

Abstract
Breast cancer is a major health problem for women worldwide. Although in vitro culture of established breast cancer cell lines is the most widely used model for preclinical assessment, it poorly represents the behavior of breast cancers in vivo. Acceleration of the development of effective therapeutic strategies requires a cost-efficient in vitro model that can more accurately resemble the in vivo tumor microenvironment. Here, we report the use of a thermoreversible poly(ethylene glycol)-g-chitosan hydrogel (PCgel) as an in vitro breast cancer model. We hypothesized that PCgel could provide a tumor microenvironment that promotes cultured cancer cells to a more malignant phenotype with drug and immune resistance. Traditional tissue culture plates and Matrigel were applied as controls in our studies. In vitro cellular proliferation and morphology, the secretion of angiogenesis-related growth factors and cytokines, and drug and immune resistance were assessed. Our results show that PCgel cultures promoted tumor aggregate formation, increased secretion of various angiogenesis- and metastasis-related growth factors and cytokines, and increased tumor cell resistance to chemotherapeutic drugs and immunotherapeutic T cells. This PCgel platform may offer a valuable strategy to bridge the gap between standard in vitro and costly animal studies for a wide variety of experimental designs.
Keywords: microenvironment, breast cancer, hydrogel, chitosan, malignancy
Introduction
Breast cancer is the second leading cause of cancer-related deaths in women in the United States.1 The disease is generally asymptomatic at the early stage and thus the majority of patients are diagnosed at later stages when most have lost the chance for cure.2,3 Although tumorectomy, radiotherapy, chemotherapy, and hormone replacement therapy have been used for breast cancer treatment, there is still no effective therapy for patients with metastatic disease.4 This signals an urgent need for the development of new and more precise therapies.
Traditional two-dimensional (2D) culture, which provides researchers with a convenient and easy in vitro platform, is extensively used to test novel treatments. However, successes seen in traditional 2D models are rarely effective in vivo in animals or clinical trials.5 Indeed, it has been well-documented that tumor cells cultured on flat Petri dishes display a dramatically reduced malignant phenotype when compared to their in vivo counterparts.5 Moreover, traditional in vitro 2D cultures fail to simulate the structure of the tumor microenvironment present in vivo, such as complex cell–cell and cell–extracellular matrix (ECM) interactions.6,7 Therefore, good in vitro models to bridge the gap between traditional in vitro and in vivo models are needed to reduce the costs and difficulties associated with animal experiments.8,9
An ideal in vitro tumor model should provide a platform for in vitro drug screening that will better translate to in vivo testing by mimicking both the spatial organization of cells and ECM signaling found in tumors in vivo.(6) A number of studies have demonstrated that 3D tumor cell culture in vitro promotes an increase in malignant phenotype and drug resistance compared to that of traditional 2D cultured cells.6,10,11 A variety of synthetic and natural materials, including poly(lactide-co-glycolide), collagen, fibrin, and commercially available Matrigel, have been explored to replicate the 3D tumor microenvironment in vitro.12−14 However, many synthetic materials degrade into nonbiocompatible byproducts.10,15 Matrigel, a commercially available proprietary mixture of ECM proteins and growth factors secreted by mouse tumor cells, represents the industry standard for ECM replacement.6,16 However, this mammalian-sourced natural ECM material is expensive, and it may introduce pathogens14 and cause inconsistent results from batch-to-batch.
Here, we report the development of a cost-effective hydrogel to address the limitations of existing in vitro tumor models. A series of thermally reversible poly(ethylene glycol)-g-chitosan hydrogels (PCgel) has been developed in our lab and shown to have great potential for clinical applications.17,18 PCgel functions at physiological pH and, similar to that of Matrigel, undergoes a thermoreversible transition from an injectable solution state at low temperatures to a gel state at body temperature, allowing for implantation without surgical intervention. Chitosan, a biodegradable, natural polysaccharide derived by the partial deacetylation of chitin, shares structural similarities to the glycosaminoglycans (GAG) present in the native ECM.19 GAGs perform a vital role in cell signaling and cell–cell communication because of their extracellular location and conserved structure across virtually all animal species.20 PEG is a neutral, water-soluble, and nontoxic polymer.19,21 It is one of only a small number of synthetic polymers approved by the U.S. Food and Drug Administration (FDA) for internal consumption and injection in a variety of foods, cosmetics, personal care products, pharmaceuticals, and biomedical applications.19,21
In this study, we tested the ability of PCgel to increase malignancy of cultured murine mammary carcinoma (MMC) cells compared to that of tissue culture plates and Matrigel. Our goal was to develop a safe and cost-effective in vitro 3D model that performs equivalent to or better than the gold standard 3D model, Matrigel. MMC cells cultured on these substrates were characterized for proliferation and cell morphology, and they were also assessed for the secreted growth factors involved in angiogenesis and cytokines. The cell malignancy was evaluated through their response to chemotherapy and immunotherapy.
Experimental Section
Materials
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise specified. Chitosan (85% deacetylated, MW = medium) and methoxy-poly(ethylene glycol) (PEG, MW = 2000) were used as received. Dulbecco’s modified Eagle media (DMEM), antibiotic–antimycotic (AA), fetal bovine serum (FBS), and Lipofectamine 2000 reagent were purchased from Invitrogen (Carlsbad, CA). Reduced growth factor Matrigel was purchased from BD Biosciences (San Jose, CA).
Hydrogel Preparation
Poly(ethylene glycol)-g-chitosan (PEG-g-chitosan) was prepared as previously described with slight modification.17,18,22 Briefly, PEG-aldehyde was prepared by oxidizing PEG with dimethyl sulfoxide (DMSO)/acetic anhydride. After 20 g of PEG was completely dissolved in anhydrous chloroform/DMSO (0.125, v/v), 10 mL of acetic anhydride was added into the solution. The mixture was constantly stirred for 16 h at room temperature under a nitrogen atmosphere. Then, the solution was precipitated with excess diethyl ether. The precipitate was dissolved with chloroform and then reprecipitated with diethyl ether. After drying under vacuum, white PEG-aldehyde powder was obtained.
PEG-g-chitosan was prepared by alkylation of chitosan followed by Schiff base formation.23 The mixture of chitosan and PEG-aldehyde (0.32, w/w) was added into a mixture of methanol and 2% acetic acid (0.25, v/v). A 5% cyanoborohydride (NaCNBH3) aqueous solution was then added dropwise into the mixture of chitosan and PEG-aldehyde at pH 5.5 (NaCNBH3/PEG-aldehyde, 0.2, w/w). The resultant mixture was dialyzed with a dialysis membrane (MW 12 000–14 000 cutoff) against DI water and 0.05 M NaOH and then against DI water again until neutral pH was achieved. The solution was subsequently freeze-dried. PEG-g-chitosan was obtained by removal of PEG-aldehyde with excess acetone. EtO gas was applied for the sterilization of PEG-g-chitosan powder. The grafted PEG in PEG-g-chitosan was determined to be 60 wt %, and the PEG-g-chitosan has good water solubility.
