

Abstract
Background
Obesity and dietary fat are associated with increased risk of several malignancies including pancreatic cancer. The incidence of pancreatic cancer is increased in countries that consume diets high in fat.
Aim
The purpose of this study was to assess the relationship and mechanism of action between dietary fat and endogenous cholecystokinin (CCK) on pancreatic tumor growth and metastasis in an immunocompetent animal model.
Methods
C57BL/6 mice were placed on regular, low-fat, or high-fat diets for 8 weeks before establishment of Panc-02 orthotopic pancreatic tumors. Mice were then treated with a CCK-A receptor antagonist, devazepide, or vehicle for an additional 2.5 weeks. Pancreas tumors were weighed and metastases counted. Blood CCK levels were measured by radioimmunoassay (RIA). Tissues were examined histologically and studied for genes associated with metastasis by RT-PCR array. Effects of the CCK antagonist on Panc-02 cells invasiveness was assessed in a Matrigel invasion assay.
Results
Mice that received the high-fat diet had larger tumors and tenfold higher serum CCK levels by RIA compared to normal diet controls (p < 0.01). Pancreatic tumors in high-fat diet mice treated with the antagonist had fewer intravascular tumor emboli and metastases compared to controls. The reduction in tumor emboli correlated with decreased vascular endothelial growth factor-A (VEGF-A) expression in tumors (p < 6 × 10−9). In vitro invasiveness of Panc-02 cells also was reduced by CCK-A receptor antagonist treatment (p = 1.33 × 10−6).
Conclusion
CCK is a mediator of dietary fat-associated pancreatic cancer. CCK is also involved in the invasiveness of pancreatic tumors through a mechanism involving VEGF-A.
Keywords: Pancreatic cancer, Devazepide, Obesity, High-fat diet, CCK, Metastasis
Introduction
Pancreatic cancer ranks as the fourth most common cause of cancer-related mortality with a 5-year survival rate of only 5–6 % [1]. Epidemiologic studies have reported that the incidence of pancreatic cancer is increased in countries that consume diets high in fat [2]. Several reports have found an association between obesity, dietary fat intake, and pancreatic cancer; and high body mass index (BMI) more than doubles the risk of pancreatic cancer [3–5]. Research in this area has focused on the roles of insulin, leptin, glucose, and adiposity as the most critical factors contributing to pancreatic cancer in obese individuals [3, 5, 6]. White et al. [7] described how moderate obesity accelerated the growth of subcutaneous murine Panc-02 pancreatic tumors, while Wang et al. [8] showed that orthotopically implanted human pancreatic tumor cells (MIA PaCa2) in nude mice fed a high-fat diet were significantly larger compared to those tumors of mice fed a normal diet. Although both studies suggest a connection between obesity and pancreatic cancer, neither study addressed a clear mechanism linking the two. Some researchers suggest that the mechanism implicates obesity itself and not the dietary fat [7] while others believe the etiology involves the increase in calories [9] or energy balance altogether [10]. Hyperinsulinemia and insulin-like growth factor-1 (IGF-I) signaling [10, 11] have also been reported to play a role in obesity-associated pancreatic cancer as well as hyperglycemia [12]. But other large epidemiology studies [13] have not found an association between IGF-I and pancreatic cancer. Some suggest that the inflammatory state and inflammatory cytokines induced by obesity contribute to pancreatic carcinogenesis [6] and in fact, Philip and coworkers [14] showed a reduction in pancreatic intraepithelial neoplasia (PanIN) progression in engineered KrasG12D transgenic mice crossed with a cyclooxygenase-2 (COX-2) knockout mouse while on a diet high in fat. And undeniably all these factors have a function in the etiology of pancreatic carcinogenesis.
In the normal physiologic state, ingestion of dietary fat results in the release of the gastrointestinal peptide cholecystokinin, CCK, from the duodenum I-cells [15]. CCK stimulates secretion of digestive enzymes from the pancreas, release of bile from the gall bladder, and mediates satiety in the brain [15]. In addition to its role in digestion, CCK has also been shown to be an important mediator of growth, proliferation, and regeneration of the pancreas physiologically [16, 17], after surgical resection [18] and after injury (i.e., pancreatitis) [19]. In this capacity, CCK not only increases pancreatic size and weight (i.e., hypertrophy), but this peptide also induces DNA synthesis (i.e., hyperplasia) [17, 20]. CCK has been used as an adjuvant treatment to accelerate carcinogen-induced pancreatic cancer in animal models [21] and in KrasG12D transgenic mice to hasten the progression of PanINs to pancreatic cancer development [22, 23]. In many of these experimental animal models, exogenous CCK or its related decapeptide cerulein was administered at a dose capable of inducing pancreatitis. Therefore, the authors concluded that the inflammation or resultant pancreatitis was the important mediator that accelerated carcinogenesis and not CCK itself.
Indeed, human pancreatic cancer has been shown to overexpress CCK receptors [24–26] and exogenous administration of CCK stimulates growth of this malignancy in vitro and in athymic nude mice [27, 28]. Smith et al. [29] previously showed that growth of subcutaneous xenografted SW-1990 human pancreatic cancers was significantly faster in nude mice fed a high-fat diet than mice on standard chow. This prior study [29] was important for several reasons: (1) the tumor was distant from the gastrointestinal tract implying that something in the circulation was responsible for the tumor growth effects; (2) the mice were not obese and were weight matched, so the effector was independent of obesity; (3) the mice had a deficient immune system, thereby eliminating the confounding factor of inflammation; and (4) the growth effect of the dietary fat was reversed with CCK receptor antagonist therapy, suggesting that CCK was the instigator. Based upon its known trophic effects, inflammatory properties, and its physiologic response to dietary fat ingestion, we hypothesize that endogenous CCK is a key contributor to the obesity-induced pancreatic cancer through its mitogenic actions at the CCK receptor. Because we believe the inflammatory cells have a role in obesity-related cancer development, the current experiments employed immunocompetent C57BL/6 mice. Furthermore, rather than the subcutaneous location, we implanted the pancreas cancer cells orthotopically using the syngeneic Panc-02 murine pancreatic cancer cell line in order to study the role of dietary fat on metastases. We hypothesized that dietary fat stimulates the release of endogenous CCK, which in turn promotes pancreatic hyperplasia and growth and progression of pancreatic cancer.
