

Abstract

Amantadine inhibits the M2 proton channel of influenza A virus, yet most of the currently circulating strains of the virus carry mutations in the M2 protein that render the virus amantadine-resistant. While most of the research on novel amantadine analogues has revolved around the synthesis of novel adamantane derivatives, we have recently found that other polycyclic scaffolds effectively block the M2 proton channel, including amantadine-resistant mutant channels. In this work, we have synthesized and characterized a series of pyrrolidine derivatives designed as analogues of amantadine. Inhibition of the wild-type M2 channel and the A/M2-S31N, A/M2-V27A, and A/M2-L26F mutant forms of the channel were measured in Xenopus oocytes using two-electrode voltage clamp assays. Most of the novel compounds inhibited the wild-type ion channel in the low micromolar range. Of note, two of the compounds inhibited the amantadine-resistant A/M2-V27A and A/M2-L26F mutant ion channels with submicromolar and low micromolar IC50, respectively. None of the compounds was found to inhibit the S31N mutant ion channel.
Introduction
Amantadine (Amt) and rimantadine (Rmt), two polycyclic aminoadamantane derivatives, have been in clinical use as anti-influenza virus agents for decades. However, the efficacy of these two drugs dropped sharply in recent years due to the global distribution of mutant viruses carrying Amt resistance mutations, prompting the Centers for Disease Control to recommend discontinuing the use of amantadine-based drugs.1,2 Therefore, there is an urgent need to develop novel antiviral drugs that are active against drug-resistant influenza viruses.
The mechanism of action of Amt and Rmt is based on the inhibition of the M2 proton channel of the influenza A virus. The M2 protein is a 97 residues long integral membrane protein with a transmembrane (TM) domain of 19 residues, a small ectodomain of 23 residues, and a 54 residues long cytoplasmic tail.3−5 Detailed mutational studies indicated that several point mutations of pore-lining residues of the A/M2 TM domain result in Amt-resistance.6 However, only a few of these mutations (i.e., L26F, V27A, and S31N) have been observed in transmissible viruses, with the S31N mutation being the most frequently occurring Amt-resistance mutation.7 In 2011, Wang et al. reported that spiro compound 1 is able of inhibiting the L26F and V27A M2 mutants with good efficacy in electrophysiological and plaque reduction assays.8−10 More recently, our group has reported the first non-adamantane inhibitor of the V27A mutant, the polycyclic pyrrolidine 2 (Chart 1).11
Chart 1. Structures of Amt, Rmt, and Recently Developed Compounds with Potent Activity against A/M2-V27A Mutant Channelsa.

a The IC50 values denote the reported 50% inhibitory concentrations on A/M2 wt, S31N, and V27A proton channel function.8−11
A common problem of 1 and 2 is that their syntheses involves several steps (e.g., up to 11 steps for 2),11 which means that the synthesis of novel analogues of these two compounds would be challenging. In the present study, we report novel scaffolds designed to inhibit the A/M2 channel. We have found that the wild-type (wt) channel can be inhibited by several easy-accessible pyrrolidine derivatives. Furthermore, we have identified two compounds, 18 and 19, that are capable of inhibiting the M2-V27A mutant ion channel with submicromolar IC50 values. In addition, both compounds are able to inhibit the M2 wt channel with an IC50 value similar to that of Amt, and both are also low micromolar inhibitors of the M2-L26F mutant channel.
Chemistry
During the past years, our group has synthesized several polycyclic Amt analogues containing different scaffolds, including ring-contracted, ring-rearranged, and 2,2-dialkyl derivatives of Amt.12−15 Several of them displayed similar IC50 values for wt A/M2 as Amt but, unfortunately, were inactive against the Amt-resistant S31N or V27A mutant forms of A/M2.13 More recently, we have reported on the synthesis of polycyclic pyrrolidines and on their inhibitory effect on the A/M2 proton channel activity by using the conductance assay in M2-expressing oocytes. Again, several of these novel compounds displayed similar IC50 values for wt A/M2 as Amt, and, interestingly, we found three guanidine derivatives that displayed low micromolar to submicromolar IC50 values against the V27A mutant channel.11
On the basis of our previous insights that polycyclic scaffolds other than adamantane effectively inhibit the M2 proton channel and that the synthesis of guanidine 2 and related compounds involved a very long synthetic sequence, we have searched for novel synthetic strategies able to yield polycyclic amines that are structurally diverse and easily available by short synthetic sequences.
The reduction of known imide 3, easily accessible in multigram quantities by the reaction of commercially available cycloheptatriene with maleimide,16 furnished amine 4 in 79% yield. The catalytic hydrogenation of 4 led to fully saturated compound 6 in 95% yield. Both pyrrolidines were transformed into their corresponding guanidines, 5 and 7, by reaction with 1H-pyrazole-1-carboxamidine. Taking into account that recent experimental and computational studies have shown that the M2-V27A mutant channel has a larger cavity than the wt M2 channel,9,17−19 we also synthesized larger compounds derived from 4 and 6. Thus, reductive alkylation of amine 4 with 1-Boc-4-piperidone followed by deprotection led to piperidine 8 in 75% overall yield. The catalytic hydrogenation of 8 furnished piperidine 9 in 85% yield (Scheme 1). Similarly, starting from known imide 10, easily available from cyclooctatetraene and maleimide,16 amines 11 and 12 and their guanidine derivatives 13 and 14, were synthesized in good yields (Scheme 1).
Scheme 1. Synthesis of Novel Polycyclic Pyrrolidine Derivatives.

To increase the chemical diversity of the compounds, while preserving the synthetic easiness, two more compact pyrrolidine derivatives, 18 and 19, were designed (Scheme 2). For the synthesis of these aesthetically appealing compounds that include one four-membered ring, three five-membered rings, and two six-membered rings, we started from known diacid 15, easily available from acetylene dicarboxylic acid and 2,3-dimethylbutadiene.20,21 The reaction of 15 with urea at 180 °C for 30 min yielded imide 16 in 75% yield. Photolysis of imide 16 at room temperature in acetone with a 125 W Hg lamp for 2 days gave a mixture of the starting imide and the tetracyclic imide 17. Column chromatography of this mixture through silica gel afforded pure 17 in 36% yield (based on recovered starting material, brsm). The imide was subsequently reduced using sodium bis(2-methoxyethoxy)aluminum hydride to give the secondary amine 18 in 71% yield. Finally, reaction of 18 with 1H-pyrazole-1-carboxamidine monohydrochloride in acetonitrile led to guanidine 19 in 66% yield (Scheme 2).
Scheme 2. Synthesis of Compounds 18 and 19.

Previously, we have seen that, in related polycyclic compounds, on going from secondary to tertiary amines, the inhibitory effect on either wt or V27A mutant M2 channels diminished.6,11,15 For this reason, in this work we have not synthesized alkylated derivatives of the novel secondary amines herein reported.
All the new compounds were fully characterized as hydrochlorides through their spectroscopic data and elemental analyses.
Pharmacological Activity and Structure–Activity Relationships
Inhibition of wt and Amt-Resistant A/M2 Ion Channels
The inhibitory activity of the compounds was tested on A/M2 channels expressed in Xenopus oocytes using the two-electrode voltage clamp (TEV) technique. All inhibitors were initially tested at 100 μM; those that inhibited the wt A/M2 channel activity by more than 80% were chosen for measurement of their IC50 value in an isochronic (2 min) inhibition assay. The results are given in Table 1.
Table 1. Inhibitory Effect of the Synthesized Compounds on A/M2 wt, S31N, or V27A Proton Channel Functionsa,b.
| A/M2 wt (mean ± SE) |
A/M2 S31N (mean ± SE) |
A/M2 V27A (mean ± SE) |
||||
|---|---|---|---|---|---|---|
| compd | inhibition by 100 μM for 2 min (%) | IC50 (μM) | inhibition by 100 μM for 2 min (%) | IC50 (μM) | inhibition by 100 μM for 2 min (%) | IC50 (μM) |
| amantadine | 91.0 ± 2.1 | 16.0 ± 1.2 | 35.6 ± 1.5 | 199.9 ± 13.5 | 10.8 ± 2.0 | NDe |
| 4 | 88.8 ± 1.3 | 2.92 | 0 | ND | 2.4 ± 1.3 | ND |
| 5 | 90.7 ± 0.9 | 1.50 | 0 | ND | 9.5 ± 0.6 | ND |
| 6 | 90.9 ± 1.0 | 3.38 | 5.7 ± 0.9 | ND | 3.4 ± 2.2 | ND |
| 7 | 93.4 ± 1.2 | 1.64 | 2.5 ± 0.4 | ND | 14.5 ± 1.7 | ND |
| 11 | 92.9 ± 1.2 | 5.54 | 3.9 ± 1.3 | ND | 5.5 ± 0.1 | ND |
| 12 | 94.5 ± 0.8 | 1.24 | 3.9 ± 1.8 | ND | 17.2 ± 5.2 | ND |
| 13 | 91.8 ± 0.8 | 4.93 | 14.2 ± 2.5 | ND | 33.8 ± 2.3 | ND |
| 14 | 94.5 ± 1.6 | 2.08 | 2.6 ± 1.4 | ND | 75.9 ± 1.2 | 22.61 |
| 18c | 90.2 ± 0.9 | 18.0 | 1.1 ± 1.1 | ND | 96.4 ± 0.5 | 0.70 |
| 19d | 91.7 ± 1.2 | 10.7 | 2.7 ± 1.5 | ND | 96.7 ± 0.5 | 0.50 |
The activity of the inhibitors was measured using the TEV technique on A/M2 channels expressed in Xenopus oocytes; percentage of inhibition was average of at least three experiments. For IC50 experiments, 7–9 concentrations were measured, and, at each concentration, experiments were run at least three times.