PEG-g-chitosan powder was reconstituted with DMEM to make PEG-g-chitosan hydrogel solution (PCgel, 2%, w/v). The solution was put on ice for 4 h with periodic vortexing to ensure that PEG-g-chitosan was fully dissolved. The PEG-g-chitosan is a solution when the temperature is equal to or below 10 °C, and it is a gel when the temperature reaches 32–40 °C. The PCgel has an average pore diameter of a few micrometers.
MMC and Therapeutic Cells
MMC cells were obtained and maintained in DMEM supplemented with 10% FBS and 1% AA as previously described.24−26 Briefly, neu-transgenic (neu-tg) mice (FVB/N-TgN(MMTVneu)-202Mul) were obtained from Charles River Laboratory (Bar Harbor, ME) and bred under specific pathogen-free conditions at the University of Washington (Seattle, WA). The mice harbor nonmutated, nonactivated rat neu under control of the mouse mammary tumor virus (MMTV) promoter. For serological analysis of expression cDNA libraries (SEREX) screening, serum-containing samples were collected from animals bearing spontaneous tumors and from control tumor-free female mice. Animal use and care was in accordance with University of Washington guidelines and an IACUC approved protocol. MMC cells were derived from a spontaneous tumor in a neu-tg mouse. MMC cells were transfected with pRFP-N2 using Lipofectamine 2000 reagent according to the manufacturer’s instructions. Forty-eight hours after transfection, the MMC cells were washed with PBS and supplied with fresh media containing G418 (500 μg/mL) for the selection of a stable transfected population. Two weeks after selection, the MMC cells were sorted by fluorescence activated cell sorting (FACS; Vantage SE). For simplicity, RFP-transfected MMC is abbreviated as MMC hereafter.
Following the methods reported in our previous studies,25,26 murine neu p98 specific T cells (p98 T cells) were generated from splenocytes of p98-immunized neu-tg mice [FVB/N-TgN(MMTVneu)-202Mul]. In brief, splenocytes were harvested from mice after three vaccinations with p98 (vaccines were administered 14 days apart). The p98 T cells were cultured in RPMI supplemented with 10% FBS, 1% AA, and 50 μM β-mercaptoethanol for 21 days, with periodic supplementation with IL-2 (recombinant human, 10 U/mL; Hoffman-La Roche) and IL-21 (recombinant murine, 100 ng/mL; PeproTech), and subjected to two rounds of peptide stimulation (days 0 and 9). The MMC cells were donated by Professor Nora Disis’s lab at the University of Washington.
Culture Systems
MMC Cell Growth
Matrigel and PCgel were thawed at 4 °C overnight to obtain viscous solutions. Prechilled pipet tips and 24-well tissue culture plates (TCP) were used for coating. Two-hundred microliters of Matrigel or PCgel was pipetted into the wells of the TCP, which was then transferred to 37 °C for 2 h for solidification. MMC cells (104) were seeded in uncoated, Matrigel-precoated (200 μL), and PCgel-precoated (200 μL) 24-well tissue culture plates. Fully supplemented DMEM media (800 μL) was added 2 h after seeding. MMC cell proliferation was measured with alamarBlue daily for 4 days. Briefly, media were gently aspirated and replaced with the alamarBlue solution (10× dilution with DMEM, 110 μg/mL resazurin). After 2 h of incubation, the alamarBlue solution was collected and transferred to a 96-well black bottomed plate. The fluorescence of the solution was measured on a SpectraMax M2 microplate reader (Molecular Device, Sunnyvale, CA) at 560 nm. The cell number was determined from calibration curves generated with known numbers of MMC cells. The MMC cellular aggregates were imaged at the indicated time points using a Nikon TE300 (Nikon, Japan) inverted microscope.
Cellular Protein Expression Analysis
MMC cells (104) were seeded in uncoated, Matrigel-precoated, and PCgel-precoated 24-well TCPs. After 3 days of culture, media from cell cultures was replaced with a low-serum counterpart (media containing 1% FBS and 1% AA), and cells were incubated for 24 h. The low-serum media was then collected and stored at −80 °C for future use. Supernatants from serum-starved MMC cells cultured on TCP, Matrigel, and PCgel were diluted on the basis of final cell numbers in the culture and analyzed using the Proteome Profiler angiogenesis and cytokine array kits from R&D Systems (ARY015 and ARY006, respectively) following the manufacturer’s protocol. Blots were imaged using a ChemiDoc XRS imaging system (Bio-Rad, Hercules, CA) and analyzed with the QuantityOne software package (Bio-Rad, Hercules, CA).
Cellular Response to Chemotherapy
MMC cells (3 × 103) were seeded in uncoated, Matrigel-precoated, and PCgel-precoated 96-well TCPs. After 3 days of culture, media from cell cultures was replaced with fully supplemented cell culture media containing various concentrations of doxorubicin (0, 0.001, 0.01, 0.1, 1, 10, and 100 μg/mL). Cells were exposed to the doxorubicin-containing media for 48 h, after which cell viability was assessed using alamarBlue. Cell viability is reported as the percentage of viable cells relative to that of untreated controls. LD50 was estimated using the 50% cell viability point on the cell kill curves.
Cellular Response to Immunotherapy
On the basis of our previous studies, a ratio of 100:1 for effector (p98 T cells) to target (MMC cells) was selected for this study.25,26 For fluorescence imaging, p98 T cells were labeled with Green Cell Tracker dye (Invitrogen) according to the manufacturer’s protocol. After 72 h of MMC cell culture in uncoated, Matrigel-precoated, and PCgel-precoated 24-well TCPs, 106 p98 T cells (Green Cell Tracker-labeled) were added. After 30 h of treatment, dead p98 T cells were washed away from MMC cells using PBS. Dead MMC cells were stained with SYTOX Blue nucleic acid stain (Invitrogen) according to the manufacturer’s instructions. Cells were imaged using an inverted fluorescence microscope (Nikon TE 300, Japan). To quantify the dead MMC cells after the treatment with labeled p98 T cells, the signals from the images were quantified through ImageJ. The dead cell percentage was calculated from the following equation:
| 1 |
For SEM analysis, samples were fixed with a 4% formaldehyde aqueous solution for 30 min at room temperature. After the fixation and dehydration in a series of ethanol washes (70, 85, 95, and 100%), the samples were dried with a critical point dryer (Denton DCP-1, Cherry Hill, NJ). The samples were mounted on SEM pin stub, sputter-coated with platinum, and then imaged with a JSM-7000F SEM (JEOL, Tokyo, Japan).
Statistical Analysis
The results are presented as mean values ± standard deviation (mean ± SD). The statistical difference was determined by one-way analysis of variance (ANOVA) and unpaired, two-tailed Student’s t-test. Values were considered to be statistically significant at p < 0.05 (*).
Results
In Vitro Cell Response
In vitro tumor microenvironment models for breast cancer were generated by culturing MMC cells on TCP, Matrigel, or PCgel. The cellular proliferative response to the different microenvironments was assessed using alamarBlue. Figure 1 shows the cellular proliferation on all three substrates from 104 seeded MMC cells during a culture period of 4 days. Cell growth was observed for TCP and PCgel throughout the culture period, whereas the cell population on Matrigel slightly decreased after day 3. In general, MMC cellular growth on Matrigel and PCgel was slower than that in 2D (TCP).
Figure 1.
