

Abstract
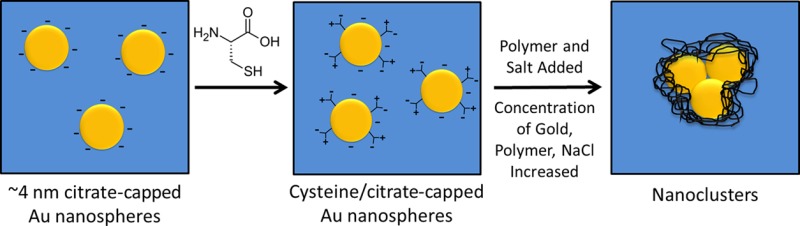
Gold nanospheres coated with a binary monolayer of bound citrate and cysteine ligands were assembled into nanoclusters, in which the size and near-infrared (NIR) extinction were tuned by varying the pH and concentration of added NaCl. During full evaporation of an aqueous dispersion of 4.5 ± 1.8 nm Au primary particles, the nanoclusters were formed and quenched by the triblock copolymer polylactic acid (PLA)(1K)-b-poly(ethylene glycol) (PEG)(10K)-b-PLA(1K), which also provided steric stabilization. The short-ranged depletion and van der Waals attractive forces were balanced against longer ranged electrostatic repulsion to tune the nanocluster diameter and NIR extinction. Upon lowering the pH from 7 to 5 at a given salinity, the magnitude of the charge on the primary particles decreased, such that the weaker electrostatic repulsion increased the hydrodynamic diameter and, consequently, NIR extinction of the clusters. At a given pH, as the concentration of NaCl was increased, the NIR extinction decreased monotonically. Furthermore, the greater screening of the charges on the nanoclusters weakened the interactions with PLA(1K)-b-PEG(10K)-b-PLA(1K) and thus lowered the amount of adsorbed polymer on the nanocluster surface. The generalization of the concept of self-assembly of small NIR-active nanoclusters to include a strongly bound thiol and the manipulation of the morphologies and NIR extinction by variation of pH and salinity not only is of fundamental interest but also is important for optical biomedical imaging and therapy.
Introduction
Gold nanoparticles which absorb light strongly in the near-infrared (NIR) wavelength region from 700 to 1100 nm, where blood and tissue absorb weakly,1 are of great utility in biomedical imaging modalities such as photoacoustic imaging.2−7 Strong NIR extinction is often produced from nanoclusters of closely spaced primary gold spheres, which can be assembled in vivo(8,9) or in vitro.10−12 Here, the close spacings of individual nanoparticles within the clusters produce dipoles, quadrupoles, and higher-order multipoles that shift the surface plasmon resonance (SPR) to the NIR region.13 Gold nanoparticles have been assembled with organic templates such as polymers, proteins, and DNA.14 The interparticle spacing may be controlled in order to tune the NIR extinction properties of the assemblies.15 In most cases, substantial amounts of inactive templating agent were required which may limit the spectral properties or functionality of the active material by affecting, for example, the spacing between gold plasmonic nanoparticles. Primary nanoparticles may be assembled into clusters of controlled sizes with small amounts of structure-directing agents by properly balancing the relevant colloidal forces.16,10,11,17,18,12,19 Clusters of nanoparticles have also been formed during synthesis of primary particles from precursors in both organic20 and aqueous solvents in the presence of various stabilizers.21−23 In these cases, the kinetics of primary particle formation must be synchronized properly relative to the kinetics of nucleation and growth of the primary particles into clusters to prevent excessive growth and precipitation. The reversibility of nanoclusters to dissociate back into individual primary particles has received relatively little attention. In many cases, bridges between particles formed from soluble precursors will fuse the clusters together permanently.21
A highly versatile synthetic concept has been presented to assemble presynthesized primary particles into clusters by tuning equilibrium and nonequilibrium colloidal interactions, even for small amounts of stabilizers.16,10,11,17,18,12 For example, primary iron oxide particles (∼6 nm in diameter) have been assembled into clusters of ∼100 to ∼500 nm by controlling solvophobic interactions between oleic acid ligands on the primary particles and the ethylene glycol solvent.16 Additionally, poly(ethylene glycol) (PEG)-capped gold nanoparticles have been assembled into clusters by modulating interparticle steric repulsions via an alkanethiol addition in water.17 Recently, our group has developed a “quenched equilibrium” assembly method to assemble small (∼4 nm) gold nanoparticles into clusters of controlled size and intense NIR extinction which reversibly dissociate to monomer.10−12 The nanocluster diameter was predicted semiquantitatively with a free energy equilibrium model whereby attractive, short-ranged van der Waals and depletion interactions were balanced against repulsive, longer-ranged electrostatic interactions. Nanoclusters were “quenched” at a metastable equilibrium size by the adsorption of a biodegradable triblock copolymer, PLA(1K)-b-PEG(10K)-b-PLA(1K), to the gold surface. At the minimum free energy, the equilibrium number of particles n* in a nanocluster for primary particles each with charge q is24,25,12
