

Abstract
Low molecular weight heparin (LMWH) is widely used in anticoagulation therapies and for the prevention of thrombosis. LMWH is administered by subcutaneous injection usually once or twice per day. This frequent and invasive delivery modality leads to compliance issues for individuals on prolonged therapeutic courses, particularly pediatric patients. Here, we report a long-term delivery method for LMWH via subcutaneous injection of long-lasting hydrogels. LMWH is modified with reactive maleimide groups so that it can be crosslinked into continuous networks with four-arm thiolated polyethylene glycol (PEG-SH). Maleimide-modified LMWH (Mal-LMWH) retains bioactivity as indicated by prolonged coagulation time. Hydrogels comprising PEG-SH and Mal-LMWH degrade via hydrolysis, releasing bioactive LMWH by first-order kinetics with little initial burst release. Separately dissolved Mal-LMWH and PEG-SH solutions were co-injected subcutaneously in New Zealand White rabbits. The injected solutions successfully formed hydrogels in situ and released LMWH as measured via chromogenic assays on plasma samples, with accumulation of LMWH occurring at day two and rising to near-therapeutic dose equivalency by day 5. These results demonstrate the feasibility of using LMWH-containing, crosslinked hydrogels for sustained and controlled release of anticoagulants.
INTRODUCTION
Cardiovascular disease is a global health issue, affecting nearly 17 million people per year.1 In particular, venous and arterial thromboembolisms are major problems. A significant proportion of myocardial infarctions and strokes, which are collectively the most common cause of death in the industrialized world2, are precipitated by arterial thromboembolism.3 Venous thromboembolism is the third leading cause of cardiovascular associated death after myocardial infarction and stroke.3 Anticoagulants are standard treatments to reduce the risk of these thromboembolisms, and currently include heparins (both unfractionated and low molecular weight derivatives)4, vitamin K antagonists (coumarins) such as oral warfarin5, protein-derived thrombin inhibitors (hirudin, lepirudin, desirudin)6,7, small-molecule thrombin inhibitors (argatroban and oral dabigatran)8, and factor Xa inhibitors (oral rivaroxaban and apixaban).9,10 Of the long list of anticoagulant therapies, heparin formulations have the longest history of clinical use.11 Unfractionated and low molecular weight heparin (LMWH) are standard treatments for myocardial infarction, cardiovascular surgery, coronary angioplasty, coronary stents and numerous other conditions.4 LMWH, which can be delivered subcutaneously, offers advantages over unfractionated heparins, which require IV delivery, such as reduced risk of hemorrhage,12 improved clearance rates,13 and more predictable pharmacokinetics.14 Perhaps most importantly, subcutaneous injections of LMWH can safely be administered at home without the need to monitor activated partial-thromboplastin time that is necessary with unfractionated heparin.15
There has been significant effort to develop oral formulations for heparin delivery, owing to compliance issues with subcutaneous injections in outpatient populations.16,17 The first systems to show limited clinical success combined heparin into oral syrups18,19 and later into solid gel capsules20,21 by combination with N-[8-(2-hydroxybenzoyl) amino]caprylate (SNAC), whereby SNAC acts as a carrier molecule via weak association of heparin, increasing the gastrointestinal absorption followed by dissociation once absorbed.22 In addition, a wide range of other oral delivery strategies have been proposed (although not yet employed clinically) including covalent modification, emulsification, nanoparticle formulation, polyion complexes, and others.23–26 Unfortunately, oral delivery approaches are hampered by the fact that heparin is not readily or consistently absorbed by the gastrointestinal tract27, thus injection remains the primary route of administration. Unfortunately, compliance with daily injection regimens is a serious problem; in one study lack, of compliance resulted in uncertain prophylaxis in 28% of adults and 100% of patients under 20 years of age.28,29 Accordingly, both oral and recurrent subcutaneous delivery modalities have drawbacks in the maintenance of consistent drug concentrations within therapeutic ranges, which is a critical difficulty for disease prevention.30 The development of time-released formulations would be a significant improvement, especially in pediatrics.
In children, similar to adults, conventional antithrombotic therapies have included primarily heparin and the oral anticoagulant warfarin; however, there are a number of important physiologic and practical considerations that make the use of these drugs in pediatric patients very different and more problematic than use in adults. First, the epidemiology of thromboembolic events in pediatric patients is different from that observed in adults.31 In addition, maturation of the clotting system, developmental changes in the distribution, binding, and metabolism of drugs, and the presence of inter-current illness or concurrent medications may affect pediatric antithrombotic therapy.32,33 It is significant that there are no specific pediatric formulations of antithrombotic drugs. Furthermore, pediatric compliance issues render current formulations especially problematic in, for example, infants who cannot comprehend the need for therapy, adolescents who comprehend but are emotionally unwilling to cooperate, and children of dysfunctional families who may experience neglect.31 In addition, there has been a dramatic rise in the use of anticoagulant treatments in children, and LMWH has become widely used to control thromboembolic complications in pediatrics. Other oral anticoagulants such as direct thrombin inhibitors, though successful for short term treatment, have yet to be proven efficacious for long-term treatment given continued concerns over hepatic toxicity and the need for routine patient monitoring.6,34,35
The combinations of therapeutic and compliance challenges associated with current treatments indicates the need for more effective long-term anticoagulant therapy. Such therapy would include facile and infrequent administration, maintenance of therapeutic drug concentration in circulation, and long-term elimination of any drug carriers. Hydrogels offer such a solution, and have been commonly used for controlled release of drugs in many applications. They are composed mostly of water and maintain their self-supporting and elastic nature by a network of hydrophilic polymers that are chemically, physically, or ionically cross-linked, leading to a material that swells to high degrees yielding mechanical and/or chemical properties similar to that of biological tissues.36 Hydrogels are particularly conducive to releasing water-soluble drugs via hydrolysis due to their highly hydrophilic environment, rapid mass transport, and homogeneous and predictable degradation conditions.36 Polysaccharides, specifically polysaccharide-modified synthetic polymers, have long been used in biomaterials for tissue engineering and drug delivery because of their functional structure, hydrophilicity and general biocompatibility.37 In accord, heparin has been primarily used as a component of growth factor-delivering systems to bind proteins, thereby both sustaining the activity of the protein and controlling the delivery.38–40 Although a bulk of these hydrogel systems have contained heparin, they have not been investigated as anti-thrombotic delivery agents. Furthermore, only a few researchers have described heparin-containing hydrogels for potential antithrombotic therapy,41–43 though their efficacy and feasibility of use was not determined in animal models.
Here we describe the development of a subcutaneously injectable hydrogel system for long-term release of LMWH. The hydrogels are prepared by in situ crosslinking of injected maleimide-functionalized LMWH (Mal-LMWH) and four-arm star, thiolated polyethylene glycol (PEG-SH). Maleimide-thiol Michael type additions are highly specific for thiols over other nucleophiles and react with fast reaction kinetics, producing no byproducts44; thus, two mutually reactive solutions of LMWH and PEG can be co-injected subcutaneously with immediate in situ formation of hydrogels. Subsequent hydrolysis of the ester linkages of the PEG-SH component results in release of bioactive LMWH that can bind antithrombin III (ATIII) and factor Xa (fXa) (Figure 1). The in vivo application of this material is possible and permits accumulation of therapeutically-relevant levels of LMWH.
FIGURE 1.
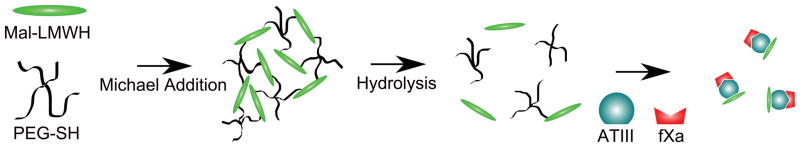
In situ crosslinking via Michael-type addition of maleimide-functionalized LMWH (Mal-LMWH) and four-arm star thiolated PEG (PEG-SH), with subsequent ester hydrolysis of the network and delivery of active LMWH that binds antithrombin III and factor Xa.