Isochronic (2 min) values for IC50 are given. See text and Supporting Information for details.
The inhibition by 100 μM for 2 min of 18 on A/M2 L26F mutant channel was 90.6% ± 1.1 with an IC50 of 8.6 μM.
The inhibition by 100 μM for 2 min of 19 on A/M2 L26F mutant channel was 88.4% ± 0.6 with an IC50 of 7.5 μM.
ND, not determined.
Amt inhibited wt A/M2 channel with an IC50 of 16.0 μM while being inactive against the V27A mutant channel. Unfortunately, piperidine derivatives 8 and 9 did not display any activity. Tetracyclo amines 4, 6, 11, and 12 and their corresponding guanidines 5, 7, 13, and 14 were potent low-micromolar inhibitors of the wt channel. Regarding the activity against the V27A M2 mutant channel, three trends were found, i.e., guanidine performed better than its corresponding amine (e.g., 7 vs 6 or 13 vs 11), the fully saturated compounds were more potent than their corresponding unsaturated analogues (e.g., 7 vs 5 or 14 vs 13), and the cyclobutane/cyclobutene analogues were more potent than the cyclopropane derivatives (e.g., 12 vs 6 or 14 vs 7). Altogether, guanidine 14 emerged as the more promising compound, being more potent than Amt against the wt M2 channel and endowed with fair activity against the V27A mutant channel. None of the compounds was found to inhibit the S31N mutant ion channel. Finally, amine 18 and guanidine 19 also proved to be active against the wt channel, with similar IC50 values as Amt (18.5 and 10.7 μM, respectively) while being endowed with submicromolar IC50 against the V27A mutant (0.7 and 0.5 μM, respectively). Moreover, both compounds were low micromolar inhibitors of the L26F mutant (8.6 and 7.5 μM, respectively).
Molecular Modeling
To gain insight into the inhibitory data of the more compact pyrrolidine derivatives, the interaction of 18 with the wt M2 channel and its V27A variant was examined by means of molecular dynamics (MD) simulations and compared with the trends found for the binding of Amt.
The analysis of the three replicas run for 18 bound to the wt channel shows a consistent binding mode, where the center-of-mass (COM) of the inhibitor is located close to the plane formed by the Cα atoms of Ser31, the amine nitrogen is pointing toward the plane formed by the His37 residues (average distance of 6.1 Å), and the tilt angle (i.e., the deviation of the amine nitrogen from the pore axis) is 16.1°, which partly reflects the twisted geometry of the pyrrolidine ring (Figure 1; see also Supporting Information Figure S1). This orientation (denoted down hereafter) is also found for Amt bound to the wt channel (Supporting Information Figure S2). In two replicas of the V27A–18 complex, the inhibitor adopts the down orientation, but in the third replica it turns over at the beginning of the trajectory and the inhibitor adopts the opposite (up) orientation (the distance of the amine nitrogen to the His37 plane and the tilt angle are, on average, 14.9 Å and 145.3°; Figure 1 and Supporting Information Figure S3). Remarkably, Amt was found in the up orientation in the three replicas run for the V27A channel (Supporting Information Figure S4), and even in one case Amt was released to the solvent after few nanoseconds.
Figure 1.

Representation of compound 18 bound to (left) the wild-type M2 channel and its V27A variant (down and up orientations shown in middle and right panels, respectively). The dashed line indicates the distance (Å) from the center-of-mass of the inhibitor to the plane formed by the Cα atoms of His37.
To explore the molecular basis of the exchange between down and up orientations found in the V27A channel, additional MD simulations were run for the apo forms of both wt and V27A channels. The root-mean-square deviation (RMSD) of the protein backbone for the transmembrane helices between the wt channel and its V27A variant amounts to 1.4 Å, which compare with the typical range of RMSD values obtained for the ligand-bound forms of the two channels (between 1.2 and 1.7 Å; Supporting Information Table S1). This suggests that the gross structural features of the channel are not drastically altered by the Val27 → Ala mutation, in agreement with previous studies.9,18 Nevertheless, inspection of the interhelical cross-diagonals distances (measured from either the Cα or the Cβ atoms) for distinct tetrads of residues shows that the mutation widens the pore at the Ala27 plane, an effect associated with the narrowing of the pore at the His37 plane (Table 2). This structural rearrangement likely contributes to the displacement of the amine nitrogen of compound 18 in the V27A channel, which is pushed away from the His37 plane by 1.6 Å (from 6.1 Å in the wt channel to 7.7 Å in the V27A variant for the inhibitor in the down orientation) and might facilitate the exchange between down and up arrangements of the inhibitor. Compared to compound 18, the lower, more compact size of Amt should likely enhance the exchange rate between the two ligand orientations, eventually leading to an easier release from the pore that could account for the reduction in inhibitory potency against the V27A channel (Table 1).
Table 2. Cross-Diagonal Distances (Å) between Helices A–C and B–D in the Apo Form of the wt and V27A Channelsa.
| wt |
V27A |
||||
|---|---|---|---|---|---|
| d1 | d2 | d1 | d2 | Δb | |
| Val27 | 10.3 ± 0.5 (8.3 ± 0.6) | 10.6 ± 0.5 (8.6 ± 0.6) | 11.6 ± 0.7 (10.0 ± 0.9) | 11.5 ± 0.8 (9.9 ± 0.9) | +1.1 (+1.5) |
| Ser31 | 11.0 ± 0.4 (11.2 ± 0.5) | 10.7 ± 0.4 (10.8 ± 0.5) | 11.4 ± 0.6 (12.0 ± 0.7) | 11.3 ± 0.6 (12.0 ± 0.7) | +0.5 (+1.0) |
| Gly34 | 10.1 ± 0.4 | 9.3 ± 0.5 | 9.6 ± 0.6 | 9.4 ± 0.6 | –0.2 |
| His37 | 13.4 ± 0.5 (10.6 ± 0.5) | 12.3 ± 0.8 (9.9 ± 0.9) | 12.3 ± 0.6 (9.6 ± 0.6) | 11.8 ± 0.8 (9.2 ± 0.9) | –0.8 (−1.0) |
Values measured from the Cα atoms of residues in the tetrad; values determined from the Cβ atoms are given in parentheses.
Difference between cross-diagonals in wt and V27A channels (positive values imply a widening of the pore in the V27A channel).
The lack of inhibitory potency of compound 18 against the S31N variant can be realized from the comparison of the inhibitor bound to the wt M2 channel and the solid-state NMR-derived structural data available for the complex of the S31N channel bound to the 5-thienyl isoxazole derivative containing a 1-(1-adamantylamino)-methylene group in position 3 (PDB entry 2LY0).22 It was noted that the structure of the S31N variant closely resembled previous structures of the wt M2 channel, showing most similarity to the Amt-bound channel structure solved by solid-state NMR.23 Similar structural resemblance is also found upon comparison with the snapshots sampled for the wt channel bound to compound 18 (see Supporting Information Figure S6), which rules out the existence of drastic structural alterations in the arrangement of the transmembrane helices triggered upon binding of the two compounds. Both NMR and MD simulations revealed that binding of the isoxazole derivative involved water-mediated contacts between the protonated amine group with two Asn31 side chains as well as by a direct interaction between the amide unit of a third Asn31 and the charged amine group. This network of interactions was facilitated by the fact that the adamantylamino group was oriented with the amine facing away the His37 plane. However, the position of the protonated amine unit in the isoxazole derivative is replaced by the hydrophobic cage of compound 18 (see Supporting Information Figure S6), which would thus be unable of forming a similar pattern of stabilizing interactions. On the other hand, adoption of the reverse arrangement of compound 18 (i.e., adopting an amine-up orientation) should not only alter the contacts with Asn31 due to the distinct orientation of the amine group but would also trigger unfavorable clashes between the terminal methyl groups with the tetrad of Gly37 residues.