| 1 |
where a is the attractive short-ranged interaction energy between particles (in kT units), R is the primary particle radius (in nm), and λb is the Bjerrum length (in nm).12 Previously, this model has been used to predict the size of equilibrium Au nanoclusters semiquantitatively, whereby short-ranged van der Waals and depletion interactions were tuned by varying Au and polymer concentrations, respectively.12 The clusters, moreover, reversibly dissociated to monomer upon degradation of the polymeric quencher.10−12
While recent studies10,12 provided insight into the mechanism of formation of reversible gold nanoclusters with high NIR extinction, the weak binding of the lysine ligands on the Au surfaces may have limited the nanocluster stability. In the protonated form, the amine group in lysine is known to bind very weakly to Au,26 although it forms ion pairs with citrate ligands on Au surfaces.27 A weakly bound ligand may be displaced in vivo or in cells by free thiols such as glutathione (GSH), as has been observed in related systems.28,29 To overcome this stability limitation, many strongly bound ligands, for example, thiolated or zwitterionic molecules, may be investigated. Although nanoclusters with NIR absorbance have been formed with PEG-SH, the size was not reported.10
Recently, we reported ∼4 nm Au primary particles capped with a binary monolayer of cysteine and citrate ligands at a ratio of 1.6/1 that do not adsorb any protein when incubated in fetal bovine serum, despite a moderate surface charge (zeta potential of −22 mV).19 We hypothesize that the zwitterionic tips on the cysteine facilitate the weak protein adsorption by shielding the protein from the buried carboxylate charged groups of citrate. However, it remains unknown whether nanoclusters could be formed from these primary particles. Furthermore, the effect of the charge of the primary particle on the nanocluster morphology has received little attention, and the roles of pH and salinity have not been investigated.12,19
Herein, we extend the concept of colloidal assembly of quenched nanoclusters to the case of primary Au nanospheres capped with a mixed monolayer containing a zwitterionic strongly bound thiol ligand, cysteine, along with citrate. Furthermore, the nanocluster size and UV–vis–NIR extinction are shown to be tunable by varying the pH and/or the concentration of added NaCl, which modulate the particle charge and degree of polymer adsorption. The compositions of the mixed ligand monolayers on the surface of primary gold nanoparticles are controlled via a place exchange reaction of anionic citrate ligands with zwitterionic cysteine ligands (see Figure 1). A dispersion of cysteine/citrate-capped Au nanospheres was assembled into nanoclusters upon mixing with an aqueous solution of the triblock copolymer PLA(1K)-b-PEG(10K)-b-PLA(1K) and subsequent complete evaporation of the solvent.12 The nanocluster size and NIR extinction are shown to increase with a decrease in surface charge (electrostatic repulsion) upon lowering the pH. At a given pH, an increase in salinity is used to vary the NIR extinction. Furthermore, the Debye screening weakens the interactions between the Au surfaces and the copolymer, resulting in a decrease in polymer content in the nanoclusters. The ability to further tailor the morphology and NIR properties of Au nanoclusters with the variables of pH and salinity and to advance the understanding of the kinetic and equilibrium aspects of the self-assembly mechanism is not only of scientific interest but also of practical interest for optical biomedical imaging and therapy.
Figure 1.
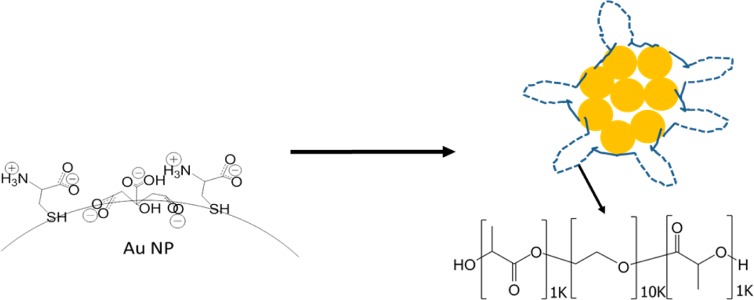
Schematic of nanoclusters assembled from cysteine/citrate-capped Au nanospheres and stabilized with PLA(1K)-b-PEG(10K)-b-PLA(1K). The nanoclusters are formed upon mixing Au dispersions with polymer solutions, in some cases containing NaCl, and then full evaporation of the solvent. The dashed lines for the adsorbed polymers on the clusters represent PEG loops and the solid lines the PLA end groups.
Experimental Section
Materials
HAuCl4·3H2O was purchased from MP Biomedicals LLC (Solon, OH). Na3C3H5O(COO)3·2H2O, NaBH4 from Fisher Scientific (Fair Lawn, NJ), and l-cysteine from Acros Chemicals (Morris Plains, NJ). PLA(1K)-b-PEG(10K)-b-PLA(1K) was acquired from Sigma-Aldrich (St. Louis, MO). Phosphate-buffered saline (PBS) was purchased from Gibco (Grand Island, NY).