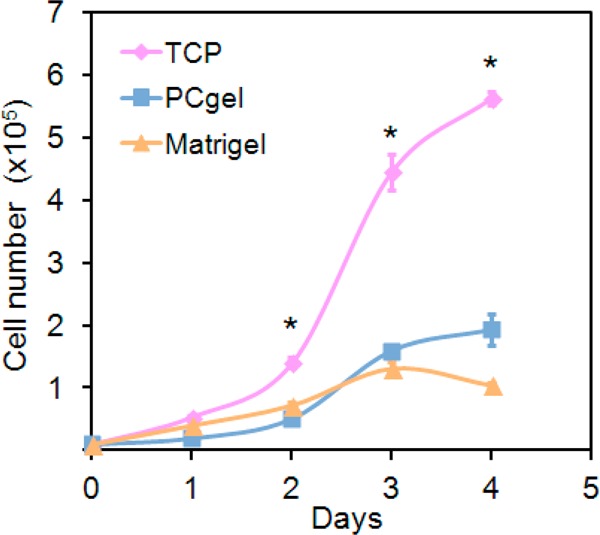
Effect of culture enivroments on MMC cell proliferation. Populations of MMC cells at seeding density of 104 cells/well grown on TCP, Matrigel, and PCgel over a culture period of 4 days. Cellular proliferation was determined by alamarBlue. Results are expressed as the mean ± SD, and * indicates statistical significance, as determined by an unpaired, two-tailed Student’s t-test, p < 0.05, n = 6.
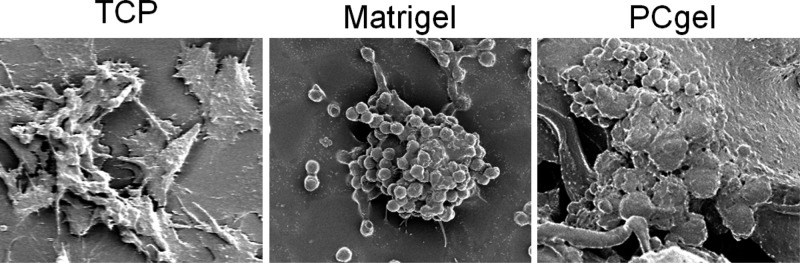
Three-dimensional culture environments have been shown to allow for cell clusters to form en mass, which promote cell–cell and cell–ECM interactions not available in 2D TCP culture.7 These interactions are essential to cell differentiation, proliferation, and gene expression27 and have been shown to generate cells that are functionally distinct from their monolayer counterparts.7 In fact, one of the important features of metastatic cancer cells is multicellular aggregate formation, which directly correlates with their increased survival potential in vitro and metastatic propensity in vivo.7 Here, the effect of the three culture conditions on cellular morphology and organization was revealed by fluorescence imaging. Figure 2 shows the cellular morphological response to the different culture environments during the culture period of 4 days. MMC cells had elongated and flat morphology and were evenly distributed across the 2D TCP with strong adhesion (Figure 2, first row). However, MMC cells grown on both Matrigel (Figure 2, second row) and PCgel (Figure 2, third row) formed clearly discernible multicellular spheroids/aggregates. Moreover, the organization of MMC cells was different between Matrigel and PCgel. The MMC cells formed multiple spheroids across the Matrigel (Figure 2, second row), whereas they formed scattered large and dense aggregates on PCgel (Figure 2, third row).
Figure 2.
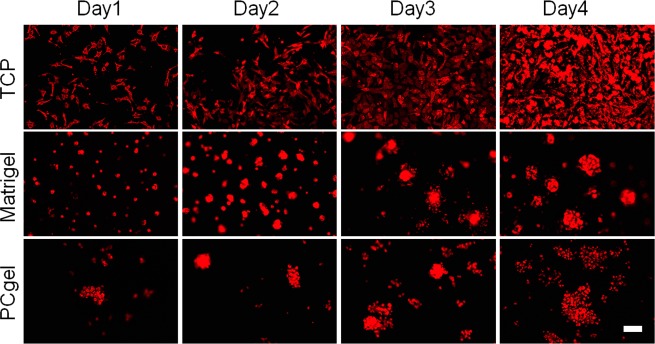
Effect of culture environments on MMC cell morphology and organization. Fluorescence images of MMC aggregates cultured on TCP, Matrigel, and PCgel for 4 days. Scale bar = 100 μm.
Cellular Secreted Protein Expression
The secreted protein expression profiles of the cultured cells were examined to determine if culture on PCgel would promote a more malignant phenotype. The expansion of malignant tumors has been shown to be dependent on the development and maintenance of the surrounding vascular network in vivo.28,29 Therefore, we evaluated the secretion of angiogenesis-related growth factors after 24 h of serum starvation through dot blot arrays. Figure 3 shows the expression of factors that have been suggested to be markers of promoting or inhibiting angiogenesis.30−32 Among the 44 factors evaluated, IP-10, PDGF-AA, HB-EGF, and Platelet Factor 4 were secreted at higher levels in PCgel- and Matrigel-cultured cells compared to that from TCP culture. IP-10 was reported to have proangiogenic activity.33 IP-10 expression was upregulated in cells on PCgel 4.44- and 1.31-fold compared to that of cells cultured on TCP and Matrigel, respectively. PDGF-AA is involved in angiogenesis through VEGF.32,34 PDGF-AA expression was upregulated in PCgel 2.66- and 2.09-fold compared to that of cells cultured on TCP and Matrigel, respectively. HB-EGF expression has been implicated progression and angiogenesis in breast cancer.35 HB-EGF expression was upregulated in cells on PCgel 1.75- and 1.51-fold compared to that of cells cultured on TCP and Matrigel, respectively. Platelet Factor 4 was reported to be involved in the neovasculaturization of breast cancer carcinoma,36 and it also reported to be a potential tumor biomarker.37 Platelet Factor 4 expression was upregulated in PCgel 3.97-fold and 1.06-fold compared to that of cells cultured on TCP and Matrigel, respectively.
Figure 3.

Expression profiles of secreted angiogenesis-related growth factors by serum-starved MMC cells in vitro cultured in three different microenvironments, TCP, Matrigel, and PCgel, as determined by the Proteome Profiler angiogenesis array kit.
