

Abstract
Autophagy is a highly conserved intracellular catabolic pathway that degrades cellular long-lived proteins and organelles. Autophagy is normally activated in response to nutrient deprivation and other stresses as a cell survival mechanism. Accumulating evidence indicates that autophagy plays a critical role in liver pathophysiology, in addition to maintain hepatic energy and nutrient balance. Alcohol consumption causes hepatic metabolic changes, oxidative stress, accumulation of lipid droplets and damaged mitochondria, all of these can be regulated by autophagy. This review summarizes the recent findings about the role and mechanisms of autophagy in alcoholic liver disease, and the possible intervention for treating alcoholic liver disease by modulating autophagy.
Keywords: autophagy, mitophagy, lipophagy, alcoholic liver disease
Introduction
Autophagy is a genetically programmed, evolutionarily conserved catabolic process that degrades cellular proteins and damaged and/or excess organelles, as a cell survival mechanism in response to stress. Almost 40 years ago, Drs. de Duve and Wattiaux first described autophagy based on the morphological changes observed as the cytoplasm is sequestered into closed, membrane-delimited vacuoles 1. The “autophagy” described by Dr. de Duve is most likely macroautophagy which is the focus of this review. There are two other forms of autophagy called microautophagy and chaperone-mediated autophagy (CMA), which differs in how the cytoplasmic materials are delivered to the lysosome in mammalian cells. Microautophagy, which is poorly understood in mammalian cells, results in the direct uptake of cytoplasm by invagination of the sequestering lysosomal membrane. CMA differs from the other two autophagy processes in that only a particular group of cytosolic molecules are degraded in the lysosomes. These proteins possess a KFERQ motif that is recognized by a molecular chaperone complex. This chaperone complex includes a heat-shock protein of 70 kDa (hsp70) and its co-chaperones are present in the cytosol and on the lysosomal membrane where the complex binds to a CMA receptor, the lysosome-associated membrane protein type-2A (LAMP-2A) 2, 3. Among the three different modes of autophagy, macroautophagy (hereafter referred to as autophagy) is thought to play a major role in intracellular degradation.
Autophagy is activated in response to adverse environments, such as the deprivation of nutrients or growth factors, as a survival mechanism 4–7. It is a highly conserved intracellular degradation pathway by which bulk cytoplasm and superfluous or damaged organelles are enveloped by double membrane structures termed autophagosomes 4–7. The contents of the autophagosomes are degraded after fusion with lysosomes, which are then called autolysosomes. However, autophagy can also occur under basal conditions, which is called basal autophagy. In addition to its role in maintaining cellular homeostasis, autophagy also plays a role in development 8, defending against microbial infections 9, and clearance of misfolded proteins. Therefore, dysfunction of autophagy can lead to the pathogenesis of numerous human diseases including cancer, neurodegenerative diseases, diabetes, infectious diseases and muscle atrophy 10, 11.
Alcohol consumption and abuse are major causes of liver disease which is a major health problem in the United States. Alcohol binge drinking induces dramatic metabolic changes, mitochondrial damage, disruption of lipid homeostasis, oxidative stress and cell death in hepatocytes. All of these can be regulated by autophagy. In this review, we will summarize the emerging role of autophagy in alcoholic liver disease.
Autophagy machinery
So far, more than 30 different autophagy related genes (Atg) have been identified in yeast and most of them have mammalian homologues that participate in autophagy or an autophagy-related process 12. Several multi-molecular complexes have been found to contribute to autophagosome formation including: (1) ULK1 protein-kinase complex, (2) VPS34-Beclin1 class III PI3-kinase complex, (3) Atg9-Atg2-Atg18 complex, and (4) the Atg5-Atg12-Atg16 and Atg8/LC3 conjugation systems (Figure 1).
Figure 1. Autophagy machinery.

Autophagy involves the formation of double membrane autophagosomes which fuse with lysosomes to form autolysosomes for the degradation of intracellular proteins and organelles. At least four important functional groups of Atg proteins are required for autophagy: (1) ULK1 protein-kinase complex and (2) VPS34-Beclin 1 class III PI3-kinase complex regulate autophagy initiation, (3) Atg9-Atg2-Atg18 complex regulates expansion of PAS by carrying lipids, and (4) the Atg5-Atg12-Atg16 and LC3 conjugation systems regulate the elongation of autophagosome membranes. Phosphatidylethanolamine (PE)-conjugated LC3 (called LC3-II) remains on the isolation membranes and autophagosome membranes, whereas the Atg12-Atg5-Atg16 complex transiently associates with the isolation membranes and dissociates from the autophagosome membranes. Once autophagosomes fuse with lysosomes to form autolysosomes, the inner membrane LC3-II is degraded by lysosomal enzymes whereas the outer membrane LC3-II is de-conjugated and recycled by Atg4. Pharmacological autophagy inhibitors such as 3-methyladenine (3-MA) and chloroquine are also highlighted. For more details, see the text.
The ULK complex in mammalian cells is composed of ULK1 (which is a yeast Atg1 homologue), FIP200 (a yeast Atg17-like molecule), Atg13 and Atg101 13–15. This complex is mainly localized in the cytosol and associates with the autophagy isolation membrane upon autophagy induction to regulate the early stage autophagosome formation. This complex also works downstream of mTOR and serves as the cellular sensor for nutrient status to initiate autophagy by recruiting downstream autophagy proteins to the autophagosomes. During starvation or rapamycin treatment, mTOR is suppressed which leads to the dephosphorylation of ULK1. ULK1 is a serine/threonine protein kinase and dephosphorylated ULK1 is actually enzymatically active leading to phosphorylation of Atg13 and FIP200 15. The kinase activity of ULK1 is thought to be important for the recruitment of other downstream Atg proteins such as Atg16L and the subsequent autophagosome formation 16.
Atg6 and its mammalian homologue, Beclin 1, is important for the initiation and regulation of autophagy 17. Beclin 1/Atg6 forms a complex with VPS34, VPS15 and Atg14. VPS34 is a class III PI-3-kinase that acts as an essential regulator of autophagy by producing phosphatidylinositol-3-phosphate (PI-3-P). The VPS34 activity is regulated by the Beclin 1/Atg6 complex. Bcl-2 and Bcl-xL interact with Beclin1 to keep autophagy in check by dissociating the interaction of Beclin 1 and VPS34. Beclin 1 also binds to several other proteins that induce autophagy including ambra-1 18 and UVRAG 19. Bif-1/endophilin B1 interacts with Beclin 1 via UVRAG acting as a regulator on the Atg6-VPS34 complex 20. Rubicon (Run domain protein as Beclin 1 interacting and cysteine-rich containing) interacts with VPS34 and acts as a negative regulator of autophagy 21, 22. 3-methyladenine (3MA), a widely used autophagy inhibitor, inhibits class III PI-3-kinase and in turn, inhibits autophagosome formation 23.