Synthesis of Citrate-Capped Au Nanospheres and Cysteine Ligand Exchange
The ∼4 nm citrate-capped Au nanospheres were synthesized by the NaBH4 reduction of HAuCl4 and purified by tangential flow filtration, in an identical method to previous studies.12 Cysteine place exchange was conducted according to a previously described method.19 Briefly, a solution of 1% (w/v) cysteine in deionized water was freshly prepared. In a typical experiment, 3.2 μL of this solution was added to 0.6 mL of a 3 mg/mL citrate-capped Au nanosphere dispersion, and the mixture was stirred at room temperature for 15 min. To test exchange stability, a single nanosphere sample was reacted for 24 h at room temperature as well. Immediately after the 15 min reactions, nanospheres were either used for nanocluster formation or diluted for characterization.
Nanosphere Characterization
Following cysteine ligand exchange, nanosphere samples were either diluted to a concentration of ∼0.04 mg/mL Au in DI water for dynamic light scattering (DLS) or UV–visible–NIR (UV–vis–NIR) analysis or diluted to ∼0.04 mg/mL Au in 1 mM KCl for zeta potential analysis. DLS measurements were taken on these diluted samples using a Brookhaven ZetaPALS analyzer with a scattering angle of 90° as reported previously.12,19 Each sample was filtered through a 200 nm poly(ether sulfone) filter prior to testing. The data were analyzed using the CONTIN method (Brookhaven DLS software version 3.34), and the Stokes–Einstein equation was used to obtain intensity-weighted distribution of hydrodynamic diameters. The data were fit using a 32-channel autoslope analysis option on each sample without specifying a minimum or maximum bound. UV–vis–NIR spectroscopy of diluted nanocluster samples was obtained using a Varian Cary 60 spectrophotometer with a path length of 1 cm. Unless otherwise noted, the extinction was normalized as unity at the peak wavelength longer than 500 nm. Unless specified otherwise, zeta potential measurements were performed using a Brookhaven ZetaPALS analyzer with an applied electric field of 5 V/cm, in which 10 runs of 30-cycle measurements were taken. In a few samples, zeta potentials taken of the 24 h cysteine place exchanged nanospheres were obtained using a ZetaPlus analyzer in high precision mode of 30 single-cycle measurements under an applied electric field of 15 V/cm. The average and standard deviations of these measurements are reported. Because of the nanocluster size (see Results), the Hückel model was used to relate the measured electrophoretic mobility to a zeta potential.30 Au dispersion concentrations were determined by flame atomic absorption spectroscopy (FAAS), utilizing a GBC 908AA analyzer (GBC Scientific Equipment Pty Ltd.) with an air/acetylene flame at a wavelength of 242.8 nm. Citrate ligand weight percents were measured on an OI Analytical 1030 TOC analyzer run in wet oxidation mode with 20 wt % sodium persulfate (Acros Chemical, Morris Plains, NJ). A sample of citrate-capped Au nanoparticles was diluted in DI water to ∼4 ppm Au and run in nonpurgeable organic carbon (NPOC) mode with a reaction time of 3 min.
Nanocluster Formation
Immediately after the completion of the cysteine place exchange reaction, cysteine/citrate-capped Au nanospheres were used to synthesize nanoclusters in a manner adapted from a previously reported method.12 A 30 mg/mL solution of PLA(1K)-b-PEG(10K)-b-PLA(1K) was freshly prepared, with salt concentrations ranging from 0 to 100 mM. 250 μL of a 30 mg/mL polymer solution was added to 0.5 mL of 3 mg/mL cysteine/citrate-capped nanospheres under vigorous stirring in order to achieve a 5/1 polymer/Au ratio. The polymer–gold mixture was then adjusted to the desired pH, as measured by a Mettler Toledo InLab Micro pH probe, using either 0.1 M HCl or 0.1 M NaOH and added to a 15 mm × 45 mm glass vial, which was placed in a 40 °C water bath and stirred vigorously. Dried air was subsequently blown over the sample through a small tube inserted into the vial at a flow rate of approximately 26 L/min. In all cases, the aqueous solvent was fully evaporated, which typically took ∼35 min to form a film. After solvent evaporation, the nanoclusters were dispersed by adding 30 mL of deionized water to the sample. The resulting solution was then centrifuged in a 50 mL polystyrene centrifuge tube at 9700 rpm for 10 min, resulting in ∼1 mL of a lower colloidal phase of densely packed nanoclusters, which was collected. An upper colloidal phase (∼29 mL) of monomer Au and highly fractal smaller aggregates was discarded. A light meniscus was observed between the dense lower phase and the upper phase.
