

Abstract
pH-Responsive hydrogels comprised of itaconic acid copolymerized with N-vinylpyrrolidone (P(IA-co-NVP)) were synthesized and tested as carriers for the oral delivery of high isoelectric point (pI) exhibiting therapeutic proteins. Swelling studies show that P(IA-co-NVP) hydrogels exhibit significantly greater and faster pH-responsive swelling than previously studied methacrylic acid-based hydrogels, achieving up to 68% greater equilibrium swelling and 10.4 times greater swelling in time-limited experiments. Using salmon calcitonin as a model high pI protein therapeutic, we show that P(IA-co-NVP) hydrogels exhibit significantly greater delivery potential than methacrylic acid-based hydrogels. Additionally, we show that utilizing a lower ionic strength solution during drug loading significantly improves drug delivery potential for high pI therapeutics. By using a 1.5 mM PBS buffer rather than the standard 150 mM PBS buffer during loading, up to 83 times as much calcitonin can be delivered in neutral conditions, with up to a 9.6 fold improvement in percent release. Using P(IA-co-NVP) hydrogel microparticles and a low ionic strength loading solution, up to 48 μg calcitonin/mg hydrogel can be delivered in small intestinal conditions. Based on expected absorption in the small intestine, this is sufficient delivery potential for achieving therapeutic dosage via a single, regularly-sized pill taken daily.
Keywords: Oral drug delivery, pH-responsive hydrogels, Protein therapeutics, Ionic strength, Itaconic acid, Isoelectric point
1. Introduction
Protein therapy offers a number of advantages in disease treatment that cannot be matched by traditional therapy with small molecule drugs. The complexity of macromolecules affords protein therapeutics high specificity to their target that prevents widespread systemic side effects, yielding well-tolerated, highly active, and effective treatment options for a variety of diseases (Leader et al., 2008). Because of these benefits, protein-based drugs have remained one of the most common classes of newly FDA-approved drugs in recent years, accounting for 23% of new drugs in 2012 (Mullard, 2013) and 20% of new drugs in 2011 (U.S. Food and Drug Administration, 2012). Since recombinant insulin was first approved in 1982, the field has expanded to over 150 different FDA-approved protein therapeutics that accounted for $108 billion in sales in 2010 (Dimitrov, 2012).
Unfortunately, therapeutic proteins are primarily administered via injection only. Because protein therapeutics most frequently require repeated administrations, the cumulative frustration with a painful and inconvenient administration method often causes patients to intentionally skip injections, leading to less effective treatment (Peyrot et al., 2010). Oral delivery is a more desirable route of administration due to its ease, familiarity, and avoidance of chronic irritation (as experienced with injection or intranasal delivery methods). Additionally, oral delivery often offers lower cost, as orally-delivered drugs need not be produced in the highly sterile cleanroom environments required for injectables (Salama et al., 2006). Development of an oral delivery strategy for protein therapeutics would therefore be a boon to both patients and the protein therapeutics industry by offering improved patient quality of life and reduced cost.
Despite significant interest, the oral route has not yet been widely adopted with protein-based drugs because of the human body's natural mechanisms for breaking down ingested protein into substituent amino acids. The drug must first retain its structure and integrity through the highly acidic and proteolytic environment of the stomach, in which it spends an average time of around 3.5 hours (Dressman and Kramer, 2005). The drug then passes on to the small intestine, where it must remain stable in neutral conditions (pH 6.8 - 7.4), survive attack from additional proteolytic enzymes, pass through the mucosal lining, and cross the epithelial cell layer by either paracellular transport through the tight junctions or transcellular transport through the cells. The drug then enters the bloodstream, where it will be distributed throughout the body to perform its function. Because of this series of barriers, protein therapeutics exhibit extremely low bioavailability via the oral route without some means of protection (Morishita and Peppas, 2006; Renukuntla et al., 2013; Gupta et al., 2013).
Studies seeking to overcome these delivery barriers using pH-responsive hydrogels such as poly(methacrylic acid-grafted-poly(ethylene glycol)) (P(MAA-g-EG)) or poly(methacrylic acid-co-N-vinylpyrrolidone) (P(MAA-co-NVP) to deliver proteins such as insulin and human growth hormone have been relatively successful, but still suffer from low bioavailability compared to injection, resulting in wasted drug and therefore higher cost (Lowman et al., 1999; Carr et al., 2010; Carr and Peppas, 2010; Foss and Peppas, 2004; Kamei et al., 2009; Kavimandan et al., 2006). Additionally, studies seeking to deliver proteins exhibiting high isoelectric points (pI) have been hampered by coulombic interactions in the small intestine between the anionic hydrogel and cationic protein, resulting in binding rather than release for uptake into the bloodstream (Carr et al., 2010).