Atg9 is the sole transmembrane protein among the core autophagy machinery proteins that is conserved in all species 24. It has six proposed transmembrane domains with its carboxyl termini exposed in the cytosol 25. There are two functional orthologues of Atg9 in mammalian cells: Atg9L1 and Atg9L2. Atg9L1 is ubiquitously expressed whereas Atg9L2 is only expressed in the placenta and pituitary gland 26. Atg9 knockout mice die immediately after birth, similar to other autophagy machinery protein knockout mice such as Atg3, Atg5, Atg7 and Atg16 27. In yeast, Atg9 interacts with Atg11 and Atg17, and this interaction is required for regulating the cytoplasm-to-vacuole (Cvt) pathway and autophagy 28. Atg9 is localized on the phagophore assembly site (PAS) and interacts with Atg18, a PI3-P binding protein, and a peripheral membrane protein, Atg2. Atg9 is proposed to cycle between the trans-Golgi network, late endosomes and the PAS to bring in the additional membrane sources necessary for the growth of the autophagosomal membranes, depending on the cellular nutrient status 24, 25.
The core autophagy machinery is then built around two ubiquitin-like conjugation systems to regulate the elongation of the pre-autophagosome structures 29. In one system, the ubiquitin-like protein, Atg12, is first activated by Atg7, an ubiquitin-activating enzyme (E1)-like protein. The activated Atg12 is then transferred by Atg10, an ubiquitin carrier (E2)-like protein, to Atg5 where a covalent bond is formed. The Atg12-Atg5 complex interacts with Atg16 to form a multimer complex, which is localized to the early autophagosome membrane. The Atg12-Atg5-Atg16 complex appears to provide the necessary platform for autophagy activation. In the other system, a ubiquitin-like protein, Atg8, or one of its mammalian orthologs, the microtubule-associated protein 1 light chain 3 (LC3), is first cleaved by Atg4 to expose the conserved Gly120 at its C-terminus. Atg8/LC3 is then conjugated to phosphatidylethanolamine (PE), also via Atg7, and Atg3, another ubiquitin carrier (E2)-like protein 29, 30. The unconjugated form of Atg8/LC3 (called LC3-I) is in the cytosol, whereas the conjugated form (called LC3-II) targets the autophagosomal membrane following the Atg12-Atg5-Atg16 complex. This association of Atg8/LC3-PE to the autophagosomes is considered important for the membrane extension of the autophagosome and the eventual closure of the membrane to form a complete vesicle. Atg8/LC3 is thus widely used as a marker for monitoring the autophagy process.
In mammalian cells, the newly formed autophagsomes are randomly distributed in the cytoplasm 31. During their maturation, autophagosomes move along the microtubules towards the microtubule organizing center, where the lysosomes are enriched. The autophagosomes then fuse with lysosomes to form autolysomes possibly through the small GTPase Rab7 and/or two lysosomal membrane proteins, Lamp1/2A 32–34. Chemicals that disrupt the microtubule structures such as vinblastine can inhibit autophagy 35. In addition to disruption of the fusion machinery, chemicals that increase lysosomal pH such as chloroquine and bafilomycin A 1 or lysosomal protease inhibitors such as leupeptin, pepstatin A and E64D, all are potent autophagy inhibitors 7, 31.
Signaling pathways regulating mammalian autophagy
Class I PI3K-Akt-mTOR
Several signaling pathways appear to be involved in the regulation of autophagy in mammalian cells. Among them, the inhibition of the mammalian target of rapamycin mTOR) has been identified as a key signaling pathway for the regulation of autophagy induction 36, 37. Using isolated rat hepatocytes, it was first discovered by Meijer et al that rapamycin, a mTOR inhibitor, significantly activates autophagy 38. In addition to regulating autophagy, it should be noted that the serine/threonine kinase mTOR also controls many aspects of cellular physiology, including transcription, translation, cell size and cytoskeletal organization. mTOR exists in two heteromeric complexes, mTOR1 and mTOR2. However, mTOR1, which is rapamycin sensitive, plays a major role in the regulation of autophagy and cell growth. The activity of mTOR1 is regulated by the integration of various signals, including growth factors, insulin, nutrients, energy availability and cellular stressors such as hypoxia, osmotic stress, reactive oxygen species and viral infection. In response to growth factors, class-I phosphatidylinosital-3 kinase (PI3K) catalyzes the production of phosphatidylinosital-3 phosphate leading to the activation of PI3K-protein kinase B (PKB, also known as Akt). Akt further activates mTOR through the tuberous sclerosis complex (TSC). There are two TSC genes (TSC1 and TSC2), which form a functional complex to negatively regulate mTOR 39. TSC1 functions to stabilize the complex. TSC2 exerts GTPase activating protein (GAP) activity towards downstream effectors such as Rheb protein, a small GTPase. TSC2 can accelerate the intrinsic rate of GTP hydrolysis of Rheb, converting Rheb from the GTP-bound (active) to the GDP-bound (inactive) form, which in turn inhibits mTOR 40. Akt phosphorylates TSC2 to disrupt its complex with TSC1 and thus activates mTOR. In contrast to mTOR1, mTOR2 was originally thought to mainly regulate cellular polarization and cytoskeletal reorganization 41. However, recent evidence suggests that mTOR2 may also negatively regulate autophagy because the full activation of Akt requires mTOR2. In starved mouse skeletal muscle, inhibition of mTOR2 leads to autophagy induction which is mediated by forkhead box O (FoxO3) transcription factor, a downstream target negatively regulated by Akt 42. Although mTOR plays a central role in regulating autophagy in mammalian cells, autophagy can also be activated in mTOR-independent pathways. Drugs such as lithium, carbamazepine, and valproic acid reduce intracellular inositol and inositol 1,4,5-trisphosphate (IP3) levels, and induce autophagy independent of mTOR activity 43. In addition, calpain inhibitors and L-type Ca2+ channel agonists as well as certain chemicals decrease intracellular cAMP, all of them have been shown to induce autophagy independent of mTOR 44.
