name. Six distinct functional domains have been defined within the p62 protein structure and include the following: Phox/Bemp1 (PB1) domain, ZZ zinc finger region, tumor necrosis factor (TNF) associated receptor-6 (TRAF6) binding (TB) domain, LIR, Kelch-like ECH-associated protein 1 (Keap1) interacting region (KIR) and ubiquitin binding domain (UBA) 4, 25. p62 also possesses two regions that are required for interaction with p38 mitogen-activated protein kinase (MAPK), which are within residues 173–182 and 335–344, respectively 26, 27. The p62 protein has two nuclear localization signal (NLS) sequences, a nuclear export signal (NES) sequence 28, and two PEST sequences rich in proline (P), glutamic acid (E), serine (S) and threonine (T) with putative phosphorylation sites 13, 15, 17, which are hypothesized to act as signaling peptides for protein degradation. The nuclear shuttling of p62 allows p62 to localize to nuclear protein aggregates such as promyelocytic leukemia bodies and may recruit proteasomes to the nuclear inclusions for their degradation 28. These various domains reveal that p62 has multiple roles in signaling amplification and suppression, protein and organelle quality control, and response to oxidative stress. As discussed above, p62 was originally found to interact with aPKC through its N-terminal PB1 domain 6, 17, 29. The PB1 domain also interacts with other protein kinases including ERK, MEKK3, and MEK5 1, 9, 30, 31 and plays a role in p62 self- and hetero-oligomerization with other PB1 domain containing proteins, such as the neighbor of BRCA gene 1 protein (NBR1) 9. Moreover, PB1-mediated p62 self-oligomerization is essential for its localization to the autophagosome formation site, a process that is independent of LC3 localization 32. It has been suggested that p62 may serve as an adaptor protein to recruit Atg proteins for initiation of autophagosome formation. However, this is controversial because starvation-induced autophagosome formation is normal in p62 KO cells 21.
Figure 1. p62 structure, binding partners and functions.

p62 has Six distinct functional domains: PB1, ZZ, TB, LIR, KIR and UBA. PB1 domain self- and hetero- oligomerizes with other PB1 containing proteins, such as NBR1, ERK and aPKC. p62 binds with RIP at ZZ zinc finger region, and TRAF6 at TB domain, which regulates NF-κB activation. At residues 173–182, p62 interacts with p38 MAPK. p62 interacts with LC3 through the LIR, and keap1 through the KIR. The c-terminal UBA domain of p62 binds to ubiquitin. p62 has two PEST sequences that are targets for post translation modifications and degradation. p62 is phosphorylated by CDK1 at T269 and S272 that regulates cell cycle. p62 is phosphorylated by CK2 at S403 that enhances autophagic degradation of ubiquitinated proteins. p62 also contains two NLS sequences and a NES sequence which allow p62 shuttling into and out of nucleus.
The ZZ zinc finger region is located next to the PB1 domain and is responsible for binding to the receptor interacting protein (RIP), a TNF-α signaling adaptor protein 33, 34. RIP is a serine/threonine kinase that regulates TNF-α receptor 1-mediated activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) through its interaction with TNF receptor type 1-associated death domain protein (TRADD) 33. In addition to the ZZ domain, the TB domain interacts with TRAF-6, a lysine 63 (K63) E3 ligase that also regulates NF-κB activation. In response to interleukin-1 (IL-1) and Receptor Activator of NF-κB ligand (RANKL), p62 undergoes self-oligmerization through its PB1 domain, which promotes K63 ubiquitination of TRAF-6 and results in NF-κB activation and a subsequent inflammatory response 6, 29.
Pankiv et al. 20 and Ichimura et al. 19 independently identified the autophagy effecter protein LC3 and other members of this family of proteins, such as gamma-aminobutyric acid receptor-associated protein (GABARAP) and GABARAP-like molecules, as binding partners for p62. The LIR is a short acidic 11 peptide sequence located within amino acids 337–343 that is required for p62-LC3B interaction. The LIR sequence was further characterized and found to contain conserved aspartic acid and tryptophan residues, which are required for interaction with LC3B. This LC3B binding motif was defined as D(D/E) (D/E)WT and is located near the C-terminus. The LIR is required for autophagic degradation of p62, and impairment of p62-LC3 interaction leads to formation of ubiquitin and p62 positive aggregates 19, 20.
Three independent groups reported that p62 directly interacts with the Kelch-repeat domain of Keap1, and the term KIR was coined by Komatsu’s group 19, 35, 36. The KIR contains the specific conserved amino acid sequence, DPSTGE, and it is located immediately following the LIR in the C-terminus between amino acids 347–352. The DPSTGE sequence is very similar to the DEETGE sequence in nuclear factor (erythroid-derived 2)-like 2 (Nrf2), which is responsible for interaction with Keap1’s kelch domain. Under normal conditions, Keap1 negatively regulates Nrf2 by promoting its ubiquitination, resulting in the rapid degradation of Nrf2 by the ubiquitin proteasome system 37. p62 competes with Nrf2 for binding to Keap1, and interaction of p62 with Keap1 results in dissociation of Nrf2 from Keap1, which allows for Nrf2 translocation to the nucleus where it induces transcription of detoxification enzymes and oxidative stress response genes 19, 35, 36. Therefore, p62 serves as a positive regulator for Nrf2 activation. The role of p62 in regulation of Nrf2 is discussed in more detail in a later section.
Vadlamudi et al. 38 first demonstrated that p62 interacts with ubiquitin using a yeast two-hybrid system and ubiquitin-conjugated Sepharose beads. Between amino acids 386–434 within its C-terminus, p62 binds non-covalently to both mono- and poly-ubiquitinated proteins via its UBA domain. Because of its unique binding to ubiquitinated proteins together with its ability to bind LC3, p62 has been demonstrated to be an important receptor protein for selective autophagic clearance of ubiquitinated protein aggregates and organelles 5, 9. The importance of the UBA domain of p62 has also been underscored by the findings that mutations of the UBA domain are frequently associated with familial and sporadic Paget’s disease of bone, which is a chronic and metabolic disorder characterized by increased bone turnover 39–41. It is well known that RANKL and its receptor RANK play an important role in regulating osteoclast differentiation, activity, and survival by activating NF-κB 6. It has been suggested that p62 forms a complex with TRAF-6 and aPKC, which is critical for RANKL-induced NF-κB activation 6, 42, 43. Mutations in the UBA domain results in loss of UBA function and also activate TRAF6–NF-kB signaling, which results in increased osteoclastogenesis 8, 39, 44. p62 KO mice also show a similar phenotype reminiscent of Paget’s disease with impaired NF-κB activation 6, further supporting that p62 is an important mediator regulating bone homeostasis.
Transcriptional and Post-translational Regulation of p62
Sqstm1 has been shown to be transcriptionally regulated by Nrf2 35, 45 and Farnesoid X Receptor (FXR) 46. It has also been suggested that Sqstm1 is a target gene of NF-kB and AP-1 38. Nrf2 binds to an antioxidant response element (ARE) within the Sqstm1 gene promoter region and induces its transcription during oxidative stress 35, 45. In addition to regulation of Sqstm1 gene expression by Nrf2, p62 protein accumulation stabilizes Nrf2 activation, which allows for nuclear translocation and induction of target gene transcription by Nrf2, as previously discussed. Therefore, p62 establishes a positive feedback loop for regulating its own gene transcription through stabilizing Nrf2 activation 35.
The nuclear receptor FXR also regulates Sqstm1 gene transcription. We previously found that FXR binds to the Sqstm1 gene in both the liver and ileum using chromatin immunoprecipitation (ChIP). In addition, luciferase activity was enhanced when HepG2 cells were transfected with a Sqstm1 fragment containing the FXR binding site, and this luciferase activity was lost when cells were transfected with a Sqstm1 fragment containing a mutant FXR binding site. However, the FXR agonist GW4064 induced Sqstm1 mRNA and protein expression in the ileum but not in the liver, which suggests that FXR regulates Sqstm1 in a tissue-specific manner 46.
The Sqstm1 gene has been shown to contain binding sites for several other transcription factors involved in mitogen signaling, monocyte differentiation, and inflammation. For example, the Sqstm1 gene contains binding sites for c-myc and Sp1 47. The promoter region also contains three TPA response elements (TRE) for binding of Fos- and Jun-related bZip proteins or Jun homodimers along with two distinct binding sites for Ets-1 family transcription factors, which are induced by mitogenic signal transduction pathway proteins or serum, as well as by monocyte differentiation pathway factors 38. However, transcriptional activation of Sqstm1 by these transcription factors has not been shown, and it would be interesting to investigate if these transcription factors also regulate expression of Sqstm1/p62 in the liver.
Expression of Sqstm1/p62 mRNA and protein is also induced pharmacologically. For example, proteasome inhibitors and hydrogen peroxide have both been shown to induce Sqstm1 expression in Neuro-2a cells and murine macrophages, respectively 48, 49. Moreover, it was reported that Resveratrol increases the expression of Sqstm1 by activating the JNK pathway in chronic myelogenous leukemia (CML) cells, although it is not clear how JNK increased Sqstm1 expression 50.
In addition to transcriptional regulation, p62 has also been found to be posttranslationally regulated. For example, p62 has been found to be phosphorylated by casein kinase 2 (CK2) on S403 within the UBA domain, which enhances the binding of p62 to ubiquitin chains and leads to increased autophagic clearance of ubiquitinated proteins 51. Moreover, p62 is phosphorylated by cyclin- dependent kinase 1 (CDK1) at T269 and S272, which is necessary to maintain appropriate cyclin B1 levels for cell cycle regulation. In response to Ras-induced transformation, the nonphosphorylatable mutant of p62 leads to a faster exit from mitosis and enhanced cell proliferation and tumorigenesis 52.