In the present work, we focus on enabling oral delivery of salmon calcitonin as a model for high pI therapeutic proteins. Our group has previously synthesized and characterized pH-sensitive copolymer hydrogels comprised of poly(itaconic acid-grafted-poly(ethylene glycol)) (P(IA-g-EG)) which showed potential as drug delivery carriers due to their favorable equilibrium swelling behavior in acidic and neutral pH environments (Betancourt et al., 2010). The additional carboxylic acid residue present in itaconic acid compared to methacrylic acid can yield superior swelling behavior and drug delivery capability that would assist in delivery of high pI proteins. However, these hydrogels were initially studied on their own and not compared directly against previous P(MAA-g-EG) or P(MAA-co-NVP) systems, and were not tested in drug loading and release experiments, so we herein extend work with these systems by determining their swelling behavior in time-limited, dynamic-pH swelling experiments, by comparing against P(MAA-g-EG) and P(MAA-co-NVP) systems, and by utilizing them in in vitro drug loading and release experiments.
Furthermore, our group has previously demonstrated improved drug delivery behavior resulting from use of N-vinylpyrrolidone as a hydrogen-bond accepting comonomer instead of poly(ethylene glycol), as it offers stronger complexation behavior for improved protein protection and faster diffusive drug release (Carr and Peppas, 2009; Carr et al., 2010). Therefore, we also synthesize and test P(IA-co-NVP) hydrogels alongside P(MAA-g-EG), P(MAA-co-NVP), and P(IA-g-EG) hydrogels with the expectation that the stronger complexation behavior will also assist in enabling the oral delivery of high pI proteins.
In this work, we demonstrate improved material responsiveness and high pI protein delivery capability resulting from use of itaconic acid as the pH-responsive monomer, as well as improved delivery capability from reducing the ionic strength of the solution used in loading the drug into the microparticles.
2. Materials and Methods
2.1. Synthesis of Hydrogels
Seven different hydrogels were formed via UV-initiated free radical polymerization. Each hydrogel was comprised of a pH-responsive moiety—either methacrylic acid (MAA) (Sigma-Aldrich, St. Louis, MO) or itaconic acid (IA) (Acros Organics, Fair Lawn, NJ)—copolymerized with a hydrophilic comonomer containing a hydrogen bond-acceptor for complexation behavior—either N-vinyl pyrrolidone (NVP) (Sigma-Aldrich) or poly(ethylene glycol) monomethyl ether monomethacrylate (PEGMMA, molecular weight 1000) (Poly-sciences, Warrington, PA). Methyl methacrylate (MMA) (Sigma-Aldrich) was also incorporated as a third monomer in one formulation. Hydrogels were crosslinked using tetra(ethylene glycol) dimethacrylate (TEGDMA) (Sigma-Aldrich) or poly(ethylene glycol) dimethacrylate (PEGDMA, molecular weight 1000) (Sigma-Aldrich). Irgacure 2959 (Ciba Specialty Chemicals Corp., Tarrytown, NY) was used as the UV-initiator.
All polymerizations were carried out in a 50:50 w/w mixture of aqueous sodium hydroxide (NaOH) and ethanol. Sodium hydroxide was prepared at a concentration such that there was a 1:2 molar ratio of NaOH to IA. No NaOH was required for MAA-based gels. Within this cosolvent, monomers were added at various molar ratios as shown in Table 1, along with crosslinker (3-5 mol% for IA-based gels; 1 mol% for MAA-based gels) and 1 mol% initiator. Molar percent is defined with regard to the total moles of monomers and crosslinker.
Table 1.
Hydrogel Feed Compositions.