Nanocluster Characterization
Nanocluster morphology was assessed by transmission electron microscopy (TEM), which was performed on an FEI TECNAI G2 F20 X-TWIN TEM using a high-angle annular dark-field detector. Samples were prepared by first dipping a 200 mesh copper-coated carbon type-A TEM grid (Electron Microscopy Sciences, Hatfield, PA) into liquid nitrogen. A 3 μL drop of dilute nanocluster dispersion was then pipetted onto the grid, which was then subsequently dried using a VirTis Advantage tray lyophilizer (VirTis, Gardiner, NY). DLS, UV–vis–NIR, and zeta potential measurements were performed on the nanoclusters in an identical manner to the primary nanospheres at the same mg/mL concentration of Au. The total carbon weight percent was measured using the same OI Analytical 1030 TOC analyzer used for citrate analysis above. The weight percent of carbon was then derived from the concentration of CO2 detected, in ppm, for samples with a known Au concentration by UV–vis measurements at 400 nm.
Results and Discussion
Synthesis of Cysteine/Citrate Nanospheres
The cysteine/citrate-capped nanospheres used in this study were synthesized identically to nanospheres reported in a previous study.19 The UV–vis–NIR spectra and DLS Dh distribution of these nanospheres (Figure S1) at ∼pH 7 were determined to be essentially the same as those in the previous study19 Also, the zeta potential of the nanospheres at pH 7 and a concentration of 1 mM KCl was −22.5 ± 0.6 mV, similar to the earlier value of −21.6 ± 1.7 mV.19 With 25 mM acetic acid buffer at pH 7 a similar value of −22.3 ± 2.3 mV was measured. The zeta potentials were not measured at a salinity of 100 mM NaCl at pH 7 as the primary particles underwent aggregation. Moreover, in order to verify the degree of the cysteine ligand exchange with time, the zeta potential of the primary particles in 1 mM KCl was measured after 24 h of exchange. Here, the zeta potential after 24 h was −19.6 ± 2.4 mV, similar to the zeta potential of −22.5 ± 0.6 mV after 15 min. Thus, we conclude that the cysteine ligand exchange reaction reached a steady state equilibrium value after ∼15 min. On the basis of these measurements and expected electrostatic screening with added salt, the changes in pH and salinity in this study are sufficient to manipulate the nanoparticle surface charge.
Effect of pH on Nanocluster Formation
For pH values of 5 and 7, the nanocluster properties were determined either without added salt or with initial salinities of 17 or 33 mM (Table 1). Each sample is referred to by the notation of “pH-initial salinity”, i.e., 7–17 for pH 7 and 17 mM initial NaCl after polymer addition. The TEMs indicated a high polydispersity in the cluster size as shown in Figure 2b,c, where sizes were on the order of ∼20 to ∼100 nm. Given the high polydispersity, DLS intensity-weighted size distributions are reported throughout this paper, and it was not attempted to determine volume distributions. It is however important to recognize that the mean size would be much smaller for the volume distribution. For samples made with no added NaCl, the hydrodynamic diameter of the clusters in the range of 40–250 nm shifted to smaller sizes with a pH increase from 5 to 7. This size decrease is consistent with the free energy model (eq 1), as the charged ligands on the gold surface become more deprotonated, which increases the magnitude of the charge. Thus, the resulting stronger electrostatic repulsion between primary particles would limit growth of clusters as observed. In this self-limited growth model (eq 1), as the overall charge in the cluster increases with each added primary particle, the cluster will eventually repel the addition of another charged particle. At pH 7 with larger charges on the primary particles, this condition will be realized at a smaller cluster size as observed.
Table 1. Effects of Solution pH and Salinity on Intensity Weighted Hydrodynamic Diameter Size Distribution, UV–Vis–NIR Extinction Ratios, and Weight Percent of Organic Carbon on the Nanoclusters.
| pH–[NaCl] | initial NaCl conc (mM) | DLS intensity weighted size distributions (nm) (cumulative size, %) | A800/A525 | wt % organic carbon (from TOC) |
|---|---|---|---|---|
| 5–0 | 0 | 5–40 (23) | 0.825 | 26.9 ± 0.6 |
| 40–100 (25) | ||||
| 100–250 (52) | ||||
| 5–17 | 17 | 5–40 (18) | 0.635 | 20.7 ± 0.9 |
| 40–100 (22) | ||||
| 100–250 (60) | ||||
| 5–33 | 33 | 5–40 (12) | 0.619 | 17.9 ± 0.7 |
| 40–100 (19) | ||||
| 100–250 (69) | ||||
| 7–0 | 0 | 5–40 (9) | 0.728 | 24.3 ± 0.5 |
| 40–100 (62) | ||||
| 100–250 (29) | ||||
| 7–17 | 17 | 5–40 (20) | 0.501 | 20.2 ± 0.5 |
| 40–100 (52) | ||||
| 100–250 (28) | ||||
| 7–33 | 33 | 5–40 (20) | 0.419 | 19.4 ± 0.3 |
| 40–100 (4) | ||||
| 100–250 (76) |
Figure 2.

(a) UV–vis–NIR extinction spectra and TEM images for nanoclusters made with 0 mM NaCl at (b) pH 5 and (c) pH 7. The syntax "5−0" denotes pH 5 and 0 mM NaCl, and so on.