Alternatively, markers including TIMP-1, MMP-3, and MMP-9 are known to be associated with inhibiting angiogenesis in breast cancer.38−40 Among the three substrates, cells grown on Matrigel showed the highest expression of TIMP-1, which is reported to be antiangiogenic.38 Cells grown on Matrigel also showed increased expression of MMP-3 and MMP-9, which are reported to be inhibitors of angiogenesis in breast cancer.39,40
On the other hand, the cytokines produced by cancer cells represent a network with a large variety of molecularly and functionally different members that act as a tumor growth-promoting or -inhibiting factors.41−43 As they affect the growth and function of immune-competent cells, they can activate or modulate specific or nonspecific antitumor responses. Therefore, we evaluated the expression of cytokines after 24 h of serum starvation through dot blot arrays, as illustrated in Figure 4. Among the 16 cytokines evaluated, TNF-α, M-CSF, I-309, JE, RANTES, sICAM-1, and MIP-2 are known to be associated with metastasis and immune resistance.44−49 TNF-α is also reported to promote tumor development.44 TNF-α expression was upregulated by MMC cells cultured on PCgel 6.21- and 4.51-fold compared to that of cells cultured on TCP and Matrigel, respectively. M-CSF has been demonstrated to have the ability to regulate metastasis to bones.45 M-SCF expression was upregulated in cells on PCgel 4.42- and 1.11-fold compared to that of cells cultured on TCP and Matrigel, respectively. I-309 was reported to be involved in angiogenesis and tumoral processes.46 I-309 expression was upregulated in cells on PCgel 6.03- and 1.93-fold compared to that of cells cultured on TCP and Matrigel, respectively. JE and RANTES are reported to act directly on breast cancer cells to promote their malignant phenotype and are involved in breast cancer metastasis.47 JE expression was upregulated in cells on PCgel 2.93- and 1.62-fold compared to that of cells cultured on TCP and Matrigel, respectively. RANTES secretion could not be detected from MMC cells cultured on TCP, but it was upregulated in cells on PCgel by 22.17-fold compared to that of cells cultured on Matrigel. sICAM-1 has been reported to be involved in the occurrence of metastases in human breast carcinoma.48 sICAM-1 expression was upregulated in cells on PCgel 4.05- and 1.48-fold compared to that of cells cultured on TCP and Matrigel, respectively. Increased expression of MIP-2 has been reported to play an important role in breast cancer metastasis and chemotherapy resistance.49 MIP-2 expression was upregulated in cells on PCgel 6.89- and 8.4-fold compared to that of cells cultured on TCP and Matrigel, respectively.
Figure 4.
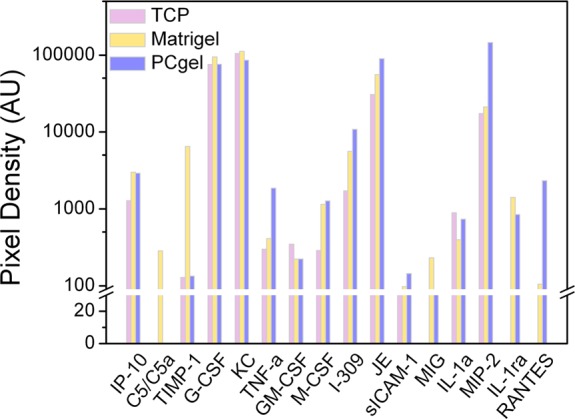
Expression profiles of secreted cytokines by serum-starved MMC cells in vitro cultured in three different microenvironments, TCP, Matrigel, and PCgel, as determined by the Proteome Profiler cytokine array kit.
Alternatively, a high mRNA expression level of MIG has been reported to correlate with an increased number of infiltrating lymphocytes.50 MIG secretion could not be detected from MMC cells cultured on TCP, but it was upregulated in cells on Matrigel by 6.48-fold compared to that of cells cultured on PCgel.
Cellular Response to Chemotherapy
Cell viability in response to doxorubicin treatment was evaluated to determine if the in vitro microenvironment is capable of inducing an environment-mediated drug response in our model. Figure 5 shows the MMC cellular viability determined by alamarBlue after the cells were exposed to doxorubicin for 48 h. Viability measurements of doxorubicin-treated cells revealed significantly different cytotoxic responses of cells under different culture conditions, particularly at high doses of doxorubicin. Forty-eight hours after drug exposure, a dose-dependent survival response was observed in which the viability of TCP-cultured MMC cells was significantly lower than that of either Matrigel- or PCgel -cultured cells when treated with doxorubicin at doses of 1, 10, and 100 μg/mL.
Figure 5.
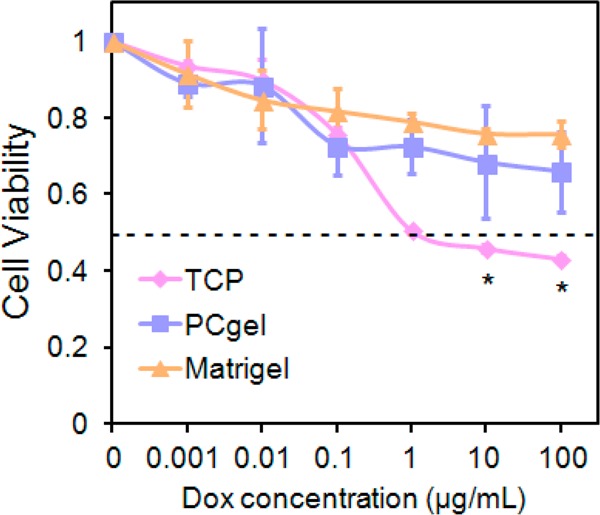
Assessment of drug resistance of MMC cells cultured in three different microenvironments. Viability of MMC cells cultured on TCP, Matrigel, or PCgel relative to that of untreated cells as determined by alamarBlue after doxorubicin exposure for 48 h. Results are the mean ± SD, and * indicates statistical significance, as determined by an unpaired, two-tailed Student’s t-test, p < 0.05, n = 4.
The LD50 is defined as the median lethal dose, which is commonly applied as a measurement of the effectiveness of a drug at inhibiting biological or biochemical function.6 The LD50 of doxorubicin under each of three culture conditions was evaluated, where MMC cells displayed a significant difference in cell viability across culture conditions (Figure 5). The LD50 of doxorubicin was 1 μg/mL for MMC cells cultured on 2D TCP, >100 μg/mL for Matrigel-cultured MMC cells, and >100 μg/mL for PCgel-cultured MMC cells, as determined at 48 h post-treatment. Both Matrigel- and PCgel-cultured MMC cells exhibited higher drug resistance toward doxorubicin, an indication of higher MMC malignancy.
Cellular Response to Immunotherapy
To assess the potential immune response by the differently cultured MMC cells in our tumor models, the cell–cell interaction was investigated after MMC cells were exposed to p98 T cells. The cell–cell interaction was first analyzed with SEM imaging. Figure 6 shows that p98 T cells could readily interact with tumors on all three substrates at the 30 h time point. In SEM images, the p98 T cells were distinguishable from tumor cells by their morphology and size, as is further detailed in SEM images acquired from monoculture samples (Supporting Information Figure 1). Physical interaction of p98 T cell with MMC cells was observed on all three substrates: TCP (Figure 6a,d), Matrigel (Figure 6b,e), and PCgel (Figure 6c,f). These images show that in vitro culture of MMC on PCgel is sufficient to allow immune cells to home to tumor aggregates developed within the 3D structure.
Figure 6.

SEM image of p98 T cells and MMC cells cocultured in three different microenvironments: TCP (left column), Matrigel (middle column), and PCgel (right column). The framed boxes in the low-magnification images (1st row, scale bar = 10 μm) are shown in the images at higher magnification (2nd row, scale bar = 2 μm).
The cell–cell interaction was also analyzed with fluorescence imaging (Figure 7). Living tumor cells were indicated by RFP signal, T cells were shown by Cell Tracker Green labeling, and dead cells were stained blue with live-cell-impermeable SYTOX nucleic acid stain. A higher number of living tumor cells were observed on PCgel among the three substrates. This indicates that the MMC cells cultured on PCgel were more immunoresistant compared to that of TCP and Matrigel cultures. The percentage of dead MMC cells in response to p98 T cell treatment was further quantified using ImageJ (Figure 8). Fewer dead MMC cells were observed on PCgel than on Matrigel or TCP. This may be an indication that the cells cultured on PCgel showed more immunoresistance.