| Hydrogel Formulation | Monomer Feed Ratio | Crosslinker | Molar % Crosslinker |
|---|---|---|---|
| 1:1 P(IA-co-NVP) | 1:1 IA:NVP | TEGDMA | 5 |
| 1:2 P(IA-co-NVP) | 1:2 IA:NVP | TEGDMA | 3 |
| 1:9 P(IA-co-NVP) | 1:9 IA:NVP | TEGDMA | 4 |
| P(IA-co-NVP-co-MMA) | 45:45:10 IA:NVP:MMA | PEGDMA | 5 |
| P(IA-g-EG) | 1:1 IA:PEGMMA | PEGDMA | 4 |
| P(MAA-co-NVP) | 2:1 MAA:NVP | TEGDMA | 1 |
| P(MAA-g-EG) | 1:1 MAA:PEGMMA | TEGDMA | 1 |
The monomer solution was then introduced into a nitrogen environment in an MBraun Labmaster 130 glove box (MBraun, Garching, Germany) and purged with nitrogen for 10 minutes to remove oxygen, a free radical scavenger. The solution was pipetted between two quartz glass plates (15 × 15 × 0.3 cm) separated by a 0.7 mm thick Teflon spacer, and then polymerized for 75 minutes in 35 mW/cm2 UV light using an IntelliRay 600 UV flood source (Uvitron International, West Springfield, MA). Disks 12 mm in diameter were collected from the resulting films immediately following polymerization for swelling studies. The remaining hydrogel was then removed from the glass plates and washed in 1 L of 18.2 MΩ-cm deionized water, changed daily for 10 days to remove unreacted monomers. Following washing, the films were dried under vacuum at 29 °C for at least 2 days, crushed into microparticles with a mortar and pestle, and sieved to 90–150 μm in size.
2.2. Swelling Studies
2.2.1. Equilibrium Swelling Studies
Hydrogel disks acquired from the 1:1 P(IA-co-NVP), 1:9 P(IA-co-NVP), P(IA-co-NVP-co-MMA), and P(MAA-co-NVP) formulations were washed in deionized water for 10 days and dried under vacuum. The dry weight of each disk was determined with an Ohaus Analytical Plus scale (Ohaus, Parsippany, NJ). Disks were then placed in either a standard PBS buffer (0.150 M, pH 7.4) (Fisher Scientific, Fair Lawn, NJ) to simulate neutral small intestine conditions or a 0.01 N hydrochloric acid (HCl) solution (Sigma-Aldrich) (pH 2.0) to simulate acidic stomach conditions at 37 °C for 72 h, allowing the disks to swell to equilibrium. The swelled disks were then removed from solution and weighed in air.
2.2.2. Dynamic Swelling Studies
Dry disks were weighed in air and subsequently swelled at 37 °C for 3 h in 0.01N HCl (pH 2.0). Ten dimethylglutaric acid (DMGA) buffers spanning a pH range from 3.2 to 7.6 were prepared and preheated to 37 °C. Following swelling in HCl, the disks were moved into the first of these ten buffers (pH 3.2), allowed to swell for 7 minutes, removed from the buffer, and weighed in air. The disks were then moved to the next buffer (in order of increasing pH), and the swelling and weighing process was repeated through all 10 buffers with 7 minute swelling intervals.
2.3. Cytotoxicity Studies
Caco-2 human colorectal adenocarcinoma cells (ATCC, Manassas, VA) were cultured normally up to a passage number of 60. The cells were then plated into a 96-well plate at an initial concentration of 1.0×104 cells/well in Dulbecco's Modified Eagle's Medium (DMEM) with 10% v/v fetal bovine serum, 1% v/v penicillin-streptomycin, and 1% v/v L-glutamine added. Each well received 200 μL DMEM (5.0 × 104 cells/mL). Cells were incubated for 72 h at 37 °C. Microparticles of the 1:1 P(IA-co-NVP), 1:1 P(IA-g-EG), and 1:1 P(MAA-g-EG) hydrogels ranging from 90-150 μm in size were sterilized by exposure to ultraviolet light and then suspended in DMEM at concentrations of 5.000, 2.500, 1.250, 0.625, 0.312, 0.156, 0.078, and 0.039 mg/mL. DMEM was removed from all wells by vacuum aspiration and replaced with 120 μL of microparticle suspension (n=3 for each concentration of each hydrogel formulation), 1.5% v/v bleach in DMEM (negative control, n=8), or fresh DMEM (positive control, n=16). Cells were incubated in presence of hydrogel microparticles at 37 °C for 2 h. The microparticle suspensions were then removed from all wells by vacuum aspiration, and all wells were washed twice with 120 μL sterile phosphate-buffered saline. Cell viability was determined using an MTS assay: 20 μL (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) (Promega, Madison, WI) and 100 μL colorless DMEM were added to each well and incubated for 3 h before absorbance was measured at both 690 nm and 490 nm.