The increase in the magnitude of charge from pH 5 to 7 may be shown to influence the decrease in the NIR extinction in two ways. Without added NaCl, the extinction ratio A800/A525 was observed to decrease from 0.825 to 0.728 (Figure 2). For a given spacing between primary particles, the extinction ratio is known to decrease with a decrease in the nanocluster size, as the SPR becomes less polarized for a smaller cluster diameter.10,12 Also, the greater electrostatic repulsion has the potential to increase the spacing between Au primary particles, which would also contribute to a decrease in A800/A525. Each of these trends with pH was also observed for 17 mM initial salinity. Here, the size distribution shifted to smaller sizes at pH 7. Furthermore, the extinction ratio dropped from 0.635 to 0.501 and, for 33 mM NaCl, from 0.619 to 0.419 (Figure 3). The only exception to this similar behavior was the unusually high amount of nanoclusters in the 100−250 nm size range for the 33 mM initial salinity.
Figure 3.
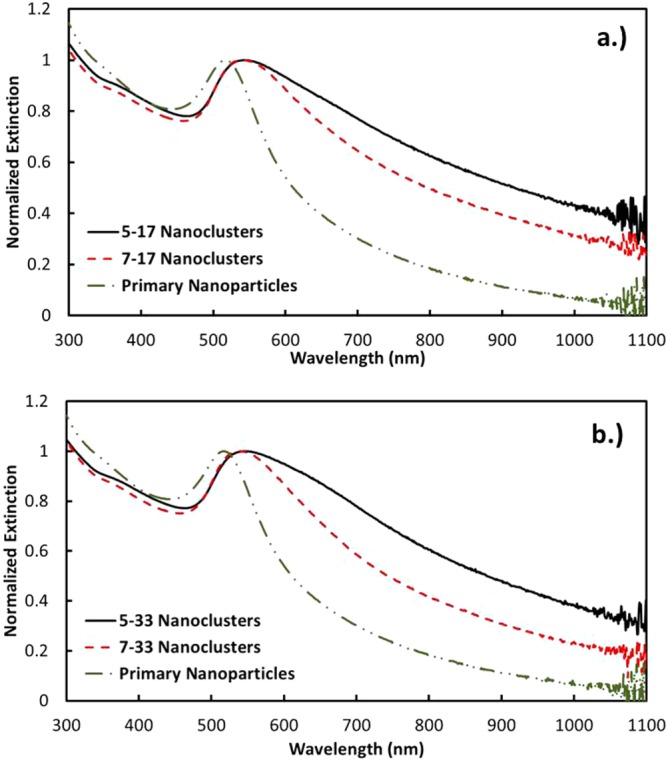
UV–vis–NIR extinction spectra of nanoclusters made with (a) 17 mM and (b) 33 mM initial NaCl concentrations at pH 5 and 7.
The total organic carbon (TOC) was measured for pH values of 5 and 7 at each salinity. For citrate-capped particles alone, the weight % carbon was determined to be 9.68 ± 0.46%. As shown in Table 1 for samples 5–0 and 7–0, the weight % of carbon on the nanoclusters decreased slightly from 26.9 ± 0.6% to 24.3 ± 0.5%, respectively. For the other two salinities the changes in TOC were even smaller. The weight % of the mixed citrate/cysteine monolayer is the same for all samples, as each nanocluster was composed of one type of primary nanoparticle. Thus, the changes in weight % C will only reflect changes in the weight % of adsorbed polymer. Since the pH had little effect on the C weight % at a given salinity, it also did not influence the polymer adsorption.
Effect of Salt at Constant pH
At each pH, the nanoclusters were assembled with the addition of 30 mg/mL polymer solutions containing 0, 17, or 33 mM NaCl. As the salinity increased, the TOC results indicate a decrease in weight % C and thus polymer adsorption. For the triblock PLA-b-PEG-b-PLA copolymer, the addition of NaCl will raise the chemical potential of the polymer in water due to desolvation of PEG,31−33 which alone would enhance the driving force for polymer adsorption. However, salt also screens the charges on bound ligands and thus weakens the charge-dipole interactions and hydrogen bonding with the polymer, which disfavors polymer adsorption. Therefore, the decrease in polymer adsorption indicates that charge screening is the dominant effect. In contrast, the polymer adsorption did not change much with an increase in pH at a given salinity despite the greater magnitude of the Au surface charge. Thus, some compensating effect was present such as the interaction of H+, OH–, Na+, and Cl– ions with the Au surfaces and ether oxygens in the PEG blocks.