Materials and Methods
Reagents and Cell Lines
Panc-02 cells (called Pan02 by some investigators), a murine pancreatic cancer cell line which is syngeneic to C57BL/6 mice [30], were a gift from professor Corbett (Wayne State University, MI, USA). Growth of this murine pancreatic cancer cell line in vivo has been recently characterized [31]. The Panc-02 cells were cultured in DMEM:F12 media with 10 % FBS. Six- to eight-week-old male C57BL/6 mice were purchased from Jackson Labs (Bar Harbor, ME, USA). All institutional guidelines for the care and use of laboratory animals were followed throughout the study in accordance with protocols approved by the Penn State Hershey Institutional Animal Use and Care Committee.
The high-fat diet chow (TestDiet #58Y1), low-fat diet chow (TestDiet #58Y2), and normal chow were obtained from VanHeek Series from Test Diet (St. Louis, MO, USA), and each diet composition is shown in Table 1. High-fat chow contained 61.6 % energy (kcal/g) from fat and approximately equal amounts of saturated and monounsaturated fatty acids, while low-fat chow contained 10.2 % energy from fat and approximately equal amounts of saturated, monounsaturated, and polyunsaturated fatty acids. Normal chow contained 17 % energy from fat and equal amounts of saturated and unsaturated fatty acids. All diets contained equal overall calorie content.
Table 1.
Composition of mouse diets
| Composition of diet | Normal diet | Low-fat diet | High-fat diet |
|---|---|---|---|
| Protein (%) | 18.8 | 17.3 | 23.1 |
| Fat (%) | 6.0 | 4.3 | 34.9 |
| Fiber (%) | 3.8 | 4.7 | 6.5 |
| Carbohydrates (%) | 50.0 | 67.4 | 25.9 |
| Energy (kcal/g) | 3.4 | 3.78 | 5.10 |
| Calories from | Calories from | Calories from | |
|---|---|---|---|
| Protein (%) | 23 | 18.3 | 18.1 |
| Fat (%) | 17 | 10.2 | 61.6 |
| Carbohydrates (%) | 60 | 71.5 | 20.3 |
Both the high-fat diet and the low-fat diet were adjusted to have a total caloric intake equal to that of normal diet
The CCK-A receptor antagonist (L-364,718), devazepide, was purchased from Tocris Biosciences (Bristol, UK).
CCK Receptor Immunofluorescence of Panc-02 Cells
Panc-02 cells were grown on glass coverslips and fixed in ice-cold methanol and acetone for 20 min each. After 3 washes in PBS, cells were permeabilized with 0.2 % Triton X-100 in PBS for 1 h at room temperature. Nonspecific binding was blocked in PBS containing 5 % goat serum and 0.1 % Triton X-100 for 1 h. Cells were subsequently incubated in PBS with 5 % FBS, 0.05 % Triton X-100 with either a rabbit polyclonal CCK-A receptor antibody (PA-3116, Pierce, Rockford, IL, USA) or a CCK-B receptor antibody (#9491, CURE, Los Angeles, CA, USA) at a titers of 1:400 overnight at 4 °C. After three PBS washes, the cells were incubated in PBS containing 1 % FBS, 0.05 % Triton X-100, and a secondary goat anti-rabbit AlexaFluor 488-labeled antibody (Amersham Biosciences, Waltham, MA, USA) at a titer of 1:2,000 for 1 h at 4 °C in the dark. The slides were washed thrice with PBS, counterstained with Hoechst 33342 (0.5 μg/mL; AnaSpec, Fremont, CA, USA), and mounted with Aqua Poly/Mount solution (Polysciences, Warington, PA, USA). Images were captured on a Leica DM IRE2 confocal microscope.
Orthotopic Panc-02 Tumor Treatment Groups
At 6 weeks of age, C57BL/6 male mice were sorted into one of three dietary groups such that the mean total body weight in each group was equivalent. Groups were then placed on a normal, high-fat, or low-fat diet ad libitum. Food intake per cage and individual animal body weight were measured 3 times weekly. After 8 weeks, when the animals on the high-fat diet had reached a weight of at least 35 g, consistent with obesity, 106 Panc-02 cells were surgically implanted orthotopically into the pancreas as previously described [32]. Beginning 1 week after tumor cell implantation, mice were treated thrice weekly with vehicle [DMSO, 5 % in PBS, intraperitoneal (i.p.)] or with the CCK receptor antagonist devazepide (4 mg/kg/day in PBS, i.p.); a dose consistent with that used in several previously reported studies [33, 34]. Devazepide injections were given intraperitoneally to prevent the systemic effects of the CCK receptor antagonist on the brain and eliminate the influence this compound could have on feeding behaviors, i.e., satiety [35]. Twenty days after the CCK receptor antagonist treatments were initiated the mice were humanely euthanized and necropsied. Animals were anesthetized and blood collected by intracardiac puncture. Tumors were excised; fresh weight was recorded, and samples were preserved in RNAlater (Qiagen, Valencia, CA, USA) for RNA extraction, fixed in formalin, or were flash frozen in liquid nitrogen. The number and size of surface metastases to the liver and mesentery were visually assessed, and the tissues were then formalin-fixed for verification of the lesions by histopathology.
Tumor Histopathology
Histopathology was performed by an veterinary pathologist blinded to the treatment groups and diet to assess tumor grade, local invasion, and inflammation. Both flash-frozen and formalin-fixed tumor tissue were sectioned and processed for hematoxylin and eosin (H&E) staining. The degree of inflammatory cell infiltration and adiposity was scored according to the following criteria: 0 = normal pancreas, 1 = minimal (5–10 % affected), 2 = mild (10–25 % affected), 3 = moderate (25–50 % affected), 4 = marked (>50 % affected). The indices of pathology were combined to give a composite score of severity for each mouse and averaged for each diet group. For confirmation of metastases, tissues were fixed, embedded, sectioned, and hematoxylin stained as described above. Metastatic lesions were assessed as single or multiple lesions and scored for percent of section involved by a pathologist blinded to treatment groups.
CCK Peptide Radioimmunoassay and Blood Glucose Determination
Mouse blood was incubated on ice for 30 min, and serum was collected after low-speed centrifugation to remove cells and debris. Plasma samples were standardized by protein concentration using a Micro BCA protein assay with albumin as the standard (Pierce). CCK peptide radioimmunoassay (RIA) was performed in duplicate using rabbit antiserum to synthetic CCK 26-33 sulfate and 125I-BH CCK-8 (ALPCO Diagnostics, Salem, NH, USA). Specificity for CCK is based on the fact that the antiserum has 0.5 % cross-reactivity to sulfated gastrin-17 and <0.01 % cross-reactivity to nonsulfated gastrin-17.
At necropsy, blood glucose levels were determined using a Nova Max PLUS glucometer.