MATERIALS AND METHODS
Materials
Four-arm hydroxyl functionalized PEG (Mn 10,000 g mol−1) was purchased from JenKem Technology USA Inc. (Allen, TX, US). Lovenox low molecular weight heparin (LMWH, Mw 4500 g mol−1) was purchased from Sanofi-aventis (Bridgewater, NJ, USA). N-(2-aminoethyl)maleimide, trifluoroacetate salt (AEM), 1-hydroxybenzotriazole hydrate (HOBT), 2-(N-morpholino)ethanesulfonic acid (MES), 3-mercaptopropionic acid (MP), and p-toluenesulfonic acid monohydrate (PTSA) were purchased from Sigma-Aldrich (St. Louis, MO, US). ACTICHROME® Heparin (anti-fXa) and HEPTEST® assays and control plasma were purchased from American Diagnostica (Stamford, CT, US). 2.5ml double-barrel mixing syringes and 4.5cm luer lock mixing tips were purchased from Medmix Systems (Risch, CH). N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl), and all other reagents and materials were purchased from Fisher Scientific (Pittsburgh, PA, US). Phosphate buffered saline (PBS) was composed of 10mM sodium phosphate and 150mM sodium chloride at a pH of 7.4 unless noted otherwise.
Synthesis of four-arm thiolated PEG (PEG-SH)
The synthesis of thiolated four-arm PEG was performed as previously reported.38 In short, PEG (1meq.), MP (40meq.), and PTSA (0.4meq.) were dissolved in toluene. Under a flow of nitrogen, the reaction was refluxed with stirring for 48hrs (Scheme 1). Water was collected by using a Dean-Stark trap. Toluene was removed under reduced pressure and the polymer was precipitated 3 times in cold ether. The polymer was reduced by dissolving 1meq in methylenechloride with DTT (1meq) and triethylamine (1meq) under nitrogen for 5hrs. The reaction was acidified with trifluoroacetic acid (1.1meq) and precipitated in ether and rinsed with copious amounts of 2-propanol and hexane. Functionality was determined via 1H NMR spectroscopy and was approximately four (>95%). 1H NMR (CDCl3): δ = 4.28 (8H, CH2-O-C(O), m), 3.74-3.50 (900H (CH2-CH2-O)n, bs), 2.84-2.64 (16H, CH2-CH2-SH, m) (Figure S1).
SCHEME 1.
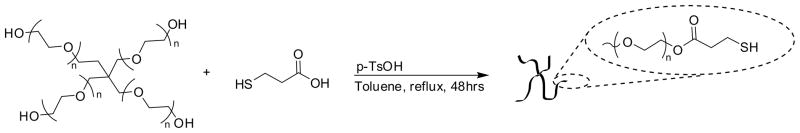
Synthesis of thiolated four-arm PEG (PEG-SH).
Synthesis of maleimide-functionalized LMWH (Mal-LMWH)
The synthesis of maleimide-functionalized heparin was performed as previously described with slight modification to yield the desired number of maleimide groups per LMWH.38 Briefly, 2g LMWH (0.44mmol) was dissolved with 0.5g AEM (2mmol) in the presence of 0.5g EDC·HCl (2.7mmol) and 0.5g HOBT (3.3mmol) in 200ml of 0.1M, pH 6.0 MES (Scheme 2). The reaction proceeded overnight at room temperature with stirring. The product was purified by dialysis (MWCO 1000) against 1M NaCl solution and deionized water. The freeze-dried sample was characterized via 1H nuclear magnetic resonance spectroscopy (NMR) indicating a degree of functionalization of 1.7. Higher functionalities were obtained by increasing the concentration of reactants. 1H NMR (400 MHz, D2O): δ = 6.84 (2H, CO-CH=CH-CO, m), δ = 5.60-5.05 (15H, heparin anomeric proton, m) (Figure S2).
SCHEME 2.
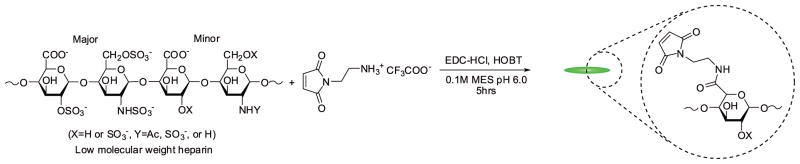
Synthesis of maleimide-functionalized LMWH (Mal-LMWH).
HEPTEST® anticoagulation time
Clotting time assessments were measured using slightly modified HEPTEST® protocols for unmodified LMWH and Mal-LMWH as well as for the supernatants from in vitro degradation experiments described below. For increased accuracy of clotting time measurements, the viscosity of the mixed HEPTEST® samples was monitored continuously using an AR-2000 rheometer (TA Instruments, New Castle, DE, US) with a 25mm parallel plate geometry with a 400μm gap and temperature-controlled lower Peltier plate set at 37°C. Hydrated solutions were kept on ice before use. 50μL plasma, 50μL fXa, and 50μL LMWH in PBS were added to the rheometer Peltier plate and incubated for 30s with shearing at 2 rad·s−1 to ensure adequate mixing. 50μL of RecalMIX was added to the stage and sheared for an additional 8s at 2 rad·s−1 and 2s at 0.1rad·s−1. A constant 0.1Pa shear stress was then applied to the solution. Clot times were recorded when head velocities decreased to zero.
Hydrogel formation
Hydrogel formation was accomplished by dissolving PEG-SH and Mal-LMWH separately in buffer with equal molar ratios with respect to functionalities. The Michael-type addition of Mal-LMWH and PEG-SH and further crosslinking is depicted in Scheme 3. Self-supporting hydrogels were formed upon mixing the two solutions. Gels cast for in vitro degradation and rheology experiments were mixed in microcentrifuge tubes and immediately vortexed for approximately 2s to ensure solution homogeneity without disrupting the evolving crosslinked network structure. The solution was then transferred to its final form before an elastic solid was formed. Subcutaneous injections were accomplished by loading each chamber of an ethylene-oxide sterilized MedMix double-barrel syringe, assembled with a mixing tip and 25G × 5/8″ needle, with the appropriate solution of either functionalized PEG or functionalized LMWH. Prior to loading, polymer solutions were filter-sterilized using 0.22μm polyethersulfone filters. Precursor hydrogel solutions were injected at rates such that gelation did not occur in the mixing tip. All hydrogels used in this study had a total solids content of 10w/v%. (Higher concentration hydrogels (up to 20w/v%) were easily formed in practice but not implemented in these experiments, thus hydrogel volumes could be reduced if necessary for future studies).
SCHEME 3.
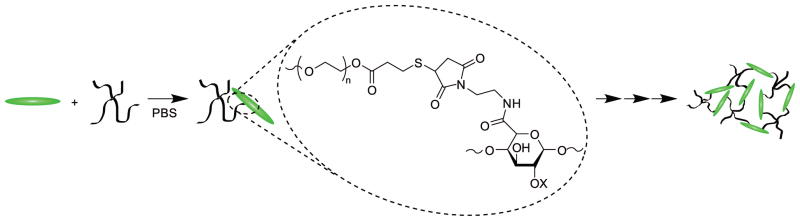
Michael-type addition reaction of Mal-LMWH and PEG-SH, shown with further reaction to form a crosslinked network.
Oscillatory rheology
Samples were gelled in situ by co-injection of the separately dissolved functionalized materials onto the rheometer Peltier plate mixed as described above. An AR-2000 rheometer was used at 25°C in a constant (1.0%) strain mode. A 20mm, 1° 56″ cone plate geometry with a 33μm truncation was used in all experiments at a constant oscillatory frequency of 6 rad s−1. Gelation kinetics were determined by measuring the storage (G′) and loss (G″) moduli as a function of time after co-injection of the materials on the rheometer stage. A gelation time was defined as the time of crossover of G′ and G″.
Heparin release in vitro
100μL hydrogels were formed in 1ml syringes (with the tips removed), with mixing as described above. Syringes were sealed with Parafilm® to minimize evaporation during gelation. Hydrogels were incubated at room temperature for 10 minutes before they were ejected from the syringe by advancing the plunger. It was determined via oscillatory rheology experiments that hydrogels reached an equilibrium plateau modulus significantly before 10 minutes and therefore could be displaced into a bath without disruption. The cast hydrogels were then each swelled separately in 14ml of PBS. The vials were placed on a test tube rocker in a 37°C incubator. 12ml samples of the supernatant were taken, and the sample replenished with fresh buffer at time points of 0, 2, 9 and 24 hours, then daily until the 7th day, and then every two days until the end of the experiment. The hydrolysis of the esters in these networks, which here liberates a succinimide thiopropionic acid-functionalized LMWH, is depicted in Scheme 4.
SCHEME 4.