Furthermore, intracellular p62 protein levels can be regulated by two intracellular protein degradation pathways: autophagy and the ubiquitin proteasome system (UPS). As discussed above, p62 directly interacts with LC3 through its LIR and is degraded during autophagy induction 18, 20. Moreover, p62 protein levels are accumulated in autophagy-deficient mouse livers and neurons, indicating that basal autophagy may constitutively degrade p62 21, 53. In the UPS pathway, p62 recruits ubiquitinated proteins and targets them for degradation by proteasomes, and p62 is also degraded in this process. p62 shuttles ubiquitinated proteins to the proteasome, and a decrease in p62 levels leads to impaired proteasomal degradation and subsequent accumulation of ubiquitinated proteins 54. These data argue that p62 plays a role in recruiting ubiquitinated proteins not only to autophagosomes, but also to proteasomes. Taken together, intracellular p62 expression levels are regulated transcriptionally and posttranslationally, and posttranslational modifications of p62 can further enhance p62-mediated removal of ubiquitinated proteins via autophagy. In addition, p62 protein levels are regulated by both autophagy and the proteasome.
p62 Serves as a Receptor for Protein Aggregates and Damaged or excess Organelles
Autophagy was generally thought to be a non-selective lysosomal degradation pathway; however, accumulating evidence now supports that autophagy can be selective. Selective autophagic degradation of intracellular misfolded proteins and damaged/or excess organelles plays an important role in regulating cellular homeostatic function 5, 9, 10, 55. Selective autophagy is mediated by autophagy receptors. An ideal autophagy receptor is able to interact directly with both the cargo that is targeted for degradation and the autophagy machinery. Increasing evidence now demonstrates that protein ubiquitination is a prerequisite for cargo to be “tagged” for autophagic recognition. An autophagy receptor protein is defined as a protein that is able to bind to ubiquitinated cargos, such as protein aggregates, and LC3 protein through its LIR 9. p62 has been identified as one of many autophagy receptors and p62 may promote autophagic degradation of cargos in at least three mechanisms. First, p62 is directly associated with early autophagosome formation by recruiting Atg proteins to the autophagosome formation site through its PB1 domain independent of its LIR 32. Second, p62 binds to ubiquitinated proteins to form protein aggregates via its UBA domain. Third, p62 also directly interacts with LC3 via its LIR motif. Therefore, p62 may facilitate delivery of autophagic cargos to the autophagosome formation site due to its association with the early autophagosome membrane. p62 may also further recruit ubiquitinated cargos to the autophagosome via its interaction with both ubiquitin and LC3 for selective degradation 9, 20. These selective cargos include protein aggregates, peroxisomes, mitochondria and invading intracellular bacteria 9, 56, 57.
How are misfolded/mutant proteins and protein aggregates selectively removed by autophagy? It has been proposed that a cargo receptor complex, which includes the tagged substrate, a receptor, a scaffold protein and an Atg8 family protein, is involved in the selective removal of misfolded/mutant protein aggregates 9. p62 has been demonstrated to concentrate ubiquitinated proteins to form aggregates before they become degraded by autophagy. LC3 and p62 form a shell around huntingtin protein aggregates, suggesting that p62 may serve to mark these aggregates for autophagic degradation 18. Indeed, over-expression of p62 reduced TAR DNA binding protein-43 (TDP-43) protein aggregates, a signature protein in inclusions seen in neurodegeneration related diseases. In contrast, siRNA knockdown of p62 levels or over-expression of a p62 deletion mutant lacking the UBA domain increased apoptosis in cells expressing mutant human huntingtin protein 18. In addition to huntingtin protein, it is well-known that p62 is associated with MDBs, the most common inclusion bodies in the liver as discussed above. However, the role of p62 in regulating the formation of MDBs is not clear. Experiments using p62 KO mice would help to elucidate whether p62 may also help remove protein aggregates in the liver as a protective mechanism.
In addition to p62, two other p62 interacting partners, the neighbor of Brca1 gene (NBR1) and autophagy linked FYVE protein (ALFY), are also needed for constitutive autophagic degradation of ubiquitinated protein aggregates 9, 58. NBR1 is a 966-amino acid protein that has similar domain organization to p62, which also has an N-terminal PB1 domain, a ZZ-type zinc finger, and a C-terminal UBA domain. Unlike p62, NBR1 cannot polymerize via its PB1 domain, but it binds to p62 via this domain. Endogenous NBR1 was found in the same complex with p62 and Atg8 family proteins, and it also accumulates in Atg7 and Atg7/p62-double KO (DKO) mouse livers as well as in human alcoholic Mallory bodies 59. ALFY is a giant 400 kDa scaffold protein that contains a FYVE domain, which is ubiquitously expressed and conserved from yeast, nematodes, and flies to mammals 60. While ALFY binds with PtdIns(3)P, which is similar to other FYVE-domain proteins, ALFY was not found on endosomes and was found to be redistributed from the nucleus to autophagic membranes in response to starvation or proteasome inhibition 60. Unlike p62 and NBR1, ALFY does not accumulate in autophagy-deficient cells, suggesting that ALFY is not a selective autophagy substrate. ALFY is highly expressed in the brain and has been shown to play a role in the clearance of mutant α-synuclein. Drosophila lacking the ALFY homolog Blue Cheese (bchs) accumulate ubiquitin positive inclusions and have a neurodegenerative phenotype and reduced life span 61. Both p62 and ALFY are found in promyelocytic leukemia nuclear bodies (PML-NBs), and the localization of ALFY to PML-NBs is dependent on p62 and vice versa 61. Although no LIR region was identified in ALFY, ALFY interacts with the Atg5-Atg12 complex and may stimulate LC3-PE conjugation. Moreover, the FYVE domain on ALFY may also recognize PI3P membranes and perhaps plays a role in autophagosome membrane recruitment 58. Therefore, it has been suggested that ALFY might work as a scaffold protein to facilitate the assembly of p62 positive aggregates and target these protein aggregates for autophagic degradation 58, 61. The role of ALFY in the formation of liver protein aggregates is currently unclear, but ALFY is expressed in the liver 60, indicating that it may indeed play a role in liver protein aggregate formation.
Histone deacetylase 6 (HDAC6) mainly resides in the cytoplasm and is another crucial component required for selective protein aggregate clearance 62–64. HDAC6 possesses two functional deacetylase domains. It also contains a ubiquitin-binding zinc-finger domain of the BUZ class that binds with ubiquitin and is essential for HDAC6-mediated selective degradation of protein aggregates 63, 64. Ubiquitin-positive protein aggregates are readily found in HDAC6-deficient mouse brains leading to neurodegeneration in these animals, indicating that HDAC6 plays an important role in removing cellular protein aggregates 63. It is well known that lysine (K) 48-linked polyubiquitination directs proteins for proteasome-mediated degradation, whereas K63-linked polyubquitination is recognized by both HDAC6 and p62 and is preferentially degraded by autophagy 5, 64, 65. Unlike p62 and NBR1, HDAC6 does not contain a LIR but can interact with microtubules and F-actin cytoskeleton via its ubiquitin binding domain 64. It has been shown that the interaction of HDAC6 with the cytoskeleton plays a critical role for transporting aggregated proteins to the peri-nuclear microtubule-organizing center (MTOC), which is where most lysosomes are located 63. While HDAC6 per se is not essential for autophagy induction, it controls the fusion of autophagosomes with lysosomes by regulating the actin network 63. Therefore, it seems that HDAC6 regulates protein aggregate degradation by not only transporting protein aggregates to the MTOC, but also by regulating autophagy at the late autophagosome maturation step 63, 64. Taken together, it seems that p62 can coordinately work with ALFY and HDAC6 to regulate selective clearance of protein aggregates by autophagy (Figure 2).
Figure 2. Autophagy receptor complex for the degradation of protein aggregates.
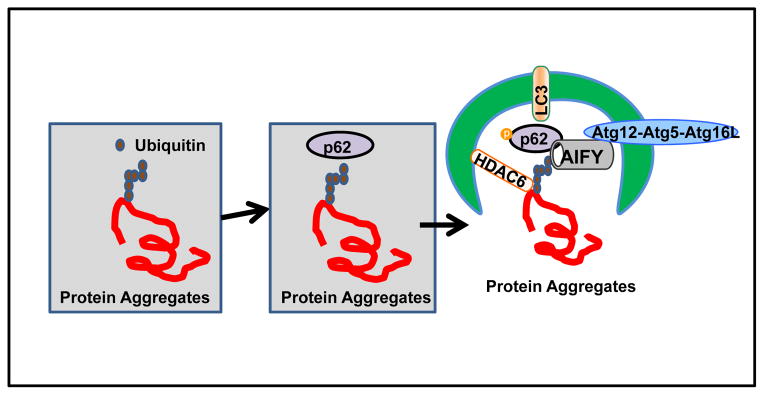
Specific ubiquitin (ub) chains are incorporated into the protein aggregates and recruit p62 via its UBA domain, which further delivers ubiquitinated protein aggregates to autophagosomes by interacting with LC3 via its LIR domain. Ubiquitin also recruits HDAC6, which facilitates the transportation of protein aggregates to the MTOC as well as fusion of autophagosomes with lysosomes for the subsequent degradation of protein aggregates. ALFY functions as a scaffold protein for p62 positive protein aggregates that targets them for degradation by autophagy via interaction with the Atg5–Atg12 complex.