Figure 7.

Fluorescence and bright-field images of p98 T cells targeting MMC cells cultured in three different environments: TCP (left column), Matrigel (middle column), and PCgel (right column). The living MMC cells are indicated by RFP signal (red), T cells are shown by Cell Tracker Green labeling, and dead MMC cells are stained blue with SYTOX nucleic acid stain. Scale bar = 10 μm.
Figure 8.
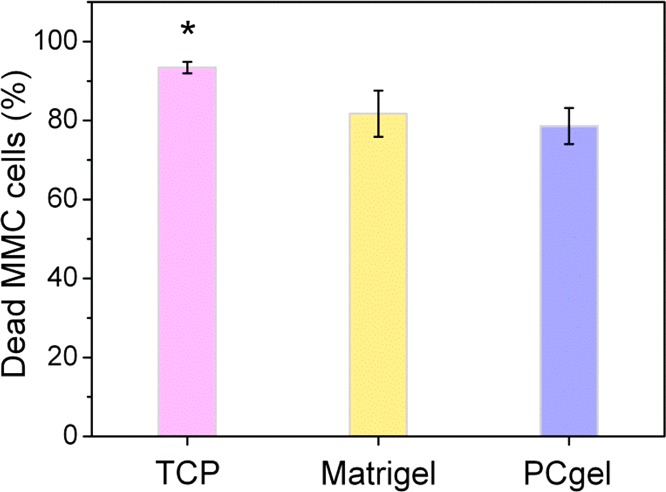
Quantification of dead MMC cells using ImageJ after the treatment by p98 T cells for 30 h in three different culture environments. Results are expressed as the mean ± SD; * indicates statistical significance as determined by an unpaired, two-tailed Student’s t-test, p < 0.05, n = 4.
Discussion
Tumor cells cultured on traditional 2D TCP are subjected to an altered microenvironment compared to that of in vivo tumors, which results in discordant cell functions. A good tumor microenvironment model that closely simulates the real tumor construct would dramatically improve the translation of novel chemotherapeutics and immunotherapeutics from in vitro to in vivo testing. To this end, we used 3D thermoreversible PCgel as a platform for modeling the breast cancer microenvironment. The incorporation of hydrophilic PEG on hydrophobic chitosan increased the water solubility of chitosan in physiological pH environment to promote cellular adhesion. It is generally recognized that cells adhere better on hydrophilic surfaces than on hydrophobic surfaces.51−53 A higher proliferation of cells cultured on TCP compared to that on Matrigel and PCgel was observed (Figure 1). The different proliferation observed between 2D and 3D culture conditions can be attributed both to the different diffusion of nutrients and oxygen to the cells in the matrix interior and to cellular acclimation to the new environment.6,10,11 A major limitation of 2D monolayers is the lack of cell–cell and cell–matrix interactions,7 which makes the in vivo tumor microenvironment inherently heterogeneous.16,54 The cells at the periphery of a tumor mass receive the most nutrients and oxygen, whereas the cells closer to the center are typically hypoxic;12 2D monolayer-cultured cells have no barrier to this exchange. Therefore, Matrigel or PCgel may be closer to an in vivo setting by showing the retarded in vitro growth rate of MMC cells compared to that on TCP.
The different stiffnesses between Matrigel and PCgel may govern different MMC cellular organizations.56 It is known that tumor stroma are stiffer than normal stroma and that cell migration ability is affected by the stiffness of the ECM.57 For breast cancer, diseased tissue can be 10 times stiffer than normal breast tissue in terms of its elastic modulus.58,59 Moreover, soft tissues are viscoelastic in nature, and their mechanical properties are described by a frequency-dependent complex shear modulus (G*), which is the combination of the storage modulus (G′) and the loss modulus (G″).60 The viscosity of breast cancer is reported to be 2.4 ± 1.7 Pas (2400 ± 1700 cP)61 and the stiffness of breast cancer is reported to be 2900 ± 300 Pa in terms of shear storage modulus.61 The viscosity of Matrigel is 10–15 cP62 and the stiffness of Matrigel is 10–50 Pa in terms of storage modulus (G′).57 The viscosity of PCgel is 0.5–4.5 Pas (500–4500 cP)17,18 and the stiffness of PCgel is 1–1000 Pa in terms of storage modulus (G′) (our unpublished result). The stiffer PCgel demonstrated greater MMC cellular organization by increasing cell–cell interactions rather than cell–matrix interactions, which resulted in the rapid formation of cell clusters (Figure 2). The stiffer PCgel may provide the right cues relative to that of the softer Matrigel to build an in vitro tumor microenvironment that better simulates the in vivo tumor environment. These MMC aggregates formed in PCgel may provide a spatially and physiologically more relevant breast tumor phenotype distinct from their monolayer conterparts.7
Further analysis of cultured MMC cells revealed that the expression of the proangiogenic factors IP-10, PDGF-AA, HB-EGF, and Platelet Factor 4 was elevated in cells grown on PCgel compared to TCP- and Matrigel-cultured cells (Figure 3). Also, MMC cells cultured on Matrigel expressed elevated antiangiogenic factors TIMP-1, MMP-3, and MMP-9 compared to their levels in those cultured on PCgel. This suggests that the cell–cell and cell–ECM interactions created upon culture on PCgel more faithfully mimicked the native tumor microenvironment that regulates angiogenic factor secretion. Moreover, TIMP-1 plays a potential role in chemoresistance by inhibiting apoptosis.63 This is consistent with our chemotherapy study that demonstrated that Matrigel- and PCgel-cultured MMC cells showed more doxorubicin resistance than TCP-cultured MMC cells (Figure 5). Doxorubicin is a cytotoxic agent commonly incorporated in catheter-based therapies for metastatic disease,6 and it killed Matrigel- or PCgel-cultured MMC cells less effectively than it did 2D TCP-cultured ones. This suggests that a Matrigel or PCgel microenvironment induced greater resistance to chemotherapy, consistent with many studies on environment-mediated, multicellular drug resistance.64
The immune response against tumors involves both lymphocytes and lymphocyte-derived mediators.65,66 The presence of tumor-infiltrating lymphocytes has been shown to be a favorable prognostic in patients.67 Additionally, analysis of the tumor microenvironment in patients with a variety of solid tumors has revealed that a major subset of tumors shows evidence of a T cell-infiltrated phenotype.68 Cancer progression is aided by the ability of tumors to evade recognition by the immune system.69 Investigating the specific components of the tumor microenvironment that promote or inhibit immune cell activity will help to design better, more effective immunotherapies. The analysis of MMC cells revealed that expression of metastasis-related cytokines TNF-α, M-CSF, I-309, JE, RANTES, sICAM-1, and MIP-2 was elevated in cells cultured on PCgel compared to that of TCP and Matrigel cultured cells (Figure 4). However, MIG has been reported to increase chemotactic T cell recruitment and to impair tumor growth,50,70 which is consistent with our immunotherapy study that showed more T cells bound to MMC cells (Figures 6 and 7) and more dead MMC cells (Figure 8) on Matrigel culture compared with that of cultures on PCgel. This suggests that the microenvironment created by our PCgel induced significant changes in cellular behavior compared to that induced by TCP or Matrigel.