Antiviral Activity
The anti-influenza virus activity of the compounds was first assessed in a relatively stringent three-day cytopathic effect (CPE) reduction assay in MDCK cells (Table 3). All the guanidines showed to be inactive, probably reflecting a permeability problem or, particularly for 13, 14, and 19, as a consequence of their cytotoxicity. Compounds 4 and 6 displayed favorable activity (i.e., antiviral EC50 values in the range of 3–8 μM) against the A/HK/7/87 virus strain, which possesses a wt M2 channel. Three other compounds, i.e., 8, 11, and 18, showed strong activity against the A/PR/8/34 strain, an A/H1N1 virus with two mutations (S31N and V27T) in the M2 protein. We previously found that A/PR/8/34 appears particularly sensitive to a slight increase in endosomal pH as induced by some polycyclic amine compounds.15 This effect may explain, at least in part, the antiviral activity of these compounds, although they were devoid of activity against the S31N M2 mutant channel. A similar behavior has very recently been observed by Kolocouris et al. in a set of novel aminoadamantanes.24
Table 3. Antiviral Activity in Influenza Virus-Infected MDCKa Cells.
| antiviral activity (μM) |
|
||||||
|---|---|---|---|---|---|---|---|
| A/PR/8/34 |
A/HK/7/87 |
cytotoxicity (μM) |
|||||
| compd | EC50 (CPE)b | EC50 (MTS)b | EC50 (CPE)b | EC50 (MTS)b | EC99 (virus yield)c | CC50d | MCCe |
| 4 | >100 | >100 | 6.2 | 8.0 | 20 | 54 | 100 |
| 5 | >100 | >100 | >100 | >100 | ND | 39 | 100 |
| 6 | >100 | >100 | 6.0 | 2.7 | <0.4 | 49 | 100 |
| 7 | >100 | >100 | >100 | >100 | ND | 41 | 100 |
| 8 | ≤0.80 | <0.80 | >100 | >100 | ND | 46 | 100 |
| 9 | >100 | >100 | >100 | >100 | ND | 9.0 | 20 |
| 11 | 1.3 | <0.80 | >100 | >100 | >10 | 4.9 | 15 |
| 12 | >100 | >100 | >100 | >100 | <0.08 | 1.9 | 4 |
| 13 | >100 | >100 | >100 | >100 | >10 | 9.4 | 20 |
| 14 | >100 | >100 | >100 | >100 | 4.6 | 1.9 | 4 |
| 18 | 1.8 | 3.0 | >100 | >100 | >50 | 49 | 100 |
| 19 | >100 | >100 | >100 | >100 | >10 | 15 | ≥4 |
| amantadine | 30 | 34 | 1.4 | 1.4 | 1.1 | >500 | ≥500 |
| rimantadine | 7.6 | 5.1 | 0.81 | 0.15 | >4 | 230 | 500 |
MDCK: Madin–Darby canine kidney cells; virus strains: A/PR/8/34 (A/H1N1) and A/HK/7/87 (A/H3N2).
50% effective concentration, or compound concentration producing 50% inhibition of virus replication, as determined by microscopic scoring of the CPE at 72 h post infection or by the MTS cell viability test.
EC99: compound concentration giving 2 log10 reduction in virus yield, as determined by quantifying the virus in the supernatant at 24 post infection, using an qRT-PCR based virus yield assay.25
CC50: 50% cytotoxic concentration, as determined by the MTS cell viability test. Values shown are the mean of 2–3 determinations.
MCC: minimum cytotoxic concentration, or concentration producing minimal alterations in cell morphology after 72 h incubation with compound. ND, not determined.
We subsequently performed a 24 h virus yield assay and a plaque reduction assay, both using the WT-M2 A/HK/7/87 virus. These experiments confirmed the activity of 4 and 6 (Table 3 and Figure 2), which, in terms of cell culture activity, emerged as the two most promising molecules among the synthesized series. Compounds 12 and 14 were shown to reduce virus yield, with EC99 values of <0.08 μM (12) and 4.6 μM (14). These compounds caused a clear (12) or modest (14) reduction in plaque size or number at a compound concentration of 0.5 μM (Figure 2). However, because both were visibly toxic at 2–8 μM, their selectivity index (i.e., ratio of cytotoxic to antivirally effective concentration) appears limited. Finally, 11, 13, and 18 caused visible inhibition of plaque formation (Figure 2) at 0.5–2 μM, but the activity of these three compounds was not confirmed in the CPE or virus yield assay (Table 3). As expected, neither the novel compounds nor the reference agents Amt and Rmt were active against influenza B virus (data not shown).
Figure 2.
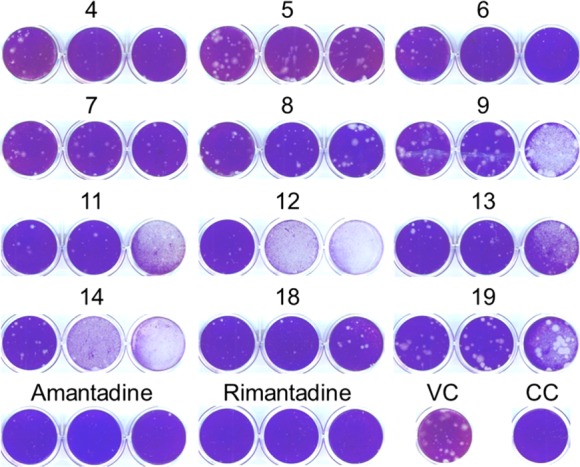
Activity of the compounds in an influenza virus plaque reduction assay. MDCK cells were infected with influenza virus (strain A/HK/7/87; 25 PFU per well) in the presence of the test compounds (concentrations, from left to right: 0.5, 2, and 8 μM). After 72 h incubation, plaques were visualized by crystal violet staining. VC: mock-treated virus control. CC: uninfected cell control.
Conclusions
The present work shows the feasibility of designing easily accessible compounds able to successfully inhibit the wt and the V27A and L26F variants of the A/M2 channels of influenza A virus. In fact, some of the newly designed compounds inhibit the three channels similarly or even more effectively than Amt inhibits the wt proton channel. In particular, amine 18 and guanidine 19 emerged as promising compounds, being low micromolar inhibitors against the wt channel and the L26F mutant, while being endowed with submicromolar IC50 against the V27A variant. Furthermore, compound 18 showed strong activity against the A/PR/8/34 strain, an A/H1N1 virus with two mutations (S31N and V27T) in the M2 protein. Overall, these results suggest that compounds 18 and 19 are suitable templates to explore novel candidates against influenza virus.
Experimental Section
Plasmid, mRNA Synthesis, and Microinjection of Oocytes
The cDNA encoding the influenza A/Udorn/72 (A/M2) was inserted into pGEM3 vector for the expression on oocyte plasma membrane. A/M2 S31N and A/M2 V27A mutants were generated by QuikChange site-directed mutagenesis kit (Agilent Technologies). The synthesis of mRNA and microinjection of oocytes have been described previously.26
Two-Electrode Voltage Clamp Analysis
Macroscopic membrane current was recorded 48–72 h after injection as described previously.8 The tested compounds were applied at pH 5.5 at various concentrations when the inward current reaches maximum. The compounds were applied for 2 min, and residual membrane current was compared with the membrane current before the application of compounds. Membrane currents were analyzed with pCLAMP 10.0 software package (Axon Instruments, Sunnyvale, CA).
Antiviral Assays
To determine the anti-influenza virus activity in Madin–Darby canine kidney (MDCK) cells, CPE reduction and virus yield assays were performed as described.25,27 Briefly, MDCK cells grown in 96-well plates were infected with influenza virus (strains A/PR/8/34 (A/H1N1); A/HK/7/87 (A/H3N2) or B/HK/5/72), and at the same time the test compounds were added in serial dilutions. After 72 h incubation at 35 °C, microscopy was performed to score the compounds’ inhibitory effect on the viral cytopathic effect (CPE) or cytotoxicity. These data were verified by measuring the cell viability with the formazan-based MTS assay. In separate experiments, supernatants were harvested from the infected and compound-treated cells at 24 h post infection, and virus yield in these samples was determined by qRT-PCR. To perform plaque reduction assays, influenza virus A/HK/7/87 was incubated (1 h at 4 °C) with different concentrations of the compounds and then added to confluent MDCK cells in 12-well plates. After 1 h incubation at 35 °C, excess virus was removed and replaced by fresh medium containing the compounds and 0.8% agarose. After 72 h incubation, plaques were visualized by cell fixation with 3.7% formaldehyde and staining with 0.1% crystal violet.
Chemical Synthesis. General Methods
Melting points were determined in open capillary tubes with a MFB 595010 M Gallenkamp. 400 MHz 1H/100.6 MHz 13C NMR spectra, and 500 MHz 1H NMR spectra were recorded on Varian Gemini 300, Varian Mercury 400, and Varian Inova 500 spectrometers, respectively. The chemical shifts are reported in ppm (δ scale) relative to internal tetramethylsilane, and coupling constants are reported in hertz (Hz). Assignments given for the NMR spectra of the new compounds have been carried out on the basis of DEPT, COSY 1H/1H (standard procedures), and COSY 1H/13C (gHSQC and gHMBC sequences) experiments. IR spectra were run on PerkinElmer Spectrum RX I spectrophotometer. Absorption values are expressed as wavenumbers (cm–1); only significant absorption bands are given. The GC/MS analysis was carried out in an inert Agilent Technologies 5975 gas chromatograph equipped with an Agilent 122-5532 DB-5MS 1b (30 m × 0.25 mm) capillary column with a stationary phase of phenylmethylsilicon (5% diphenyl–95% dimethylpolysiloxane), using the following conditions: initial temperature of 50 °C (1 min), with a gradient of 10 °C/min up to 300 °C, and a temperature in the source of 250 °C, solvent delay (SD) of 4 min and a pressure of 7,35 psi. Column chromatography was performed on silica gel 60 AC.C (35–70 mesh, SDS, ref 2000027). Thin-layer chromatography was performed with aluminum-backed sheets with silica gel 60 F254 (Merck, ref 1.05554), and spots were visualized with UV light and 1% aqueous solution of KMnO4. The analytical samples of all of the new compounds which were subjected to pharmacological evaluation possessed purity ≥95% as evidenced by their elemental analyses.