AMPK
The role of another upstream mTOR regulator, the AMP-activated protein kinase (AMPK), is still controversial in autophagy activation. A high AMP/ATP ratio reflects low-energy status, which can be monitored by AMPK. Under such energy-stress conditions, AMPK is activated by direct AMP binding. The active form of AMPK then phosphorylates TSC2 and stimulates TSC2 GAP activity toward Rheb. Subsequently, mTOR activity is down-regulated, leading to the dephosphorylation of p70 ribosomal S6 kinase (p70S6K) and eukaryote initiation factor 4E binding protein 1 (4EBP1), two downstream substrates of mTOR, resulting in the inhibition of protein translation and activation of autophagy 45. Indeed, inhibition of AMPK activity by transfection with a dominant negative form of AMPK almost completely inhibited autophagy in hepatocytes, HT-29 cells and Hela cells 46. In contrast, Seglen et al reported that adenosine analogue adenosine, 5-amino-4-imidazole carboxamide riboside (AICAR), an AMPK activator, suppresses autophagy in hepatocytes 47. Furthermore, compound C, an AMPK inhibitor, induces autophagy in cancer cells by AMPK-independent down regulation of Akt/mTOR pathway 48. Further studies are needed to clarify these controversial findings. One possibility is that the pharmacological inhibitors or activators of AMPK may have off-target effects. To further clarify the role of AMPK in autophagy, it will be more helpful using genetic models in the future.
Bcl-2 family proteins
In addition to regulating cell death, Bcl-2 family proteins play a dual role in autophagy regulation. Anti-apoptotic proteins, such as Bcl-2, Bcl-xL, and Mcl-1, can suppress autophagy by directly binding to Beclin 1 49, 50. The binding of Bcl-2 to Beclin 1 dissociates the Beclin 1-VPS34 complex which decreases VPS34 kinase activity and thereby inhibits autophagy. Interestingly, it should be noted that only the endoplasmic reticulum-Bcl-2 (ER-Bcl-2) protein suppresses autophagy 49, 51. Accumulating evidence supports that the ER membrane could be the membrane source for the initiation of autophagosome formation. A small cradle structure which is PI-3-P-enriched subdomains of the ER (omegasomes) has been shown to be related to the initiation of autophagosome formation 52, 53. Moreover, NAF-1 (nutrient-deprivation autophagy factor-1), an ER membrane protein, has been reported to interact with Bcl-2 on the ER to functionally antagonize Beclin 1-dependent autophagy 51. In addition to binding with Beclin 1, Bcl-2 and Bcl-xL may also negatively regulate the autophagic response by controlling the ER calcium homeostasis 54, 55. In contrast to anti-apoptotic Bcl-2 family proteins, pro-apoptotic BH3-only proteins such as Bnip3, Bnip3L/Nix, Bad, Bik, Noxa, Puma and Bim can induce autophagy through their binding to Bcl-2 and subsequent displacement of the inhibitory Bcl-2 from the Beclin 1 complex. We recently found that Nix also promotes autophagy by inducing mitochondria depolarization and reactive oxygen species production 56. Moreover, Nix also plays an important role in the specific mitochondrial autophagy (mitophagy) during the maturation of erythroid cells 57, 58. Unlike the BH3-only proteins, Bax and Bak contain three BH domains (BH1-BH3). Bax is normally located in the cytosol of healthy cells whereas Bak resides in the outer membrane of mitochondria. Either of the two pro-apoptotic multi-domain proteins is required for the mitochondria membrane permeabilization leading to the release of mitochondrial apoptotic factors, and in turn resulting in the activation of caspases to trigger apoptosis 59. Because Bax has a BH3 domain and also interacts with Bcl-2, one would imagine that Bax may also activate autophagy by competitively binding to and disrupting Bcl-2-Beclin-1 complex like other BH3-domain Bcl-2 family proteins. However, autophagy is induced by etoposide in Bax/Bak double knockout mouse embryonic fibroblasts and by ER stress inducers in Bax-deficient HCT116 cells 60, 61, suggesting that Bax and Bak could be dispensable for autophagy induction. More interestingly, a recent study found that Bax may actually inhibit autophagy because overexpression and activation of Bax leads to caspase-mediated Beclin-1 cleavage resulting in the inhibition of Beclin-1 complex 62. In general, it seems that anti-apoptotic Bcl-2 family proteins inhibit autophagy whereas pro-apoptotic BH3-only Bcl-2 family proteins promote autophagy.
ER stress
ER is a complicated intracellular membranous structure with the major function being the synthesis of macromolecules, proper folding of newly synthesized proteins and the post-translational modifications of proteins 63, 64. Misfolded proteins due to either genetic mutations or ER dysfunction could be therefore accumulated within the ER. Normally, the degradation of these abnormal proteins is mainly through the ER-associated degradation (ERAD) pathway mediated by the ubiquitin proteasome system 63, 64. ER stress can be caused by such agents as A23187, a calcium ionophore, thapsigargin (TG), an inhibitor of the sarcoplasmic/endoplasmic Ca2+-ATPase resulting in ER Ca2+ depletion, tunicamycin which blocks N-linked protein glycosylation or brefeldin A which inhibits the transportation of ER vesicles to the Golgi apparatus 65. The increase of misfolded/unfolded proteins can also lead to ER stress, which initiates cellular protective mechanisms against the stress. The major protective and compensatory mechanism for ER function during ER stress is the so-called unfolded protein response (UPR) 63, 64. Mammalian cells have developed a complicated UPR, involving at least three different pathways, the ATF6 pathway, the PERK-eif-2α pathway and the IRE1-XBP1 pathway 63–65. All are initiated by the reduction of an ER-lumen chaperone-like protein, Bip/GRP78, which is consumed by the unfolded/misfolded proteins. We recently found that inhibition of proteasome activity, another intracellular degradation pathway, can also trigger ER stress and subsequent autophagy 66. ER stress induces autophagy through the activation of IRE1 and c-Jun N-terminal protein kinase (JNK) 66–68. JNK may further phosphorylate Bcl-2 to dissociate its binding to Beclin 1 and in turn activates autophagy 69. ER stress-induced autophagy can help relieve ER stress and protect ER stress-induced cell death by removing misfolded proteins 61, 66.