In summary, PCgel provided a convenient and valuable in vitro model for studying clinically relevant improvements to breast cancer therapy. Our results show that PCgel was compatible with MMC cells, and the cells adhered and proliferated well in the gel. More importantly, PCgel promoted tumor aggregate formation, increased the secretion of growth factors and cytokines associated with angiogenesis and metastasis, and increased tumor cell resistance to chemotherapeutic drugs and immunotherapeutic T cells. PCgel has an average pore diameter of a few micrometers and is suitable for transportation and permeability of biomolecules such as drugs and proteins. Unlike Matrigel that lacks well-defined components and ingredients, PCgel has a well-defined composition and thus has consistent properties from batch-to-batch. Unlike expensive Matrigel, the cost of chitosan and PEG as well as the cost of making PCgel is very low. PCgel has a large number of functional groups (namely, amine groups on chitosan) that can be easily modified for many desired applications. PCgel is clinically preferable over Matrigel and any other animal-sourced gels because of its excellent biocompatibility, biodegradability, low immunogenicity, and low potential for pathogen transfer. For a more thorough understanding of the entire microenvironment on breast cancer malignancy, further studies involving coculture of other stromal cells in vitro are needed to assess potential mechanisms.25,26,71 However, our study shows the utility of PCgel as an in vitro platform to bridge the gap between traditional in vitro and in vivo models, and it lays the groundwork for future studies.
Acknowledgments
This work was supported in part by NIH grant NIH/NCI R01CA172455 and Kyocera Professor Endowment to M.Z. C.T. acknowledges support by the National Science Council, Taiwan. F.M.K. acknowledges support from the American Brain Tumor Association Basic Research Fellowship in Honor of Susan Kramer. K.W. acknowledges support from the University of Washington College of Engineering Dean’s Fellowship. A.E.E. acknowledges support from Ruth L. Kirschstein NIH training grant T32CA138312. We acknowledge the use of resources at the Center for Nanotechnology and Department of Immunology at the University of Washington.
Supporting Information Available
Morphology of MMC cells and p98 T cells on TCP characterized by SEM, which showed the heterogeneously spread MMC cells (6–15 μm in diameter) and the round–small p98 T cells (1–3 μm in diameter). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- DeSantis C.; Ma J.; Bryan L.; Jemal A. Breast cancer statistics, 2013. Ca–Cancer J. Clin. 2013, 64, 52–62. [DOI] [PubMed] [Google Scholar]
- Mouawad R.; Spano J. P.; Khayat D. Lymphocyte Infiltration in Breast Cancer: A Key Prognostic Factor That Should Not Be Ignored. J. Clin. Oncol. 2011, 29, 1935–1936. [DOI] [PubMed] [Google Scholar]
- DeNardo D. G.; Coussens L. M. Inflammation and Breast Cancer. Balancing Immune Response: Crosstalk between Adaptive and Innate Immune Cells during Breast Cancer Progression. Breast Cancer Res. 2007, 9, 212–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Zhong Y. Breast Cancer Immunotherapy. Cell Mol. Immunol. 2004, 1, 247–255. [PubMed] [Google Scholar]
- Li L.; Lu Y. Optimizing a 3D Culture System to Study the Interaction between Epithelial Breast Cancer and Its Surrounding Fibroblasts. J. Cancer 2011, 2, 458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung M.; Kievit F. M.; Florczyk S. J.; Veiseh O.; Wu J.; Park J. O.; Zhang M. Chitosan-Alginate Scaffold Culture System for Hepatocellular Carcinoma Increases Malignancy and Drug Resistance. Pharm. Res. 2010, 27, 1939–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. B.; Stein R.; O’Hare M. J. Three-Dimensional in Vitro Tissue Culture Models of Breast Cancer–A Review. Breast Cancer Res. Treat. 2004, 85, 281–291. [DOI] [PubMed] [Google Scholar]
- Yamada K. M.; Cukierman E. Modeling Tissue Morphogenesis and Cancer in 3D. Cell 2007, 130, 601–610. [DOI] [PubMed] [Google Scholar]
- Kim J. B.; O’Hare M. J.; Stein R. Models of Breast Cancer: Is Merging Human and Animal Models the Future?. Breast Cancer Res. 2004, 6, 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kievit F. M.; Florczyk S. J.; Leung M. C.; Veiseh O.; Park J. O.; Disis M. L.; Zhang M. Chitosan-Alginate 3D Scaffolds as a Mimic of the Glioma Tumor Microenvironment. Biomaterials 2010, 31, 5903–5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florczyk S. J.; Wang K.; Jana S.; Wood D. L.; Sytsma S. K.; Sham J. G.; Kievit F. M.; Zhang M. Porous Chitosan-Hyaluronic Acid Scaffolds as a Mimic of Glioblastoma Microenvironment ECM. Biomaterials 2013, 34, 10143–10150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach C.; Chen R.; Matsumoto T.; Schmelzle T.; Brugge J. S.; Polverini P. J.; Mooney D. J. Engineering Tumors with 3D Scaffolds. Nat. Methods 2007, 4, 855–860. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Basu S.; Tomasko D. L.; Lee L. J.; Yang S. T. Fabrication of Well-Defined PLGA Scaffolds Using Novel Microembossing and Carbon Dioxide Bonding. Biomaterials 2005, 26, 2585–2594. [DOI] [PubMed] [Google Scholar]
- Lutolf M. P.; Hubbell J. A. Synthetic Biomaterials as Instructive Extracellular Microenvironments for Morphogenesis in Tissue Engineering. Nat. Biotechnol. 2005, 23, 47–55. [DOI] [PubMed] [Google Scholar]
- Tsao C. T.; Chang C. H.; Lin Y. Y.; Wu M. F.; Han J. L.; Hsieh K. H. Tissue Response to Chitosan/Gamma-PGA Polyelectrolyte Complex Using a Rat Model. J. Bioact. Compat. Polym. 2011, 26, 191–206. [Google Scholar]
- Smalley K. S.; Herlyn M. Towards the Targeted Therapy of Melanoma. Mini-Rev. Med. Chem. 2006, 6, 387–393. [DOI] [PubMed] [Google Scholar]
- Bhattarai N.; Ramay H. R.; Gunn J.; Matsen F. A.; Zhang M. PEG-Grafted Chitosan as an Injectable Thermosensitive Hydrogel for Sustained Protein Release. J. Controlled Release 2005, 103, 609–624. [DOI] [PubMed] [Google Scholar]