4-Azatetracyclo[5.3.2.02,6.08.10]dodec-11-ene Hydrochloride, 4·HCl
A solution of imide 3 (8.00 g, 42.3 mmol) in dry and degassed toluene (280 mL) was cooled down to 5 °C with an ice bath. Sodium bis(2-methoxyethoxy) aluminum hydride (64.5 mL, 65% solution in toluene, 211.4 mmol) was added dropwise, and the resulting solution was heated to reflux and stirred for 2 days. The solution was allowed to cool down to room temperature, and 30% aqueous KOH (300 mL) was added dropwise. The organic phase was separated and the aqueous layer was extracted with dichloromethane (3 × 250 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuo to give a red oil. This oil was dissolved in EtOAc and treated with an excess of a 1.2 N ethereal solution of HCl and was allow to stand at 0 °C for 24 h. The suspension was filtered to get 4·HCl as a white solid (6.54 g, 79%). An analytical sample of 4·HCl was obtained by crystallization from MeOH/Et2O, mp 197–198 °C. IR (KBr) ν 3430, 2932, 2894, 2765, 2519, 2640, 1569, 1466, 1450, 1422, 1395, 1383, 1355, 1301, 1281, 1245, 1170, 1132, 1068, 1026, 1016, 936, 902, 841, 824, 758, 731, 649, 591, 580 cm–1. 1H NMR (400 MHz, CD3OD) δ 0.23 (complex signal, 2 H, 9-H2), 1.05 [m, 2 H, 8(10)-H], 2.75 [m, 2 H, 2(6)-H], 2.82 [dm, J = 12.0 Hz, 2 H, 3(5)-Ha], 2.95 [m, 2 H, 1(7)-H], 3.38 [dd, J = 12.0 Hz, 2 H, 3(5)-Hb], 5.91 [m, 2 H, 11(12)-H]. 13C NMR (100.6 MHz, CD3OD) δ 5.5 (CH2, C9), 11.1 [CH, C8(10)], 35.5 [CH, C1(7)], 44.8 [CH, C2(6)], 50.4 [CH2, C3(5)], 130.7 [CH, C11(12)]. MS (DI), m/e (%). Main ions: 161 (M•+, 5), 94 (14), 93 (16), 92 (19), 91 (47), 81 (12), 80 (20), 68 (100), 67 (35).
4-Azatetracyclo[5.3.2.02,6.08.10]dodecane Hydrochloride, 6·HCl
To a solution of 4·HCl (200 mg, 1.00 mmol) in absolute EtOH (35 mL), Pd on activated charcoal (5%, 40 mg) was added. The black suspension was set in a hydrogenator at 150 psi of H2 and was stirred at room temperature overnight. The black suspension was filtered, and 6·HCl was recovered as an off-white solid (190 mg, 95%). An analytical sample was obtained by crystallization from 2-propanol, mp 227–228 °C. IR (KBr) ν 3421, 3073, 2935, 2755, 2646, 2585, 2460, 2363, 1590, 1447, 1423, 1347, 1100, 1058, 1028, 999, 902, 881, 812, 798, 752, 723, 662, 526 cm–1. 1H NMR (400 MHz, CD3OD) δ 0.59 (dt, J = 6.0 Hz, J′ = 7.6 Hz, 1 H, 9-Ha), 0.97 (dt, J = 6.0 Hz, J′ = 3.6 Hz, 1 H, 9-Hb), 1.04 [m, 2 H, 8(10)-H], 1.25 [broad d, J = 8.8 Hz, 2 H, 11(12)-Ha], 1.42 [broad d, J = 8.8 Hz, 2 H, 11(12)-Hb], 1.96 [broad s, 2 H, 1(7)-H], 2.68 [m, 2 H, 2(6)-H], 3.16 [dd, J = 12.0 Hz, J′ = 6.8 Hz, 2 H, 3(5)-Ha], 3.42 [dd, J = 12.0 Hz, J′ = 8.6 Hz, 2 H, 3(5)-Hb]. 13C NMR (100.6 MHz, CD3OD) δ 6.9 [CH2, C9], 16.7 [CH, C8(10)], 17.9 [CH2, C11(12)], 28.1 [CH, C1(7)], 41.2 [CH, C2(6)], 48.6 [CH2, C3(5)]. MS (DI), m/e (%). Main ions: 163 (M•+, 35), 162 (15), 134 (73), 105 (12), 94 (14), 93 (13), 92 (12), 91 (35), 80 (20), 79 (30), 77 (25), 70 (100), 68 (62), 67 (21).
t-Butyl 4-[4-Azatetracyclo[5.3.2.02,6.08.10]dodec-11-ene]piperidine-1-carboxylate
To a solution of 4·HCl (1.15 g, 5.8 mmol) in water (30 mL) was added a 10 N aqueous solution of NaOH. It was then extracted with EtOAc (3 × 50 mL), and the organic phase was dried over Na2SO4, filtered, and concentrated under vacuo (yellowish oil, 945 mg, quantitative yield). This oil was dissolved in MeOH (20 mL), and sodium cyanoborohydride (1.06 g, 16.9 mmol), 1-Boc-4-piperidone (1.39 g, 7.0 mmol), and glacial acetic acid (0.67 mL) were added to the solution. The solution was stirred at room temperature for 8 h, and a second portion of sodium cyanoborohydride (1.06 g, 16.9 mmol) and 1-Boc-4-piperidone (1.39 g, 7.0 mmol) were added. The yellow solution was further stirred at room temeprature overnight. It was concentrated under vacuo, and the yellow oil was redissolved in water (50 mL) and extracted with EtOAc (3 × 50 mL). The organic phase was dried over Na2SO4, filtered, and concentrated under vacuo (clear sticky solid, 3.90 g). Column chromatography of the solid (silica gel, EtOAc) gave the pure carbamate as an off-white solid (1.52 g, 76%). An analytical sample was prepared by crystallization from MeOH/Et2O, mp 176–177 °C. IR (KBr) ν 3004, 2980, 2683, 2612, 2330, 2203, 2163, 1684, 1455, 1420, 1364, 1264, 1170, 1150, 1113, 1068, 1047, 1016, 974, 944, 866, 841, 814, 768, 733 cm–1. 1H NMR (500 MHz, CDCl3) δ 0.21 (dt, J = 5.5 Hz, J′ = 3.0 Hz, 1 H, 9-Hb), 0.27 (dt, J = 5.5 Hz, J′ = 7.5 Hz, 1H, 9-Ha), 1.01 [m, 2 H, 8(10)-H], 1.44 [s, 9 H, C(CH3)3], 1.65 [dq, J = 12.5 Hz, J′ = 2.5 Hz, piperidine-C3(5)-Hax], 1.94 [dm, J = 12.5 Hz, 2 H, piperidine-C3(5)-Heq], 2.39 [broad s, 2 H, 3(5)-Ha], 2.68 [m, 2 H, piperidine-C2(6)-Ha], 2.81 [m, 2 H, 2(6)-H], 2.93 [complex signal s, 3 H, 1(7)-H and piperidine-C4-H], 3.60 [broad s, 2 H, 3(5)-Hb], 4.21 [broad s, 2 H, piperidine-C2(6)-Hb], 5.86 [m, 2 H, 11(12)-H]. 13C NMR (125.6 MHz, CDCl3) δ 5.4 (CH2, C9), 9.9 [CH, C8(10)], 28.0 [broad CH2, C3(5)-piperidine], 28.3 [CH3, C(CH3)3], 33.3 [CH, C1(7)], 41.7 [broad CH2, C2(6)-piperidine], 42.1 [CH, C2(6)], 54.6 [CH2, C3(5)], 62.2 (CH, C4-piperidine), 80.4 [C, C(CH3)3], 129.4 [CH, C11(12)], 154.1 [C=O, CO2C(CH3)3]. MS (DI), m/e (%). Main ions: 344 (M•+, 26), 287 [(M-C4H9)+, 35], 271 (12), 243 [(M-CO2C4H9)+,12], 207 (14), 195 (100), 188 (14), 151 (15), 94 (19), 57 (27). HRMS-ESI+ m/z [M + H]+ calcd for [C21H32N2O2 + H+], 345.2537; found, 345.2533.
4-(4-Piperidinyl)-4-azatetracyclo[5.3.2.02,6.08.10]dodec-11-ene Dihydrochloride, 8·2HCl
To a solution of the carbamate previously reported (760 mg, 2.20 mmol) in methanol (25 mL), a 0.8 N HCl solution in methanol (30 mL) was added. The resulting clear solution was heated to reflux and stirred for 30 min. It was then allowed to cool down to room temperature and was concentrated under vacuo to give a white solid that was crystallized from MeOH/Et2O to give 8·2HCl as a white solid (700 mg, quantitative), mp >250 °C (dec.). IR (KBr) ν 3479, 3405, 2945, 2804, 2733, 2642, 2585, 2496, 2363, 1630, 1458, 1437, 1239, 1157, 1044, 974, 842, 766, 727, 679, 583 cm–1. 1H NMR (400 MHz, CD3OD) δ 0.28 (m, 2 H, 9-H2), 1.04 [m, 2 H, 8(10)-H2], 2.00 [dm, J = 13.6 Hz, 2 H, piperidine-C3(5)-Hax], 2.35 [d, J = 13.6 Hz, 2 H, piperidine-C3(5)-Heq], 2.59 [broad s, 2 H, 3(5)-Ha], 2.76 [broad s, 2 H, 2(6)-H], 2.96 [broad s, 2 H, 1(7)-H], 3.07 [td, J = 13.2 Hz, J′ = 2.8 Hz, 2 H, piperidine-C2(6)-Hax], 3.40 (tt, J = 13.0 Hz, J′ = 4.0 Hz, 1 H, piperidine–C4-H), 3.54 [dm, J = 13.2 Hz, 2 H, piperidine-C3(5)-Heq], 3.78 [broad s, 2 H, 3(5)-Hb], 5.90 [m, 2 H, 11(12)-H]. 13C NMR (100.6 MHz, CD3OD) δ 5.8 (CH2, C9), 10.9 [CH, C8(10)], 26.7 [CH2, C3(5)-piperidine], 34.4 [CH, C1(7)], 43.5 [CH2, C2(6)-piperidine], 43.8 [CH, C2(6)], 56.6 [CH2, C3(5)], 60.1 (CH, C4-piperidine), 130.5 [CH, C11(12)]. MS (DI), m/e (%). Main ions: 244 (M•+, 23), 200 (14), 189 (27), 188 (25), 164 (18), 163 (57), 162 (33), 160 (14), 152 (100), 151 (43), 120 (12), 108 (43), 107 (26), 97 (35), 96 (15), 95 (15), 94 (50), 91 (35), 86 (15), 85 (58), 84 (50), 83 (41), 82 (20), 69 (27), 68 (44), 58 (16), 57 (28), 55 (26).