Real-Time qRT-PCR
RNA was extracted from Panc-02 cells and tumors using an RNAeasy kit (Qiagen), and cDNA was produced using a cDNA Reverse Transcriptase Kit (ABI, New York, NY, USA) with 2 μg of RNA per reaction. For Panc-02 cells, qRT-PCR was carried out with 100 ng of cDNA per reaction using CCK-BR primers (Mm00432329_m1) and CCK-AR primers (Mm00438060_m1). For tumor RNA samples, qRT-PCR analysis was done using VEGFA primers (Mm01281449_m1). Both analyses used murine HPRT (Mm00446968_m1) as the internal control. RNA levels were calculated from the mean relative quantity (RQ = 2−ΔΔCT) with a 95 % confidence interval (CI; RQ = 2−(ΔΔCT±CI)). Multiple markers of metastatic tumor progression were initially assessed in tumors from vehicle-treated, high-fat diet mice versus devazepide-treated, high-fat diet mice using the mouse tumor metastasis RT2 Pro-filer™ PCR Array kit (Qiagen).
In Vitro Matrigel Invasion Assay
Panc-02 cell invasion was evaluated using BioCoat Matrigel Invasion Chambers (24-well cell culture inserts containing an 8.0 μm PET membrane with a uniform layer of BD Matrigel Basement Membrane Matrix (Becton–Dickinson, San Jose, CA, USA). Panc-02 cells were detached with a nonenzymatic cell stripper solution (Cellgro, Manassas, VA, USA), resuspended in serum-free medium, and pre-treated for 10 min with an equal volume of either vehicle (0.1 % ethanol) or devazepide to a final concentration of 100 nM. Cells were then seeded at 2.5 × 104 cells/well into the upper chambers in serum-free media containing either vehicle or 100 nM devazepide. The lower chambers were filled with media containing 10 % fetal bovine serum as a chemoattractant and either vehicle or devazepide. After a 24 h incubation at 37 °C, residual cells were carefully and thoroughly wiped from the top surface of the filter and invasion was quantified by HEMA3 (Fisher Scientific, Pittsburgh, PA, USA) staining of the cells that had migrated to the underside. Cells were counted in six fields/filter; six filters were counted per assay, and the assays were independently repeated at least three times.
Statistical Analysis
The results are expressed as mean ± standard error of the mean (SEM), and comparisons were made by ANOVA and two sample Student’s t test with a Bonferroni correction for multiple comparisons.
Results
Panc-02 Murine Pancreatic Cancer Cells Express the CCK-A Receptor
Quantitative RT-PCR analysis of Panc-02 cell mRNA demonstrated that these cells expressed only the CCK-A receptor isoform and not the CCK-B receptor (Fig. 1a). Immunoreactivity for the CCK-A receptor protein expression on the surface of Panc-02 cells was confirmed by staining with a CCK-A receptor antibody (Fig. 1b). Immunoreactivity with the selective CCK-B receptor antibody was negative (Fig. 1c). Therefore, further studies using Panc-02 to evaluate the effects of CCK receptor blockade in vitro and in vivo, only employed the CCK-A receptor-specific antagonist.
Fig. 1.

Panc-02 cells express only CCK-A receptor mRNA and protein. a mRNA for only the CCK-A receptor is detected in Panc-02 cells not the CCK-B receptor by quantitative RT-PCR. Receptor mRNA levels are standardized to murine HPRT mRNA levels as an internal control. N.D. not detected. Columns represent the fold change in mRNA levels calculated from the mean relative quantity and bars represent a 95 % CI (RQ = 2−(ΔΔCT±CI)). b CCK-A receptor protein is present in Panc-02 cells by immunofluorescence. c No immunoreactivity is detected using a CCK-B receptor antibody indicating that Panc-02 cells express the CCK-A receptor and not the CCK-B receptor
Effects of Diet on Mouse Body Weight
Although no difference in total food intake was recorded between diet groups; the mice on the high-fat diet had a steady increase in body weight compared to mice on both the normal diet and low-fat diet (Fig. 2a). At the time when tumor cells were orthotopically injected (Day 54), the mean body weight for the high-fat cohort had exceeded the 35 g, a value established as obese in mice [36]. This weight was significantly higher than the mice consuming the normal and low-fat diets (p = 2.67 × 10−6). In contrast, the mean body weight for mice on the low-fat diet was not different from the weight of the normal diet mice (p = 0.5). The slight decrease in body weight observed in all groups at Day 54 was due to the post-surgery recovery period. After the CCK-A receptor antagonist treatments were initiated, no differences in body weight or food and water intake were recorded between treatments (vehicle vs. devazepide) within the diet groups.
Fig. 2.

Effects of diet and a CCK-A receptor antagonist on body weight, serum CCK levels, and serum glucose levels. a Mean weight gain of mice fed a low-fat diet, a high-fat diet, or normal diet. Animals on the high-fat diet (solid inverted triangles) gained significantly more weight than animals on a normal diet (solid circles) or low-fat diet (open circles), ***p = 2.67 × 10−6. No differences in food or water intake were noted among treatment groups. A slight decrease in body weight was observed in all the diet groups on Day 54 due to surgery to implant pancreatic cancers cells (n = 10–12 mice per diet group). b Serum CCK levels were increased tenfold in animals on the high-fat diet (gray bars, **p <0.01), whereas CCK serum levels were tenfold decreased in animals on the low-fat diet (white bars, *p < 0.05) compared to animals on a normal diet (black bar). Treatment with the CCK-A receptor antagonist had no effect on serum CCK levels independent of diet (bars with crosshatches, n = 5–6 animals per treatment group). c Serum glucose levels in mice fed a normal diet (black bar), high-fat diet with vehicle treatment (gray bar), or high-fat diet with CCK receptor antagonist treatment (gray bar with cross-hatches). Although a high-fat diet caused an increase in serum glucose (**p = 0.010), CCK receptor antagonist treatment had no significant effect on glucose levels
Circulating Serum CCK Levels and Glucose Levels Are Impacted by Dietary Fat
Compared to mice fed a normal diet, animals on the high-fat diet had nonfasting serum CCK levels that were tenfold higher (Fig. 2b; **p < 0.01). Conversely, mice fed a low-fat diet had significantly reduced levels of serum CCK (Fig. 2b; *p < 0.05). Since the receptor antagonist only blocks the interaction of CCK with its receptor and has no effect on production, it is no surprise that CCK levels were also increased in all mice on the high-fat diet regardless of antagonist treatment.