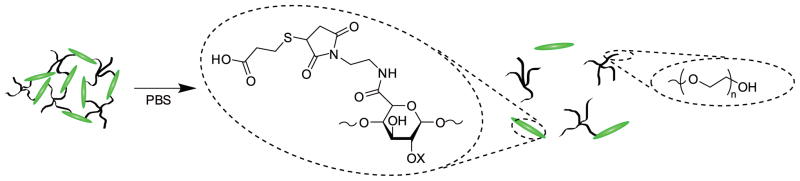
Degradation of hydrogel via ester hydrolysis.
The concentration of LMWH in solution was determined using the established toluidine blue assay, with slight modification.45 Briefly, 200μL of LMWH-containing supernatants were added to 200μL 0.005% toluidine blue solution in 0.01N HCl, and 400μL dichloromethane, in a 1.5ml polypropylene centrifuge tube. Tubes were vortex mixed and incubated overnight at room temperature and finally centrifuged for 10 minutes at 20,000 × g. 100μL of aqueous solutions were added to a 96-well plate and absorbance values at 590nm measured on a Fusion microplate reader (Perkin Elmer, Waltham, MA, US). Concentrations were derived from a calibration curve constructed of eight samples with LMWH concentrations ranging from zero to 0.035mg/ml. It was determined that the conjugation of polymer to the LMWH did not impact the measurements, via comparisons of LMWH with mono-PEGylated LMWH. Calibration curves for unfunctionalized LMWH and Mal-LMWH were also identical, indicating that the measurement of LMWH in this assay was not affected by the functionalization of the LMWH.
Subcutaneous injection of LMWH in vivo
11 healthy New Zealand White rabbits (Covance Research, Princeton, NJ, USA) of 5–9 months of age and a weight range from 2.7–3.4 kg were selected for subcutaneous injection of heparin solutions. All animal procedures were approved by the A.I. duPont Hospital for Children Institutional Animal Care and Use Committee and overseen by a staff veterinarian. Rabbits were purchased with femoral venous access ports (VAPs) implanted for ease of blood draws during experiments. The animals were divided into three groups: 4 animals received unmodified LMWH, 3 animals received Mal-LMWH, and 4 animals received hydrogel precursor solutions for in situ hydrogel formation. General analgesia (0.05 mg/kg buprenorphine) was administered to all animals prior to subcutaneous injections.
Unmodified LMWH was diluted to 10mg/ml and injected daily based on the weight of the rabbit (1mg LMWH/kg). Mal-LMWH (f=1.7) was injected daily at 2.5mg Mal-LMWH/kg, to account for the decrease in fXa activity indicated via HEPTEST® assays as described above. The amount of hydrogel necessary to deliver a therapeutic level of LMWH was determined based on an estimated degradation time of approximately 14 days with an assumed near linear release of LMWH, for a total injection of 35mg Mal-LMWH/kg, 32.7mg PEG-SH/kg, for an injection volume of 680μl/kg. All LMWH solutions were injected subcutaneously via 25G × 5/8″ needles into the animal’s dorsum on the right side of the midline, just below the level of the scapula. Hydrogel injections were injected as described above.
Blood draws were taken from VAPs for pre-procedure baseline, 20, 40, 60 and 180 minutes after day 1 injections for all samples. Subsequent blood draws were taken 30 minutes after injection for later daily injections of LMWH and Mal-LMWH. Hydrogel animal blood draws were collected at 20, 40, 60 and 180 minutes after day 1 injections as well as daily thereafter to a total of 10 time points over 5 days. To prevent coagulation or bacterial infection within the VAP, 5 mL of sterile saline was flushed through the catheter after each blood draw, followed by 1 mL of 3 mg/mL minocycline with 30 mg/mL ethylenediaminetetraacetic acid (EDTA).46 Collected blood samples were diluted with 1/10 volume of 3.8% trisodium citrate and centrifuged at 4°C for 20 minutes at 5,000 × g and stored at −80°C as per standard ACTICHROME® collection procedures. All recommended procedures were followed for assay calibration and measurement. Animals were euthanized at the end of the experiment with 1 mL beuthanasia. NIH guidelines for the care and use of laboratory animals (NIH Publication #85-23 Rev. 1985) were observed.
RESULTS AND DISCUSSION
Anticoagulant activity
Maleimides were coupled to uronic acid residues of LMWH via carbodiimide coupling of the amine group of the maleimide and carboxylates of LMWH (Scheme 2). The number of functional maleimide groups appended to the LMWH was controlled to be around 2; a minimum number of maleimides (2) is required to permit the formation of percolated networks when Mal-LMWH is reacted with four-arm star, thiolated PEG. Two maleimides per LMWH was targeted in order to minimize the impact on antithrombin III (ATIII) binding and to thus preserve anticoagulant activity. Two different average functionality values were tested; these values were determined by proton NMR spectroscopy to be 1.7 and 2.4 maleimides per LMWH by integration of the maleimide peak (6.82ppm) and the anomeric protons of the polysaccharide (5.60-5.05 ppm) assuming the average number of saccharides to be 15 (Figure S2).
The anticoagulant activity of the modified Mal-LMWH was quantified with a standard coagulation assay (HEPTEST®, American Diagnostica). Coagulation curves (clotting time versus heparin concentration) were constructed by measuring the clotting time of various known concentrations of each modified heparin in normal human plasma (Figure 2A). The positive slopes of these curves illustrate that the modified heparins retained anticoagulant activity, and the slope obtained from the data via linear regression was interpreted to be an indication of this anticoagulant activity for each heparin (plotted in Figure 2B). The slopes were statistically different based on a 2-sample t-test (p < 0.01). Addition of 1.7 maleimide groups per LMWH reduced the activity 2.5-fold (relative to unmodified LMWH, ~40% activity), while addition of 2.4 maleimides per LMWH chain reduced the activity by 6-fold. Comparatively, oral LMWH and unfractionated heparin, though markedly different in terms of absorption and delivery routes, have not achieved perfect bioavalability either, with maximum bioavailability ranging from ~10% to a maximum of ~60% depending on the co-delivery agent or vehicle.22,25,47 The impact of covalent modification on bioactivity was not unexpected given that ATIII is thought to bind to a pentasaccharide unit in heparin4, and that the average number of saccharides in LMWH (enoxaparin) is approximately 15, thus yielding only 3 pentasaccharide units. Modification of 2 of the 3 pentasaccharides could be expected to interfere with binding of ATIII. It has also been shown that chemical modification of high molecular weight heparin can impact the binding efficiency to proteins, though to a lesser degree than measured here.48 Typical side reactions of maleimide ring opening44 and carbodiimide rearrangement49 were detected via proton NMR at low conversions (affecting fewer than 0.2 carboxylic acids per chain for each functionality, see Supporting Information). Further minimization of these side reactions would most likely increase the anticoagulant activity for a given maleimide functionality. Considering this, Mal-LMWH with a functionality of 1.7 was used for all subsequent experiments as this provided sufficient anticoagulant activity and enabled formation of fully crosslinked hydrogels. Any further decrease in maleimide functionality prevented the formation of crosslinked hydrogels.
FIGURE 2.
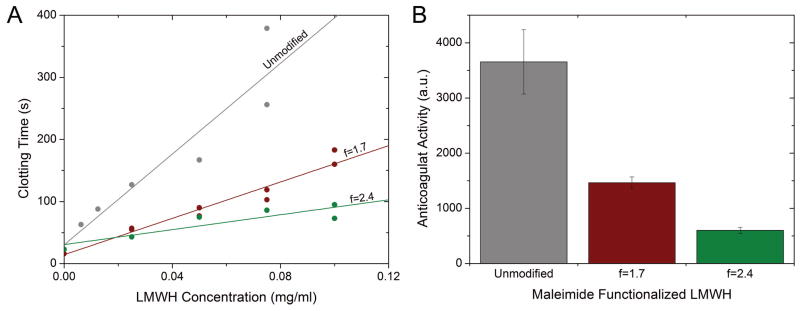
Impact of heparin functionality on anticoagulant activity. (A) Clotting times for various concentrations of modified Mal-LMWH as measured via HEPTEST®; (B) Comparison of the anticoagulant activities, determined from the slopes from the HEPTEST® results. Error bars indicate standard error of the regression, and all slopes are statistically different (p<0.01, 2 sample t-test).