Reactive oxygen species and autophagy
Accumulating evidence now supports that reactive oxygen species (ROS) induce autophagy. In mammalian cells, one of the major sources of ROS is the mitochondrial electron transport chain, where about 1–3% of the oxygen used by mitochondria is converted to ROS 70. Indeed, mitochondrial electron transport chain inhibitors such as rotenone (complex I inhibitor) or thenoyl trifluoroacetone (TTFA, complex II inhibitor) induces autophagy 71, 72. We also found that carbonyl cyanide m-chlorophenylhydrazone (CCCP), an uncoupler of mitochondria, also induces autophagy in various mammalian cell lines. More importantly, CCCP-induced autophagy can be almost completely suppressed by the anti-oxidant, N-acetylcysteine (NAC) 56. Starvation, a well-documented autophagy inducer, increases ROS level and treatment with antioxidants inhibits starvation-induced autophagy 73. It is still not clear how ROS activates autophagy in mammalian cells. One of the possible mechanisms is that ROS may induce autophagy by modulating the action of Atg4B on LC3. Atg4B is a cysteine protease which not only cleaves the full length LC3 to generate the LC3-I but also delipidates LC3-II from the autophagosomal outer membrane to allow the recycling of LC3. It has been reported that ROS modulates Atg4B activity by directly oxidizing a specific cysteine residue, Cys81 on Atg4B 73. It has been suggested that the inactivation of Atg4B by ROS allows LC3 to lipidate and thereby initiate autophagy. In addition, ROS may increase the protein level of Beclin 1 to promote autophagy 74, although it is not known whether ROS promotes the gene expression or the post-translational modification of Beclin 1.
Ethanol consumption and the activation of autophagy
As one of the most active organs, the liver plays a central role in regulating the overall organism energy balance by controlling carbohydrate and lipid metabolism. The Liver functions as a major buffering system to maintain the homeostasis of macro- and micronutrients to allow other tissues to function normally under physiological stress 75. Liver-targeted autophagy deficiency (Atg7 knockout) results in accumulation of ubiquitin positive protein aggregates, damaged mitochondria, steatosis and liver injury 76. These findings support a pro-survival role of autophagy in maintaining protein, lipid and organelle quality control by eliminating damaged proteins and organelles as well as excessive lipid droplets in the liver during stress. In addition, accumulating evidence now indicates that autophagy is also involved in hepatocyte cell death, steatohepatitis, hepatitis virus infection, and hepatocellular carcinoma 77–79.
Alcoholic liver disease (ALD) is a major cause of death in the United States. Characteristics of ALD are fatty liver, inflammation, fibrosis, and cirrhosis. Alcohol abuse and alcoholism account for approximately 50% of all death induced by liver cirrhosis. Besides cirrhosis, ALD can progress to hepatocellular carcinoma. Although many treatments are being used to ameliorate alcoholic hepatitis, few are successful. It is estimated that 28% of the adult population has a high-risk drinking pattern, such as binge drinking. However, less attention has been paid to acute alcoholic liver injury, especially by binge alcohol drinking, even though binge drinking is more common than chronic alcoholism, especially among young people 80. Alcohol binge drinking can result in mitochondrial damage, inhibition of insulin signaling, steatosis, apoptotic and necrotic cell death, all of which can be regulated by autophagy. Indeed, we recently found that ethanol treatment induces autophagy in primary cultured mouse hepatocytes and in hepatoma cells expressing alcohol dehydrogenase (ADH) and cytochrome P450 2E1 (Cyp2E1), as well as in acute alcohol binge-treated mouse liver 81. The induction of autophagy was demonstrated by increased number of GFP-LC3 positive autophagosomes and autophagic flux. As we discussed above, upon autophagy activation, LC3 is processed and conjugated with phosphatidyethanolamine (PE), and the PE-conjugated form of LC3 (LC3-II) translocates to the autophagosomal membrane. In non-treated hepatocytes, most GFP-LC3 signals display a diffuse pattern because unconjugated GFP-LC3 is found only in the cytosol (Figure 3a). In contrast, there is an increase in the number of GFP-LC3 puncta structures, which represent the membrane targeted PE-conjugated GFP-LC3 (Figure 3b). We further confirmed that there was an increased autophagosome number in ethanol-treated hepatocytes by electron microscopy (Figure 3d). Interestingly, we found that ethanol-induced autophagy seems to selectively target damaged mitochondria and accumulated lipid droplets but not long-lived proteins. Ethanol-induced GFP-LC3 positive autophagosomes specifically envelop mitochondria (Figure 4a) and lipid droplets (Figure 4b). The selective mitophagy (autophagy for mitochondria) and lipophagy (autophagy for lipid droplets) induced by acute ethanol treatment appears to play a role in attenuating ethanol-induced liver injury, because damaged mitochondria and steatosis are two well-known key changes in the liver after alcohol binge drinking 82, 83. Indeed, we found that suppression of autophagy with pharmacological inhibitors or small interference RNA for Atg7 increased mitochondria-mediated apoptosis, steatosis and liver injury. In contrast, induction of autophagy by rapamycin reduced ethanol-induced steatosis and liver injury associated with acute ethanol exposure 81. In accordance with our findings, several recent studies also found that ethanol induces autophagy in human hepatoma cells that express ethanol metabolism enzymes, and in chronic ethanol-fed mouse liver 84, 85. The finding that ethanol induces autophagy as a protective mechanism is important because it is known that only about 25 percent of excessive alcohol drinkers develop ALD. It is also generally known that ethanol-induced cell death in vivo is mild, resulting in a modest increase of blood alanine aminotransferase (ALT) in either acute or chronic ethanol-treated animals.
Figure 3. Ethanol induces autophagy in primary mouse hepatocytes.

Mouse hepatocytes were infected with Adenovirus-GFP-LC3 (10 viral particles per cell) for 16 hrs, and then either untreated (a) or treated with ethanol (80 mM) for 6 hrs (b) followed by confocal microscopy for GFP-LC3 or electron microscopy (c–d). N: nuclei, M: mitochondria. Arrow heads: autophagosomes; arrows: autolysosomes.
Figure 4. Ethanol induces mitophagy and lipophagy in hepatocytes.