In addition, nonfasting blood glucose levels were increased by 60 % in mice on the high-fat diet (Fig. 2c; *p = 0.010). Treatment with the CCK receptor antagonist did not significantly alter glucose levels (p = 0.13).
Dietary Fat Stimulates the Growth and Metastases of Orthotopic Pancreatic Tumors
Mice on a high-fat diet developed larger pancreatic tumors compared to normal controls or to low-fat diet counterparts. Tumors from mice on the high-fat diet weighed 63 and 133 % more than mice on the regular and low-fat diets, respectively (Fig. 3a; *p < 0.05). Tumors from mice on the low-fat diet were slightly smaller than those in mice fed a normal diet; however, this difference was not statistically significant. Mean tumor mass in all the CCK-A receptor antagonist treatment groups was unchanged compared to the corresponding vehicle control groups.
Fig. 3.

Role of dietary fat and a CCK-A receptor antagonist on Panc-02 primary pancreatic tumor growth and metastases in vivo. a Panc-02 tumor mass was significantly increased (*p < 0.05) in mice fed a high-fat diet (blue bars) compared to tumors grown in mice given a normal diet (black bar) or a low-fat diet (red bars). No difference in tumor weight was found between mice fed a normal diet or low-fat diet. Tumor mass was not significantly affected by CCK-AR antagonist (hatched bars) compared to their respective vehicle controls. b The total number of tumor metastases was significantly increased in mice on the high-fat diet compared to normal or low-fat diet mice (**p <0.003). Compared to vehicle treatments, the CCK-AR antagonist decreased the number of tumor metastases in high-fat diet fed animals (*p = 0.05). Antagonist = CCK-A receptor antagonist, devazepide; vehicle = DMSO. Bars represent the standard error of the mean of 5–7 mice per treatment group
In addition to stimulating Panc-02 primary tumor growth, the number of metastases was also significantly increased in mice on a high-fat diet compared with normal diet animals (Fig. 3b; **p < 0.003). The most frequent sites of metastases were liver and mesentery.
CCK Receptor Antagonism Decreases Pancreatic Cancer Metastases
The total number of metastases was decreased in the high-fat diet group with the CCK-A receptor antagonist treatment compare to the high-fat diet vehicle treatment group (Fig. 3b; *p = 0.05). Interestingly, there was a similar decrease in the total number of surface metastases in both the high-fat diet cohort (80 %) and the low-fat diet cohort (73 %) in response to the CCK-A receptor antagonist treatment (Fig. 3b, hatched bars). In both dietary groups, CCK receptor antagonist treatment decreased the number of observed liver metastases.
Tumor-Associated Vascular Emboli Are Decreased by CCK-A Receptor Antagonist Treatment
Excised Panc-02 tumors were further assessed in a blinded fashion for histopathological changes in inflammation, presence of fat, and presence of emboli in tumor vessels. Orthotopic Panc-02 tumors grown in animals on a high-fat diet had significant immune cell infiltrate and were highly vascularized and locally invasive. Pancreata from control high-fat diet mice that were not injected with tumor cells (diet-only control) showed little inflammation or fat infiltrate (Fig. 4a) suggesting that the inflammation was not due to dietary fat but related to the cancer. This paucity of intrapancreatic fat in mice fed a high-fat diet has recently been shown by others (16). However, the livers from high-fat diet mice that were not injected with tumor cells (diet-only control) showed steatosis (Fig. 4b).
Fig. 4.

Histopathological characteristics of Panc-02 tumors and noncancerous tissues from mice. a H&E of a normal pancreas (without a tumor) from a mouse on a high-fat diet showing a lack of inflammatory or fat infiltrate. b H&E of a liver from a mouse on the high-fat diet but without a tumor (control) showing extensive steatosis. c H&E staining of a liver section from a representative tumor-bearing mouse given a high-fat diet/vehicle treatment. Multiple metastatic lesions (black arrows) and tumor emboli occluding the portal vein (white arrow) are shown. d Metastatic lesion to the mouse mesentery is shown with a tumor emboli in the center (black circle). e Tumor emboli in a small artery (arrow) from a representative vehicle-treated mouse on a high-fat diet. f Tumor vessels from a representative mouse on the high-fat diet treated with CCK-AR antagonist contain blood cells but lack tumor cell emboli. Scale bars are shown on each figure
To be certain that these lesions were true metastases, the livers were then assessed histologically. A representative liver from a tumor-bearing mouse that received a high-fat diet showed significantly increased number of metastases infiltrating the liver parenchyma and numerous tumor emboli in the portal vein (Fig. 4c). In contrast, the liver tissue from a mouse in the high-fat/CCK-AR antagonist treatment group had infrequent metastases and no visible tumor emboli (not shown). The mesenteric metastases were also confirmed histologically (Fig. 4d). The most striking difference between tumors from high-fat diet/vehicle-treated mice and tumors from the high-fat diet/CCK-A receptor antagonist-treated mice was the complete lack of tumor emboli in the tumor vasculature of CCK-A receptor antagonist-treated animals. Animals with tumors from both the normal diet/vehicle and high-fat diet/vehicle treatment groups were equally likely to have emboli in tumor vessels (Fig. 4e). In contrast, emboli were completely absent from the tumor vasculature of all mice treated with the CCK-A receptor antagonist (Fig. 4f). The absence of emboli in tumor vessels and the portal vein as well as fewer metastases in mice treated with the CCK-A receptor antagonist suggested that CCK receptor blockade reduced the spread of tumor cells beyond the primary tumor. And since the antagonist did not affect the primary tumor mass in this experiment, the significant differences in metastatic lesions were not a result of primary tumor size.
No overall differences in pancreatic or tumor inflammation were noted between the normal diet and high-fat/vehicle treatment group (Table 2). Although there was a trend for a reduction in the inflammation in the mice in the high-fat/CCK-A receptor antagonist treatment group compared to the high-fat vehicle group, this difference was not significant (Table 2). The most striking finding of the blinded histologic scoring assessment was the complete lack of tumor emboli in the CCK antagonist-treated group.
Table 2.