Gelation kinetics
Oscillatory rheology was used to measure the kinetics of gel formation as well as the elastic modulus of the hydrogel. The gelation kinetics are important for the proposed system so that gel formation is sufficiently rapid to prevent dilution of the precursor solutions in the subcutaneous space, but not so rapid that gelation occurs in the mixing syringe, preventing homogeneous mixing or an increased solution viscosity such that the injection cannot be made through a small-gauge needle.50 Furthermore, any fracture of the forming crosslinked network in the syringe tip would potentially alter the LMWH release profile and make release profiles variable from gel to gel. In order to control the gelation time, the pH of the solutions was altered, as the Michael-type addition of thiols to maleimides is known to be dependent on the thiolate ion concentration which is directly dependent on solution pH.51 Variation of the pH is thus a facile means to control gelation times for this and other potential applications.
Figure 3A shows the evolution of the elastic moduli (G′) over time for 10w/v% hydrogels formed at different pH values. These gels were formed by mixing solutions of Mal-LMWH and PEG-SH, at a stoichiometric concentration of maleimide to thiol groups. The most rapid gelation occurred at pH 7.4, which was the upper limit tested in these studies owing to the inactivation of the maleimide via ring opening at higher pH values.44 Much slower gelation occurred for solutions at pH 5.5. The time when the mixed solution would form an elastic solid with no fluidity, or critical gelation time, was assessed when G′ was greater than the loss modulus (G″) and plotted against pH (Figure 3B). A pH of 7.4 was optimal for injection with the epoxy style mixing tip employed in our in vivo studies (Figure S3), allowing sufficient time for all the solution to pass through the syringe mixing tip and needle without ring-opening of the maleimide (data not shown). Figure 3C shows a typical time-sweep for a 10w/v% hydrogel formed at pH 7.4, showing that the length of time necessary to reach equilibrium is on the order of a few minutes under the conditions employed in the in vivo experiments. Notably, a critical gelation time within 20 seconds would permit the hydrogels to remain intact upon injection in the subcutaneous space, while complete gelation occurred within 10 minutes. Comparatively, other injectable hydrogels crosslinked by addition chemistries at physiological pH exhibit slower critical gelation times (ca. 15 minutes or longer) for four-arm PEG vinyl sulfone crosslinked with thiolated peptides.52 Increasing the functionality of the crosslinking macromer (i.e., functionality much greater than 4) has reduced the critical gelation time to 3–5 minutes for acrylate-thiol polysaccharide hydrogels40,53 and 0.5–7 minutes for vinyl sulfone-modified dextran and PEG-thiol hydrogels.54 Although the system described here is composed of low functionalities (Mal-LMWH ~2, and PEG-SH ~4), short gelation times were achieved, owing to the faster reaction kinetics of the maleimide-thiol addition reactions compared to acrylate-thiol or vinyl sulfone-thiol additions. Additionally, the final modulus of ca. 2.3 kPa is in the range of what typically would be observed for subcutaneous tissues indicating that these injected hydrogels would have mechanical properties similar to the surrounding normal tissue.55
FIGURE 3.

Oscillatory rheology characterization of hydrogels. (A) Impact of solution buffer pH on gelation kinetics plotted as storage modulus (G′) against gelation time; decreasing the solution pH increased the time of the onset of gelation. (B) Critical gel point time as a function of pH. (C) Typical oscillatory storage (●) and loss (○) modulus for gels cast at room temperature in pH 7.4 PBS, achieving a measureable modulus within 20 seconds and final modulus within 10 minutes.
Release of LMWH from hydrogels in vitro
The esterification reaction of 4-mercaptopropionic acid with hydroxyl-terminated, four-arm star PEG (Scheme 3) provided a facile method for thiolating PEG. The introduction of the ester linkages also imparted a strategy for controlling hydrolytic degradation rates of the hydrogels, and in turn controlling the release of LMWH. The rate of chemical hydrolysis depends mainly on pH and temperature, as well as on the hydrophobicity of the environment around the hydrolytically labile group.56 Degradation of 10w/v% hydrogels, immersed in an excess of PBS at 37°C, was quantified by measurement of released LMWH by established toluidine blue measurements.45 Figure 4 displays the average cumulative release of LMWH over time. An initial burst release of 14.8± 0.9% was measured at two hours, followed by a near linear release until day 16 when accelerated degradation was observed. The accelerated degradation has been attributed to the point at which the de-percolation of the continuous hydrogel network occurs.57 It was further shown that de-percolation occurs at variable mass loss percent dependent on the length between crosslinks as well as hydrolysis rates using model hydrogels composed of linear PEG-acrylate functionalized with hydrolyzable lactide groups and crosslinked with multifunctional thiol molecules.58 Although our system is composed of LMWH and PEG rather than PEG alone, a similar onset of depercolation is exhibited at mass loss of 62%. However, the onset of de-percolation in the experimental PEG systems and predicted models resulted in instant degradation of the hydrogel network; whereas, a gradual increase in degradation rate was measured at the onset of de-percolation rather than complete degradation in our present studies. This effect could be attributed to the larger molecular weight distribution of LMWH (ca 1.359) versus the PEG (<1.1). Furthermore, the functionalization of LMWH produces variable molecular weights between crosslinks. The variable molecular weight would broaden the complete de-percolation of the network when compared to the model system in which the molecular weight between crosslinks is more uniform.
FIGURE 4.

Cumulative LMWH release (left axis) and log of remaining LMWH (right axis) from 10wt% hydrogels in PBS incubated at 37°C. Concentrations of LMWH were determined by toluidine blue assays; error bars indicate standard error of three separate sample measurements. The linear portion of the log of remaining LMWH was fit to first-order kinetic release equation (k=0.0483 ± 0.004 days−1; R̄2=0.994).
Hydrolysis of esters in a buffered solution is pseudo first-order; therefore, the cumulative release of LMWH (Figure 4, left axis) can be fit to known release equations by plotting the log of the amount remaining versus time (Figure 4, right axis).60 Discarding the first data points, which are from the release of entrapped (and not covalently reacted) LMWH, as well as the final data points that are indicative of hydrogel de-percolation, the linear section of the data in Figure 4 can be fit to a line with high-adjusted coefficients of determination (R̄2=0.994) yielding a pseudo first-order rate constant, k′, of 0.0483 ± 0.004 days−1 and a half life, t1/2, of 14.4±0.1 days for the pseudo first-order portion of the curve (see Supporting Information). This calculation of release ignores other factors such as enzymatic depolymerization of heparins, and it is therefore assumed that in vivo hydrolysis may be accelerated to some degree over these values. Other hydrogels such as implantable heparin release systems have obtained variable results for controlled release. Thermosensitive hydrogels composed of N-isopropyl acrylamide (NiPAAm) copolymerized with butyl methacrylate (BMA) or acrylic acid (AAc) comonomers had limited success controlling the burst release as the delivery mechanism was based on the de-swelling of the implantable gel. Both materials showed 80% heparin released within 2hrs of incubation with PBS with 100% release over 10hrs for NiPAAm/BMA and ~5 days for NiPAAm/AAc.41. Other attempts with NiPAAm methacrlyic acid were less successful with near 100% release within 10 minutes, indicating that pore sizes of the gel were too large to reduce free diffusion.43 Co-matrices of chitosan and polyethylene vinyl acetate with some hydrogel-like properties have also been reported for controlled release of aspirin (acetylsalicylate) and heparin.42 These implantable systems exhibited near zero-order release of heparin over extended periods of time (measured up to 60 days), limiting the diffusion-controlled release by addition of an insoluble surface coating of polystyrene-butadiene.42 Clearly, the use of diffusion-based control for heparin release without the use of barrier layers has proven difficult as heparin has a relatively small hydrodynamic radius and freely diffuses in swollen polymeric networks.43 Here, we obtain intermediate release rates with minimal burst release, as control of release was dependent on the hydrolysis of labile esters.
We also sought to determine if the LMWH released from hydrogels during in vitro degradation experiments maintained bioactivity, specifically because some LMWH may retain single or multiple PEG branches not removed via hydrolysis. The toluidine blue test is insensitive to the presence of PEG (data not shown), therefore the precise concentration of LMWH in the supernatants collected could be calculated and tested using the standard fXa anticoagulant test described above. Figure 5 shows the clotting time versus concentration for unconjugated LMWH (solid) and a random sampling of LMWH released from hydrogels (open). The coagulation time points determined for the released LMWH fall within the 95% predictive interval for the fitted data, and are statistically similar, indicating that the LMWH released during hydrogel degradation retains the same bioactivity (2-sample t-test of slopes, p = 0.55).
FIGURE 5.