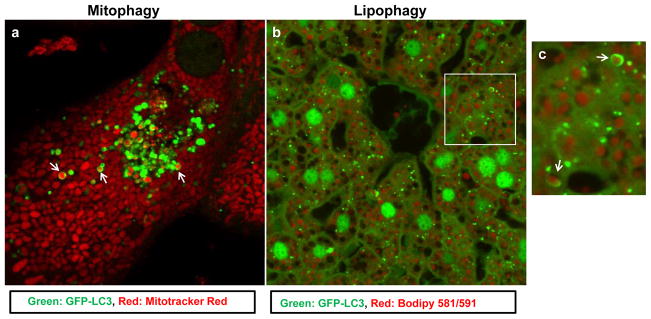
(a) Mouse hepatocytes were infected with Adenovirus-GFP-LC3 (10 viral particles per cell) for 16 hrs, and then loaded with Mitotracker Red (50 nM) for 15 min followed by ethanol (80 mM) treatment for another 6 hrs and confocal microscopy. Arrows: GFP-LC3 positive ring-like structures enveloped mitochondria (mitophagy). (b) GFP-LC3 transgenic mice were treated with ethanol (4.5 g/kg) through acute gavage for 16 hrs. Liver cryosections were prepared and stained with Bodipy 581/591-C11 for lipid droplets followed by confocal microscopy. Panel c was the enlarged photograph from boxed area in panel b. Arrows: GFP-LC3 positive ring-like structures enveloped lipid droplets (lipophagy).
How is it possible that alcoholism induces liver injury and even death due to alcoholic liver cirrhosis if alcohol-induced autophagy plays a protective role? Increasing evidence now support the notion that autophagy and apoptosis mutually regulate each other 86. Autophagy not only provides nutrients for the cell survival during starvation but also selectively removes damaged organelles including damaged mitochondria 87. Mitochondria are the central executioners for apoptosis and many times damaged mitochondria trigger cell death. Many apoptotic stimuli induce apoptosis but also induce autophagy at the same time, indicating that the cell’s fate is decided by the balance of autophagy versus the accumulation of damaged mitochondria. If damaged mitochondria are limited to a faction of mitochondria within the capacity of autophagic removal, cell death will be avoided. Nevertheless, cells may eventually die if the number of damaged mitochondria is too overwhelming to be removed by autophagy. Thus, autophagy may increase the threshold for the death stimuli. Accumulating evidence demonstrates that pharmacologic or genetic inhibition of autophagy greatly enhanced cell death 61, 66, 88. Our previous studies also clearly indicate that suppression of autophagy exacerbates alcohol-induced liver injury 81. Therefore, cell death may still occur even in the presence of autophagy induction if the death signals are too strong. Furthermore, cell death can be significantly increased in the absence of the protection of autophagy. This notion is particularly important, because many alcoholic patients have other pathophysiological conditions which may impair autophagy such as diabetes, hyperinsulinemia, obesity and hepatitis C virus (HCV), which increase the detrimental effects of alcohol toxicity 89–91.
Mechanisms of ethanol-induced autophagy
Several mechanisms have been identified to contribute to acute ethanol-induced autophagy including ROS production, ethanol metabolism and the suppression of mTOR 81. Ethanol-induced ROS production mainly derived from its metabolism and the induction of cytochrome P450 2E1 (Cyp2E1) as well as damaged mitochondria. We found that ethanol-induced ROS production is required for autophagy induction because antioxidants suppress ethanol-induced autophagy 81. As we discussed above, ROS can also activate autophagy by the inactivation of Atg4B. However, whether ethanol-induced ROS also inactivate Atg4B to promote autophagy in hepatocytes remains unknown. The metabolism of ethanol also seems play a critical role in ethanol-induced autophagy based on two observations. First, ethanol-induced autophagy is blocked by 4-methyl-pyrazole (4-MP), an inhibitor of ethanol oxidation by inhibiting alcohol dehydrogenase (ADH) and Cyp2E1. Second, ethanol only induces autophagy in HepG2 cells that are expressing ADH and Cy2E1 but not in normal HepG2 cells that lack the ability to metabolize ethanol 81. In addition to ROS and ethanol metabolism, suppression of Akt and mTOR could be another mechanism by which ethanol induces autophagy. Indeed, both acute and chronic ethanol treatment suppress Akt in vitro and in mouse liver, through the up-regulation of the PTEN phosphatase 92, 93. We also reported that ethanol induces suppression of mTOR in primary cultured mouse hepatocytes and in acute alcohol-treated mouse liver, which is mediated by ROS production 81 and decreased Akt phosphorylation (Ding et al., unpublished observation). Suppression of mTOR leads to the activation of the downstream ULK1 complex to trigger autophagy. It remains to be seen whether alcohol would affect the ULK1 complex. In addition to mTOR, ethanol-induced autophagy also requires the activation of Beclin 1/ VPS34 PI-3 kinase complex because 3MA, a PI-3 kinase inhibitor, suppresses acute ethanol-induced autophagy 81. Furthermore, ethanol has been shown to suppress proteasome activity, induce ER stress and activate JNK in hepatocytes, and all of these mechanisms have been shown to induce autophagy in non-hepatocyte models 66, 69, 94–96. Whether proteasome inhibition, ER stress and JNK activation play a role in ethanol-induced autophagy in hepatocytes needs to be further studied.
It has been speculated that ethanol may inhibit autophagy because chronic ethanol consumption reduces AMPK activity in the liver 97. However, as we discussed above, the role of AMPK in autophagy is still controversial, and inhibition of AMPK can also induce autophagy 98. In addition, AMPK induces autophagy mainly by inhibiting mTOR. Because mTOR is activated by Akt, the status of mTOR in ethanol-treated hepatocytes relies on the extent of impaired Akt and AMPK by ethanol. The finding that acute ethanol suppresses mTOR suggests that impaired Akt could play a more dominant role than impaired AMPK in ethanol-induced autophagy in hepatocytes 79. Therefore, it is possible that activation of AMPK, such as by treating the animals with AMPK agonists, may further enhance ethanol-induced autophagy by maximally inhibiting mTOR. It has been reported that several AMPK agonists such as AICAR and metformin significantly protect against ethanol-induced liver injury in animal models 99, 100. However, it remains to be determined whether these protective effects are also associated with the induction of autophagy in these models.
It should also be noted that although a variety of criteria for assessing autophagy indicate that acute ethanol conditions lead to autophagy induction in hepatocytes and in mouse liver, the evidence to support such an induction in the chronic alcohol consumption model is relatively scarce. Data from two abstracts presented in 2010 The Liver Meeting suggested that autophagy may be elevated in the mouse liver when they were fed with Lieber-DeCarli diet for 4 weeks 84, 85. However, it is not clear whether autophagic flux assays were conducted in these studies. Therefore, the relevance of autophagy in chronic alcohol consumption still needs to be determined. Finally, it would be interesting to see whether autophagy would also be altered in ALD patients. The Liver Center at the University of Kansas Medical center has collected more than 100 human liver specimens. We are currently planning to examine autophagy in some of the specimens from alcoholic patients.