As the NaCl concentration increases, the electrostatic repulsion between primary particles decreases, which would be expected to shift the nanocluster size distribution to large sizes according to eq 1. In all but a single case this behavior was observed at each pH except for sample 7–17, which showed a modest gain in the smallest part of the distribution. With greater attraction between the primary particles, the primary particles could potentially become spaced more closely together, which would increase the NIR extinction. Instead, the opposite behavior was observed whereby the NIR extinction decreased with salinity (Figure 4). Perhaps the steric effects of the citrate and cysteine ligands limited the minimum spacing between the primary particles. Another possibility is that the NIR extinction is also influenced by the kinetics of assembly, which may influence the fractal dimensions.34 In this scenario, the clusters may form more rapidly at higher salt concentrations due to the weaker electrostatic repulsion. Thus, the more strongly interacting nanoparticles will have less time to explore the interior of growing clusters and will have a smaller driving force to relax to form more dense aggregates. The behavior would be more indicative of diffusion limited cluster aggregation, resulting in looser, more fractal aggregates which would inherently yield lower NIR extinction. However, any changes in the fractal dimension were not discernible in the TEM images.
Figure 4.

(a) UV–vis–NIR extinction and TEM images of clusters made at pH 7 and (b) 0 mM and (c) 17 mM initial NaCl concentrations, respectively.
Figure 5.

(a) UV–vis–NIR extinction and TEM images of clusters made at (b) 0 mM and (c) 33 mM initial NaCl concentrations, respectively, at pH 5.
Comparison to Nanoclusters Made with Lysine/Citrate Nanospheres
The cysteine/citrate-capped primary Au nanospheres used in this study have been previously shown to completely resist protein adsorption in undiluted fetal bovine serum.19 Interestingly, however, in the present study these primary nanospheres were found to aggregate and settle immediately after incubation in both 1× phosphate buffered saline (PBS) or 150 mM NaCl at a gold concentration of ∼0.04 mg/mL (data not shown), which is approximately the same salinity as FBS. Here charge screening weakens the electrostatic repulsion. The stability in FBS but not in NaCl nor PBS may be attributed to a potential “depletion stabilization” mechanism, whereby the proteins present in FBS impart stability to the primary nanospheres through creating a stabilizing osmotic pressure gradient without physically adsorbing to the nanosphere surfaces.35 This mechanism has been previously proposed for the stabilization of Au nanoparticles in solutions of high salinity with PEG as well as iron oxide nanoparticles in fetal calf serum (FCS)-supplemented cellular growth media.36,37
The use of zwitterionic cysteine instead of cationic lysine as a ligand along with citrate in the mixed monolayer-capped primary nanospheres also significantly affected the nanocluster formation process. The addition of zwitterionic cysteine reduced the charge on the primary particle to −21.6 ± 1.7 mV compared to −16.1 ± 2.9 mV with the addition of cationic lysine,12,19 resulting in a higher q value in eq 1 for cysteine/citrate-capped nanospheres. Thus, in order to obtain similar nanocluster sizes and morphologies, the attraction a had to be increased to overcome the excess repulsion. Thus, higher evaporation extents (∼100% vs 50%) were used to achieve nanoclusters with the closely packed Au nanospheres necessary for intense NIR extinction. Moreover, the use of salt and pH modulation in the formation of cysteine/citrate nanoclusters allowed for lower polymer/Au ratios (5/1 compared with 20/1 for lysine/citrate), as a lower polymer/Au was required in order to achieve a similar level of quenching.
Conclusions
The concept of the colloidal assembly of gold nanoclusters quenched with a polymer stabilizer10−12,19 has been generalized to include primary particles capped with a strongly binding zwitterionic thiol, cysteine, to form a binary monolayer with citrate. Furthermore, the solution pH and salt concentration have been utilized as new variables to tune the particle size, NIR extinction, and degree of polymer adsorption by manipulating the colloidal interactions, particularly the Au surface charge on the primary nanospheres. At each salinity (0–33 mM), the hydrodynamic diameter and NIR extinction increased as the solution pH was decreased from 7 to 5, consistent with weaker electrostatic repulsion, as would be expected from the equilibrium free energy model. Furthermore, at a given pH, the size distribution shifted to larger clusters with salt addition, again as may be expected with weakened electrostatic repulsion. Therefore, the free energy model may be used to provide a unified understanding of the size data in this study over a wide range of conditions in pH and salinity. Finally, the polymer adsorption decreased as the screening of the charges on the Au surfaces with salt weakened the interactions with the polar groups on PLA(1K)-b-PEG(10K)-b-PLA(1K). The ability to further tailor the morphology and NIR properties of Au nanoclusters with the variables of pH and salinity, resulting from the emerging understanding of the kinetic and equilibrium aspects of the self-assembly mechanism in the current and previous studies,11,17,22,12,19 will be highly beneficial for practical application to optical biomedical imaging and therapy.
Acknowledgments
K.P.J. and K.V.S. acknowledge support from NSF (CBET-0968038) and NIH (CA143663). K.P.J. and T.M.T acknowledge support from the Welch Foundation (F-1319 and F-1696, respectively) and NSF (CBET-1247945).