Histologic characteristics of Panc-02 tumors based upon diet
| Normal diet | High-fat diet + vehicle | High-fat diet + antagonist | |
|---|---|---|---|
| Tumor inflammation | 2.00 ± 0.17 | 1.90 ± 0.28 | 1.56 ± 0.29 |
| % of tumors with vascular emboli | 33 | 33 | 0 |
| Inflammation in adjacent normal pancreas | 1.45 ± 0.25 | 1.83 ± 0.17 | 1.40 ± 0.40 |
| Tumor vascularity | 1.91 ± 0.09 | 1.80 ± 0.13 | 1.89 ± 0.20 |
CCK receptor antagonism completely prevented tumor emboli
High-Fat Diet Increases and Treatment with a CCK-A Receptor Antagonist Decreases VEGF-A Expression in Panc-02 Tumors
When RNA from Panc-02 tumors of the vehicle-treated, high-fat diet mice and from tumors of mice in CCK receptor antagonist-treated, high-fat diet groups were compared using a mouse tumor metastasis PCR array, a total of 12 genes were identified that were significantly up or down-regulated in response to CCK receptor antagonist treatment. Of the genes with a greater than twofold difference between vehicle and antagonist treatment groups, VEGF-A (vascular endothelial growth factor-A) exhibited the greatest change in gene expression in response to antagonist treatment. The increase in VEGF-A mRNA was verified using an individual qPCR assay (Fig. 5a, black bar vs. gray bar; p < 0.0002). In addition, VEGF-A mRNA levels from tumors of high-fat diet/CCK receptor antagonist-treated mice were significantly reduced compared to high-fat/vehicle mice (Fig. 5a, gray bar vs. hatched bar; p < 6 × 10−9). Furthermore, the VEGF-A mRNA levels were restored to the same levels as that of the control mice which received a normal diet as a result of treatment with the CCK receptor antagonist. Other genes initially indicated as differentially expressed by array analysis, including MMP-7, 10, and 13, were not different between treatment groups when assessed by individual qPCR assays. The marked change in tumor VEGF-A expression, which was significantly increased in tumors from high-fat diet animals and completely returned to normal levels in high-fat diet/CCK-A receptor antagonist-treated mice, indicates that VEGF-A expression is linked to CCK receptor signaling.
Fig. 5.
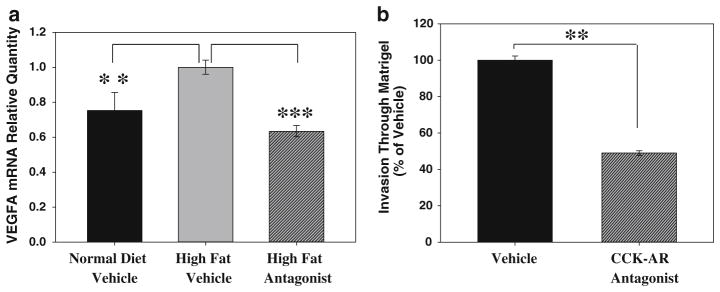
CCK-AR antagonist treatment alters VEGF-A expression in vivo and decreases tumor cell invasiveness in vitro. a Panc-02 tumors from mice on the high-fat diet (gray bar) had increased expression of VEGF-A mRNA when compared to tumors from mice fed normal chow (black bar)(**p < 0.0002). This increase in VEGF-A expression was reversed with the CCK-AR antagonist treatment (hatched bar, ***p < 6 × 10−9). Tumors from 5 to 6 mice were analyzed for each treatment group. b Vehicle-treated Panc-02 cells (black bar) clearly moved through a Matrigel-coated filter in vitro. However, in the presence of the antagonist (100 nM, hatched bar), the number of invasive Panc-02 cells moving through Matrigel was reduced by more than 50 % (**p = 1.33 × 10−6). Error bars denote the standard error of the mean of the individual assays normalized to vehicle treatments
CCK-A Receptor Blockade Reduced In Vitro Invasiveness of Panc-02 Cells
A reduction in the number of vascular emboli in tumors from mice treated with the CCK-A receptor antagonist could also be due to a decrease in the ability of the cancer cells to pass through basement membrane and into circulation. Indeed, CCK receptor signaling is linked to increased invasiveness [37]. To directly assess the effect of the CCK-A receptor antagonist on the ability of Panc-02 cells to migrate through extracellular matrix, an in vitro Matrigel invasion assay was used. Vehicle-treated Panc-02 cells freely moved through the Matrigel-coated filter, while Panc-02 cells treated with the CCK-A receptor antagonist were 50 % less invasive (Fig. 5b, p = 1.33 × 10−6). Thus, even in the absence of dietary influences, treatment of Panc-02 cells with a CCK-A receptor antagonist affects tumor cell invasiveness, a critical characteristic associated with pancreatic tumor progression.
Discussion
In the current study, we demonstrated that dietary fat induces growth and metastasis of pancreatic cancer. The mechanism through which a fatty diet accelerates tumor progression is in part mediated by the interaction of CCK with its receptor. A new finding reported in this investigation was that CCK receptor blockade could completely prevent tumor emboli and metastasis through a mechanism involving VEGF-A.
Epidemiologic studies have shown that countries that consume high-fat diets have an increased prevalence of pancreatic cancer [2]. We clearly show herein the relationship between chronic fat consumption and markedly increased blood levels of the trophic peptide CCK. Our studies revealed that a high-fat diet increases circulating CCK peptide to a level ten times that in mice fed a normal diet. Nonfasting, normal diet mice had an average of 4–5 pg/mL serum CCK (4–5 pM) which is consistent with reported values. The tenfold increase in serum CCK in the high-fat diet mice in this study was even greater than the sevenfold–eightfold increase in serum CCK reported in rats after a short-term administration of fatty acids [38] and was much greater than the increase in serum glucose in high-fat diet animals (a 60 % increase). This finding suggests that prolonged exposure to diets high in fat has a significant effect on circulating CCK levels. And since CCK stimulates pancreatic hyperplasia and growth [39], this peptide is a mediator of fat and obesity-associated cancer progression.
Based on previous work done by Smith et al. [29], we would have predicted that dietary fat would stimulate growth of orthotopic Panc-02 pancreatic tumors and the antagonist reverse the growth. Indeed, tumor mass from animals on the high-fat diet was much greater than normal diet controls; however, unlike the previous study [29], tumor mass was unaffected in our current experiments by antagonist treatment. Several differences in design of the two studies may account for the lack of antagonist on primary tumor growth in the present investigation. These differences include the frequency of antagonist dosing in the current study (three times per week vs. twice daily), time at which the antagonist treatment was initiated (1 week after tumor cell injection vs. concurrent with tumor cell injection), differences in the strain of mice used (immunocompetent C57BL/6 mice vs. athymic nu/nu mice), the tumor implantation site (orthotopic vs. subcutaneous), and the differences in growth rates of the two cell lines (murine Panc-02 cells grow faster than human SW-1990 cells). In spite of these variations, our report is the first to link increased endogenous CCK blood levels to accelerated pancreatic cancer growth and metastases. Knowing that chronic ingestion of a diet high in fat increases CCK blood levels and because CCK is known to stimulate growth of normal pancreas tissue [16] and pancreatic cancer [27, 28], this study supports the role of CCK in cancer risk associated with dietary fat. Since patients with chronic pancreatitis may also have increased blood levels of CCK [40], the risk of developing pancreatic cancer in this group may also be in part related to the endogenous increase in CCK.