Potential therapeutic approaches to treat ALD by modulating autophagy
It has been well recognized that ALD is a major cause of morbidity and mortality in the world. However, there are few other successful treatments for ALD in addition to abstinence from drinking. While it is difficult to develop an effective treatment for chronic alcohol exposure associated liver pathogenesis, it would be more applicable for developing treatments for hospitalized patients with acute alcohol intoxication associated liver injury.
One of the early features of ALD is fatty liver due to the accumulation of lipid droplets. Alcoholic fatty liver has been thought to be a benign condition; however, recent evidence suggests that it is a potentially pathological condition which can progress to steatohepatitis, fibrosis, cirrhosis, and even hepatocelluar carcinoma, in particular, if co-existing with other factors such as hepatitis virus infection, diabetes, and the use of cigarettes 101. Moreover, increased oxidative stress resulting from ethanol metabolism in the liver can damage mitochondria and induce cell death leading to the elevation of serum ALT levels 101. Autophagy regulates lipid and mitochondria homoeostasis, and therefore, it is a very attractive therapeutic approach for treating ALD. Indeed, rapamycin, a widely used immunosuppressor in clinical transplant patients 102, has been shown to improve alcohol-induced liver pathogenesis by inducing autophagy through its inhibition of mTOR. Using an acute ethanol binge mouse model, we recently found that rapamycin treatment significantly decreased the number of ethanol-induced lipid droplets and total hepatic triglyceride levels 81. This is most likely due to the enhanced autophagy in the mouse liver because rapamycin further increased the number of ethanol-induced autophagosomes. It is well-known that alcohol abuse can cause protein aggregates in the liver called Mallory-Denk bodies (MDB). These cytosolic inclusion bodies are enriched with cytokeratin 8 and 18, and in association with other proteins including ubiquitin and p62. Keratin 8-overexpressing transgenic mice are predisposed to MDB formation. Interestingly, treatment with rapamycin decreased the number of MDB in these mice significantly 103. In another classical protein aggregates-induced liver injury model, the α1-antitrypsin deficiency, carbamazepine (CBZ) decreased the hepatic load of α1-antitrypsin mutant proteins as well as hepatic fibrosis by inducing autophagy 104. In contrast to rapamycin, CBZ induces autophagy in the liver independent of mTOR, suggesting that various autophagy pathways could be targeted to treat liver diseases by inducing autophagy. Several high-throughput screenings for autohagy inducers have been conducted recently, which have identified several potential therapeutic compounds 105–107. It would be of great interest to determine the therapeutic effects of these newly identified agents on ALD through the modulating of autophagy.
Conclusions
Recent studies have indicated that alcohol may activate autophagy as a protective mechanism against alcoholic liver injury by selectively removing damaged mitochondria and hepatic lipid droplets. Alcohol-induced autophagy requires alcohol metabolism and production of ROS. In addition, alcohol may also induce autophagy by impairing the Akt-mTOR pathway. Alcohol-induced proteasome inhibition and ER stress, as well as AMPK- and mTOR-independent pathways, could also be involved in alcohol-induced autophagy in the liver. Modulating autophagy could provide novel therapeutic approaches for treating ALD (Figure 5).
Figure 5. Schematic diagram of the major pathways and roles of alcohol-induced autophagy in hepatocytes.

Ethanol-induced autophagy could be mediated by multiple mechanisms. (1) Ethanol-induced autophagy requires ethanol metabolism and ROS production. ROS may activate autophagy by further suppressing mTOR, Atg4B and proteasome activity. Proteasome inhibition may further trigger ER stress and JNK activation to activate autophagy. (2) Ethanol may also suppress Akt and mTOR through the upregulation of PTEN. (3) Ethanol-induced impaired AMPK and Akt may counteract to each other on mTOR, and impaired Akt plays a dominant role toward the inhibition of mTOR. (4) Other AMPK-and mTOR-independent pathways remain to be determined in alcohol-induced autophagy. (5) Ethanol-induced autophagy selectively removes damaged mitochondria (mitophagy) and lipid droplets (lipophagy) to protect against ethanol-induced steatosis and liver injury.
Figure 2. Molecular signaling pathways regulating autophagy.

Autophagy in mammalian cells can be activated by various stimuli. Although the suppression of the mammalian target of rapamycin (mTOR) is a major signaling pathway regulating autophagy, autophagy can also be activated independent of mTOR by various stimuli. Two major pathways that regulate mTOR in mammalian cells are the PI3K-Akt and AMPK pathways. The PI3K-Akt pathway is triggered by the binding of insulin growth factors (IGF or other growth factors) to its receptor, thereby activating PI3K. Activated PI3K converts PIP2 to PIP3 to activate Akt. Akt then phosphorylates and inactivates the TSC1/TSC2 complex resulting in activation of Rheb and mTOR. AMPK is normally activated by its upstream kinase LKB-1 or by an increased intracellular ratio of AMP/ATP. AMPK can be suppressed by chemical inhibitors such as compound C. Active AMPK then directly phosphorylates TSC2 and inhibits mTOR to activate autophagy. mTOR also phosphorylates two downstream targets 4E-BP1 and p70S6K that control protein translation.
Acknowledgments
We thank Ms Abigail Bockus for critical reading of this manuscript. This work was supported by NIH R21 AA017421& P20 RR021940& P20 RR016475 from the INBRE program of the National Center for Research Resources (W.X.D).