Supporting Information Available
Additional UV-vis-NIR and DLS data for nanospheres. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
A.K.M and R.J.S. contributed equally to this manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Weissleder R. A clearer vision for in vivo imaging. Nat. Biotechnol. 2001, 19, 316–317. [DOI] [PubMed] [Google Scholar]
- Agarwal A.; Huang S. W.; O’Donnell M.; Day K. C.; Day M.; Ashkenazi S. Targeted gold nanorod contrast agent for prostate cancer detection by photoacoustic imaging. J. Appl. Phys. 2007, 102, 0647011–0647014. [Google Scholar]
- Mallidi S.; Larson T.; Tam J.; Joshi P. P.; Karplouk A.; Sokolov K.; Emelianov S. Multiwavelength photoacoustic imaging and plasmon resonance coupling of gold nanoparticles for selective detection of cancer. Nano Lett. 2009, 9, 2825–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K. H.; Kim C.; Cobley C. M.; Xia Y.; Wang L. V. Near-infrared gold nanocages as a new class of tracers for photoacoustic sentinel lymph node mapping on a rat model. Nano Lett. 2009, 9, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa S.; Agrawal A.; Rodríguez-Lorenzo L.; Pastoriza-Santos I.; Alvarez-Puebla R. n. A.; Kornowski A.; Weller H.; Liz-Marzán L. M. Tuning size and sensing properties in colloidal gold nanostars. Langmuir 2010, 26, 14943–14950. [DOI] [PubMed] [Google Scholar]
- Yoon S. J.; Mallidi S.; Tam J. M.; Tam J. O.; Murthy A.; Johnston K. P.; Sokolov K. V.; Emelianov S. Y. Utility of biodegradable plasmonic nanoclusters in photoacoustic imaging. Opt. Lett. 2010, 35, 3751–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreaden E. C.; Alkilany A. M.; Huang X.; Murphy C. J.; El-Sayed M. A. The golden age: gold nanoparticles for biomedicine. Chem. Soc. Rev. 2012, 41, 2740–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aaron J.; Nitin N.; Travis K.; Kumar S.; Collier T.; Park S. Y.; Jose-Yacaman M.; Coghlan L.; Follen M.; Richards-Kortum R.; et al. Plasmon resonance coupling of metal nanoparticles for molecular imaging of carcinogenesis in vivo. J. Biomed. Opt. 2007, 12, 1–11. [DOI] [PubMed] [Google Scholar]
- Aaron J.; Travis K.; Harrison N.; Sokolov K. Dynamic imaging of molecular assemblies in live cells based on nanoparticle plasmon resonance coupling. Nano Lett. 2009, 9, 3612–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam J. M.; Murthy A. K.; Ingram D. R.; Nguyen R.; Sokolov K. V.; Johnston K. P. Kinetic assembly of near-IR active gold nanoclusters using weakly adsorbing polymers to control the size. Langmuir 2010, 26, 8988–8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam J. M.; Tam J. O.; Murthy A.; Ingram D. R.; Ma L. L.; Travis K.; Johnston K. P.; Sokolov K. V. Controlled assembly of biodegradable plasmonic nanoclusters for near-infrared imaging and therapeutic applications. ACS Nano 2010, 4, 2178–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy A. K.; Stover R. J.; Borwankar A. U.; Nie G. D.; Gourisankar S.; Truskett T. M.; Sokolov K. V.; Johnston K. P. Equilibrium gold nanoclusters quenched with biodegradable polymers. ACS Nano 2013, 7, 239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halas N. J.; Lal S.; Chang W.-S.; Link S.; Nordlander P. Plasmons in strongly coupled metallic nanostructures. Chem. Rev. 2011, 111, 3913–3961. [DOI] [PubMed] [Google Scholar]
- Ofir Y.; Samanta B.; Rotello V. M. Polymer and biopolymer mediated self-assembly of gold nanoparticles. Chem. Soc. Rev. 2008, 37, 1814–1825. [DOI] [PubMed] [Google Scholar]
- Frankamp B. L.; Boal A. K.; Rotello V. M. Controlled interparticle spacing through self-assembly of Au nanoparticles and poly(amidoamine) dendrimers. J. Am. Chem. Soc. 2002, 124, 15146–15147. [DOI] [PubMed] [Google Scholar]
- Zhuang J.; Wu H.; Yang Y.; Cao Y. C. Controlling colloidal superparticle growth through solvophobic interactions. Angew. Chem., Int. Ed. 2008, 47, 2208–2212. [DOI] [PubMed] [Google Scholar]
- Larson-Smith K.; Pozzo D. C. Scalable synthesis of self-assembling nanoparticle clusters based on controlled steric interactions. Soft Matter 2011, 7, 5339–5347. [Google Scholar]
- Lu Z.; Yin Y. Colloidal nanoparticle clusters: functional materials by design. Chem. Soc. Rev. 2012, 41, 6874–6887. [DOI] [PubMed] [Google Scholar]