In the normal physiologic state, CCK is synthesized in the I-cells of the duodenum [15]; however, in conditions associated with obesity, CCK mRNA expression has also been reported within the pancreas. Lavine et al. [41] reported that CCK mRNA in the pancreas of obese mice (C57BL/6-Leptin ob/ob mice) was up-regulated 500-fold compared to lean mice; the most up-regulated gene in the obese mice pancreas. While the CCK peptide was undetectable in the pancreas of lean mice, obese mice had high levels of the bioactive form of CCK. The effect of obesity on CCK expression was specific to the pancreas; levels of CCK mRNA in brain and intestine were identical in lean and obese animals. Indeed Zyromski et al. [42] recently reported that a murine pancreatic cancer cell line, Pan02, formed larger tumors when grown in either ob/ob or db/db obese mice.
An advantage regarding the design of this study with an orthotopic model was that it allowed us to examine the effect of CCK-A receptor blockade on tumor metastasis, which had not previously been done. Despite a similar primary tumor size, animals given the CCK-A receptor antagonist had fewer metastatic lesions than vehicle-treated animals. Similarly, the frequency of emboli in the tumor vasculature and in the portal vein was significantly reduced by the antagonist. Our data indicate that these findings were due to a CCK-A receptor-dependent reduction in the expression of VEGF-A in pancreatic tumors. CCK receptor blockade reduced VEGF-A levels in animals given a high-fat diet to levels similar to those seen in normal diet animals. Although CCK-B receptor activity has been linked to increased VEGF-A expression in colon cancer [43], this is the first report of such an association in pancreatic cancer and the first to connect CCK-A receptor activation to VEGF-A expression. Our findings are also consistent with clinical data showing that the level of VEGF, regulated mainly by HIF-1α and NF-κB, increases with high BMI [44].
It has been previously reported that VEGF receptors are widely expressed on pancreatic cancer cell lines and that VEGF receptor signaling promotes migration and invasion [45], and the diet-induced increase in VEGF may activate tumor cells that express VEGF receptor-1 via an autocrine pathway. Because VEGF-A increases the permeability of tumor vasculature, promotes EMT and tumor cell migration, and influences endothelial barrier function [43, 46–48], many of the changes in tumor histopathology in the CCK receptor antagonist-treated animals could be a direct result of reduced VEGF-A expression. The complete lack of emboli in the tumor vasculature of CCK receptor antagonist-treated mice clearly indicates that extravasation of tumor cells into circulation was affected by the antagonist treatment. The significant reduction in VEGF-A mRNA levels seen in tumors from antagonist-treated animals could result in a decrease in the permeability of tumor vessels, making it less likely for emboli to penetrate those vessels. Altered VEGF-A levels due to CCK receptor blockade also could affect the invasive capacity of tumor cells, as was demonstrated in the in vitro Matrigel invasion assay, and the decrease in VEGF-A in antagonist-treated mice could contribute to the reduced infiltration of inflammatory cells into the tumors of CCK-A receptor antagonist-treated mice. Previous work correlated high levels of VEGF in human pancreatic ductal adenocarcinoma specimens with increased liver metastasis and significantly shorter survival [49]. The lack of portal vein emboli and the reduction in liver metastases in CCK receptor antagonist-treated mice support the important role these receptors play in metastatic tumor spread.
Overall this pre-clinical study demonstrates the important role of endogenous CCK and its receptor in tumor progression and metastasis of pancreatic cancer, particularly in the context of high dietary fat. These findings suggest a link between dietary fat and tumor progression through the action of CCK and CCK receptors. This work also provides a new insight into the mechanism of pancreatic tumor metastasis and suggests a potential therapeutic approach to reduce disease progression in pancreatic cancer patients. CCK receptor blockade may impair the ability of human pancreatic cancer cells to spread beyond the primary tumor, through the basement membrane and into the tumor vasculature. In short, we were able to show that a CCK-A receptor antagonist blocks the progression of established pancreatic tumors in vivo. Because pancreatic cancer is rarely detected in the early stages and metastatic disease is often present at diagnosis, any treatment that slows the invasion or metastatic progression of tumors could extend patient survival. CCK receptor antagonists have been tested in human clinical trials for other conditions and are safe, well tolerated, and have few side effects [50]. Thus, it may be possible in the future to combine the use CCK receptor antagonists with chemotherapeutic agents to reduce the risk of cancer progression and metastases.
Acknowledgments
This work was funded by the NIH R01 CA117926 grant to JPS. We recognize additional support from The V-Foundation for Cancer Research to M.K. and the Robert Sullivan Foundation to J.P.S. We appreciate the technical services of Michael Stephan and Evan Shirey from Messiah College, Grantham, PA. The expert technical assistance of Weifang Lin, Department of Comparative Medicine, Rob Brucklacher in the Functional Genomics Core, Wade Edris in the Microscopy Imaging Core, and Dr. Bruce Stanley, Director of Scientific Programs, Section of Research Resources, Pennsylvania State University College of Medicine, are also appreciated. Core Facility services and instruments used in this project were funded, in part, under a grant with the Pennsylvania Department of Health using Tobacco Settlement Funds. This department specifically disclaims responsibility for any analyses, interpretations, or conclusions.
Abbreviations
- BMI
Body mass index
- BSA
Bovine serum albumin
- CCK
Cholecystokinin
- DMEM/F12
Dulbecco’s modified Eagle’s medium/Ham’s F12
- DMSO
Dimethyl sulfoxide
- FBS
Fetal bovine serum
- H&E
Hematoxylin and eosin
- i.p
Intraperitoneal
- kcal/g
Kilocalories per gram
- NCI
National Cancer Institute
- PBS
Phosphate-buffered saline
- qRT-PCR
Quantitative real-time polymerase chain reaction
- RIA
Radioimmunoassay
- RQ
Relative quantity
- VEGF-A
Vascular endothelial growth factor-A
- VEGFR
Vascular endothelial growth factor receptor
Footnotes
Conflict of interest None.