Abbreviations
- AMPK
AMP-activated protein kinase
- ALD
Alcoholic liver disease
- CMA
Chaperone-mediated autophagy
- 4E-BP1
Eukaryote initiation factor 4E binding protein 1
- ER
Endoplastic reticulum
- ERAD
ER-associated degradation
- LDs
Lipid droplets
- LAMP2A
Lysosome-associated membrane protein type-2A
- LC3
Microtubule-associated protein 1 light chain 3
- JNK
c-Jun N-terminal protein kinase
- MDB
Mallory-Denk bodies
- mTOR
Mammalian target of rapamycin
- PAS
Phagophore assembly site
- p70-S6K
p70 ribosomal S6 kinase 1 (S6K)
- PI-3-P
Phosphatidylinositol-3-phosphate
- ROS
Reactive oxygen species
- UPR
Unfolded protein response
Footnotes
References
- 1.De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–92. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 2.Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990;15:305–9. doi: 10.1016/0968-0004(90)90019-8. [DOI] [PubMed] [Google Scholar]
- 3.Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–3. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- 4.Kamada Y, Sekito T, Ohsumi Y. Autophagy in yeast: a TOR-mediated response to nutrient starvation. Curr Top Microbiol Immunol. 2004;279:73–84. doi: 10.1007/978-3-642-18930-2_5. [DOI] [PubMed] [Google Scholar]
- 5.Poole B, Ohkuma S. Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J Cell Biol. 1981;90:665–9. doi: 10.1083/jcb.90.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 2004;7:167–78. doi: 10.1016/j.devcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–36. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–82. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirkegaard K, Taylor MP, Jackson WT. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat Rev Microbiol. 2004;2:301–14. doi: 10.1038/nrmicro865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–5. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edinger AL, Thompson CB. Defective autophagy leads to cancer. Cancer Cell. 2003;4:422–4. doi: 10.1016/s1535-6108(03)00306-4. [DOI] [PubMed] [Google Scholar]
- 12.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–21. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13. FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 22:132–9. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 15.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, Guan JL, Oshiro N, Mizushima N. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan EY, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol. 2009;29:157–71. doi: 10.1128/MCB.01082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–72. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–5. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 19.Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–99. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ, Pledger WJ, Wang HG. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–51. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11:468–76. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Q, Zhang J, Fan W, Wong KN, Ding X, Chen S, Zhong Q. The run domain of rubicon is important for HVPS34 binding, lipid kinase inhibition and autophagy suprression. J Biol Chem. 2010 doi: 10.1074/jbc.M110.126425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889–92. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Webber JL, Tooze SA. New insights into the function of Atg9. FEBS Lett. 2010;584:1319–26. doi: 10.1016/j.febslet.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 25.Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, Hailey DW, Lippincott-Schwartz J, Tooze SA. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006;119:3888–900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 26.Yamada T, Carson AR, Caniggia I, Umebayashi K, Yoshimori T, Nakabayashi K, Scherer SW. Endothelial nitric-oxide synthase antisense (NOS3AS) gene encodes an autophagy-related protein (APG9-like2) highly expressed in trophoblast. J Biol Chem. 2005;280:18283–90. doi: 10.1074/jbc.M413957200. [DOI] [PubMed] [Google Scholar]
- 27.Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, Yamamoto N, Kawai T, Ishii K, Takeuchi O, Yoshimori T, Akira S. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A. 2009;106:20842–6. doi: 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki K, Ohsumi Y. Current knowledge of the pre-autophagosomal structure (PAS) FEBS Lett. 2010;584:1280–6. doi: 10.1016/j.febslet.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–6. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- 30.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- 32.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–60. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 33.Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117:4837–48. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- 34.Saftig P, Beertsen W, Eskelinen EL. LAMP-2: a control step for phagosome and autophagosome maturation. Autophagy. 2008;4:510–2. doi: 10.4161/auto.5724. [DOI] [PubMed] [Google Scholar]
- 35.Kovacs AL, Reith A, Seglen PO. Accumulation of autophagosomes after inhibition of hepatocytic protein degradation by vinblastine, leupeptin or a lysosomotropic amine. Exp Cell Res. 1982;137:191–201. doi: 10.1016/0014-4827(82)90020-9. [DOI] [PubMed] [Google Scholar]
- 36.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–95. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem. 1995;270:2320–6. doi: 10.1074/jbc.270.5.2320. [DOI] [PubMed] [Google Scholar]
- 39.Yang Q, Guan KL. Expanding mTOR signaling. Cell Res. 2007;17:666–81. doi: 10.1038/cr.2007.64. [DOI] [PubMed] [Google Scholar]
- 40.Kono H, Arteel GE, Rusyn I, Sies H, Thurman RG. Ebselen prevents early alcohol-induced liver injury in rats. Free Radic Biol Med. 2001;30:403–11. doi: 10.1016/s0891-5849(00)00490-1. [DOI] [PubMed] [Google Scholar]
- 41.Schmidt A, Bickle M, Beck T, Hall MN. The yeast phosphatidylinositol kinase homolog TOR2 activates RHO1 and RHO2 via the exchange factor ROM2. Cell. 1997;88:531–42. doi: 10.1016/s0092-8674(00)81893-0. [DOI] [PubMed] [Google Scholar]
- 42.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–71. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 43.Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170:1101–11. doi: 10.1083/jcb.200504035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 45.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 46.Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;281:34870–9. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- 47.Samari HR, Seglen PO. Inhibition of hepatocytic autophagy by adenosine, aminoimidazole-4-carboxamide riboside, and N6-mercaptopurine riboside. Evidence for involvement of amp-activated protein kinase. J Biol Chem. 1998;273:23758–63. doi: 10.1074/jbc.273.37.23758. [DOI] [PubMed] [Google Scholar]
- 48.Vucicevic L, Misirkic M, Kristina J, Vilimanovich U, Sudar E, Isenovic E, Prica M, Harhaji-Trajkovic L, Kravic-Stevovic T, Vladimir B, Trajkovic V. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy. 2010;7:40–50. doi: 10.4161/auto.7.1.13883. [DOI] [PubMed] [Google Scholar]
- 49.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 50.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 51.Chang NC, Nguyen M, Germain M, Shore GC. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J. 2009;29:606–18. doi: 10.1038/emboj.2009.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–7. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 53.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brady NR, Hamacher-Brady A, Yuan H, Gottlieb RA. The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. Febs J. 2007;274:3184–97. doi: 10.1111/j.1742-4658.2007.05849.x. [DOI] [PubMed] [Google Scholar]