In addition to the removal of ubiquitinated protein aggregates, p62 also plays an important role in selective removal of mitochondria 66, 67, peroxisomes 56, midbody ring structures 68, and bacteria 57, 69. When mitochondria are depolarized by uncouplers such as CCCP, the ubiquitin E3 ligase Parkin is recruited from the cytosol to damaged depolarized mitochondria 66, 70. The recruitment of Parkin to mitochondria also requires the PTEN-induced putative protein kinase 1 (PINK1), a serine/threonine kinase that contains a mitochondrial targeting sequence, which is degraded in healthy mitochondria by PARL, a mitochondria intramembrane protease 71–73. Although it remains unclear how PINK1 recruits Parkin to mitochondria, it seems that the kinase activity of PINK1 is required for Parkin mitochondrial translocation and for increasing ubiquitination activity of Parkin 74, 75. Following its mitochondrial translocation, Parkin induces two types of polyubiquitin chains, which appear to have different functions. Parkin-mediated K48 chains on specific outer mitochondrial membrane proteins trigger proteasome-dependent degradation of these proteins, which include Miro, Mitofusin1/2, hFis1, and Tom20 as well as others 76, 77. The Parkin-mediated proteasome-dependent degradation of these outer mitochondrial membrane proteins seems to be required, but it is not sufficient for mitophagy. For example, proteasomal degradation of Mitofusin1/2 causes mitochondrial fragmentation, which may make them more readily enveloped by autophagosomes and thus, promote their clearance by mitophagy 55, 76. In a second model, Parkin also induces K63 and K27 ubiquitin chains that initiate a signaling cascade to activate mitophagy, which may involve an autophagy receptor such as p62 67. VDAC1, a component of the mitochondrial permeability transition pore, has been reported to be a target for Parkin-mediated K63 and K27 poly-ubiquitination and promotes mitophagy 67. However, the role of VDAC1 ubiquitination in mitophagy is controversial because it was found that VDAC1 is dispensable for mitophagy by another study 78. It is possible that the presence of ubiquitination on the mitochondria, rather than a particular ubiquitinated protein on the mitochondria, is important and sufficient for the induction of mitophagy. This notion is supported by observations that p62 is recruited to depolarized mitochondria in a Parkin-dependent manner, while VDAC1 and VDAC3 are dispensable for p62 mitochondrial translocation 78. Once on the mitochondria, p62 serves as a receptor to further recruit LC3-positive autophagosomes to p62-ubiquitin decorated mitochondria for their degradation. However, the role of p62 in mitophagy is still controversial. While studies from siRNA-mediated knockdown of p62 suggested that it plays a role in Parkin-mediated mitophagy, studies using genetic p62 KO cells suggest that p62 is required for Parkin-induced mitochondrial clustering, but not for mitophagy 78. The discrepancy could be due to the lack of reliable quantitative assays for mitophagy because immunostaining for the mitochondrial outer membrane protein Tom20 was used for a marker for mitophagy in these studies 66, 67, 78. It is now known that Tom20 is largely degraded through Parkin-mediated proteasome-dependent degradation and not by autophagy 76, 77. While more reliable quantitative assays for mitophagy are desperately needed, it seems that the degradation of mitochondria matrix proteins, such as heat shock protein (HSP60), is more dependent on autophagy 76. Moreover, a recent study using adipocyte-specific p62 KO mice suggested that p62 may regulate mitochondrial homeostasis through a p38-mediated transcriptional program 7. More studies are definitely needed to further understand the role of p62 in mitophagy, preferably when more reliable quantitative assays are available in vivo. Moreover, we recently demonstrated that Parkin is ubiquitously expressed in many tissues including liver in mice 55, but the exact role of Parkin-p62 in mitophagy in the liver is not clear.
p62 in Liver physiology and Pathology
Liver is one of the most dynamic and metabolically active organs, and increasing evidence indicates that autophagy in the liver plays an important role in maintaining the levels of blood glucose and amino acids. As discussed above, p62 was found to be a major component of the liver protein aggregates, MDBs, which are the signature manifestation of alcoholic liver disease 22, 79. Thus, p62 may play an important role in the regulation of protein aggregates in the liver 80. As a major signaling adapter protein, p62 has also been shown to regulate the Nrf-2-mediated antioxidant response, as previously discussed. In addition, p62 has been shown to regulate NF-κB activation, mTOR and MAPK signaling pathways. As a result, p62 has emerged as a major factor in regulating obesity, insulin sensitivity, energy homeostasis, cholestasis and liver tumorigenesis. The role of p62 in these individual pathologies is discussed in more detail below.
p62 Regulation of Protein Aggregates in the Liver: MDBs and IHBs
MDBs are intracellular protein aggregates that are composed mainly of p62, ubiquitin, chaperones, proteasomal subunits, and intermediate filaments 24, 80. MDBs are hallmarks of liver pathogenesis, such as chronic alcoholic steatohepatitis and non-alcoholic steatophepatitis 23. Furthermore, other cytoplasmic inclusions, such as IHBs, are the hallmark of hepatocellular carcinoma, and sometimes exist alongside MDBs 22. MDBs are also a manifestation of copper toxicity diseases including Wilson’s disease, Indian childhood cirrhosis, and idiopathic copper toxicosis. These diseases are also characterized by hepatocyte ballooning, inflammation, and cytoskeletal alternations resulting in development of micronodular cirrhosis 81.
It has been suggested that MDBs are degraded by both the ubiquitin-proteasome system (UPS) and autophagy, and p62 may play a role in this process 23, 24. Proteasome inhibitors induce MDB-like structures in cultured cells and in mouse liver 82, whereas liver-specific Atg7 KO mice have increased ubiquitin and p62 positive protein aggregates 21. These findings support the notion that both proteasome and autophagy regulate MDB formation. Indeed, it has been shown that induction of autophagy by rapamycin significantly suppresses MDB formation both in vitro and in vivo83. Currently, the role of p62 in liver pathogenesis associated with MDBs is not clear. p62 plays a role in reducing protein misfolding caused by copper toxicity induced-oxidative stress by sequestering the misfolded protein into MDBs 81. This suggests that p62 likely participates in cellular rescue mechanisms during oxidative hepatocyte injury exerted by copper toxicity 81. Moreover, it has also been suggested that soluble protein aggregates are more detrimental to cells, and p62 may play a protective role by packing these aggregates into insoluble and less toxic “aggresomes” 64, 65, 84. However, this theory does not explain why Atg7/p62 double KO (DKO) mice are actually protected against the liver injury found in Atg7 liver-specific KO mice 21. It is possible that p62-mediated persistent activation of Nrf2 plays a more important role in this liver injury than soluble protein aggregates because Atg7/Nrf2 DKO mice also have less liver injury than Atg7 liver-specific KO mice 85.
Cholestasis
Immunohistochemistry analysis of liver tissues from primary biliary cirrhosis (PBC) patients showed increased LC3 expression and p62 positive aggregates in biliary epithelial cells in damaged small bile ducts, which indicates that p62 may have a role in cholestasis 86, 87. Moreover, MDBs have also been found in chronic cholestatic conditions, particularly in patients with PBC and primary sclerosing cholangitis 80, 88, 89. It has also been suggested that elevated levels of bile acids plays a role in MDB formation. For example, cholic acid feeding and common bile duct ligation result in formation of MDBs in drug-primed mice, suggesting that bile acids may drive the formation of MDBs in cholestatic liver disease 88. Indeed, MDBs in PBC livers were found in acinar zone 1, where bile acids levels are highest 89. Furthermore, drug-induced MDBs by diethyl 1,4,-dihydro-2,4,6,-trimethyl-3,5-pyridine dicarboxylate (DDC) or griseofulvin also display cholestasis and elevated serum bile acids 88, 90, 91. We recently found that there was a significant increase of p62 protein levels in FXR-deficient mouse livers compared to wild type mice (Manley et al., manuscript in preparation), and FXR-KO mice also have increased hepatic bile acid levels. Based on the above evidence, it is possible that p62 is involved in the pathogenesis of cholestasis, although further investigation is definitely needed.
Obesity and Insulin Resistance
Obesity is a global-wide health issue that is implicated in metabolic syndrome, which includes insulin resistance, type II diabetes and hepatocellular carcinoma (HCC) 92, 93. p62 KO mice develop mature-onset obesity in addition to impaired glucose and insulin tolerance 94. Studies from genetic p62 KO mice suggest that p62 plays an important role in metabolic syndrome development and obesity pathogenesis by regulating ERK, JNK, and Akt signaling pathways.
The ERK1/2 signaling pathway is a subgroup of MAPK signaling cascades responsible for adipogenesis 95, 96. Mice fed with high fat diet have increased ERK activity in their adipose tissue. Moreover, patients with type II diabetes display enhanced ERK activity in adipocytes 96. Furthermore, increased basal ERK activity is observed in fat tissues of both obese and non-obese p62 KO mice, suggesting that p62 could be a negative regulator of ERK activity 94. Indeed, p62 interacts with and sequesters ERK through its PB1 domain and subsequently suppresses ERK enzymatic activity and function. Consistent with this notion, p62 is induced during adipocyte differentiation as a negative feedback mechanism to down regulate ERK mediated adipogenesis by inhibiting expression of downstream targets of ERK, C/EBPβ and PPARγ 94. ERK1/p62 DKO mice do not have p62-deficiency-induced adipogenesis, indicating that the increased adipogenesis seen in p62 KO mice is due to the lack of negative regulation of ERK activity by p62 97. Interestingly, adipocyte-specific, but not hepatocyte-specific, p62 KO mice have increased body fat, hepatic steatosis, impaired glucose tolerance and decreased insulin sensitivity 98, which suggests a tissue-specific role for p62 in regulating adipogenesis and energy expenditure.
c-Jun-N-terminal kinase (JNK) is another subgroup of the MAPK pathway that is activated in response to diverse stimuli including cytokines, reactive oxygen species, xenobiotics, and metabolic changes. JNK also plays a role in hepatic steatosis, fibrosis and HCC. Both mice fed with high-fat diet (HFD) and genetically obese (ob/ob) mice display enhanced JNK activity 99, 100. Deletion of JNK1 prevents diet-induced obesity and increases insulin sensitivity in mice fed with HFD 99, 101, 102. JNK has also been shown to have a role in activation of autophagy. JNK is required for induction of proteasome inhibitor induced-autophagy downstream of IRE-1 103. Moreover, JNK is also involved in starvation-induced autophagy through JNK-mediated phosphorylation of Bcl-2 to release Beclin-1 104 or through c-jun-mediated transcription of Beclin-1 expression 105. While it is not clear if p62 regulates JNK activation, JNK may regulate p62 protein indirectly through activation of autophagy, which causes p62 degradation. In addition, we can speculate that JNK-deficiency may lead to increased p62 levels due to impaired autophagy. If this is true, increased p62 levels may contribute to the decreased lipogenesis and steatosis observed in JNK1 KO mice fed with HFD by down regulating ERK and p38 activity, as previously discussed.