- Murthy A. K.; Stover R. J.; Hardin W. G.; Schramm R.; Nie G. D.; Gourisankar S.; Truskett T. M.; Sokolov K. V.; Johnston K. P. Charged gold nanoparticles with essentially zero serum protein adsorption in undiluted fetal bovine serum. J. Am. Chem. Soc. 2013, 135, 7799–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J.; Hu Y.; Biasini M.; Beyermann W. P.; Yin Y. Superparamagnetic magnetite colloidal nanocrystal clusters. Angew. Chem., Int. Ed. 2007, 46, 4342–4345. [DOI] [PubMed] [Google Scholar]
- Ma L. L.; Feldman M. D.; Tam J. M.; Paranjape A. S.; Cheruki K. K.; Larson T. A.; Tam J. O.; Ingram D. R.; Paramita V.; Villard J. W.; et al. Small multifunctional nanoclusters (nanoroses) for targeted cellular imaging and therapy. ACS Nano 2009, 3, 2686–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y. S.; Nguyen T. D.; Yang M.; Lee B.; Santos A.; Podsiadlo P.; Tang Z. Y.; Glotzer S. C.; Kotov N. A. Self-assembly of self-limiting monodisperse supraparticles from polydisperse nanoparticles. Nat. Nanotechnol. 2011, 6, 580–587. [DOI] [PubMed] [Google Scholar]
- Ma L. L.; Borwankar A. U.; Willsey B. W.; Yoon K. Y.; Tam J. O.; Sokolov K. V.; Feldman M. D.; Milner T. E.; Johnston K. P. Growth of textured thin Au coatings on iron oxide nanoparticles with near infrared absorbance. Nanotechnology 2013, 24, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenewold J.; Kegel W. K. Anomalously large equilibrium clusters of colloids. J. Phys. Chem. B 2001, 105, 11702–11709. [Google Scholar]
- Johnston K. P.; Maynard J. A.; Truskett T. M.; Borwankar A. U.; Miller M. A.; Wilson B. K.; Dinin A. K.; Khan T. A.; Kaczorowski K. J. Concentrated dispersion of equilibrium protein nanoclusters that reversibly dissociate into active monomers. ACS Nano 2012, 6, 1357–1369. [DOI] [PubMed] [Google Scholar]
- Joshi H.; Shirude P. S.; Bansal V.; Ganesh K. N.; Sastry M. Isothermal titration calorimetry studies on the binding of amino acids to gold nanoparticles. J. Phys. Chem. B 2004, 108, 11535–11540. [Google Scholar]
- Kondoh H.; Kodama C.; Sumida H.; Nozoye H. Molecular processes of adsorption and desorption of alkanethiol monolayers on Au(1 1 1). J. Chem. Phys. 1999, 111, 1175–1184. [Google Scholar]
- Hong R.; Han G.; Fernandez J. M.; Kim B.; Forbes N. S.; Rotello V. M. Gluathione-mediated delivery and release using monolayer protected nanoparticle carriers. J. Am. Chem. Soc. 2006, 128, 1078–1079. [DOI] [PubMed] [Google Scholar]
- Chompoosor A.; Han G.; Rotello V. M. Charge dependent of ligand release and monolayer stability of gold nanoparticles by biogenic thiols. Bioconjugate Chem. 2008, 19, 1342–1345. [DOI] [PubMed] [Google Scholar]
- Hiemenz P. C.; Rajagopalan R.. Principles of Colloid and Surface Chemistry, 3rd ed.; Taylor & Francis: New York, 1997; pp 546–556. [Google Scholar]
- Tadros T. F.; Vincent B. Influence of temperature and electrolytes on the adsorption of poly(ethylene oxide)-poly(propylene oxide) block copolymer on polystyrene latex and on the stability of the polymer-coated particles. J. Phys. Chem. 1980, 84, 1575–1580. [Google Scholar]
- Alexandridis P.; Holzwarth J. F. Differential scanning calorimetry investigation of the effect of salts on aqueous solution properties of an amphiphilic block copolymer (Poloxamer). Langmuir 1997, 13, 6074–6082. [Google Scholar]
- Su Y.-L.; Wei X.-F.; Liu H.-Z. Effect of sodium chloride on association behavior of poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) block copolymer in aqueous solutions. J. Colloid Interface Sci. 2003, 264, 526–531. [DOI] [PubMed] [Google Scholar]
- Weitz D. A.; Huang J. S.; Lin M. Y.; Sung J. Limits of the fractal dimension for irreversible kinetic aggregation of gold colloids. Phys. Rev. Lett. 1985, 54, 1416–1419. [DOI] [PubMed] [Google Scholar]
- Lekkerkerker H. N. W.; Tuinier R.. Colloids and the Depletion Interaction; Springer: New York, 2011; Vol. 833, pp 12–15. [Google Scholar]
- Petri-Fink A.; Steitz B.; Finka A.; Salaklang J.; Hofmann H. Effect of cell media on polymer coated superparamagnetic iron oxide nanoparticles (SPIONs): Colloidal stability, cytotoxicity, and cellular uptake studies. Eur. J. Pharm. Biopharm. 2008, 68, 129–137. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Servos M. R.; Liu J. Ultrahigh nanoparticle stability against salt, pH, and solvent with retained surface accessibility via depletion stabilization. J. Am. Chem. Soc. 2012, 134, 9910–9913. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.