4-(4-Piperidinyl)-4-azatetracyclo[5.3.2.02,6.08.10]dodecane Dihydrochloride, 9·2HCl
To a solution of 8·2HCl (200 mg, 0.63 mmol) in absolute EtOH (35 mL), Pd on charcoal (40 mg, ca. 10% Pd) was added and the resulting suspension was hydrogenated at room temperature and at 150 psi of H2 overnight. The black suspension was filtered, and 9·2HCl was recovered as a white solid (170 mg, 85%). An analytical sample was obtained by crystallization from MeOH, mp >300 °C (dec.). IR (KBr) ν 3422, 2929, 2889, 2840, 2779, 2726, 2646, 2608, 2495, 2453, 2419, 2371, 1609, 1473, 1399, 1364, 1288, 1098, 1078, 879, 841, 800, 610, 510 cm–1. 1H NMR (400 MHz, CD3OD) δ 0.59 (td, J = 7.6 Hz, J′ = 6 Hz, 1 H, 9-Ha), 1.00 (m, 1 H, 9-Hb), 1.04 [m, 2 H, 8(10)-H], 1.26 [broad d, J = 8.8 Hz, 2H, 11(12)-Ha], 1.55 [broad d, J = 8.8 Hz, 2 H, 11(12)-Hb], 1.98 [s, 2 H, 1(7)-H], 2.07 [m, 2 H, piperidine-3(5)-Hax], 2.45 [d, J = 13.2 Hz, 2 H, piperidine-3(5)-Heq], 2.69 [broad s, 2 H, 2(6)-H], 3.07 [broad s, 2 H, 3(5)-Ha], 3.12 [td, J = 13.2 Hz, J′ = 2.8 Hz, 2H, piperidine-2(6)-Hax], 3.58 [d, J = 13.2 Hz, 2 H, piperidine-2(6)-Heq], 3.63 (m, 1 H, piperidine-4-H), 3.78 [broad s, 2 H, 3(5)-Hb]. 13C NMR (100.6 MHz, CD3OD) δ 7.0 (CH2, C9), 16.7 [CH, C8(10)], 17.7 [CH2, C11(12)], 26.9 [CH2, C3(5)-piperidine], 27.0 [CH, C1(7)], 40.4 [CH, C2(6)], 43.6 [CH2, C2(6)-piperidine 54.5 [CH2, C3(5)], 60.2 (CH, C4-piperidine). MS (DI), m/e (%). Main ions: 205 (47), 204 (28), 112 (100), 111 (36), 110 (30), 91 (19), 79 (16), 72 (17), 68 (41).
4-Amidino-4-azatetracyclo[5.3.2.02,6.08.10]dodec-11-ene Hydrochloride, 5·HCl
A solution of 4·HCl (1.15 g, 5.83 mmol) in water (30 mL) was basified to pH = 14 with a 10 N aqueous solution of NaOH. It was then extracted with EtOAc (3 × 50 mL), and the joined organic phase was dried over Na2SO4, filtered off, and concentrated under vacuo (yellow oil, 900 mg, 5.55 mmol, 95% yield). To a suspension of this oil in acetonitrile (35 mL), 1H-pyrazole-1-carboxamidine monohydrochloride (1.08 g, 7.40 mmol) was added. The suspension was heated to reflux for 6 h. The yellow precipitate was filtered to give 5·HCl as yellow crystals (1.13 g, 85%). An analytical sample was obtained by crystallization from MeOH/Et2O, mp 248–249 °C (dec.). IR (KBr) ν 3468, 3410, 3327, 3135, 3047, 3025, 2997, 2948, 2880, 2414, 1669, 1620, 1527, 1470, 1434, 1374, 1363, 1281, 1169, 1085, 1045, 1015, 973, 854, 819, 766, 710, 645 cm–1. 1H NMR (400 MHz, CD3OD) δ 0.15–0.23 (complex signal, 2 H, 9-H2), 1.02 [m, 2 H, 8(10)-H], 2.81 [m, 2 H, 2(6)-H], 2.92 [broad s, 2 H, 1(7)-H], 3.12 [dd, J = 10.8 Hz, J′ = 3.4 Hz, 2 H, 3(5)-Ha], 3.52 [m, 2 H, 3(5)-Hb], 5.85 [m, 2 H, 11(12)-H]. 13C NMR (100.6 MHz, CD3OD) δ 4.7 (CH2, C9), 10.7 [CH, C8(10)], 37.0 [CH, C1(7)], 45.1 [CH, C2(6)], 52.4 [CH2, C3(5)], 130.1 [CH, C11(12)], 155.5 (C, CNH). MS (DI), m/e (%). Main ions: 203 (M•+, 15), 126 (13), 125 (32), 124 (28), 115 (15), 114 (16), 113 (50), 112 (78), 111 (55), 110 (14), 92 (25), 91 (70), 77 (14), 69 (43), 68 (100), 67 (18), 65 (13).
4-Amidino-4-azatetracyclo[5.3.2.02,6.08.10]dodecane Hydrochloride, 7·HCl
To a solution of 6·HCl (377 mg, 1.57 mmol) in absolute EtOH (35 mL), Pd on charcoal (78 mg, ca. 10% Pd) was added, and the resulting suspension was hydrogenated at room temperature and at 150 psi of H2 overnight. The black suspension was filtered to furnish 7·HCl as an off-white powder (290 mg, 77%). An analytical sample was obtained by crystallization from MeOH/Et2O, mp 208–209 °C. IR (KBr) ν 3311, 3124, 3007, 2993, 2949, 2924, 2907, 1653, 1602, 1529, 1484, 1465, 1378, 1352, 1337, 1299, 1283, 1208, 1185, 1171, 1136, 1071, 1024, 936, 805, 642, 539, 487 cm–1. 1H NMR (400 MHz, CD3OD) δ 0.51 [dt, J = 6.2 Hz, J′ = 7.8 Hz, 2 H, 9(10)-Ha], 0.87 [dt, J = 6.2 Hz, J′ = 3.6 Hz, 2 H, 9(10)-Hb], 1.02 [m, 2 H, 8(10)-H], 1.18 [broad d, J = 10.0 Hz, 2 H, 11(12)-Ha], 1.29 [broad d, J = 10.0 Hz, 2 H, 11(12)-Hb], 1.92 [broad s, 2 H, 1(7)-H], 2.70 [m, 2 H, 2(6)-H], 3.48 [m, 2 H, 3(5)-Ha], 3.54 [dt, J = 11.2 Hz, J′ = 2.0 Hz, 2 H, 3(5)-Hb]. 13C NMR (100.6 MHz, CD3OD) δ 5.4 (CH2, C9), 15.8 [CH, C8(10)], 18.4 [CH2, C11(12)], 30.4 [CH, C1(7)], 40.9 [CH, C2(6)], 51.0 [CH2, C3(5)], 156.1 (C, CNH). MS (DI), m/e (%). Main ions: 205 (M•+, 46), 204 (29), 138 (11), 112 (100), 111 (36), 110 (30), 91 (19), 79 (17), 72 (17), 68 (42).
4-Azatetracyclo[5.4.2.02,6.08.11]trideca-9,12-diene Hydrochloride, 11·HCl
From a solution of imide 10 (1.46 g, 7.25 mmol) and sodium bis(2-methoxyethoxy)aluminum hydride (15.8 mL, 65% solution in toluene, 51.7 mmol) in dry and degassed toluene (45 mL) and following the same procedure that the one reported for compound 4, diene 11 was obtained as an oil (876 mg, 70%). An analytical sample of 11·HCl was obtained by solubilization of 9 in diethyl ether and addition of an excess of a 1.2 N ethereal solution followed by filtration of the solid, mp 206–207 °C. IR (ATR) ν 2924, 2482, 2340, 2158, 2016, 1716, 1540, 1049, 878, 824, 617 cm–1. 1H NMR (400 MHz, CD3OD) δ 2.55 [m, 2 H, 2(6)-H], 2.74 [broad d, J = 11.6 Hz, 2 H, 1(7)-H], 2.76 [broad s, 2 H, 8(11)-H], 2.88 [m, 2 H, 3(5)-Ha], 3.43 [m, 2 H, 3(5)-Hb], 5.87 [s, 2 H, 9(10)-H], 6.04 [dd, 2 H, J = 4.4 Hz, J′ = 3.2 Hz, 12(13)-H]. 13C NMR (100.6 MHz, CD3OD) δ 39.2 [CH, C1(7)], 42.9 [CH, C2(6)], 46.2 [CH, C8(11)], 51.5 [CH2, C3(5)], 131.4 [CH, C12(13)], 138.9 [CH, C9(10)]. MS (DI), m/e (%). Main ions: 173 (M•+, 13), 128 (20), 120 (37), 115 (16), 106 (31), 91 (29), 80 (19), 78 (22), 68 (100), 67 (15).