LMWH released from in-vivo degradation retained its ability to delay coagulation time as determined by the fXa HEPTEST®. (●) Calibration curve based on functionalized LMWH and (○) LMWH released from hydrogels in in vitro experiments. The 95% confidence interval (···) and prediction interval (---) are displayed, determined from the linear regression fit of the slopes of the calibration curves. Concentrations of LMWH were determined via toluidine blue tests.
In vivo experiments
Animal studies were conducted using New Zealand White rabbits to determine the utility of the PEG-LMWH hydrogels for longer-term release of LMWH in vivo. Animals were assigned to three groups and received either daily subcutaneous injections of soluble unmodified LMWH, daily subcutaneous injections of soluble maleimide-modified LMWH, or a single subcutaneous injection of PEG-LMWH hydrogel. The delivery of LMWH to the blood over time was estimated using chromogenic assays (ACTICHROME®/anti-fXa, American Diagnostics) with unmodified LMWH as a calibration standard. Figures 6A-C display the determined concentration of LMWH in blood over the first three hours after each daily injection (average of 5 daily injections for 4 unmodified LMWH injected animals and 3 Mal-LMWH injected animals). Figure 6D displays the concentration of LMWH in blood over five days after a single hydrogel injection (with subsequent blood draws every 24 hrs after the first day). All concentrations presented in Figure 6 represent equivalents of unmodified LMWH. The concentration of unmodified LMWH in the blood (Figure 6A) is consistent with standard heparin dosage curves previously described in the literature, with an initial delay from the subcutaneous injection, followed by a maximum and gradual decrease to baseline shortly thereafter.61 In our experiments, peak concentrations of LMWH were detected within the first hour of injection. In comparison, the subcutaneous injection of maleimide-modified LMWH produced a different concentration curve (Figure 6B). It was expected that Mal-LMWH would have similar pharmacokinetics as unmodified LMWH after accounting for its decreased anticoagulant activity. A peak dose was detected with high variability approximately 20 minutes after injection, but a rapid decline to baseline was measured afterward, an effect most likely caused by reaction of the maleimide functional groups. Maleimides typically react with thiols, but are also known to react with amines with slower kinetics.44 Without pre-reaction or co-injection with exogenous thiols, the Mal-LMWH may have covalently reacted with subcutaneous tissues, reducing the available amount of LMWH in the blood.
FIGURE 6.

LMWH release in vivo versus time after injection, measured by the ACTICHROME® chromogenic assay for injections of (A) unmodified LMWH (B) Mal-LMWH and (C, D) Hydrogels. Top panels (A,B) are averages of 4 and 3 animals for LMWH and Mal-LMWH respectively with measurement after each daily injection for 5 days. Bottom panels (C,D) are an average of 4 animals with a single injection of a hydrogel at time 0, and LMWH concentration monitored over 5 days after injection. All LMWH concentrations represent equivalent doses of unmodified LMWH. Error bars indicate standard error of sample sets.
The amount of LMWH introduced in the subcutaneous space in PEG-LMWH hydrogels was at a targeted, higher mass of LMWH per kilogram relative to the injection amounts for free LMWHs (35mg/kg gel-LMWH vs 1mg/kg LMWH and 2.5mg/kg Mal-LMWH). This elevated amount was based on the fact that the LMWH-containing gel would be injected only once (followed by its controlled release) and was adjusted to account for the reduced specific activity of Mal-LMWH compared to unmodified LMWH. Sufficient LMWH to last for 14 days was administered to rabbits in the hydrogel form (2.5mg·14 days, owing for the decreased activity and the number of days targeted for release). As shown in Figure 6C, an initial increase of LMWH was measured in the blood, at similar concentrations to those measured for unmodified LMWH (Figure 6A). The initial increase in LMWH may be attributed to the release of unmodified LMWH from the hydrogel, consistent with our observations during in vitro tests (see above). Due to the large polydispersity of LMWH, it is conceivable that a small percentage of LMWH would not carry maleimide functionality, and therefore would not be covalently attached to the hydrogel network. The amounts of LMWH detected in the blood from the PEG-LMWH samples are greater than those detected for the Mal-LMWH samples (Figure 6C versus 6B). Although the LMWH in both cases is modified with maleimide, in the case of the PEG-LMWH, all maleimides are reacted with the thiols of the PEG, thus no LMWH would be expected to be ‘lost’ by covalent attachment to tissue.
As shown in Figure 6D, after the clearance of the free LMWH from the blood of the PEG-LMWH gel-injected animals, there was a trough in LMWH detected at 48 hrs followed by a sustained increase in LMWH concentration thereafter. The accumulation of LMWH in the blood beginning 48 hrs post-injection is consistent with the release of LMWH from the hydrogel at a rate greater than that of clearance of heparin from the blood, allowing the concentration to return to near the peak level of LMWH (0.002 mg/ml in Figure 6A) seen after injection of a standard therapeutic dose. The area under the curve (AUC) of plasma levels over time estimates total drug exposure. AUC can be determined for each individual animal (Figure 7), with 0–24 hr AUCs shown for all three injection types, and subsequent 24 hr AUC time intervals for the hydrogel injection. The boxplot displays the median response (solid horizontal), with the box extents representing standard error and whiskers representing 10th and 90th percentile range. As shown, high variability is observed in the data, although statistical significance was exhibited for time intervals of 24–48 hrs, and 48–72 hrs after the hydrogel injection versus the standard injection of LMWH. Thus, the AUC data are in agreement with the hydrogel release data, in that the cumulative amount of LMWH in the blood reaches a minimum at 24–48 hrs, with a subsequent increase in LMWH concentrations at later time points, in contrast to the profile at longer time points observed for the unmodified LMWH. Although the hydrogel release profiles show relatively low anticoagulation activities from 24–120 hrs, the AUC for the hydrogel reached a value similar to standard injections by day 4, suggesting that a similar level of exposure had been reached with the two delivery systems.
FIGURE 7.

Comparison of area under curve (AUC) results for 24hr segments for LMWH, Mal-LMWH and hydrogel injections. Horizontal lines represent the medians, box extents represent standard error, whiskers represent 10th and 90th percentile range. AUC were statistically similar except for 24–48, and 48–72hr intervals of LMWH release from hydrogel (2 sample t-test), whereby the release of LMWH was not sufficient to maintain an adequate level concentration in the blood. Subsequent 24hr AUC intervals return to the reference range established by standard LMWH injections.
Hydrogels were excised at day five of the experiment (Figure 8). All hydrogels, which formed ovoid masses during their injection, remained intact during the length of the study, also indicating that additional LMWH was available for release over longer timescales. The volume of the hydrogel after excision was also similar to the injected volume, as might be expected given that hydrolytic degradation is a bulk phenomenon, with degradation occurring homogeneously throughout the hydrogel’s crosslinked network so that the initial shape is conserved and devoid of surface erosion.62
FIGURE 8.
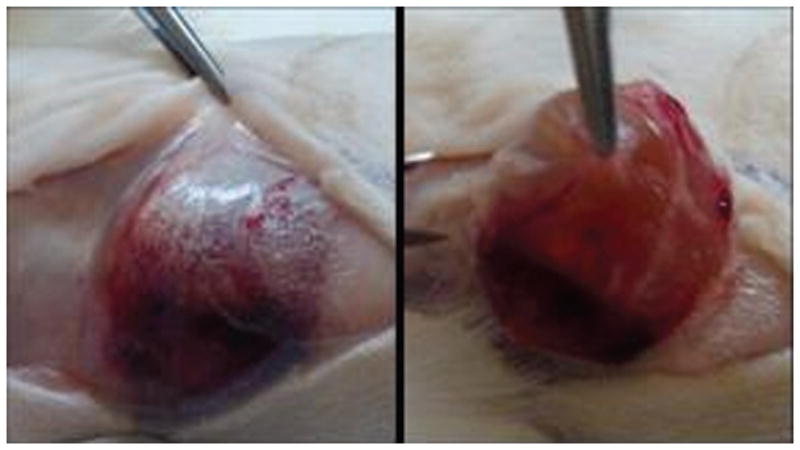
Excised hydrogel 5 days after implantation. The hydrogel remained intact over the study, indicating that release of LMWH over longer time periods would be possible.