- Bhattarai N.; Matsen F. A.; Zhang M. PEG-Grafted Chitosan as an Injectable Thermoreversible Hydrogel. Macromol. Biosci. 2005, 5, 107–111. [DOI] [PubMed] [Google Scholar]
- Chen S. H.; Tsao C. T.; Chang C. H.; Lai Y. T.; Wu M. F.; Chuang C. N.; Chou H. C.; Wang C. K.; Hsieh K. H. Synthesis and Characterization of Reinforced Poly(ethylene glycol)/Chitosan Hydrogel as Wound Dressing Materials. Macromol. Mater. Eng. 2013, 298, 429–438. [Google Scholar]
- Linhardt R. J.; Toida T. Role of Glycosaminoglycans in Cellular Communication. Acc. Chem. Res. 2004, 37, 431–438. [DOI] [PubMed] [Google Scholar]
- Chen S. H.; Tsao C. T.; Chang C. H.; Lai Y. T.; Wu M. F.; Chuang C. N.; Chou H. C.; Wang C. K.; Hsieh K. H. Assessment of Reinforced Poly(ethylene glycol) Chitosan Hydrogels as Dressings in a Mouse Skin Wound Defect Model. Mater. Sci. Eng., C 2013, 33, 2584–2594. [DOI] [PubMed] [Google Scholar]
- Bhattarai N.; Gunn J.; Zhang M. Chitosan-Based Hydrogels for Controlled, Localized Drug Delivery. Adv. Drug Delivery Rev. 2010, 62, 83–99. [DOI] [PubMed] [Google Scholar]
- K K. Chitin and Chitosan: Functional Biopolymers from Marine Crustaceans. Mar. Biotechnol. 2006, 8, 203–226. [DOI] [PubMed] [Google Scholar]
- Lu H.; Knutson K. L.; Gad E.; Disis M. L. The Tumor Antigen Repertoire Identified in Tumor-Bearing Neu Transgenic Mice Predicts Human Tumor Antigens. Cancer Res. 2006, 66, 9754–9761. [DOI] [PubMed] [Google Scholar]
- Phan-Lai V.; Florczyk S. J.; Kievit F. M.; Wang K.; Gad E.; Disis M. L.; Zhang M. Three-Dimensional Scaffolds to Evaluate Tumor Associated Fibroblast-Mediated Suppression of Breast Tumor Specific T Cells. Biomacromolecules 2013, 14, 1330–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan-Lai V.; Kievit F. M.; Florczyk S. J.; Wang K.; Disis M. L.; Zhang M. CCL21 and IFNgamma Recruit and Activate Tumor Specific T Cells in 3D Scaffold Model of Breast Cancer. Anti-Cancer Agents Med. Chem. 2013, 14, 204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F.; Burg K. J. Three-Dimensional Polymeric Systems for Cancer Cell Studies. Cytotechnology 2007, 54, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh D. J.; Wilson C. The Interleukin-8 Pathway in Cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [DOI] [PubMed] [Google Scholar]
- Lee E.; Koskimaki J. E.; Pandey N. B.; Popel A. S. Inhibition of Lymphangiogenesis and Angiogenesis in Breast Tumor Xenografts and Lymph Nodes by a Peptide Derived from Transmembrane Protein 45A. Neoplasia 2013, 15, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiji H.; Gomez D. E.; Shibuya M.; Thorgeirsson U. P. Expression of Vascular Endothelial Growth Factor, Its Receptor, and Other Angiogenic Factors in Human Breast Cancer. Cancer Res. 1996, 56, 2013–2016. [PubMed] [Google Scholar]
- Relf M.; LeJeune S.; Scott P. A.; Fox S.; Smith K.; Leek R.; Moghaddam A.; Whitehouse R.; Bicknell R.; Harris A. L. Expression of the Angiogenic Factors Vascular Endothelial Cell Growth Factor, Acidic and Basic Fibroblast Growth Factor, Tumor Growth Factor Beta-1, Platelet-Derived Endothelial Cell Growth Factor, Placenta Growth Factor, and Pleiotrophin in Human Primary Breast Cancer and Its Relation to Angiogenesis. Cancer Res. 1997, 57, 963–969. [PubMed] [Google Scholar]
- Heinzman J. M.; Brower S. L.; Bush J. E. Comparison of Angiogenesis-Related Factor Expression in Primary Tumor Cultures under Normal and Hypoxic Growth Conditions. Cancer Cell Int. 2008, 8, 11-1–11-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F.; Mantovani A. Inflammation and Cancer: Back to Virchow?. Lancet 2001, 357, 539–5345. [DOI] [PubMed] [Google Scholar]
- Madsen C. V.; Steffensen K. D.; Olsen D. A.; Waldstrom M.; Sogaard C. H.; Brandslund I.; Jakobsen A. Serum Platelet-Derived Growth Factor and Fibroblast Growth Factor in Patients with Benign and Malignant Ovarian Tumors. Anticancer Res. 2012, 32, 3817–3825. [PubMed] [Google Scholar]
- Ongusaha P. P.; Kwak J. C.; Zwible A. J.; Macip S.; Higashiyama S.; Taniguchi N.; Fang L.; Lee S. W. HB-EGF is a Potent Inducer of Tumor Growth and Angiogenesis. Cancer Res. 2004, 64, 5283–5290. [DOI] [PubMed] [Google Scholar]
- Borgstrom P.; Discipio R.; Maione T. E. Recombinant Platelet Factor 4, an Angiogenic Marker for Human Breast Carcinoma. Anticancer Res. 1998, 18, 4035–4041. [PubMed] [Google Scholar]
- Cervi D.; Yip T. T.; Bhattacharya N.; Podust V. N.; Peterson J.; Abou-Slaybi A.; Naumov G. N.; Bender E.; Almog N.; Italiano J. E. Jr.; Folkman J.; Klement G. L. Platelet-Associated PF-4 as a Biomarker of Early Tumor Growth. Blood 2008, 111, 1201–1207. [DOI] [PubMed] [Google Scholar]
- Thorgeirsson U. P.; Yoshiji H.; Sinha C. C.; Gomez D. E. Breast Cancer; Tumor Neovasculature and the Effect of Tissue Inhibitor of Metalloproteinases-1 (TIMP-1) on Angiogenesis. In Vivo 1996, 10, 137–144. [PubMed] [Google Scholar]
- Bendrik C.; Dabrosin C. MMP-3 and MMP-9 Gene Transfer Decrease Growth and Angiogenesis in Breast Cancer Xenografts in Vivo. Cancer Res. 2009, 69, 4164. [Google Scholar]
- Bendrik C.; Robertson J.; Gauldie J.; Dabrosin C. Gene Transfer of Matrix Metalloproteinase-9 Induces Tumor Regression of Breast Cancer in Vivo. Cancer Res. 2008, 68, 3405–3412. [DOI] [PubMed] [Google Scholar]
- Nicolini A.; Carpi A.; Rossi G. Cytokines in Breast Cancer. Cytokine Growth Factor Rev. 2006, 17, 325–337. [DOI] [PubMed] [Google Scholar]
- Rollins B. J. Inflammatory Chemokines in Cancer Growth and Progression. Eur. J. Cancer. 2006, 42, 760–767. [DOI] [PubMed] [Google Scholar]
- Tanaka T.; Bai Z.; Srinoulprasert Y.; Yang B. G.; Hayasaka H.; Miyasaka M. Chemokines in Tumor Progression and Metastasis. Cancer Sci. 2005, 96, 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M.; Zhou X.; Niu L.; Lin G.; Huang J.; Zhou W.; Gan H.; Wang J.; Jiang X.; Yin B.; Li Z. Targeting Transmembrane TNF-alpha Suppresses Breast Cancer Growth. Cancer Res. 2013, 73, 4061–4074. [DOI] [PubMed] [Google Scholar]