p62 also regulates the insulin signaling pathway by interacting with insulin receptor substrate-1 (IRS-1) 106. Over-expression of p62 increases phosphorylation of Akt at S473 and T308, which is a downstream target of IRS-1, and promotes the translocation of GLUT4 to the cell membrane for glucose uptake 106. Conversely, tyrosine phosphorylation of both IRS-1 and its downstream target Akt in response to insulin injection was severely decreased in both muscle and fat tissues of p62 KO mice 94. These results suggest that p62 may play a positive role in insulin sensitivity by enhancing the insulin signaling pathway, and p62 could be a potential target for diabetes.
p62 Regulates Liver Tumorigenesis by Activating Multiple Signaling Pathways
Increasing evidence now supports a strong association of p62 protein levels with cancer. Increased expression of p62 has been found in various cancer tissues, including HCC 22, 107, breast cancer 108, lung cancer 109 and colon cancer 110. The level of p62 has also been found to be associated with poor prognosis for patients with lung carcinoma. Mice with specific deletion of either Atg7 or Atg5 in the liver have spontaneous development of liver adenoma, and high levels of p62 protein have been found in these mouse livers 52, 107. More importantly, Atg7/p62 DKO mice have significantly decreased liver tumorigenesis 52, which convincingly support the notion that p62 promotes tumorigenesis. As discussed above and below, p62 has multiple functions and sits at the crossroads for several signaling pathways including NF-κB, Nrf2, mTOR and autophagy, which are all important for cell proliferation and tumoral transformation.
p62 Regulation of NF-κB and its Role in Liver Tumorigenesis
p62 regulates NF-κB by interacting with multiple players through its different domains, including interaction with aPKC through its PB1 domain, RIP through its ZZ domain and TRAF6 through its TB domain 111. As discussed above, p62 interacts with RIP and bridges RIP to aPKCs resulting in the activation of NF-κB by the TNF-α signaling pathway 33. Inhibition of aPKC and down-regulation of p62 decreases NF-κB activation by IL-1, indicating that p62 promotes NF-κB activation 29. In addition, p62 interacts with TRAF6 to induce TRAF6 polyubiquitination, and the formation of a p62-TRAF6-IKKβ-aPKC signaling complex activates NF-κB by nerve growth factor 43. Deletion or mutation of the UBA domain, deletion of the N-terminal dimerization PB1 domain, and elimination of the TRAF6 binding site all induce abolishment of TRAF6 polyubiquitination 43. Furthermore, the rear end acidic cluster region of the p62 PB1 domain binds the front end basic region of MAPK kinase kinase 3 (MEKK3) to promote NF-κB activation 112. Conversely, the PB1 domain of p62 also interacts with CYLD, a deubiquitinase, which inhibits TRAF6 polyubiquitination and serves as a negative regulator for RANK-mediated NF-κB activation and osteoclastogenesis 113. Although it is not clear how p62 exactly balances positive and negative regulators of NF-κB activation, it appears that p62 modulates multiple pathways to exquisitely regulate activation of NF-κB.
HCC is one of the most common cancers worldwide and is frequently linked with chronic hepatitis characterized by liver inflammation, hepatocyte apoptosis and compensatory liver regeneration 114. However, the mechanisms underlying these sequential events are poorly understood. Recently, accumulating evidence has suggested that NF-κB signaling plays an important role in the development of liver tumors. Depending on the model used, NF-κB activation has been shown to inhibit or promote liver tumorigenesis. Mdr2-deficient mice develop HCC in the context of chronic bile duct inflammation. Tumor progression in Mdr2-deficient mice was inhibited by overexpression of a non-degradable IκBα super-repressor that blocks NF-κB activation 115. In contrast, liver-specific KO of IKKβ increased tumor formation in a mouse model of chemical carcinogen-induced HCC 116. Moreover, mice with conditional ablation of NEMO in parenchymal liver cells develop spontaneous liver cancer, suggesting that NF-κB inhibits liver tumorigenesis 117. The contradicting findings from these studies could be due to the use of different tumor models and different approaches for blocking NF-κB. Therefore, the function of NF-κB in liver cancer seems to be complex, acting either as a tumor promoter or as a tumor suppressor.
While NF-κB has been implicated in liver cancer development, the role of p62-mediated NF-κB activation in liver tumorigenesis is still elusive. In a Ras-induced mouse lung adenocarcinoma model, p62 was found to be induced by Ras and activated IKKβ by polyubiquitination of TRAF6. More importantly, p62 KO mice were resistant to Ras-induced lung adenocarcinoma due to increased ROS levels as a result of decreased NF-κB activation 42. This has been thought to lead to enhanced cell death and subsequent decreased tumorigenicity of Ras. In contrast, using autophagy-and apoptosis-defective immortalized baby mouse kidney (iBMK) cells, it was found that overexpression of p62 inhibited NF-κB activation and promoted tumorigenesis 118. These findings are similar to the previously discussed carcinogen-induced liver tumorigenesis in IKKβ- and NEMO-deficient hepatocytes, suggesting that defective autophagy and deregulation of p62 can be associated with suppression of the canonical NF-κB pathway. This NF-κB suppression may in turn promote tumorigenesis through the non-canonical pathway. The discrepancy regarding the role of p62 in NF-κB activation has been suggested to be due to the metabolic stress conditions used for culture of the iBMK cells, in which overexpression of p62 may drive the NF-κB components into huge p62 positive aggregates. It would be interesting to determine the NF-κB status in Atg7 or Atg5 liver-specific KO mouse livers, which have accumulated p62 levels in their hepatocytes and are more physiologically relevant than cultured iBMK cells. Nevertheless, even though there is unresolved controversy regarding the role of p62 in NF-κB activation, the evidence that accumulation of p62 promotes tumorigenesis in the liver, as well as other tissues, is consistent.
p62 Regulation of the Non-Canonical Nrf2 Pathway
Nrf2, a basic leucine zipper (bZIP) transcription factor, is activated during stress and induces expression of antioxidant enzymes 37, 119. In the canonical pathway, Nrf2 is negatively regulated by Keap1, an adaptor for the cullin-3-type ubiquitin ligase, which targets Nrf2 for its ubiquitination and proteasome-mediated degradation. In response to oxidative stress, the activity of this E3 ubiquitin ligase is inhibited due to modification of cysteine residues in Keap1, which results in stabilization and activation of Nrf2 37.
As discussed above, p62 can interact with Keap1 through its KIR motif, which releases Keap1 from Nrf2 and, in turn, promotes Nrf2 stabilization and activation. As a result, Nrf2 is persistently activated in the autophagy-deficient mouse liver. Activation of Nrf2 leads to the expression of a number of antioxidants, phase II detoxifying enzymes, and hepatic transporters, which are essential for cytoprotection against oxidative stress 37, 120, 121. Paradoxically, autophagy-deficient mice show severe liver injury as demonstrated by hepatomegaly, accumulation of damaged mitochondria and elevated serum hepatic enzymes, even though these mice have persistent activation of Nrf2 due to the aberrant accumulation of p62 21. The liver injury in liver-specific Atg7 KO mice was rescued by the deletion of either p62 or Nrf2, suggesting that both p62 and Nrf2 could be responsible for the liver injury found in the autophagy-deficient mice 21, 85. More recently, Yamamoto’s group generated Atg7/keap1 DKO mice, which exhibited more severe liver injury than Atg7 liver-specific single KO mice 122. Interestingly, further deletion of Nrf2 in Atg7/keap1 DKO mice was more protective against liver injury compared to further deletion of p62 in these mice 122. While these results suggest that Nrf2 activation as a result of p62 accumulation plays a dominant role in liver injury in autophagy-deficient mice, it is possible that other mechanisms could also be involved.
In addition to activating Nrf2, p62 has also been shown to increase apoptosis by promoting caspase-8 activation. It was shown that cullin3 (CUL3) E3 ligase-mediated caspase-8 polyubiquitination is required for the activation of caspase-8 in response to death receptor activation, and this process was promoted by p62 123. However, recent evidence suggests that caspase-8 forms a complex with Atg5 and colocalizes with LC3 and p62. Moreover, the LC3 positive autophagosome membrane serves as a platform for caspase-8 aggregation and activation 124, 125. LC3 positive membranes are likely more important than the presence of p62 because the activation of caspase-8 was significantly reduced in Atg5-deficient cells in which p62 levels were accumulated, but no LC3 positive autophagosome membranes were formed 126. However, it should also be noted that these studies were conducted in vitro in cultured cells. The role of p62 for the activation of caspase-8 in vivo is not clear yet. We found that both caspase-8 and caspase-3 are activated in liver-specific Atg5 KO mice, which have aberrant p62 accumulation without LC3-positive autophagosomes (Ni et al., Manuscript in preparation). These results suggest that p62 protein alone may be sufficient to induce caspase-8 activation in autophagy-deficient livers, although more work is needed to further elucidate whether this would involve CUL3-mediated caspase-8 polyubiquitination. Currently, it is not clear how Nrf2 activation increases liver injury. A recent study suggests that the Nrf2-dependent stress response, which results in accumulation of ubiquitin conjugates, may be involved 127. Further studies are needed to further elucidate the role of Nrf2 in the liver injury observed in autophagy-deficient mice.