Contributor Information
Gail L. Matters, Department of Biochemistry and Molecular Biology, College of Medicine, The Pennsylvania State University, Hershey, PA 17033, USA
Timothy K. Cooper, Department of Comparative Medicine, College of Medicine, The Pennsylvania State University, Hershey, PA 17033, USA. Department of Pathology, College of Medicine, The Pennsylvania State University, Hershey, PA 17033, USA
Christopher O. McGovern, Department of Medicine, College of Medicine, The Pennsylvania State University, Hershey, PA 17033, USA
Evan L. Gilius, Department of Medicine, College of Medicine, The Pennsylvania State University, Hershey, PA 17033, USA
Jiangang Liao, Department of Public Health Sciences, College of Medicine, The Pennsylvania State University, Hershey, PA 17033, USA.
Brian M. Barth, Department of Pharmacology, College of Medicine, The Pennsylvania State University, Hershey, PA 17033, USA
Mark Kester, Department of Pharmacology, College of Medicine, The Pennsylvania State University, Hershey, PA 17033, USA.
Jill P. Smith, Email: jsmith2@psu.edu, Department of Medicine, College of Medicine, The Pennsylvania State University, Hershey, PA 17033, USA. Department of Medicine, Georgetown University Hospital, 3800 Reservoir Rd, NW, 2 Main, 2nd Floor, Washington, DC 20007, USA
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144:1252–1261. doi: 10.1053/j.gastro.2013.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bracci PM. Obesity and pancreatic cancer: overview of epidemiologic evidence and biologic mechanisms. Mol Carcinog. 2012;51:53–63. doi: 10.1002/mc.20778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li D, Morris JS, Liu J, et al. Body mass index and risk, age of onset, and survival in patients with pancreatic cancer. JAMA. 2009;301:2553–2562. doi: 10.1001/jama.2009.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts DL, Dive C, Renehan AG. Biological mechanisms linking obesity and cancer risk: new perspectives. Annu Rev Med. 2010;61:301–316. doi: 10.1146/annurev.med.080708.082713. [DOI] [PubMed] [Google Scholar]
- 6.Aleman JO, Eusebi LH, Ricciardiello L, et al. Mechanisms of obesity-induced gastrointestinal neoplasia. Gastroenterology. 2014;146:357–373. doi: 10.1053/j.gastro.2013.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White PB, Ziegler KM, Swartz-Basile DA, et al. Obesity, but not high-fat diet, promotes murine pancreatic cancer growth. J Gastrointest Surg. 2012;16:1680–1685. doi: 10.1007/s11605-012-1931-5. [DOI] [PubMed] [Google Scholar]
- 8.Wang F, Kumagai-Braesch M, Herrington MK, et al. Increased lipid metabolism and cell turnover of MiaPaCa2 cells induced by high-fat diet in an orthotopic system. Metabolism. 2009;58:1131–1136. doi: 10.1016/j.metabol.2009.03.027. [DOI] [PubMed] [Google Scholar]
- 9.Dawson DW, Hertzer K, Moro A, et al. High-fat, high-calorie diet promotes early pancreatic neoplasia in the conditional KrasG12D mouse model. Cancer Prev Res (Phila) 2013;6:1064–1073. doi: 10.1158/1940-6207.CAPR-13-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lashinger LM, Harrison LM, Rasmussen AJ, et al. Dietary energy balance modulation of Kras− and Ink4a/Arf+/−-driven pancreatic cancer: the role of insulin-like growth factor-I. Cancer Prev Res (Phila) 2013;6:1046–1055. doi: 10.1158/1940-6207.CAPR-13-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pisani P. Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch Physiol Biochem. 2008;114:63–70. doi: 10.1080/13813450801954451. [DOI] [PubMed] [Google Scholar]
- 12.Stattin P, Bjor O, Ferrari P, et al. Prospective study of hyperglycemia and cancer risk. Diabetes Care. 2007;30:561–567. doi: 10.2337/dc06-0922. [DOI] [PubMed] [Google Scholar]
- 13.Stolzenberg-Solomon RZ, Limburg P, Pollak M, et al. Insulin-like growth factor (IGF)-1, IGF-binding protein-3, and pancreatic cancer in male smokers. Cancer Epidemiol Biomark Prev. 2004;13:438–444. [PubMed] [Google Scholar]
- 14.Philip B, Roland CL, Daniluk J, et al. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology. 2013;145:1449–1458. doi: 10.1053/j.gastro.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dockray GJ. Cholecystokinin. Curr Opin Endocrinol Diabetes Obes. 2012;19:8–12. doi: 10.1097/MED.0b013e32834eb77d. [DOI] [PubMed] [Google Scholar]
- 16.Solomon TE, Petersen H, Elashoff J, et al. Interaction of caerulein and secretin on pancreatic size and composition in rat. Am J Physiol. 1978;235:E714–E719. doi: 10.1152/ajpendo.1978.235.6.E714. [DOI] [PubMed] [Google Scholar]
- 17.Solomon TE, Vanier M, Morisset J. Cell site and time course of DNA synthesis in pancreas after caerulein and secretin. Am J Physiol. 1983;245:G99–G105. doi: 10.1152/ajpgi.1983.245.1.G99. [DOI] [PubMed] [Google Scholar]
- 18.Lehv M, Fitzgerald PJ. Pancreatic acinar cell regeneration. IV. Regeneration after resection. Am J Pathol. 1968;53:513–535. [PMC free article] [PubMed] [Google Scholar]
- 19.Elsasser HP, Adler G, Kern HF. Time course and cellular source of pancreatic regeneration following acute pancreatitis in the rat. Pancreas. 1986;1:421–429. doi: 10.1097/00006676-198609000-00006. [DOI] [PubMed] [Google Scholar]
- 20.Mainz DL, Black O, Webster PD. Hormonal control of pancreatic growth. J Clin Invest. 1973;52:2300–2304. doi: 10.1172/JCI107418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howatson AG, Carter DC. Pancreatic carcinogenesis-enhancement by cholecystokinin in the hamster-nitrosamine model. Br J Cancer. 1985;51:107–114. doi: 10.1038/bjc.1985.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carriere C, Young AL, Gunn JR, et al. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun. 2009;382:561–565. doi: 10.1016/j.bbrc.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guerra C, Schuhmacher AJ, Canamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 24.Smith JP, Rickabaugh CA, McLaughlin PJ, et al. Cholecystokinin receptors and PANC-1 human pancreatic cancer cells. Am J Physiol. 1993;265:G149–G155. doi: 10.1152/ajpgi.1993.265.1.G149. [DOI] [PubMed] [Google Scholar]