- Mancino A. T.; Klimberg V. S.; Yamamoto M.; Manolagas S. C.; Abe E. Breast Cancer Increases Osteoclastogenesis by Secreting M-CSF and Upregulating RANKL in Stromal Cells. J. Surg. Res. 2001, 100, 18–24. [DOI] [PubMed] [Google Scholar]
- Bernardini G.; Spinetti G.; Ribatti D.; Camarda G.; Morbidelli L.; Ziche M.; Santoni A.; Capogrossi M. C.; Napolitano M. I-309 Binds to and Activates Endothelial Cell Functions and Acts as an Angiogenic Molecule in Vivo. Blood 2000, 96, 4039–4045. [PubMed] [Google Scholar]
- Soria G.; Ben-Baruch A. The Inflammatory Chemokines CCL2 and CCL5 in Breast Cancer. Cancer Lett. 2008, 267, 271–285. [DOI] [PubMed] [Google Scholar]
- Rosette C.; Roth R. B.; Oeth P.; Braun A.; Kammerer S.; Ekblom J.; Denissenko M. F. Role of ICAM1 in Invasion of Human Breast Cancer Cells. Carcinogenesis 2005, 26, 943–950. [DOI] [PubMed] [Google Scholar]
- Feliciano P. CXCL1 and CXCL2 Link Metastasis and Chemoresistance. Nat. Genet. 2012, 44, 840. [Google Scholar]
- Bronger H.; Kraeft S.; Schwarz-Boeger U.; Cerny C.; Stockel A.; Avril S.; Kiechle M.; Schmitt M. Modulation of CXCR3 Ligand Secretion by Prostaglandin E2 and Cyclooxygenase Inhibitors in Human Breast Cancer. Breast Cancer Res. 2012, 14, R30-1–R30-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagvolden G.; Giaever I.; Pettersen E. O.; Feder J. Cell Adhesion Force Microscopy. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 471–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling D. P.; Miller I. S.; Ardhaoui M.; Gallagher W. M. Effect of Surface Wettability and Topography on the Adhesion of Osteosarcoma Cells on Plasma-Modified Polystyrene. J. Biomater. Appl. 2011, 26, 327–347. [DOI] [PubMed] [Google Scholar]
- Horbett T. A.; Waldburger J. J.; Ratner B. D.; Hoffman A. S. Cell Adhesion to a Series of Hydrophilic–Hydrophobic Copolymers Studied with a Spinning Disc Apparatus. J. Biomed. Mater. Res. 1988, 22, 383–404. [DOI] [PubMed] [Google Scholar]
- Tredan O.; Galmarini C. M.; Patel K.; Tannock I. F. Drug Resistance and the Solid Tumor Microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [DOI] [PubMed] [Google Scholar]
- Discher D. E.; Janmey P.; Wang Y. L. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science 2005, 310, 1139–1143. [DOI] [PubMed] [Google Scholar]
- Zaman M. H.; Trapani L. M.; Sieminski A. L.; Mackellar D.; Gong H.; Kamm R. D.; Wells A.; Lauffenburger D. A.; Matsudaira P. Migration of Tumor Cells in 3D Matrices Is Governed by Matrix Stiffness along with Cell–Matrix Adhesion and Proteolysis. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 10889–10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P.; Weaver V. M.; Werb Z. The Extracellular Matrix: A Dynamic Niche in Cancer Progression. J. Cell Biol. 2012, 196, 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental K. R.; Yu H.; Kass L.; Lakins J. N.; Egeblad M.; Erler J. T.; Fong S. F.; Csiszar K.; Giaccia A.; Weninger W.; Yamauchi M.; Gasser D. L.; Weaver V. M. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi C. U.; Bharat Chandran R. S.; Vasu R. M.; Sood A. K. Measurement of Visco-Elastic Properties of Breast-Tissue Mimicking Materials Using Diffusing Wave Spectroscopy. J. Biomed. Opt. 2007, 12, 034035. [DOI] [PubMed] [Google Scholar]
- Sinkus R.; Tanter M.; Xydeas T.; Catheline S.; Bercoff J.; Fink M. Viscoelastic Shear Properties of in Vivo Breast Lesions Measured by MR Elastography. Magn. Reson. Imaging 2005, 23, 159–165. [DOI] [PubMed] [Google Scholar]
- Lii J.; Hsu W. J.; Parsa H.; Das A.; Rouse R.; Sia S. K. Real-Time Microfluidic System for Studying Mammalian Cells in 3D Microenvironments. Anal. Chem. 2008, 80, 3640–3647. [DOI] [PubMed] [Google Scholar]
- Wurtz S. O.; Schrohl A. S.; Sorensen N. M.; Lademann U.; Christensen I. J.; Mouridsen H.; Brunner N. Tissue Inhibitor of Metalloproteinases-1 in Breast Cancer. Endocr. Relat. Cancer 2005, 12, 215–227. [DOI] [PubMed] [Google Scholar]
- Davies G. F.; Berg A.; Postnikoff S. D.; Wilson H. L.; Arnason T. G.; Kusalik A.; Harkness T. A. TFPI1 Mediates Resistance to Doxorubicin in Breast Cancer Cells by Inducing a Hypoxic-Like Response. PLoS One 2014, 9, e84611-1–e84611-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S.; Cohen M. C. Mechanisms of Tumor Immunity. An Overview. Am. J. Pathol. 1978, 93, 449–458. [PMC free article] [PubMed] [Google Scholar]
- Drake C. G.; Jaffee E.; Pardoll D. M. Mechanisms of Immune Evasion by Tumors. Adv. Immunol. 2006, 90, 51–81. [DOI] [PubMed] [Google Scholar]
- Rahir G.; Moser M. Tumor Microenvironment and Lymphocyte Infiltration. Cancer Immunol. Immunother. 2012, 61, 751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski T. F.; Schreiber H.; Fu Y. X. Innate and Adaptive Immune Cells in the Tumor Microenvironment. Nat. Immunol. 2013, 14, 1014–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks B. A.; Holtzhausen A.; Evans K. S.; Jamieson R.; Gimpel P.; Campbell O. M.; Hector-Greene M.; Sun L.; Tewari A.; George A.; Starr M.; Nixon A.; Augustine C.; Beasley G.; Tyler D. S.; Osada T.; Morse M. A.; Ling L.; Lyerly H. K.; Blobe G. C. Type III TGF-beta Receptor Downregulation Generates an Immunotolerant Tumor Microenvironment. J. Clin. Invest. 2013, 123, 3925–3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walser T. C.; Ma X.; Kundu N.; Dorsey R.; Goloubeva O.; Fulton A. M. Immune-Mediated Modulation of Breast Cancer Growth and Metastasis by the Chemokine MIG (CXCL9) in a Murine Model. J. Immunother. 2007, 30, 490–498. [DOI] [PubMed] [Google Scholar]
- Florczyk S. J.; Liu G.; Kievit F. M.; Lewis A. M.; Wu J. D.; Zhang M. 3D Porous Chitosan-Alginate Scaffolds: A New Matrix for Studying Prostate Cancer Cell–Lymphocyte Interactions in Vitro. Adv. Healthcare Mater. 2012, 1, 590–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.