While persistent activation of Nrf2 induces liver injury in autophagy-deficient mice, p62-mediated Nrf2 activation attenuates drug or lipogenic stress-induced liver injury that involves oxidative stress 53, 128. Acetaminophen (APAP) is a safe drug at therapeutic levels, but an overdose can cause severe liver injury in animals and man by inducing necrosis 129. We found that pharmacological induction of autophagy by rapamycin significantly protects against APAP-induced liver injury 130. However, to our surprise, liver-specific Atg5 KO mice were more resistant to APAP-induced liver injury. Subsequently, we found that persistent Nrf2 activation resulted in upregulation of glutathione synthase and drug detoxification genes in liver-specific Atg5 KO mice, which was accountable for the resistance to APAP-induced liver injury 53. In another study, it was shown that sestrins, which are p53 target genes involved in protection of cells from oxidative stress, interact with Keap1, p62 and the ubiquitin ligase, Rbx1 128. It was found that the antioxidant effects of sestrins are mediated through Nrf2 activation by p62-mediated autophagic degradation of Keap1. Interestingly, sestrin-mediated Nrf2 activation protected against liver injury induced from the acute lipogenic stimulus elicited by re-feeding after food deprivation 128. These results suggest that persistent activation of Nrf2 plays a dual role in the regulation of cell injury in the liver. While persistent activation of Nrf2 itself is detrimental to the autophagy-deficient liver, it renders these mice more resistant to drug- and oxidative stress-induced further liver damage.
In addition to regulating liver injury, the non-canonical Nrf2 pathway activated by the accumulation of p62 may also play a critical role in liver tumorigenesis. Nrf2 target genes are critical to eliminate toxicants or carcinogens to maintain redox homeostasis, so it has long been thought that activation of Nrf2 would be a promising strategy to prevent carcinogenesis and tumorigenesis. Many natural dietary compounds and synthetic small molecules that activate Nrf2 have been found to have chemo preventative effects in cell culture and animal models 131. In line with this notion, Nrf2-KO mice have increased sensitivity to carcinogenesis due to blunted efficacy of chemo preventive compounds 132, 133. Paradoxically, increasing evidence also suggests that activation of Nrf-2 promotes tumorigenesis and/or tumor progression as well as chemo resistance 131, 134. It is not surprising that tumors may take advantage of the Nrf2 pathway to promote tumor cell survival by increasing the expression of antioxidant proteins as well as detoxifying enzymes. Somatic mutations of KEAP1 and Nrf2 have been found in patients with lung cancer or head and neck cancers, causing constitutive Nrf2 activation in these patients 134, 135. Up-regulation of Nrf2 target genes, including heme oxygenase-1 (HMOX-1), peroxiredoxin (Prx) and NAD(P)H quinone oxidoreductase-1 (NQO-1), has been found in many cancers and may contribute to chemo resistance and tumor progression 131. As we discussed above, autophagy-deficient mouse livers have aberrant accumulation of p62 resulting in persistent activation of Nrf2 and eventual development of liver adenoma 107. Importantly, Komatsu’s group found that more than 25% of human HCC (26 of 102 patients) had increased p62 protein levels 107. A recent cohort study of patients with lung cancer found that 34% and 37% of non-small-cell lung cancer (NSCLC) patients had an accumulation of Nrf2 and p62, respectively. Interestingly, accumulation of p62 is found to be associated with a poor diagnosis for these patients, although accumulation of Nrf2 seems to be unrelated to accumulation of p62 in these patients 109. Taken together, it is possible that p62-mediated persistent activation of Nrf2 would promote the liver tumorigenesis seen in Atg7 liver-specific KO mice. Studies on Nrf2 and Atg7 liver DKO mice would help to answer this question.
p62 Regulating mTOR Complex 1 (mTORC1)
It is well-known that autophagy is negatively regulated by the mammalian target of rapamycin (mTOR), a serine/theornine kinase 136, 137. Meijer’s group first discovered that rapamycin inhibits mTOR and activates autophagy in isolated rat hepatocytes 138. mTOR exists in two heteromeric complexes, mTOR complex 1 (mTORC1) and mTORC2. It is known that rapamycin-sensitive mTORC1 plays a major role in the regulation of autophagy and cell growth, although there is evidence that rapamycin-insensitive mTORC2 may also regulate autophagy in certain tissues 139–141. Growth factors or insulin activates class-I-phosphatidylinosital-3 kinase (PI3K) and AKT, which leads to mTOR activation through the tuberous sclerosis complex protein (TSC)-Rheb signaling cascade 142. Activation of mTOR then shuts down autophagy, which is a cellular catabolic process, by regulating the ULK1-FIP200-Atg13 complex 139. Meanwhile, activation of mTOR also activates the anabolic process of protein synthesis by directly phosphorylating the translational initiation factor 4E (eIF4E)-binding protein (4E-BP1) and S6 kinase 1 (S6K1) 143, 144. Moreover, mTORC1 also plays a role in controlling mammalian lipid metabolism, including fatty acid and cholesterol synthesis by regulating the sterol regulatory element-binding protein1/2 (SREBP1/2) transcription factor, lipid oxidation, transport, lipolysis and adipocyte differentiation 145, 146.
It should be noted that in addition to growth factors, amino acids have been known to activate mTORC1, although the mechanisms of activation has only been recently revealed. When amino acids are abundant, active mTORC1 localizes on the lysosomes and late endosomes, which is activated by a group of GTPase proteins called Ragulators 147. Thus, amino acids activate mTORC1 independent of TSC and do not involve the insulin/PI3K– AKT signaling pathway. Since activation of autophagy may transiently increase cellular amino acid levels due to lysosomal degradation of proteins, amino acids from lysosomal degradation may serve as a negative feedback mechanism to inhibit autophagy by activating mTORC1 148.
As discussed above, it is well known that p62 is an autophagy substrate and normally degraded during autophagy activation. Interestingly, p62 can also negatively regulate autophagy by activating mTORC1. p62 can promote mTORC1 activation by directly interacting with Raptor, a key component of mTORC1, but not Rictor or any other component of mTORC2 149. It was found that the region between the ZZ and TB domains (amino acids 167–230) of p62 is required for the interaction between p62 and Raptor, while the UBA, TB, PB1, and ZZ-type zinc finger domains are not essential for p62-Raptor interaction. In response to stimulation with amino acids, deletion of p62 results in reduced mTORC1 activity. Conversely, over-expression of p62 enhanced mTORC1 activation 149. In addition, p62 also activates the Ragulator complex to promote mTORC1 activation. p62 preferentially interacts with Rag C, and this interaction is further enhanced in the presence of Raptor. Moreover, p62 may serve as a docking site and is required for mTORC1 localization to the lysosomal membrane in response to amino acid stimulation 149. Therefore, p62-mTORC1 signaling may also establish another line of feedback regulating p62 levels: decreased autophagy leads to the accumulation of p62, and p62 activates mTORC1 to suppress autophagy resulting in more p62 accumulation.
It is not surprising that activation of mTORC1 promotes tumorigenesis. mTORC1 activation increases protein synthesis while blocking autophagy, and autophagy is a well-known process to suppress tumorigenesis. Indeed, liver-specific TSC1 KO mice have persistent activation of mTORC1 and develop spontaneous HCC 150. Pathological analysis indicates that these mice have the primary etiologies of HCC in humans, including liver damage, inflammation, necrosis, and regeneration 150. Therefore, p62-mediated mTORC1 activation may also contribute to tumorigenesis. Taken together, current evidence support that p62 may work as a tumor promoter by modulating multiple signaling pathways including NF-κB, Nrf2, mTOR and autophagy.
Concluding remarks and future directions
As a multifunctional adaptor protein, p62 has emerged as a critical player in liver physiology and pathology (Figure 3). Although the major physiological role of p62 is to regulate metabolic homeostasis and bone formation, p62 may also play a role in cholestasis, diabetes, obesity, insulin resistance and liver tumorigenesis.
Figure 3. Pathophysiologcal roles of p62 in the liver.

p62 negatively regulates adipogenesis and inhibits obesity. p62 also plays a positive role in insulin sensitivity. The role of p62 in regulating cholestasis is currently not clear. p62 promotes liver tumorigenesis by regulating NF-κB, Nrf2 and mTOR activation. p62-mediated Nrf2 activation may also cause resistance to drug-induced liver injury. Finally, p62-Nrf2 also forms a positive feedback loop, which accumulated p62 activates Nrf2 and Nrf2 activation increases transcription for p62. The role of p62 in regulating cholestasis is currently not clear.
p62 is an autophagy substrate and is degraded during autophagy activation, but p62 can also regulate autophagy by activating mTORC1. Suppression of autophagy leads to accumulation of p62, and p62 further inhibits autophagy, which in turn leads to more p62 accumulation. This forms a positive feedback loop for more p62 accumulation and autophagy suppression.
However, many questions remain to be answered. While it is clear that p62 may serve as a receptor for selective autophagic removal of protein aggregates, its role in selective mitophagy is still controversial due to the lack of reliable mitophagy makers and quantitative assays. Moreover, the role of p62 in mitophagy in vivo has not been explored. In addition, the mechanisms for liver toxicity induced by p62-mediated Nrf2 activation are still not clear. Nevertheless, as understanding of the pathophysiological roles of p62 increases, it seems that targeting p62 may be a promising strategy for treating diabetes, obesity and liver hepatosteatosis.
Acknowledgments
The research work in W.X Ding’s lab was supported in part by the NIAAA funds R01 AA020518-01 and National Center for Research Resources (5P20RR021940-07. J. A. Williams was supported by the “Training Program in Environmental Toxicology” [grant 5 T32 ES007079] from the National Institute of Environmental Health Sciences. Sharon Manley is a recipient of the Biomedical Research Training Program Fellowship from University of Kansas Medical Center.