4-Azatetracyclo[5.4.2.02,6.08.11]tridecane Hydrochloride, 12·HCl
To a solution of 11·HCl (2.39 g, 11.4 mmol) in absolute EtOH (170 mL), Pd on charcoal (240 mg, ca. 10% Pd) was added and the resulting suspension was hydrogenated at room temperature and at atmospheric pressure until the addition of hydrogen stopped. The black suspension was filtered to furnish 12·HCl as an off-white powder (2.30 g, 94%). An analytical sample was obtained by crystallization from MeOH/Et2O, mp 266–269 °C. IR (ATR) ν 2897, 2769, 1568, 1489, 879, 640 cm–1. 1H NMR (400 MHz, CD3OD) δ 1.55–1.59 [complex signal, 4 H, 12(13)-Ha and 1(7)-H], 1.98 [dm, 2 H, J = 8.0 Hz, 12(13)-Hb], 2.10–2.25 [complex signal, 4 H, 9(10)-H2], 2.28 [m, 2 H, 2(6)-H], 2.46 [m, 2 H, 8(11)-H], 3.18 [m, 2 H, 3(5)-Ha], 3.48 [m, 2 H, 3(5)-Hb], 4.86 (broad s, 2 H, NH2). 13C NMR (100.6 MHz, CD3OD) δ 15.5 [CH2, C12(13)], 21.3 [CH2, C9(10)], 30.2 [CH, C1(7)], 38.1 [CH, C8(11)], 39.6 [CH, C2(6)], 49.0 [CH2, C3(5)]. MS (DI), m/e (%). Main ions: 177 (M•+, 38), 176 (16), 149 (100), 91 (23), 79 (23), 77 (16), 70 (82), 68 (35), 67 (13).
4-Amidino-4-azatetracyclo[5.4.2.02,6.08.11]trideca-9,12-diene Hydrochloride, 13·HCl
To a suspension of 11·HCl (1.0 g, 4.76 mmol) in acetonitrile (20 mL), 1H-pyrazole-1-carboxamidine monohydrochloride (838 mg, 5.74 mmol), and triethylamine (1.22 mL) were added. The suspension was heated to reflux for 6 h. The precipitate was filtered and washed with cold acetonitrile to give 13·HCl as white crystals (816 mg, 68%), mp 234–237 °C; IR (ATR) ν 3120, 2926, 1657, 1644, 1593, 1534, 1475, 1294, 1195, 1030, 788, 742, 703, 624 cm–1; 1H NMR (400 MHz, CD3OD) δ 2.63 [m, 2 H, 2(6)-H], 2.70 [m, 2 H, 1(7)-H], 2.74 [broad s, 2 H, 8(11)-H], 3.20 [dd, 2 H, J = 10.8 Hz, J = 3.6 Hz, 3(5)-Ha], 3.60 [m, 2 H, 3(5)-Hb], 5.86 [s, 2 H, 9(10)-H], 5.99 [dd, 2 H, J = 4.4 Hz, J′ = 3.2 Hz, 12(13)-H]; 13C NMR (100.6 MHz, CD3OD) δ 40.9 [CH, C1(7)], 43.5 [CH, C2(6)], 46.0 [CH, C8(11)], 53.6 [CH2, C3(5)], 130.8 [CH, C12(13)], 138.9 [CH, C9(10)]. MS (DI), m/e (%); main ions: 215 (M•+, 10), 214 (18), 128 (21), 120 (17), 115 (15), 111 (100), 110 (18), 91 (24), 78 (17), 77 (15), 68 (79).
4-Amidino-4-azatetracyclo[5.4.2.02,6.08.11]tridecane Hydrochloride, 14·HCl
To a solution of 12·HCl (1.0 g, 4.68 mmol) in acetonitrile (20 mL), 1H-pyrazole-1-carboxamidine monohydrochloride (823 mg, 5.64 mmol), and triethylamine (1.19 mL) were added. The suspension was heated to reflux for 6 h. The precipitate was filtered and washed with cold acetonitrile to give 14·HCl as white crystals (1.18 g, 98%), mp 299–301 °C. IR (ATR) ν 3308, 3112, 2938, 2899, 1659, 1644, 1590, 1536, 1487, 1188, 1157, 1030, 647, 627 cm–1. 1H NMR (400 MHz, CD3OD) δ 1.47–1.60 [complex signal, 4 H, 12(13)-Ha and 1(7)-H], 1.96 [broad d, 2 H, J = 8.4 Hz, 12(13)-Hb], 2.12–2.25 [complex signal, 4 H, 9(10)-H2], 2.42–2.46 [complex signal, 4 H, 2(6)-H and 8(11)-H], 3.58 [m, 4 H, 3(5)-H2]. 13C NMR (100.6 MHz, CD3OD) δ 15.9 [CH2, C12(13)], 21.6 [CH2, C9(10)], 32.5 [CH, C1(7)], 37.5 [CH, C8(11)], 39.9 [CH, C2(6)], 51.6 [CH2, C3(5)]. MS (DI), m/e (%). Main ions: 219 (M•+, 41), 218 (21), 191 (38), 137 (24), 112 (57), 111 (100), 110 (19), 91 (20), 79 (18), 72 (15), 68 (39).
3,4,8,9-Tetramethyl-11,13-dioxo-12-azatricyclo[4.4.3]deca-3,8-diene (16)
A mixture of known diacid 15 (200 mg, 0.72 mmol)20,21 and urea (216 mg, 3.59 mmol) was heated slowly to 135 °C. When the mixture melted, it was heated to 180 °C for 30 min and cooled. Water (5 mL) was added, and the suspension was extracted with CH2Cl2 (6 × 4 mL). The combined organic extracts were washed with brine (1 × 5 mL), dried with anhyd Na2SO4, filtered and concentrated in vacuo to dryness to give imide 16 as a white solid (193 mg, 75% yield). An analytical sample of 16 was obtained by crystallization from CH2Cl2/pentane, mp 197–198 °C. IR (ATR) ν 3202, 3072, 2983, 2928, 2859, 1775, 1706, 1659, 1439, 1364, 1322, 1186, 1106, 982, 829, 739, 677 cm–1. 1H NMR (400 MHz, CDCl3) δ 1.63 [d, J = 1.2 Hz, 12 H, 3(4,8,9)-CH3], 2.07 [d, J = 14.2 Hz, 4 H, 2(5,7,10)-Ha], 2.35 [d, J = 14.2 Hz, 4 H, 2(5,7,10)-Hb], 8.23 (broad s, 1H, NH). 13C NMR (100.6 MHz, CDCl3) δ 19.1 [CH3, C3(4,8,9)-CH3], 38.7 [CH2, C2(5,7,10], 52.9 [C, C1(6)], 127.4 [CH, C3(4,8,9)], 182.8 [C=O, C11(13)]. GC/MS, m/z (%). Main ions: 259 (M•+, 49), 188 (24), 17467 (25), 173 (15), 162 (C12H18•+, 100), 144 (13), 133 (16), 132 (10), 119 (18), 91 (13). HRMS-ESI+ m/z [M + H]+ calcd for [C16H21NO2 + H+], 260.1645; found, 260.1649.
7,8,9,10-Tetramethyl-2,4-dioxo-3-azapentacyclo[7.2.1.15,8.01,5.07,10]tridecane (17)
A solution of 16 (630 mg, 2.43 mmol) and dry and degassed acetone (70 mL) in a quartz reactor was irradiated with a 125 W Hg lamp for 2 days under Ar atmosphere. The dark-yellow solution was concentrated under vacuo to give a yellow paste (1.08 g). The paste was purified by column chromatography (silica gel, hexane/EtOAc mixtures). With hexane/EtOAc (85/15) was recovered starting material (190 mg), while with hexane/EtOAc (80/20) imide 17 was obtained (160 mg, 36% brsm) as a white solid, mp 272–273 °C. IR (ATR) ν 3167, 3054, 2953, 2919, 2868, 1770, 1704, 1452, 1374, 1345, 1312, 1268, 1173, 1154, 1108, 1052, 1018, 842, 739, 628 cm–1. 1H NMR (400 MHz, CDCl3) δ 1.01 [s, 12 H, 7(8,9,10)-CH3], 1.32 [d, J = 11.4 Hz, 4 H, 6(11,12,13)-Ha], 1.83 [d, J = 11.4 Hz, 4 H, 6(11,12,13)-Hb], 8.58 (broad s, 1H, NH). 13C NMR (100.6 MHz, CDCl3) δ 15.2 [CH3, C14(15,16,17)], 40.5 [CH2, C2(6,8,10)], 46.1 [C, C7(8,9,10)], 50.6 [C, C1(5)], 180.9 [C=O, C2(4)]. GC/MS, m/z (%). Main ions: 259 (M•+, 74), 188 [(C14H20)•+, 39], 177 (36), 173 (17), 162 [(C12H18)•+, 100], 145 (13), 133 (14), 119 (17), 91 (17). HRMS-ESI+ m/z [M + H]+ calcd for [C16H21NO2 + H+], 260.1645; found, 260.1643.