Our results indicate the feasibility of this hydrogel system for the extended release of LMWH; future application will require tuning of the degradation rate to increase the LMWH blood concentration at early and intermediate time points, which could be achieved by accelerating the initial degradation rates of the hydrogel. One strategy to accelerate the degradation could include shortening the length of the alkyl segment of the esterified mercapto-acid (Scheme 1). This has been shown to be effective in modulating the hydrolysis of tethered model drugs to PEG chains63 and hydrolysis of thiol-acrylate photopolymers.56 Furthermore, mixed hydrogel formulations of different mercapto-acid functionalized PEGs could increase initial release while maintaining steady release over longer timescales. By tailoring the degradation kinetics, degradation profiles could be chosen for complete release of LMWH from days to months depending on patient need.
The controlled release of LMWH from injected hydrogels also has implications for improving subcutaneous therapies for a variety of drugs provided they are amenable to maleimide modification and that proper delivery profiles can be achieved. Recent studies support this approach for LMWH delivery as continuous intravenous infusion of LMWH has been shown to improve outcomes for children with deep vein thrombosis over subcutaneous injections of equivalent daily doses when peak therapeutic levels of LMWH were consistently maintained in the continuous infusion.64,65 Similar concepts have also been discussed in comparing continuous intravenous infusion of unfractionated heparin versus subcutaneous LMWH injections with varying results.15,66–69 In light of these recent pediatric studies and issues of non-adherence to daily drug regimens among children, a material such as suggested here may incorporate the advantages of a single subcutaneous injection with benefits of continuous infusion, provided the release profile of LMWH from the hydrogel could be tuned to provide higher LMWH blood concentrations earlier while maintaining comparable AUCs. Controlling the release of LMWH also is of key importance given that adverse effects have been reported from LMWH therapies, although as discussed above, to a lesser extent than with unfractionated heparin. There have been few reports of consequences, such as bleeding, from acute LMWH overdoses with most cases left untreated or treated with protamine.70–72 Here, a small amount of carboxylates of LMWH (~1–3) were modified to reduce the impact on bioactivity as discussed above, thus it remains feasible that the charge characteristics of the LMWH generally remained intact, possibly indicating that protamine therapy could be used to reverse anticoagulation effects if needed as with standard LMWH.73,74 Likewise, considerations would have to be made for protamine therapy to accommodate the continuous release of LMWH from the hydrogel if a complete reduction of blood LMWH concentration was desired.
CONCLUSIONS
Our experiments have shown that using a simple PEG-LMWH system to controllably release LMWH from an in situ forming PEG hydrogel is feasible with promising in vivo results. Anticoagulant properties of the modified LMWH were retained with bioactivities 40% that of unmodified LMWH; optimization of the LMWH modification may improve the activity. Mixing solutions of modified LMWH with four-arm star PEG resulted in rapidly gelling hydrogels with kinetic rates controlled by solution pH, while in vitro degradation studies showed a near-linear release of LMWH up to 21 days, controlled by bulk hydrolytic degradation of the hydrogel network.
This system builds on a well-established anticoagulant with extensive clinical history and an excellent safety profile. PEG-drug conjugates have been known to demonstrate favorable properties including prolonged residence in the body, decreased degradation by metabolic enzymes, and a reduction in antigenicity; properties which could aid in the long term delivery of LMWH.75 Lastly, the natural heterogeneous structure and polydispersity of LMWH offers significant opportunities for derivatization as taken advantage in this study; whereas, smaller molecules like fondaparinux or small-molecule direct thrombin inhibitors would be less likely to retain anticoagulant activity after modification.
Supplementary Material
Acknowledgments
This work was supported by grants from the Nemours Foundation (REA and CDD), the National Institutes of Health (COBRE P20-RR020173, REA; INBRE P20-RR016472, REA and KLK; P20-RR016472-10, KLK), and the National Science Foundation (DGE-0221651, KLK). The authors thank Anne Heseck and Dr. Gwen Talham for their expert assistance in designing the experiments and helpful discussions.
Footnotes
The contents of the manuscript are the sole responsibility of the authors and do not necessarily reflect the official views of the Nemours Foundation, the National Institutes of Health, nor the National Center for Research Resources.
References
- 1.Mackay J, Mensah GA, Mendis S, Greenlund K World Health Organization., NetLibrary Inc. The atlas of heart disease and stroke. Geneva: World Health Organization; 2004. p. 112. [Google Scholar]
- 2.Mathers CD, Loncar D. Projections of Global Mortality and Burden of Disease from 2002 to 2030. PLoS Med. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mackman N. Triggers, targets and treatments for thrombosis. Nature. 2008;451:914–918. doi: 10.1038/nature06797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirsh J, Warkentin TE, Shaughnessy SG, Anand SS, Halperin JL, Raschke R, Granger C, Ohman EM, Dalen JE. Heparin and Low-Molecular-Weight Heparin Mechanisms of Action, Pharmacokinetics, Dosing, Monitoring, Efficacy, and Safety. Chest. 2001;119:64S–94. doi: 10.1378/chest.119.1_suppl.64s. [DOI] [PubMed] [Google Scholar]
- 5.Ansell J, Hirsh J, Poller L, Bussey H, Jacobson A, Hylek E. The Pharmacology and Management of the Vitamin K Antagonists. Chest. 2004;126:204S–233S. doi: 10.1378/chest.126.3_suppl.204S. [DOI] [PubMed] [Google Scholar]
- 6.Di Nisio M, Middeldorp S, Büller HR. Direct Thrombin Inhibitors. N Engl J Med. 2005;353:1028–1040. doi: 10.1056/NEJMra044440. [DOI] [PubMed] [Google Scholar]
- 7.Weitz JI, Crowther M. Direct thrombin inhibitors. Thromb Res. 2002;106:V275–V284. doi: 10.1016/s0049-3848(02)00093-2. [DOI] [PubMed] [Google Scholar]
- 8.O’Brien PJ, Mureebe L. Direct Thrombin Inhibitors. J Cardiovas Pharmacol Ther. 2011 doi: 10.1177/1074248410395941. [DOI] [PubMed] [Google Scholar]
- 9.Eriksson BI, Quinlan DJ, Weitz JI. Comparative pharmacodynamics and pharmacokinetics of oral direct thrombin and factor Xa inhibitors in development. Clin Pharmacokinet. 2009;48:1–22. doi: 10.2165/0003088-200948010-00001. [DOI] [PubMed] [Google Scholar]
- 10.Turpie AGG. Oral, Direct Factor Xa Inhibitors in Development for the Prevention and Treatment of Thromboembolic Diseases. Arterioscler, Thromb, Vasc Biol. 2007;27:1238–1247. doi: 10.1161/ATVBAHA.107.139402. [DOI] [PubMed] [Google Scholar]
- 11.Hedenius PER, Wilander O. The Influence of Intravenous Injections of Heparin in Man on the Time of Coagulation. 1. Acta Medica Scandinavica. 1936;88:443–449. [Google Scholar]
- 12.Cade JF, Buchanan MR, Boneu B, Ockelford P, Carter CJ, Cerskus AL, Hirsh J. A comparison of the anti thrombotic and haemorrhagic effects of low molecular weight heparin fractions: The influence of the method of preparation. Thromb Res. 1984;35:613–625. doi: 10.1016/0049-3848(84)90265-2. [DOI] [PubMed] [Google Scholar]