Abbreviations
- 4E-BP1
Translational initiation factor 4E (eIF4E)-binding protein
- ALFY
autophagy linked FYVE protein
- APAP
Acetaminophen
- AP-1
Activator protein 1
- aPKC
Atypical protein kinase C
- ARE
Antioxidant response element
- bchs
Blue cheese
- bZIP
Basic leucine zipper
- C/EBPβ
CCAAT-enhancer-binding protein β
- CCCP
Carbonyl cyanide m-chlorophenyl hydrazone
- CDK1
Cyclin-dependent kinase 1
- ChIP
Chromatin immunoprecipitation
- CK2
Casein kinase 2
- CUL3
Cullin3
- DDC
Diethyl 1,4,-dihydro-2,4,6,-trimethyl-3,5-pyridine dicarboxylate
- DKO
Double knockout
- ERK
Extracellular-signal-regulated kinase
- FXR
Farnesoid X Receptor
- GABARAP
Gamma-aminobutyric acid receptor-associated protein
- HCC
Hepatocellular carcinoma
- HDAC6
Histone deacetylase 6
- HFD
High-fat diet
- HMOX-1
heme oxygenase-1
- HSP60
Heat shock protein 60
- iBMK
immortalized baby mouse kidney
- IHBs
Intracytoplasmic hyaline bodies
- IKKβ
Inhibitor of κB kinase
- IL-1
Interleukin-1
- IRE-1
Inositol-requiring protein 1
- IRS-1
insulin receptor substrate-1
- JNK
c-Jun N-terminal kinase
- K
Lysine
- K27
Lysine 27
- K48
Lysine 48
- K63
Lysine 63
- Keap1
Kelch-like ECH-associated protein 1
- KIR
Keap1 interacting region
- KO
Knockout
- LC3
Microtubule light chain 3
- LIR
LC3 interacting region
- MAPK
Mitogen-activated protein kinase
- MDBs
Mallory-Denk Bodies
- Mdr2
Multidrug resistance protein 2
- MEKK3
MAPK kinase kinase 3
- MEK5
MAPK kinase 5
- MTOC
Microtubule-organizing center
- mTOR
Mammalian target of rapamycin
- mTORC1
mTOR complex 1
- NBR1
Neighbor of BRCA gene 1
- NEMO
NF-κB essential modulator/IKKγ
- NES
Nuclear export signal
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NLS
Nuclear localization signal
- NQO-1
NAD(P)H quinone oxidoreductase-1
- Nrf2
Nuclear factor (erythroid-derived 2)-like 2
- NSCLC
non-small-cell lung cancer
- PB1
Phox/Bemp1
- PBC
Primary biliary cirrhosis
- PEST
Proline (P), glutamic acid (E), serine (S), and threonine (T) rich sequence
- PI3K
Class-I-phosphatidylinosital-3 kinase
- PINK1
PTEN-induced putative protein kinase 1
- PML-NBs
promyelocytic leukemia nuclear bodies
- PPARγ
peroxisome proliferator-activated receptor γ
- Prx
Peroxidredoxin
- RANK
Receptor Activator of NF-κB
- RANKL
Receptor Activator of NF-κB ligand
- RIP
Receptor interacting protein
- S6K1
S6 kinase 1
- SH2
Src-homology 2
- Sp1
Specificity protein 1
- SQSTM1
Sequestosome 1
- SREBP1/2
Sterol regulatory element-binding protein1/2
- TB
TRAF-6 binding
- TDP-43
TAR DNA binding protein-43
- TNF
Tumor necrosis factor
- TOM20
Translocase of outer mitochondrial membrane 20
- TRADD
TNF receptor type 1-associated death domain protein
- TRAF6
TNF-associated receptor-6
- TRE
TPA response element
- TSC
Tuberous sclerosis complex protein
- UBA
Ubiquitin-associated
- UPS
Ubiquitin proteasome system
- ZIP
Cytoplasmic zeta-interacting protein
References
- 1.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–4. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Komatsu M, Kageyama S, Ichimura Y. p62/SQSTM1/A170: physiology and pathology. Pharmacol Res. 2012;66:457–62. doi: 10.1016/j.phrs.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 3.Ichimura Y, Kominami E, Tanaka K, Komatsu M. Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy. 2008;4:1063–6. doi: 10.4161/auto.6826. [DOI] [PubMed] [Google Scholar]
- 4.Komatsu M, Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 2010;584:1374–8. doi: 10.1016/j.febslet.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 5.Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ. 2013;20:21–30. doi: 10.1038/cdd.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duran A, Serrano M, Leitges M, Flores JM, Picard S, Brown JP, Moscat J, Diaz-Meco MT. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev Cell. 2004;6:303–9. doi: 10.1016/s1534-5807(03)00403-9. [DOI] [PubMed] [Google Scholar]
- 7.Muller TD, Lee SJ, Jastroch M, Kabra D, Stemmer K, Aichler M, Abplanalp B, Ananthakrishnan G, Bhardwaj N, Collins S, Divanovic S, Endele M, Finan B, Gao Y, Habegger KM, Hembree J, Heppner KM, Hofmann S, Holland J, Kuchler D, Kutschke M, Krishna R, Lehti M, Oelkrug R, Ottaway N, Perez-Tilve D, Raver C, Walch AK, Schriever SC, Speakman J, Tseng YH, Diaz-Meco M, Pfluger PT, Moscat J, Tschop MH. p62 Links beta-adrenergic input to mitochondrial function and thermogenesis. J Clin Invest. 2013;123:469–78. doi: 10.1172/JCI64209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goode A, Layfield R. Recent advances in understanding the molecular basis of Paget disease of bone. J Clin Pathol. 2010;63:199–203. doi: 10.1136/jcp.2009.064428. [DOI] [PubMed] [Google Scholar]
- 9.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–96. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12:836–41. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 11.Moscat J, Diaz-Meco MT. Feedback on fat: p62-mTORC1-autophagy connections. Cell. 2011;147:724–7. doi: 10.1016/j.cell.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park I, Chung J, Walsh CT, Yun Y, Strominger JL, Shin J. Phosphotyrosine-independent binding of a 62-kDa protein to the src homology 2 (SH2) domain of p56lck and its regulation by phosphorylation of Ser-59 in the lck unique N-terminal region. Proc Natl Acad Sci U S A. 1995;92:12338–42. doi: 10.1073/pnas.92.26.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joung I, Strominger JL, Shin J. Molecular cloning of a phosphotyrosine-independent ligand of the p56lck SH2 domain. Proc Natl Acad Sci U S A. 1996;93:5991–5. doi: 10.1073/pnas.93.12.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shin J. P62 and the sequestosome, a novel mechanism for protein metabolism. Arch Pharm Res. 1998;21:629–33. doi: 10.1007/BF02976748. [DOI] [PubMed] [Google Scholar]
- 15.Ishii T, Yanagawa T, Kawane T, Yuki K, Seita J, Yoshida H, Bannai S. Murine peritoneal macrophages induce a novel 60-kDa protein with structural similarity to a tyrosine kinase p56lck-associated protein in response to oxidative stress. Biochem Biophys Res Commun. 1996;226:456–60. doi: 10.1006/bbrc.1996.1377. [DOI] [PubMed] [Google Scholar]
- 16.Sanchez P, De Carcer G, Sandoval IV, Moscat J, Diaz-Meco MT. Localization of atypical protein kinase C isoforms into lysosome-targeted endosomes through interaction with p62. Mol Cell Biol. 1998;18:3069–80. doi: 10.1128/mcb.18.5.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puls A, Schmidt S, Grawe F, Stabel S. Interaction of protein kinase C zeta with ZIP, a novel protein kinase C-binding protein. Proc Natl Acad Sci U S A. 1997;94:6191–6. doi: 10.1073/pnas.94.12.6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–14. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K, Komatsu M. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283:22847–57. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- 20.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 21.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–63. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 22.Stumptner C, Heid H, Fuchsbichler A, Hauser H, Mischinger HJ, Zatloukal K, Denk H. Analysis of intracytoplasmic hyaline bodies in a hepatocellular carcinoma. Demonstration of p62 as major constituent. Am J Pathol. 1999;154:1701–10. doi: 10.1016/S0002-9440(10)65426-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zatloukal K, French SW, Stumptner C, Strnad P, Harada M, Toivola DM, Cadrin M, Omary MB. From Mallory to Mallory-Denk bodies: what, how and why? Exp Cell Res. 2007;313:2033–49. doi: 10.1016/j.yexcr.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 24.Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, Kleinert R, Prinz M, Aguzzi A, Denk H. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol. 2002;160:255–63. doi: 10.1016/S0002-9440(10)64369-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seibenhener ML, Geetha T, Wooten MW. Sequestosome 1/p62--more than just a scaffold. FEBS Lett. 2007;581:175–9. doi: 10.1016/j.febslet.2006.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawai K, Saito A, Sudo T, Osada H. Specific regulation of cytokine-dependent p38 MAP kinase activation by p62/SQSTM1. J Biochem. 2008;143:765–72. doi: 10.1093/jb/mvn027. [DOI] [PubMed] [Google Scholar]
- 27.Saito A, Kawai K, Takayama H, Sudo T, Osada H. Improvement of photoaffinity SPR imaging platform and determination of the binding site of p62/SQSTM1 to p38 MAP kinase. Chem Asian J. 2008;3:1607–12. doi: 10.1002/asia.200800099. [DOI] [PubMed] [Google Scholar]
- 28.Pankiv S, Lamark T, Bruun JA, Overvatn A, Bjorkoy G, Johansen T. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J Biol Chem. 2010;285:5941–53. doi: 10.1074/jbc.M109.039925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19:1576–86. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nezis IP, Stenmark H. p62 at the interface of autophagy, oxidative stress signaling, and cancer. Antioxidants & redox signaling. 2012;17:786–93. doi: 10.1089/ars.2011.4394. [DOI] [PubMed] [Google Scholar]
- 31.Moscat J, Diaz-Meco MT, Wooten MW. Of the atypical PKCs, Par-4 and p62: recent understandings of the biology and pathology of a PB1-dominated complex. Cell death and differentiation. 2009;16:1426–37. doi: 10.1038/cdd.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itakura E, Mizushima N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol. 2011;192:17–27. doi: 10.1083/jcb.201009067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanz L, Sanchez P, Lallena MJ, Diaz-Meco MT, Moscat J. The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. The EMBO journal. 1999;18:3044–53. doi: 10.1093/emboj/18.11.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moscat J, Diaz-Meco MT, Wooten MW. Signal integration and diversification through the p62 scaffold protein. Trends in biochemical sciences. 2007;32:95–100. doi: 10.1016/j.tibs.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 35.Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576–91. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol. 2010;30:3275–85. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev. 2006;38:769–89. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- 38.Vadlamudi RK, Shin J. Genomic structure and promoter analysis of the p62 gene encoding a non-proteasomal multiubiquitin chain binding protein. FEBS Lett. 1998;435:138–42. doi: 10.1016/s0014-5793(98)01021-7. [DOI] [PubMed] [Google Scholar]