- 25.Smith JP, Liu G, Soundararajan V, et al. Identification and characterization of CCK-B/gastrin receptors in human pancreatic cancer cell lines. Am J Physiol. 1994;266:R277–R283. doi: 10.1152/ajpregu.1994.266.1.R277. [DOI] [PubMed] [Google Scholar]
- 26.Weinberg DS, Ruggeri B, Barber MT, et al. Cholecystokinin A and B receptors are differentially expressed in normal pancreas and pancreatic adenocarcinoma. J Clin Invest. 1997;100:597–603. doi: 10.1172/JCI119570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith JP, Solomon TE, Bagheri S, et al. Cholecystokinin stimulates growth of human pancreatic adenocarcinoma SW-1990. Dig Dis Sci. 1990;35:1377–1384. doi: 10.1007/BF01536744. [DOI] [PubMed] [Google Scholar]
- 28.Smith JP, Kramer ST, Solomon TE. CCK stimulates growth of six human pancreatic cancer cell lines in serum-free medium. Regul Pept. 1991;32:341–349. doi: 10.1016/0167-0115(91)90027-e. [DOI] [PubMed] [Google Scholar]
- 29.Smith JP, Kramer S, Bagheri S. Effects of a high-fat diet and L364,718 on growth of human pancreas cancer. Dig Dis Sci. 1990;35:726–732. doi: 10.1007/BF01540175. [DOI] [PubMed] [Google Scholar]
- 30.Corbett TH, Roberts BJ, Leopold WR, et al. Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res. 1984;44:717–726. [PubMed] [Google Scholar]
- 31.Nikfarjam M, Yeo D, He H, et al. Comparison of two syngeneic orthotopic murine models of pancreatic adenocarcinoma. J Invest Surg. 2013;26:352–359. doi: 10.3109/08941939.2013.797057. [DOI] [PubMed] [Google Scholar]
- 32.Matters GL, Harms JF, McGovern CO, et al. Growth of human pancreatic cancer is inhibited by down-regulation of gastrin gene expression. Pancreas. 2009;38:e151–e161. doi: 10.1097/MPA.0b013e3181a66fdc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barrachina MD, Martinez V, Wang L, et al. Synergistic interaction between leptin and cholecystokinin to reduce short-term food intake in lean mice. Proc Natl Acad Sci USA. 1997;94:10455–10460. doi: 10.1073/pnas.94.19.10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carrillo J, Garcia-Aragoncillo E, Azorin D, et al. Cholecystokinin down-regulation by RNA interference impairs Ewing tumor growth. Clin Cancer Res. 2007;13:2429–2440. doi: 10.1158/1078-0432.CCR-06-1762. [DOI] [PubMed] [Google Scholar]
- 35.Cummings DE, Overduin J. Gastrointestinal regulation of food intake. J Clin Invest. 2007;117:13–23. doi: 10.1172/JCI30227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Surwit RS, Feinglos MN, Rodin J, et al. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and A/J mice. Metabolism. 1995;44:645–651. doi: 10.1016/0026-0495(95)90123-x. [DOI] [PubMed] [Google Scholar]
- 37.Grabowska AM, Watson SA. Role of gastrin peptides in carcinogenesis. Cancer Lett. 2007;257:1–15. doi: 10.1016/j.canlet.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 38.Lewis LD, Williams JA. Regulation of cholecystokinin secretion by food, hormones, and neural pathways in the rat. Am J Physiol. 1990;258:G512–G518. doi: 10.1152/ajpgi.1990.258.4.G512. [DOI] [PubMed] [Google Scholar]
- 39.Smith JP, Solomon TE. Cholecystokinin and pancreatic cancer: the chicken or the egg? Am J Physiol Gastrointest Liver Physiol. 2014;306:G91–G101. doi: 10.1152/ajpgi.00301.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shiratori K, Takeuchi T, Satake K, et al. Clinical evaluation of oral administration of a cholecystokinin-A receptor antagonist (loxiglumide) to patients with acute, painful attacks of chronic pancreatitis: a multicenter dose-response study in Japan. Pancreas. 2002;25:e1–e5. doi: 10.1097/00006676-200207000-00003. [DOI] [PubMed] [Google Scholar]
- 41.Lavine JA, Raess PW, Stapleton DS, et al. Cholecystokinin is up-regulated in obese mouse islets and expands beta-cell mass by increasing beta-cell survival. Endocrinology. 2010;151:3577–3588. doi: 10.1210/en.2010-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zyromski NJ, Mathur A, Pitt HA, et al. Obesity potentiates the growth and dissemination of pancreatic cancer. Surgery. 2009;146:258–263. doi: 10.1016/j.surg.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 43.Weis S, Cui J, Barnes L, et al. Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J Cell Biol. 2004;167:223–229. doi: 10.1083/jcb.200408130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silha JV, Krsek M, Sucharda P, et al. Angiogenic factors are elevated in overweight and obese individuals. Int J Obes (Lond) 2005;29:1308–1314. doi: 10.1038/sj.ijo.0802987. [DOI] [PubMed] [Google Scholar]
- 45.Wey JS, Fan F, Gray MJ, et al. Vascular endothelial growth factor receptor-1 promotes migration and invasion in pancreatic carcinoma cell lines. Cancer. 2005;104:427–438. doi: 10.1002/cncr.21145. [DOI] [PubMed] [Google Scholar]
- 46.Korc M. Pathways for aberrant angiogenesis in pancreatic cancer. Mol Cancer. 2003;2:8. doi: 10.1186/1476-4598-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takahashi H, Shibuya M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin Sci (Lond) 2005;109:227–241. doi: 10.1042/CS20040370. [DOI] [PubMed] [Google Scholar]
- 48.Yang AD, Camp ER, Fan F, et al. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res. 2006;66:46–51. doi: 10.1158/0008-5472.CAN-05-3086. [DOI] [PubMed] [Google Scholar]
- 49.Seo Y, Baba H, Fukuda T, et al. High expression of vascular endothelial growth factor is associated with liver metastasis and a poor prognosis for patients with ductal pancreatic adenocarcinoma. Cancer. 2000;88:2239–2245. doi: 10.1002/(sici)1097-0142(20000515)88:10<2239::aid-cncr6>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 50.Berna MJ, Jensen RT. Role of CCK/gastrin receptors in gastrointestinal/metabolic diseases and results of human studies using gastrin/CCK receptor agonists/antagonists in these diseases. Curr Top Med Chem. 2007;7:1211–1231. doi: 10.2174/156802607780960519. [DOI] [PMC free article] [PubMed] [Google Scholar]