- 13.Frydman AM, Bara L, Le Roux Y, Woler M, Chauliac F, Samama MM. The antithrombotic activity and pharmacokinetics of enoxaparine, a low molecular weight heparin, in humans given single subcutaneous doses of 20 to 80 mg. J Clin Pharmacol. 1988;28:609–618. doi: 10.1002/j.1552-4604.1988.tb03184.x. [DOI] [PubMed] [Google Scholar]
- 14.Handeland GF, Abildgaard U, Holm HA, Arnesen KE. Dose adjusted heparin treatment of deep venous thrombosis: a comparison of unfractionated and low molecular weight heparin. Eur J Clin Pharmacol. 1990;39:107–112. doi: 10.1007/BF00280041. [DOI] [PubMed] [Google Scholar]
- 15.Koopman MMW, Prandoni P, Piovella F, Ockelford PA, Brandjes DPM, van der Meer J, Gallus AS, Simonneau Gr, Chesterman CH, Prins MH, et al. Treatment of Venous Thrombosis with Intravenous Unfractionated Heparin Administered in the Hospital as Compared with Subcutaneous Low-Molecular-Weight Heparin Administered at Home. N Engl J Med. 1996;334:682–687. doi: 10.1056/NEJM199603143341102. [DOI] [PubMed] [Google Scholar]
- 16.Arbit E, Goldberg M, Gomez-Orellana I, Majuru S. Oral heparin: status review. Thrombosis Journal. 2006;4:6. doi: 10.1186/1477-9560-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Homar M, Cegnar M, Kotnik M, Peternel L. Toward Effective Long-Term Prevention of Thromboembolism: Novel Oral Anticoagulant Delivery Systems. Semin Thromb Hemost. 2010;31:113, 122. doi: 10.1055/s-0030-1248730. [DOI] [PubMed] [Google Scholar]
- 18.Baughman RA, Kapoor SC, Agarwal RK, Kisicki J, Catella-Lawson F, FitzGerald GA. Oral Delivery of Anticoagulant Doses of Heparin: A Randomized, Double-Blind, Controlled Study in Humans. Circulation. 1998;98:1610–1615. doi: 10.1161/01.cir.98.16.1610. [DOI] [PubMed] [Google Scholar]
- 19.Rivera TM, Leone-Bay A, Paton DR, Leipold HR, Baughman RA. Oral Delivery of Heparin in Combination with Sodium N-[8-(2-Hydroxybenzoyl)amino]caprylate: Pharmacological Considerations. Pharm Res. 1997;14:1830–1834. doi: 10.1023/a:1012160703533. [DOI] [PubMed] [Google Scholar]
- 20.Majuru S. Advances in the Oral Delivery of Heparin from Solid Dosage Forms Using Emisphere’s eligen® Oral Drug Delivery Technology. Drug Delivery Technology. 2004;4:84–89. [Google Scholar]
- 21.Mousa SA, Zhang F, Aljada A, Chaturvedi S, Takieddin M, Zhang H, Chi L, Castelli MC, Friedman K, Goldberg MM, et al. Pharmacokinetics and Pharmacodynamics of Oral Heparin Solid Dosage Form in Healthy Human Subjects. J Clin Pharmacol. 2007;47:1508–1520. doi: 10.1177/0091270007307242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leone-Bay A, Paton DR, Variano B, Leipold H, Rivera T, Miura-Fraboni J, Baughman RA, Santiago N. Acylated non-[alpha]-amino acids as novel agents for the oral delivery of heparin sodium, USP. J Control Release. 1998;50:41–49. doi: 10.1016/s0168-3659(97)00101-6. [DOI] [PubMed] [Google Scholar]
- 23.Park JW, Jeon OC, Kim SK, Al-Hilal TA, Moon HT, Kim CY, Byun Y. Anticoagulant Efficacy of Solid Oral Formulations Containing a New Heparin Derivative. Mol Pharm. 2010;7:836–843. doi: 10.1021/mp900319k. [DOI] [PubMed] [Google Scholar]
- 24.Jiao Y, Ubrich N, Marchand-Arvier M, Vigneron C, Hoffman M, Lecompte T, Maincent P. In Vitro and In Vivo Evaluation of Oral Heparin-Loaded Polymeric Nanoparticles in Rabbits. Circulation. 2002;105:230–235. doi: 10.1161/hc0202.101988. [DOI] [PubMed] [Google Scholar]
- 25.Hoffart V, Lamprecht A, Maincent P, Lecompte T, Vigneron C, Ubrich N. Oral bioavailability of a low molecular weight heparin using a polymeric delivery system. J Control Release. 2006;113:38–42. doi: 10.1016/j.jconrel.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 26.Hayes PY, Ross BP, Thomas BG, Toth I. Polycationic lipophilic-core dendrons as penetration enhancers for the oral administration of low molecular weight heparin. Bioorg Med Chem. 2006;14:143–152. doi: 10.1016/j.bmc.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 27.Dal Pozzo A, Acquasaliente M, Geron MR, Andriuoli G. New heparin complexes active by intestinal absorption: I-multiple ion pairs with basic organic compounds. Thromb Res. 1989;56:119–124. doi: 10.1016/0049-3848(89)90014-5. [DOI] [PubMed] [Google Scholar]
- 28.Spahn G. Compliance with Self-Administration of Heparin Injections in Outpatients. European Journal of Trauma. 2002;28:104–109. [Google Scholar]
- 29.Weitz JI, Linkins L-A. Beyond heparin and warfarin: the new generation of anticoagulants. Expert Opin Invest Drugs. 2007;16:271–282. doi: 10.1517/13543784.16.3.271. [DOI] [PubMed] [Google Scholar]
- 30.Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM. Polymeric Systems for Controlled Drug Release. Chem Rev (Washington, DC, U S) 1999;99:3181–3198. doi: 10.1021/cr940351u. [DOI] [PubMed] [Google Scholar]
- 31.Monagle P, Chalmers E, Chan A, deVeber G, Kirkham F, Massicotte P, Michelson AD. Antithrombotic Therapy in Neonates and Children*. Chest. 2008;133:887S–968S. doi: 10.1378/chest.08-0762. [DOI] [PubMed] [Google Scholar]
- 32.Andrew M, Vegh P, Johnston M, Bowker J, Ofosu F, Mitchell L. Maturation of the hemostatic system during childhood. Blood. 1992;80:1998–2005. [PubMed] [Google Scholar]
- 33.Andrew M, Paes B, Milner R, Johnston M, Mitchell L, Tollefsen D, Powers P. Development of the human coagulation system in the full-term infant. Blood. 1987;70:165–172. [PubMed] [Google Scholar]
- 34.Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G. Pharmacology and Management of the Vitamin K Antagonists*. Chest. 2008;133:160S–198S. doi: 10.1378/chest.08-0670. [DOI] [PubMed] [Google Scholar]
- 35.White RH, Zhou H, Romano P, Mungall D. Changes in plasma warfarin levels and variations in steady-state prothrombin times. Clin Pharmacol Ther. 1995;58:588–593. doi: 10.1016/0009-9236(95)90179-5. [DOI] [PubMed] [Google Scholar]
- 36.Hoffman AS. Hydrogels for biomedical applications. Adv Drug Delivery Rev. 2002;54:3–12. doi: 10.1016/s0169-409x(01)00239-3. [DOI] [PubMed] [Google Scholar]
- 37.Baldwin AD, Kiick KL. Polysaccharide-modified synthetic polymeric biomaterials. Peptide Science. 2010;94:128–140. doi: 10.1002/bip.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nie T, Baldwin A, Yamaguchi N, Kiick KL. Production of heparin-functionalized hydrogels for the development of responsive and controlled growth factor delivery systems. J Control Release. 2007;122:287–296. doi: 10.1016/j.jconrel.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tae G, Scatena M, Stayton PS, Hoffman AS. PEG-cross-linked heparin is an affinity hydrogel for sustained release of vascular endothelial growth factor. J Biomater Sci, Polym Ed. 2006;17:187–197. doi: 10.1163/156856206774879090. [DOI] [PubMed] [Google Scholar]
- 40.Cai S, Liu Y, Zheng Shu X, Prestwich GD. Injectable glycosaminoglycan hydrogels for controlled release of human basic fibroblast growth factor. Biomaterials. 2005;26:6054–6067. doi: 10.1016/j.biomaterials.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 41.Gutowska A, Bae YH, Feijen J, Kim SW. Heparin release from thermosensitive hydrogels. J Control Release. 1992;22:95–104. [Google Scholar]
- 42.Vasudev SC, Chandy T, Sharma CP. Development of chitosan/polyethylene vinyl acetate co-matrix: controlled release of aspirin-heparin for preventing cardiovascular thrombosis. Biomaterials. 1997;18:375–381. doi: 10.1016/s0142-9612(96)00131-7. [DOI] [PubMed] [Google Scholar]
- 43.Brazel CS, Peppas NA. Pulsatile local delivery of thrombolytic and antithrombotic agents using poly(N-isopropylacrylamide-co-methacrylic acid) hydrogels. J Control Release. 1996;39:57–64. [Google Scholar]
- 44.Hermanson GT. Bioconjugate techniques. San Diego, California: Academic Press; 2008. p. 1202. [Google Scholar]