7,8,9,10-Tetramethyl-3-azapentacyclo[7.2.1.15,8.01,5.07,10]tridecane Hydrochloride (18·HCl)
From 17 (210 mg, 0.81 mmol) in dry toluene (7 mL) and sodium bis(2-methoxyethoxy)aluminum hydride (1.2 mL, 70% solution in toluene, 4.05 mmol) and following the same procedure as reported for 4·HCl, amine 18 as its hydrochloride was obtained as an off-white solid (154 mg, 71% yield). An analytical sample was obtained by crystallization from CH2Cl2/n-pentane, mp >300 °C (dec). IR (ATR) ν 2918, 2907, 2869, 2824, 2734, 2696, 2675, 2559, 2533, 2471, 1612, 1594, 1454, 1435, 1364, 1314, 1282, 1240, 1201, 1159, 1134, 1124, 1090, 1037, 1002, 979, 916 cm–1. 1H NMR (400 MHz, CD3OD) δ 0.95 [d, J = 11.0 Hz, 4 H, 6(11,12,13)-H], 1.00 [s, 12 H, C7(8,9,10)-CH3], 1.84 [d, J = 11.0 Hz, 4 H, 6(11,12,13)-H], 3.35 [s, 4 H, 2(4)-H]. 13C NMR (100.6 MHz, CD3OD) δ 15.7 [CH3, C7(8,9,10)–CH3], 42.7 [CH2, C6(11,12,13)], 46.7 [C, C7(8,9,10)], 48.8 [C1(5)], 52.7 [CH2, C2(4)]. MS, m/z (%). Main ions: 231 [(M-HCl)•+, 100], 230 (58), 216 (25), 201 (14), 188 (36), 187 (31), 186 (60), 185 (19), 173 (20), 172 (17), 171 (65), 160 (14), 159 (23), 157 (16), 149 (26), 148 (94), 146 (47), 145 (34), 134 (20), 132 (23), 119 (40), 105 (21), 93 (17), 91 (36), 79 (18), 77 (21).
3-Amidino-7,8,9,10-tetramethyl-3-azapentacyclo[7.2.1.15,8.01,5.07,10]tridecane Hydrochloride (19·HCl)
From a suspension of 18·HCl (187 mg, 0.70 mmol), Et3N (0.15 mL, 1.05 mmol), and 1H-pyrazole-1-carboxamidine hydrochloride (123 mg, 0.84 mmol) in CH3CN (7.5 mL) and following the same procedure as reported for 5·HCl, guanidine 19·HCl was obtained as a white solid (142 mg, 66% yield). An analytical sample was obtained by crystallization from t-butanol, mp >300 °C (dec). IR (ATR) ν 3178, 2854, 2914, 2868, 1771, 1704, 1697, 1651, 1555, 1455, 1374, 1347, 1312, 1269, 1173, 1154, 1052, 842, 739, 628 cm–1. 1H NMR (400 MHz, CD3OD) δ 0.95 [d, J = 11.0 Hz, 4 H, 6(11,12,13)-H], 1.00 [s, 12 H, C7(8,9,10)-CH3], 1.87 [d, J = 11.0 Hz, 4 H, 6(11,12,13)-H], 3.59 [s, 4 H, 2(4)-H]. 13C NMR (100.6 MHz, CD3OD) δ 15.8 [CH3, C7(8,9,10)-CH3], 43.9 [CH2, C6(11,12,13)], 46.6 [C, C7(8,9,10)], 49.5 [C1(5)], 54.6 [CH2, C2(4)], 156.6 (C=NH). MS, m/z (%). Main ions: 273 [(M – HCl)•+, 85), 272 (18), 192 (25), 191 (100), 190 (76), 176 (15), 171 (65), 148 (15), 132 (12), 119 (11), 105 (10), 91 (17), 72 (26).
Molecular Modeling
The binding mode of Amt and compound 18 to the wt M2 channels and its V27A variant embedded on a model bilayer of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) was studied by molecular dynamics simulations. The M2 channel was modeled from the solid-state NMR structure obtained by Sharma et al.28 (PDB entry 2L0J), and it was oriented using as template the solid state NMR structure with PDB entry 2KQT(23) as deposited in the Orientations of Proteins in Membranes (OPM) database.29 The X-ray crystallographic structure 3LBW from the Protein Data Bank was used to model the position of water molecules inside the channel.30 The initial position of the ligands was chosen to resemble the orientation of Amt in the 2KQT structure.
The CHARMM-GUI web server was used to build up the initial systems.31−33 Briefly, the complex formed by the protein, the inner lumen water molecules, and the ligand was embedded on a 100 Å × 100 Å POPC bilayer patch. A 25 Å layer of TIP3P34 water molecules was set up at both sides of the bilayer, and K+ cations and Cl– anions were added to achieve an ionic strength of 150 mM. The Parm99SB force field was used for the protein,35 the ligands were parametrized using the gaff force field36 in conjunction with RESP (HF/6-31G(d)) charges37 as implemented in the Antechamber module of AMBER12 software package,38 and the POPC molecules were parametrized according to the GAFF-lipid force field.39 Joung and Cheatham parameters were used to model the counterions.40 Each system comprised around 97000 atoms, including the protein ligand complex, 265 POPC molecules, around 58000 waters and 108 (51 K+; 57 Cl–) counterions in a simulation box of 100000 Å3.
The geometry of the system was minimized in five cycles that combined 3500 steps of steepest descent algorithm followed by 4500 of conjugate gradient. Thermalization of the system was performed in 5 steps of 1 ns, where the temperature was gradually increased from 50 K to 298 K, while the protein, ligand, and POPC molecules were restrained with a force constant of 1 kcal mol–1 Å–2. Prior to the production runs, a set of 20 ns simulations was performed to smoothly equilibrate the systems by gradually reducing the restraints first for the POPC molecules (restraints reduced by 0.1 kcal mol–1 Å–2 at each step) and then for the protein (restraints reduced by 0.2 kcal mol–1 Å–2 at each step). At this point, three different replicas were generated for each ligand–protein complex (accounting for a total of 12 different simulation systems).
We took advantage of the GPU-accelerated PMEMD module from AMBER12 software package for the production runs,41 which consisted of 50 ns trajectories (accounting for a global simulation time of 600 ns) using SHAKE for bonds involving hydrogen atoms, a time step of 2 fs, periodic boundary conditions using anisotropic constant pressure and temperature (298 K; Langevin thermostat with a collision frequency of 3 ps–1), particle mesh Ewald for long-range electrostatic interactions, and a cutoff of 10 Å for nonbonded interactions.
Acknowledgments
E.T., J.J.-J., and S.V. thank the Spanish Ministerio de Ciencia e Innovación (FPU and FIS fellowships to E.T. and J.J.-J., respectively; grant CTQ2011-22433 to S.V.; grant SAF2011-27642) and the Generalitat de Catalunya (FI fellowship to S.L.; grant 2014SGR1189) for financial support. M.R.-C. acknowledges a predoctoral grant from the Government of Andorra (ATCR2012/2013-00XX-AND). L.N. acknowledges financial support from the Geconcerteerde Onderzoeksacties (GOA/10/014), and the technical assistance from W. van Dam. F.J.L. is grateful to Icrea Academia for financial support. W.F.D. acknowledges support from GM56423 from the NIH. The Barcelona Supercomputer Center is acknowledged for computational facilities.