- 55.Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, Mathiasen IS, Jaattela M. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 56.Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW, 2nd, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010;285:27879–90. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–5. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104:19500–5. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–8. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 61.Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282:4702–10. doi: 10.1074/jbc.M609267200. [DOI] [PubMed] [Google Scholar]
- 62.Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ. 2010;17:268–77. doi: 10.1038/cdd.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–9. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 64.Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 65.Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene. 2003;22:8608–18. doi: 10.1038/sj.onc.1207108. [DOI] [PubMed] [Google Scholar]
- 66.Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–24. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–31. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4:141–50. doi: 10.4161/auto.5190. [DOI] [PubMed] [Google Scholar]
- 69.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–88. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 71.Chen Y, Gibson SB. Is mitochondrial generation of reactive oxygen species a trigger for autophagy? Autophagy. 2008;4:246–8. doi: 10.4161/auto.5432. [DOI] [PubMed] [Google Scholar]
- 72.Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J Cell Sci. 2007;120:4155–66. doi: 10.1242/jcs.011163. [DOI] [PubMed] [Google Scholar]
- 73.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–60. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008;15:171–82. doi: 10.1038/sj.cdd.4402233. [DOI] [PubMed] [Google Scholar]
- 75.Muoio DM, Newgard CB. Obesity-related derangements in metabolic regulation. Annu Rev Biochem. 2006;75:367–401. doi: 10.1146/annurev.biochem.75.103004.142512. [DOI] [PubMed] [Google Scholar]
- 76.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yin XM, Ding WX, Gao W. Autophagy in the liver. Hepatology. 2008;47:1773–85. doi: 10.1002/hep.22146. [DOI] [PubMed] [Google Scholar]
- 78.Rautou PE, Mansouri A, Lebrec D, Durand F, Valla D, Moreau R. Autophagy in liver diseases. J Hepatol. 2010;53:1123–34. doi: 10.1016/j.jhep.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 79.Ding WX. Role of autophagy in liver physiology and pathophysiology. World J Biol Chem. 2010;1:3–12. doi: 10.4331/wjbc.v1.i1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zakhari S, Li TK. Determinants of alcohol use and abuse: Impact of quantity and frequency patterns on liver disease. Hepatology. 2007;46:2032–9. doi: 10.1002/hep.22010. [DOI] [PubMed] [Google Scholar]
- 81.Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, Lu B, Stolz DB, Clemens DL, Yin XM. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–52. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lumeng L, Crabb DW. Alcoholic liver disease. Curr Opin Gastroenterol. 2000;16:208–18. doi: 10.1097/00001574-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 83.Bailey SM, Cunningham CC. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol Med. 2002;32:11–6. doi: 10.1016/s0891-5849(01)00769-9. [DOI] [PubMed] [Google Scholar]
- 84.Thomas Paul GCST, Osna Natalia A, Clemens Dahn L, thiele Geoffey M, Duryee Michael J, Fox Howard S, Haorah James, Donohue Terrence M. Proteasome activity and autophagy in liver are reciprocally affected after ethanol exposure. Hepatology. 2010;52:615A. [Google Scholar]
- 85.Lin C-W, Tsai Mei-Hsing, Chen Xiaoyun, Yin Xiao-Ming. Enhanced autohagy alleviated ethanol-induced fatty liver in mouse. Hepatology. 2010;52:379A. [Google Scholar]
- 86.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–93. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–53. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–40. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–78. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sir D, Chen WL, Choi J, Wakita T, Yen TS, Ou JH. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology. 2008;48:1054–61. doi: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, Liu Z, Cao W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem. 2009;284:31484–92. doi: 10.1074/jbc.M109.033936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yeon JE, Califano S, Xu J, Wands JR, De La Monte SM. Potential role of PTEN phosphatase in ethanol-impaired survival signaling in the liver. Hepatology. 2003;38:703–14. doi: 10.1053/jhep.2003.50368. [DOI] [PubMed] [Google Scholar]
- 93.Shulga N, Hoek JB, Pastorino JG. Elevated PTEN levels account for the increased sensitivity of ethanol-exposed cells to tumor necrosis factor-induced cytotoxicity. J Biol Chem. 2005;280:9416–24. doi: 10.1074/jbc.M409505200. [DOI] [PubMed] [Google Scholar]
- 94.Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–99. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- 95.Donohue TM, Jr, Cederbaum AI, French SW, Barve S, Gao B, Osna NA. Role of the proteasome in ethanol-induced liver pathology. Alcohol Clin Exp Res. 2007;31:1446–59. doi: 10.1111/j.1530-0277.2007.00454.x. [DOI] [PubMed] [Google Scholar]
- 96.Nishitani Y, Matsumoto H. Ethanol rapidly causes activation of JNK associated with ER stress under inhibition of ADH. FEBS Lett. 2006;580:9–14. doi: 10.1016/j.febslet.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 97.You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. 2004;127:1798–808. doi: 10.1053/j.gastro.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 98.Vucicevic L, Misirkic M, Janjetovic K, Vilimanovich U, Sudar E, Isenovic E, Prica M, Harhaji-Trajkovic L, Kravic-Stevovic T, Bumbasirevic V, Trajkovic V. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy. 2011;7 doi: 10.4161/auto.7.1.13883. [DOI] [PubMed] [Google Scholar]
- 99.Tomita K, Tamiya G, Ando S, Kitamura N, Koizumi H, Kato S, Horie Y, Kaneko T, Azuma T, Nagata H, Ishii H, Hibi T. AICAR, an AMPK activator, has protective effects on alcohol-induced fatty liver in rats. Alcohol Clin Exp Res. 2005;29:240S–5S. doi: 10.1097/01.alc.0000191126.11479.69. [DOI] [PubMed] [Google Scholar]
- 100.Bergheim I, Guo L, Davis MA, Lambert JC, Beier JI, Duveau I, Luyendyk JP, Roth RA, Arteel GE. Metformin prevents alcohol-induced liver injury in the mouse: Critical role of plasminogen activator inhibitor-1. Gastroenterology. 2006;130:2099–112. doi: 10.1053/j.gastro.2006.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Purohit V, Gao B, Song BJ. Molecular mechanisms of alcoholic fatty liver. Alcohol Clin Exp Res. 2009;33:191–205. doi: 10.1111/j.1530-0277.2008.00827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Saemann MD, Haidinger M, Hecking M, Horl WH, Weichhart T. The multifunctional role of mTOR in innate immunity: implications for transplant immunity. Am J Transplant. 2009;9:2655–61. doi: 10.1111/j.1600-6143.2009.02832.x. [DOI] [PubMed] [Google Scholar]
- 103.Harada M, Hanada S, Toivola DM, Ghori N, Omary MB. Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology. 2008;47:2026–35. doi: 10.1002/hep.22294. [DOI] [PubMed] [Google Scholar]
- 104.Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, Maurice N, Mukherjee A, Goldbach C, Watkins S, Michalopoulos G, Perlmutter DH. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–32. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- 105.Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, Zhu H, Yu AD, Xie X, Ma D, Yuan J. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci U S A. 2007;104:19023–8. doi: 10.1073/pnas.0709695104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Balgi AD, Fonseca BD, Donohue E, Tsang TC, Lajoie P, Proud CG, Nabi IR, Roberge M. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS One. 2009;4:e7124. doi: 10.1371/journal.pone.0007124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]