- 45.Macintosh FC. A colorimetric method for the standardization of heparin preparations. Biochem J. 1941;35:776–782. doi: 10.1042/bj0350776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raad I, Hachem R, Tcholakian RK, Sherertz R. Efficacy of Minocycline and EDTA Lock Solution in Preventing Catheter-Related Bacteremia, Septic Phlebitis, and Endocarditis in Rabbits. Antimicrob Agents Chemother. 2002;46:327–332. doi: 10.1128/AAC.46.2.327-332.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Motlekar NA, Srivenugopal KS, Wachtel MS, Youan B-BC. Evaluation of the oral bioavailability of low molecular weight heparin formulated with glycyrrhetinic acid as permeation enhancer. Drug Dev Res. 2006;67:166–174. doi: 10.1002/ddr.20087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Osmond RIW, Kett WC, Skett SE, Coombe DR. Protein-heparin interactions measured by BIAcore 2000 are affected by the method of heparin immobilization. Anal Biochem. 2002;310:199–207. doi: 10.1016/s0003-2697(02)00396-2. [DOI] [PubMed] [Google Scholar]
- 49.Young JJ, Cheng KM, Tsou TL, Liu HW, Wang HJ. Preparation of cross-linked hyaluronic acid film using 2-chloro-1-methylpyridinium iodide or water-soluble 1-ethyl-(3,3-dimethylaminopropyl)carbodiimide. J Biomater Sci, Polym Ed. 2004;15:767–780. doi: 10.1163/156856204774196153. [DOI] [PubMed] [Google Scholar]
- 50.Yu L, Ding J. Injectable hydrogels as unique biomedical materials. Chem Soc Rev. 2008;37:1473–1481. doi: 10.1039/b713009k. [DOI] [PubMed] [Google Scholar]
- 51.Schelté P, Boeckler C, Frisch B, Schuber F. Differential Reactivity of Maleimide and Bromoacetyl Functions with Thiols: Application to the Preparation of Liposomal Diepitope Constructs. Bioconjug Chem. 1999;11:118–123. doi: 10.1021/bc990122k. [DOI] [PubMed] [Google Scholar]
- 52.Lutolf MP, Hubbell JA. Synthesis and Physicochemical Characterization of End-Linked Poly(ethylene glycol)-co-peptide Hydrogels Formed by Michael-Type Addition. Biomacromolecules. 2003;4:713–722. doi: 10.1021/bm025744e. [DOI] [PubMed] [Google Scholar]
- 53.Zheng Shu X, Liu Y, Palumbo FS, Luo Y, Prestwich GD. In situ crosslinkable hyaluronan hydrogels for tissue engineering. Biomaterials. 2004;25:1339–1348. doi: 10.1016/j.biomaterials.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 54.Hiemstra C, van der Aa LJ, Zhong Z, Dijkstra PJ, Feijen J. Novel in Situ Forming, Degradable Dextran Hydrogels by Michael Addition Chemistry:_ Synthesis, Rheology, and Degradation. Macromolecules. 2007;40:1165–1173. [Google Scholar]
- 55.Latridis JC, Wu J, Yandow JA, Langevin HM. Subcutaneous tissue mechanical behavior is linear and viscoelastic under uniaxial tension. Connective Tissue Research. 2003;44:208–217. [PubMed] [Google Scholar]
- 56.Rydholm AE, Anseth KS, Bowman CN. Effects of neighboring sulfides and pH on ester hydrolysis in thiol-acrylate photopolymers. Acta Biomaterialia. 2007;3:449–455. doi: 10.1016/j.actbio.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reddy SK, Anseth KS, Bowman CN. Modeling of network degradation in mixed step-chain growth polymerizations. Polymer. 2005;46:4212–4222. [Google Scholar]
- 58.Rydholm AE, Reddy SK, Anseth KS, Bowman CN. Controlling Network Structure in Degradable Thiol-Acrylate Biomaterials to Tune Mass Loss Behavior. Biomacromolecules. 2006;7:2827–2836. doi: 10.1021/bm0603793. [DOI] [PubMed] [Google Scholar]
- 59.Bisio A, Vecchietti D, Citterio L, Guerrini M, Raman R, Bertini S, Eisele G, Naggi A, Sasisekharan R, Torri G. Structural features of low-molecular-weight heparins affecting their affinity to antithrombin. Thromb Haemost. 2009;102:865–873. doi: 10.1160/TH09-02-0081. [DOI] [PubMed] [Google Scholar]
- 60.Bourne DW. Pharmacokinetics. In: Banker GS, Rhodes CT, editors. Modern Pharmaceutics. New York: Marcel Dekker inc; 2002. p. 838. [Google Scholar]
- 61.Jeske WP, Walenga JM, Hoppensteadt DA, Vandenberg C, Brubaker A, Adiguzel C, Bakhos M, Fareed J. Differentiating Low-Molecular-Weight Heparins Based on Chemical, Biological, and Pharmacologic Properties: Implications for the Development of Generic Versions of Low-Molecular-Weight Heparins. Semin Thromb Hemost. 2008;34:074, 085. doi: 10.1055/s-2008-1066026. [DOI] [PubMed] [Google Scholar]
- 62.Tamada JA, Langer R. Erosion kinetics of hydrolytically degradable polymers. Proceedings of the National Academy of Sciences. 1993;90:552–556. doi: 10.1073/pnas.90.2.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schoenmakers RG, van de Wetering P, Elbert DL, Hubbell JA. The effect of the linker on the hydrolysis rate of drug-linked ester bonds. J Control Release. 2004;95:291–300. doi: 10.1016/j.jconrel.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 64.Blatný J, Fiamoli V. Treatment of deep vein thrombosis with continuous intravenous infusion of LMWH in children--an alternative to subcutaneous application when needed. Vnitr ní lékar ství. 2009;55:227. [PubMed] [Google Scholar]
- 65.Fiamoli V, Blatný J, Zapletal O, Kohlerova S, Janousova E. Treatment of Deep Vein Thrombosis with Continuous IV Infusion of LMWH: A Retrospective Study in 32 Children. Thrombosis. 2011;2011 doi: 10.1155/2011/981497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hommes DW, Bura A, Mazzolai L, Büller HR, ten Cate JW. Subcutaneous Heparin Compared with Continuous Intravenous Heparin Administration in the Initial Treatment of Deep Vein Thrombosis. Ann Intern Med. 1992;116:279–284. doi: 10.7326/0003-4819-116-4-279. [DOI] [PubMed] [Google Scholar]
- 67.Hull RD, Raskob GE, Hirsh J, Jay RM, Leclerc JR, Geerts WH, Rosenbloom D, Sackett DL, Anderson C, Harrison L, et al. Continuous Intravenous Heparin Compared with Intermittent Subcutaneous Heparin in the Initial Treatment of Proximal-Vein Thrombosis. N Engl J Med. 1986;315:1109–1114. doi: 10.1056/NEJM198610303151801. [DOI] [PubMed] [Google Scholar]
- 68.Hull RD, Raskob GE, Pineo GF, Green D, Trowbridge AA, Elliott CG, Lerner RG, Hall J, Sparling T, Brettell HR, et al. Subcutaneous Low-Molecular-Weight Heparin Compared with Continuous Intravenous Heparin in the Treatment of Proximal-Vein Thrombosis. N Engl J Med. 1992;326:975–982. doi: 10.1056/NEJM199204093261502. [DOI] [PubMed] [Google Scholar]
- 69.Prandoni P, Carta M, Cogo A, Ruol A, Vigo M, Casara D, Lensing AWA, Büller HR, ten Cate JW. Comparison of subcutaneous low-molecular-weight heparin with intravenous standard heparin in proximal deep-vein thrombosis. The Lancet. 1992;339:441–445. doi: 10.1016/0140-6736(92)91054-c. [DOI] [PubMed] [Google Scholar]
- 70.Monte AA, Bodmer M, Schaeffer TH. Low-Molecular-Weight Heparin Overdose: Management by Observation. The Annals of Pharmacotherapy. 2010;44:1836–1839. doi: 10.1345/aph.1P318. [DOI] [PubMed] [Google Scholar]
- 71.Ngo A, Tan D, Olson K. Low Molecular Weight Heparin Overdose. Clinical Toxicology. 2008;46:388. [Google Scholar]
- 72.Romagnoli F, Banfi R, Cinaglia I, Balloni L, Paciello A. A massive overdose of the low molecular weight heparin, parnaparin: a case report. European Journal of Hospital Pharmacy Practice. 2007;13:32–33. [Google Scholar]
- 73.Carr JA, Silverman N. The heparin-protamine interaction. A review. Journal of cardiovascular surgery. 1999;40:659. [PubMed] [Google Scholar]
- 74.Levy JH, Cormack JG, Morales A. Heparin neutralization by recombinant platelet factor 4 and protamine. Anesth Analg. 1995;81:35. doi: 10.1097/00000539-199507000-00007. [DOI] [PubMed] [Google Scholar]
- 75.Veronese FM, Pasut G. PEGylation, successful approach to drug delivery. Drug Discov Today. 2005;10:1451–1458. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.