- 39.McManus S, Roux S. The adaptor protein p62/SQSTM1 in osteoclast signaling pathways. J Mol Signal. 2012;7:1. doi: 10.1186/1750-2187-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet. 2002;70:1582–8. doi: 10.1086/340731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hocking LJ, Lucas GJ, Daroszewska A, Mangion J, Olavesen M, Cundy T, Nicholson GC, Ward L, Bennett ST, Wuyts W, Van Hul W, Ralston SH. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet. 2002;11:2735–9. doi: 10.1093/hmg/11.22.2735. [DOI] [PubMed] [Google Scholar]
- 42.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–54. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 43.Wooten MW, Geetha T, Seibenhener ML, Babu JR, Diaz-Meco MT, Moscat J. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J Biol Chem. 2005;280:35625–9. doi: 10.1074/jbc.C500237200. [DOI] [PubMed] [Google Scholar]
- 44.Chamoux E, McManus S, Laberge G, Bisson M, Roux S. Involvement of kinase PKC-zeta in the p62/p62(P392L)-driven activation of NF-kappaB in human osteoclasts. Biochim Biophys Acta. 2013;1832:475–84. doi: 10.1016/j.bbadis.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 45.Fujita K, Maeda D, Xiao Q, Srinivasula SM. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc Natl Acad Sci U S A. 2011;108:1427–32. doi: 10.1073/pnas.1014156108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams JA, Thomas AM, Li G, Kong B, Zhan L, Inaba Y, Xie W, Ding WX, Guo GL. Tissue specific induction of p62/Sqstm1 by farnesoid X receptor. PloS one. 2012;7:e43961. doi: 10.1371/journal.pone.0043961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okazaki M, Ito S, Kawakita K, Takeshita S, Kawai S, Makishima F, Oda H, Kakinuma A. Cloning, expression profile, and genomic organization of the mouse STAP/A170 gene. Genomics. 1999;60:87–95. doi: 10.1006/geno.1999.5902. [DOI] [PubMed] [Google Scholar]
- 48.Kuusisto E, Suuronen T, Salminen A. Ubiquitin-binding protein p62 expression is induced during apoptosis and proteasomal inhibition in neuronal cells. Biochemical and biophysical research communications. 2001;280:223–8. doi: 10.1006/bbrc.2000.4107. [DOI] [PubMed] [Google Scholar]
- 49.Ishii T, Yanagawa T, Yuki K, Kawane T, Yoshida H, Bannai S. Low micromolar levels of hydrogen peroxide and proteasome inhibitors induce the 60-kDa A170 stress protein in murine peritoneal macrophages. Biochemical and biophysical research communications. 1997;232:33–7. doi: 10.1006/bbrc.1997.6221. [DOI] [PubMed] [Google Scholar]
- 50.Puissant A, Robert G, Fenouille N, Luciano F, Cassuto JP, Raynaud S, Auberger P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010;70:1042–52. doi: 10.1158/0008-5472.CAN-09-3537. [DOI] [PubMed] [Google Scholar]
- 51.Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279–89. doi: 10.1016/j.molcel.2011.07.039. [DOI] [PubMed] [Google Scholar]
- 52.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ni HM, Boggess N, McGill MR, Lebofsky M, Borude P, Apte U, Jaeschke H, Ding WX. Liver-specific loss of Atg5 causes persistent activation of Nrf2 and protects against acetaminophen-induced liver injury. Toxicol Sci. 2012;127:438–50. doi: 10.1093/toxsci/kfs133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24:8055–68. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ding WX, Guo F, Ni HM, Bockus A, Manley S, Stolz DB, Eskelinen EL, Jaeschke H, Yin XM. Parkin and mitofusins reciprocally regulate mitophagy and mitochondrial spheroid formation. J Biol Chem. 2012;287:42379–88. doi: 10.1074/jbc.M112.413682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci U S A. 2008;105:20567–74. doi: 10.1073/pnas.0810611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ponpuak M, Davis AS, Roberts EA, Delgado MA, Dinkins C, Zhao Z, Virgin HWt, Kyei GB, Johansen T, Vergne I, Deretic V. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity. 2010;32:329–41. doi: 10.1016/j.immuni.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Filimonenko M, Isakson P, Finley KD, Anderson M, Jeong H, Melia TJ, Bartlett BJ, Myers KM, Birkeland HC, Lamark T, Krainc D, Brech A, Stenmark H, Simonsen A, Yamamoto A. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell. 2010;38:265–79. doi: 10.1016/j.molcel.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Overvatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–16. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 60.Simonsen A, Birkeland HC, Gillooly DJ, Mizushima N, Kuma A, Yoshimori T, Slagsvold T, Brech A, Stenmark H. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J Cell Sci. 2004;117:4239–51. doi: 10.1242/jcs.01287. [DOI] [PubMed] [Google Scholar]
- 61.Clausen TH, Lamark T, Isakson P, Finley K, Larsen KB, Brech A, Overvatn A, Stenmark H, Bjorkoy G, Simonsen A, Johansen T. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy. 2010;6:330–44. doi: 10.4161/auto.6.3.11226. [DOI] [PubMed] [Google Scholar]
- 62.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–92. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 63.Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, Taylor JP, Cuervo AM, Yao TP. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29:969–80. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yao TP. The role of ubiquitin in autophagy-dependent protein aggregate processing. Genes Cancer. 2010;1:779–786. doi: 10.1177/1947601910383277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4:141–50. doi: 10.4161/auto.5190. [DOI] [PubMed] [Google Scholar]
- 66.Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW, 2nd, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010;285:27879–90. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–31. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 68.Pohl C, Jentsch S. Midbody ring disposal by autophagy is a post-abscission event of cytokinesis. Nat Cell Biol. 2009;11:65–70. doi: 10.1038/ncb1813. [DOI] [PubMed] [Google Scholar]
- 69.Cemma M, Kim PK, Brumell JH. The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy. 2011;7:341–5. doi: 10.4161/auto.7.3.14046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrane J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107:378–83. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–42. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ. PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol. 2013 doi: 10.1083/jcb.201210111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep. 2012;2:1002. doi: 10.1038/srep01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–37. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286:19630–40. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090–106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stumptner C, Fuchsbichler A, Zatloukal K, Denk H. In vitro production of Mallory bodies and intracellular hyaline bodies: the central role of sequestosome 1/p62. Hepatology. 2007;46:851–60. doi: 10.1002/hep.21744. [DOI] [PubMed] [Google Scholar]
- 80.Strnad P, Zatloukal K, Stumptner C, Kulaksiz H, Denk H. Mallory-Denk-bodies: lessons from keratin-containing hepatic inclusion bodies. Biochimica et biophysica acta. 2008;1782:764–74. doi: 10.1016/j.bbadis.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 81.Muller T, Langner C, Fuchsbichler A, Heinz-Erian P, Ellemunter H, Schlenck B, Bavdekar AR, Pradhan AM, Pandit A, Muller-Hocker J, Melter M, Kobayashi K, Nagasaka H, Kikuta H, Muller W, Tanner MS, Sternlieb I, Zatloukal K, Denk H. Immunohistochemical analysis of Mallory bodies in Wilsonian and non-Wilsonian hepatic copper toxicosis. Hepatology. 2004;39:963–9. doi: 10.1002/hep.20108. [DOI] [PubMed] [Google Scholar]
- 82.Bardag-Gorce F, Riley NE, Nan L, Montgomery RO, Li J, French BA, Lue YH, French SW. The proteasome inhibitor, PS-341, causes cytokeratin aggresome formation. Exp Mol Pathol. 2004;76:9–16. doi: 10.1016/j.yexmp.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 83.Harada M, Hanada S, Toivola DM, Ghori N, Omary MB. Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology. 2008;47:2026–35. doi: 10.1002/hep.22294. [DOI] [PubMed] [Google Scholar]
- 84.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–38. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 85.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–23. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 86.Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. A possible involvement of p62/sequestosome-1 in the process of biliary epithelial autophagy and senescence in primary biliary cirrhosis. Liver Int. 2012;32:487–99. doi: 10.1111/j.1478-3231.2011.02656.x. [DOI] [PubMed] [Google Scholar]
- 87.Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Autophagy may precede cellular senescence of bile ductular cells in ductular reaction in primary biliary cirrhosis. Dig Dis Sci. 2012;57:660–6. doi: 10.1007/s10620-011-1929-y. [DOI] [PubMed] [Google Scholar]
- 88.Fickert P, Trauner M, Fuchsbichler A, Stumptner C, Zatloukal K, Denk H. Bile acid-induced Mallory body formation in drug-primed mouse liver. The American journal of pathology. 2002;161:2019–26. doi: 10.1016/S0002-9440(10)64480-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gerber MA, Orr W, Denk H, Schaffner F, Popper H. Hepatocellular hyalin in cholestasis and cirrhosis: its diagnostic significance. Gastroenterology. 1973;64:89–98. [PubMed] [Google Scholar]
- 90.Yokoo H, Craig RM, Harwood TR, Cochrane C. Griseofulvin-induced cholestasis in Swiss albino mice. Gastroenterology. 1979;77:1082–7. [PubMed] [Google Scholar]
- 91.Meerman L, Koopen NR, Bloks V, Van Goor H, Havinga R, Wolthers BG, Kramer W, Stengelin S, Muller M, Kuipers F, Jansen PL. Biliary fibrosis associated with altered bile composition in a mouse model of erythropoietic protoporphyria. Gastroenterology. 1999;117:696–705. doi: 10.1016/s0016-5085(99)70464-6. [DOI] [PubMed] [Google Scholar]
- 92.Nguyen DM, El-Serag HB. The epidemiology of obesity. Gastroenterology clinics of North America. 2010;39:1–7. doi: 10.1016/j.gtc.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Caldwell SH, Crespo DM, Kang HS, Al-Osaimi AM. Obesity and hepatocellular carcinoma. Gastroenterology. 2004;127:S97–103. doi: 10.1053/j.gastro.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 94.Rodriguez A, Duran A, Selloum M, Champy MF, Diez-Guerra FJ, Flores JM, Serrano M, Auwerx J, Diaz-Meco MT, Moscat J. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 2006;3:211–22. doi: 10.1016/j.cmet.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 95.Bost F, Aouadi M, Caron L, Even P, Belmonte N, Prot M, Dani C, Hofman P, Pages G, Pouyssegur J, Le Marchand-Brustel Y, Binetruy B. The extracellular signal-regulated kinase isoform ERK1 is specifically required for in vitro and in vivo adipogenesis. Diabetes. 2005;54:402–11. doi: 10.2337/diabetes.54.2.402. [DOI] [PubMed] [Google Scholar]