Glossary
Abbreviations Used
- Amt
amantadine
- brsm
based on recovered starting material
- BSA
bovine serum albumin
- CPE
cytopathic effect
- DMEM
Dulbecco’s Modified Eagle Medium
- DMPC
dimyristoylphosphatidylcholine
- DPC
dodecylphosphocholine
- MDCK
Madin–Darby canine kidney
- MD
molecular dynamics
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- PBS
phosphate buffered saline
- PDB
Protein Data Bank
- qRT-PCR
quantitative real-time reverse transcription polymerase chain reaction
- Rmt
rimantadine
- ssNMR
solid state nuclear magnetic resonance
- TEV
two-electrode voltage clamps
- TM
transmembrane
- wt
wild-type
Supporting Information Available
Elemental analysis data of the new compounds; modeling studies. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bright R. A.; Shay D. K.; Shu B.; Cox N. J.; Klimov A. I. Adamantane resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. JAMA, J. Am. Med. Assoc. 2006, 295, 891–894. [DOI] [PubMed] [Google Scholar]
- Fiore A. E.; Shay D. K.; Haber P.; Iskander J. K.; Uyeki T. M.; Mootrey G.; Bresee J. S.; Cox N. J. Prevention and control of influenza. Recommendations of the Advisory Committee on Immunization Practices (ACIP), 2007. MMWR Recomm. Rep. 2007, 56, 1–54. [PubMed] [Google Scholar]
- Lamb R. A.; Zebedee S. L.; Richardson C. D. Influenza virus M2 protein is an integral membrane protein expressed on the infected-cell surface. Cell 1985, 40, 627–633. [DOI] [PubMed] [Google Scholar]
- Hong M.; DeGrado W. F. Structural basis for proton conduction and inhibition by the influenza M2 protein. Protein Sci. 2012, 21, 1620–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Qiu J. X.; Soto C. S.; DeGrado W. F. Structural and dynamic mechanisms for the function and inhibition of the M2 proton cannel from influenza A virus. Curr. Opin. Struct. Biol. 2011, 21, 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balannik V.; Carnevale V.; Fiorin G.; Levine B. G.; Lamb R. A.; Klein M. L.; DeGrado W. F.; Pinto L. H. Functional studies and modeling of pore-lining residue mutants of the influenza a virus M2 ion channel. Biochemistry 2010, 49, 696–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse Y.; Suzuki A.; Oshitani H. Large-scale sequence analysis of M gene of influenza A viruses from different species: mechanisms for emergence and spread of amantadine resistance. Antimicrob. Agents Chemother. 2009, 53, 4457–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balannik V.; Wang J.; Ohigashi Y.; Jing X.; Magavern E.; Lamb R. A.; DeGrado W. F.; Pinto L. H. Design and pharmacological characterization of inhibitors of amantadine-resistant mutants of the M2 ion channel of influenza A virus. Biochemistry 2009, 48, 11872–11882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Ma C.; Fiorin G.; Carnevale V.; Wang T.; Hu F.; Lamb R. A.; Pinto L. H.; Hong M.; Klein M. L.; DeGrado W. F. Molecular dynamics simulation directed rational design of inhibitors targeting drug-resistant mutants of influenza A virus M2. J. Am. Chem. Soc. 2011, 133, 12834–12841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Ma C.; Wu Y.; Lamb R. A.; Pinto L. H.; DeGrado W. F. Exploring organosilane amines as potent inhibitors and structural probes of influenza A virus M2 proton channel. J. Am. Chem. Soc. 2011, 133, 13844–13847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey-Carrizo M.; Torres E.; Ma C.; Barniol-Xicota M.; Wang J.; Wu Y.; Naesens L.; DeGrado W. F.; Lamb R. A.; Pinto L. H.; Vázquez S. 3-Azatetracyclo[5.2.1.15,8.01,5]undecane derivatives: from wild-type inhibitors of the M2 ion channel of influenza A virus to derivatives with potent activity against the V27A mutant. J. Med. Chem. 2013, 56, 9265–9274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps P.; Duque M. D.; Vázquez S.; Naesens L.; De Clercq E.; Sureda F. S.; López-Querol M.; Camins A.; Pallàs M.; Prathalingam S. R.; Kelly J. M.; Romero V.; Ivorra D.; Cortés D. Synthesis and pharmacological evaluation of several ring-contracted amantadine analogs. Bioorg. Med. Chem. 2008, 16, 9925–9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duque M. D.; Ma C.; Torres E.; Wang J.; Naesens L.; Juárez-Jiménez J.; Camps P.; Luque F. J.; DeGrado W. F.; Lamb R. A.; Pinto L. H.; Vázquez S. Exploring the size limit of templates for inhibitors of the M2 ion channel of influenza A virus. J. Med. Chem. 2011, 54, 2646–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres E.; Vanderlinden E.; Fernández R.; Miquet S.; Font-Bardia M.; Naesens L.; Vázquez S. Synthesis and anti-influenza activity of 2,2-dialkylamantadines and related compounds. ACS Med. Chem. Lett. 2012, 3, 1065–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres E.; Duque M. D.; Vanderlinden E.; Ma C.; Pinto L. H.; Camps P.; Froeyen M.; Vázquez S.; Naesens L. Role of the viral hemagglutinin in the anti-influenza virus activity of newly synthesized polycyclic amine compounds. Antiviral Res. 2013, 99, 281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abou-Gharbia M.; Patel U. R.; Webb M. B.; Moyer J. A.; Andree T. H.; Muth E. A. Polycyclic aryl- and heteroarylpiperazinyl imides as 5-HT1A receptor ligands and potential anxiolytic agents: synthesis and structure–activity relationship studies. J. Med. Chem. 1988, 31, 1382–1392. [DOI] [PubMed] [Google Scholar]
- Pielak R. M.; Chou J. J. Solution NMR structure of the V27A drug resistant mutant of influenza A M2 channel. Biochem. Biophys. Res. Commun. 2010, 401, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu R.-X.; Liu L. A.; Wang Y.-H.; Xu Q.; Wei D. Q. Structural comparison of the wild-type and drug-resistant mutants of the influenza A M2 proton channel by molecular dynamics simulations. J. Phys. Chem. B 2013, 117, 6042–6051. [DOI] [PubMed] [Google Scholar]
- Gu R.-X.; Liu L. A.; Wang Y.-H.; Wei D.-Q. Structural and energetic analysis of drug inhibition of the influenza A M2 proton channel. Trends Pharmacol. Sci. 2013, 34, 571–580. [DOI] [PubMed] [Google Scholar]
- Mousseron-Canet M.; Mousseron M.; Brown G. Rev. Chim., Acad. Rep. Populaire Roumaine 1962, 7, 1089–1101. [Google Scholar]
- Avila W. B.; Silva R. A. 3,4,8,9-Tetramethyltetracyclo[4.4.0.03,9.04,8]decane-1,6-dioic anhydride. A Photosensitized π2s + π2s intramolecular cycloaddition. Chem. Commun. 1970, 94–95. [Google Scholar]
- Wang J.; Wu Y.; Ma C.; Fiorin G.; Wang J.; Pinto L. H.; Lamb R. A.; Klein M. L.; DeGrado W. F. Structure and Inhibition of the Drug-Resistant S31N Mutant of the M2 Ion Channel of Influenza A Virus. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady S. D.; Schmidt-Rohr K.; Wang J.; Soto C. S.; DeGrado W. F.; Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature 2010, 463, 689–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolocouris A.; Tzitzoglaki C.; Johnson F. B.; Zell R.; Wright A. K.; Cross T. A.; Tietjen I.; Fedida D.; Busath D. D. Aminoadamantanes with persistent in vitro efficacy against H1N1 (2009) influenza A. J. Med. Chem. 2014, 57, 4629–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevaert A.; Dallocchio R.; Dessì A.; Pala N.; Rogolino D.; Sechi M.; Naesens L. Mutational analysis of the binding pockets of the diketo acid inhibitor L-742,001 in the influenza virus PA endonuclease. J. Virol. 2013, 87, 10524–10538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Soto C. S.; Ohigashi Y.; Taylor A.; Bournas V.; Glawe B.; Udo M. K.; DeGrado W. F.; Lamb R. A.; Pinto L. H. Identification of the pore-lining residues of the BM2 ion channel protein of influenza B virus. J. Biol. Chem. 2008, 283, 15921–15931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderlinden E.; Göktas F.; Cesur Z.; Froeyen M.; Reed M. L.; Russell C. J.; Cesur N.; Naesens L. Novel inhibitors of influenza virus fusion: structure–activity relationship and interaction with the viral hemagglutinin. J. Virol. 2010, 84, 4277–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M.; Yi M.; Dong H.; Qin H.; Peterson E.; Busath D. D.; Zhou H. X.; Cross T. A. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science 2010, 330, 509–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize M. A.; Lomize A. L.; Pogozheva I. D.; Mosberg H. I. OPM: Orientations of Proteins in Membranes Database. Bioinformatics 2006, 22, 623–625. [DOI] [PubMed] [Google Scholar]
- Acharya R.; Carnevale V.; Florin G.; Levine B. G.; Polishchuk A. L.; Balannik V.; Samish I.; Lamb R. A.; Pinto L. H.; DeGrado W. F.; Klein M. L. Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 15075–15080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. CHARMM-GUI: A Web-based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- Jo S.; Lim J. B.; Klauda J. B.; Im W. CHARMM-GUI Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes. Biophys. J. 2009, 97, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Im W. Automated Builder and Database of Protein/Membrane Complexes for Molecular Dynamics Simulations. PLoS One 2007, 2, e880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. M.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2005, 26, 1157–1174. [DOI] [PubMed] [Google Scholar]
- Bayly C. I.; Cieplak P.; Cornell W.; Kollman P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar]
- Case D. A.; Darden T. A.; Cheatham T. E. III; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Walker R. C.; Zhang W.; Merz K. M.; Roberts B.; Hayik S.; Roitberg A.; Seabra G.; Swails J.; Goetz A. W.; Kolossváry I.; Wong K. F.; Paesani F.; Vanicek J.; Wolf R. M.; Liu J.; Wu X.; Brozell S. R.; Steinbrecher T.; Gohlke H.; Cai Q.; Ye X.; Wang J.; Hsieh M.-J.; Cui G.; Roe D. R.; Mathews D. H.; Seetin M. G.; Salomon- Ferrer R.; Sagui C.; Babin V.; Luchko T.; Gusarov S.; Kovalenko A.; Kollman P. A.. AMBER 12; University of California: San Francisco, 2012. [Google Scholar]
- Callum J. D.; Rosso L.; Betz R. M.; Walker R. C.; Gould I. R. GAFFlipid: a General Amber Force Field for the accurate molecular dynamics simulation of phospholipid. Soft Matter 2012, 8, 9617–9627. [Google Scholar]
- Joung I. S.; Cheatham T. E. Molecular dynamics simulations of the dynamic and energetic properties of alkali and halide ions using wáter-model-specific ion parameters. J. Phys. Chem. B 2009, 113, 13279–13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon-Ferrer R.; Goetz A. W.; Poole D.; Le Grand S.; Walker R. C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.