- 96.Bost F, Aouadi M, Caron L, Binetruy B. The role of MAPKs in adipocyte differentiation and obesity. Biochimie. 2005;87:51–6. doi: 10.1016/j.biochi.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 97.Lee SJ, Pfluger PT, Kim JY, Nogueiras R, Duran A, Pages G, Pouyssegur J, Tschop MH, Diaz-Meco MT, Moscat J. A functional role for the p62-ERK1 axis in the control of energy homeostasis and adipogenesis. EMBO Rep. 2010;11:226–32. doi: 10.1038/embor.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Muller TD, Lee SJ, Jastroch M, Kabra D, Stemmer K, Aichler M, Abplanalp B, Ananthakrishnan G, Bhardwaj N, Collins S, Divanovic S, Endele M, Finan B, Gao Y, Habegger KM, Hembree J, Heppner KM, Hofmann S, Holland J, Kuchler D, Kutschke M, Krishna R, Lehti M, Oelkrug R, Ottaway N, Perez-Tilve D, Raver C, Walch AK, Schriever SC, Speakman J, Tseng YH, Diaz-Meco M, Pfluger PT, Moscat J, Tschop MH. p62 Links beta-adrenergic input to mitochondrial function and thermogenesis. The Journal of clinical investigation. 2012 doi: 10.1172/JCI64209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–6. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 100.Solinas G, Naugler W, Galimi F, Lee MS, Karin M. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16454–9. doi: 10.1073/pnas.0607626103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Seki E, Brenner DA, Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307–20. doi: 10.1053/j.gastro.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Singh R, Wang Y, Xiang Y, Tanaka KE, Gaarde WA, Czaja MJ. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology. 2009;49:87–96. doi: 10.1002/hep.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. The American journal of pathology. 2007;171:513–24. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Molecular cell. 2008;30:678–88. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li DD, Wang LL, Deng R, Tang J, Shen Y, Guo JF, Wang Y, Xia LP, Feng GK, Liu QQ, Huang WL, Zeng YX, Zhu XF. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene. 2009;28:886–98. doi: 10.1038/onc.2008.441. [DOI] [PubMed] [Google Scholar]
- 106.Geetha T, Zheng C, Vishwaprakash N, Broderick TL, Babu JR. Sequestosome 1/p62, a scaffolding protein, is a newly identified partner of IRS-1 protein. J Biol Chem. 2012;287:29672–8. doi: 10.1074/jbc.M111.322404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M, Lee MS, Tanaka K, Komatsu M. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275–84. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Thompson HG, Harris JW, Wold BJ, Lin F, Brody JP. p62 overexpression in breast tumors and regulation by prostate-derived Ets factor in breast cancer cells. Oncogene. 2003;22:2322–33. doi: 10.1038/sj.onc.1206325. [DOI] [PubMed] [Google Scholar]
- 109.Inoue D, Suzuki T, Mitsuishi Y, Miki Y, Suzuki S, Sugawara S, Watanabe M, Sakurada A, Endo C, Uruno A, Sasano H, Nakagawa T, Satoh K, Tanaka N, Kubo H, Motohashi H, Yamamoto M. Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci. 2012;103:760–6. doi: 10.1111/j.1349-7006.2012.02216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Myung Park J, Huang S, Wu TT, Foster NR, Sinicrope FA. Prognostic impact of Beclin 1, p62/sequestosome 1 and LC3 protein expression in colon carcinomas from patients receiving 5-fluorouracil as adjuvant chemotherapy. Cancer Biol Ther. 2013;14:100–7. doi: 10.4161/cbt.22954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Moscat J, Diaz-Meco MT. p62: a versatile multitasker takes on cancer. Trends in biochemical sciences. 2012;37:230–6. doi: 10.1016/j.tibs.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nakamura K, Kimple AJ, Siderovski DP, Johnson GL. PB1 domain interaction of p62/sequestosome 1 and MEKK3 regulates NF-kappaB activation. J Biol Chem. 2010;285:2077–89. doi: 10.1074/jbc.M109.065102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jin W, Chang M, Paul EM, Babu G, Lee AJ, Reiley W, Wright A, Zhang M, You J, Sun SC. Deubiquitinating enzyme CYLD negatively regulates RANK signaling and osteoclastogenesis in mice. J Clin Invest. 2008;118:1858–66. doi: 10.1172/JCI34257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.He G, Karin M. NF-kappaB and STAT3 - key players in liver inflammation and cancer. Cell Res. 2011;21:159–68. doi: 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 116.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–90. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 117.Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, Roskams T, Trautwein C, Pasparakis M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–32. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 118.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–75. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, Wong PK, Zhang DD. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29:1235–43. doi: 10.1093/carcin/bgn095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kwak MK, Itoh K, Yamamoto M, Sutter TR, Kensler TW. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dimethiole-3-thione. Mol Med. 2001;7:135–45. [PMC free article] [PubMed] [Google Scholar]
- 121.Du Y, Villeneuve NF, Wang XJ, Sun Z, Chen W, Li J, Lou H, Wong PK, Zhang DD. Oridonin confers protection against arsenic-induced toxicity through activation of the Nrf2-mediated defensive response. Environ Health Perspect. 2008;116:1154–61. doi: 10.1289/ehp.11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Taguchi K, Fujikawa N, Komatsu M, Ishii T, Unno M, Akaike T, Motohashi H, Yamamoto M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc Natl Acad Sci U S A. 2012;109:13561–6. doi: 10.1073/pnas.1121572109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, Ashkenazi A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137:721–35. doi: 10.1016/j.cell.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 124.Pan JA, Ullman E, Dou Z, Zong WX. Inhibition of protein degradation induces apoptosis through a microtubule-associated protein 1 light chain 3-mediated activation of caspase-8 at intracellular membranes. Mol Cell Biol. 2011;31:3158–70. doi: 10.1128/MCB.05460-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ullman E, Pan JA, Zong WX. Squamous cell carcinoma antigen 1 promotes caspase-8-mediated apoptosis in response to endoplasmic reticulum stress while inhibiting necrosis induced by lysosomal injury. Mol Cell Biol. 2011;31:2902–19. doi: 10.1128/MCB.05452-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, Sharma AK, Amin S, Hu CD, Zhang J, Kester M, Wang HG. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem. 2012;287:12455–68. doi: 10.1074/jbc.M111.309104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Riley BE, Kaiser SE, Shaler TA, Ng AC, Hara T, Hipp MS, Lage K, Xavier RJ, Ryu KY, Taguchi K, Yamamoto M, Tanaka K, Mizushima N, Komatsu M, Kopito RR. Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J Cell Biol. 2010;191:537–52. doi: 10.1083/jcb.201005012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN, Lee HE, Kang D, Rhee SG. Sestrins Activate Nrf2 by Promoting p62-Dependent Autophagic Degradation of Keap1 and Prevent Oxidative Liver Damage. Cell Metab. 2013;17:73–84. doi: 10.1016/j.cmet.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 129.Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci. 2006;89:31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- 130.Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55:222–32. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. Dual roles of Nrf2 in cancer. Pharmacol Res. 2008;58:262–70. doi: 10.1016/j.phrs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, Kensler TW. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98:3410–5. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Iida K, Itoh K, Kumagai Y, Oyasu R, Hattori K, Kawai K, Shimazui T, Akaza H, Yamamoto M. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 2004;64:6424–31. doi: 10.1158/0008-5472.CAN-04-1906. [DOI] [PubMed] [Google Scholar]
- 134.Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–88. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 135.Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16:123–40. doi: 10.1111/j.1365-2443.2010.01473.x. [DOI] [PubMed] [Google Scholar]
- 136.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–12. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 138.Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem. 1995;270:2320–6. doi: 10.1074/jbc.270.5.2320. [DOI] [PubMed] [Google Scholar]
- 139.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–9. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 140.Gurusamy N, Lekli I, Mukherjee S, Ray D, Ahsan MK, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by resveratrol: a novel mechanism via autophagy involving the mTORC2 pathway. Cardiovasc Res. 2010;86:103–12. doi: 10.1093/cvr/cvp384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–71. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 142.Efeyan A, Zoncu R, Sabatini DM. Amino acids and mTORC1: from lysosomes to disease. Trends in molecular medicine. 2012;18:524–33. doi: 10.1016/j.molmed.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol. 2009;19:R1046–52. doi: 10.1016/j.cub.2009.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ricoult SJ, Manning BD. The multifaceted role of mTORC1 in the control of lipid metabolism. EMBO Rep. 2013 doi: 10.1038/embor.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, Hailey DW, Oorschot V, Klumperman J, Baehrecke EH, Lenardo MJ. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–6. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Molecular cell. 2011;44:134–46. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Menon S, Yecies JL, Zhang HH, Howell JJ, Nicholatos J, Harputlugil E, Bronson RT, Kwiatkowski DJ, Manning BD. Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci Signal. 2012;5:ra24. doi: 10.1126/scisignal.2002739. [DOI] [PMC free article] [PubMed] [Google Scholar]