2.4. Drug Loading Studies
A solution of salmon calcitonin (sCT) (Selleck Chemicals, Houston, TX) was prepared at a concentration of 0.40 mg/mL in 0.0150 M PBS buffer (pH 7.4), and 1.5 mL of this solution was added to each of 21 (n=3 per hydrogel formulation), 2.0 mL, low-adhesion microcentrifuge tubes. Microparticles 90-150 μm in size were added to this solution at 10 mg of dry hydrogel per tube. The mixture was agitated for 1 h using an Eppendorf Thermomixer (Eppendorf, Hauppauge, NY), allowing the sCT to diffuse into the interior of the particles. The particles were collapsed using 75 μL of 0.1 N HCl and isolated by centrifugation and decanting. The supernatant was collected for analysis. The particles were resuspended in two washes consisting of 1.0 mL of 0.01 N HCl to remove surface-bound protein. After each wash, the particles were isolated by centrifugation and decanting, and the rinse was collected for analysis. The isolated microparticles with encapsulated sCT were lyophilized until dry. Samples of the stock solution, the supernatant following particle collapse, and the two acid rinses were analyzed by a Micro BCA protein concentration assay (Thermo Fisher Scientific, Rockford, IL).
2.5. Drug Release Investigation
Once the microparticles were dry from lyophilization, 1.0 mL of 0.150 M PBS buffer at a pH of 3.0 (from adding HCl) was added to each microcentrifuge tube and agitated for 1 h at 37 °C. A 150 μL sample was removed for analysis and replaced with 150 μL fresh, pH 3.0 PBS before raising the pH back to 7.4 by addition of 1 N NaOH. The neutralized mixture was agitated for 24 h at 37 °C, with 150 μL samples collected at time points of 1, 2, 4, and 24 h, each time being replaced by 150 μL fresh PBS (pH 7.4). All samples were analyzed for sCT concentration using a Micro BCA protein concentration assay.
2.6. Ionic Strength Experiments
Three separate trials were undertaken to examine the effects of changing the ionic strength of the loading solution on salmon calcitonin delivery. In the first trial, three different concentrations of PBS buffer at pH 7.4 were used: 1.5 M (10× concentrated PBS), 150 mM (1× standard PBS), and 15 mM (0.1× concentrated PBS). In these solutions, salmon calcitonin was dissolved at a concentration of 0.1 mg/mL. Using 40 mL of each solution, 100 mg of 1:1 P(IA-co-NVP) microparticles, 90-150 μm in size, were added to each solution and mixed for 1 h. Particles were collapsed by addition of 1 N HCl to reduce pH to 2.0, isolated by decanting, and lyophilized. Samples of the stock solutions and post-collapse supernatant were analyzed for sCT concentration using high performance liquid chromatography to determine sCT loading levels. After lyophilization, 30 mg of each sample were added to 30 mL of 150 mM (1×), pH 3.0 PBS buffer and stirred for 1 h at a temperature of 37.0 °C using a Distek dissolution apparatus (Distek, North Brunswick, NJ). After 1 h, a 500 μL sample was taken for analysis and replaced with fresh, pH 3.0 PBS. The pH was then raised to pH 7.4 by addition of 1 N NaOH. Samples were collected at time points of 45, 75, and 120 minutes following neutralization. Salmon calcitonin concentration in all samples was determined by a Micro BCA protein assay.
In the second trial, a fourth solution of 1.5 mM PBS (0.01×) was added for study. The four solutions (0.01×, 0.1×, 1×, and 10× PBS) were prepared, and salmon calcitonin was dissolved in 7 mL of each solution at a concentration of 250 μg/mL. To each of these solutions, 10 mg of 1:1 P(IA-co-NVP) microparticles were added and allowed to imbibe sCT for 1 h while agitated. The particles were then collapsed by addition of 1 N HCl to reduce pH to 2.0 and were collected by centrifugation and decanting. Samples from the stock solutions and supernatant were analyzed for sCT concentration by HPLC. The collected particles were dried by lyophilization. Following drying, the microparticle samples were added to 2.0 mL microcentrifuge tubes containing 1.75 mL of 150 mM (1×), pH 3.0 PBS buffer and agitated at 37 °C using an Eppendorf Thermomixer. After 1 h, 150 μL samples of the solutions were collected for analysis and replaced with fresh PBS (pH 3.0). The solutions were then neutralized by addition of 0.2 N NaOH to a pH of 7.4. Release at neutral conditions was carried out for 2 h, with samples acquired at time points of 1 and 2 h. All samples were analyzed for sCT concentration using a Micro BCA protein concentration assay.
In the third trial, the same four solutions were used as in the second trial. Calcitonin was dissolved in 30 mL of each solution at a concentration of 0.20 mg/mL. To each solution, 100 mg of 1:1 P(IA-co-NVP) microparticles were added, and the pH was maintained at 7.0 by addition of NaOH. The solutions were mixed for 18 h at room temperature to allow for drug loading prior to collapse by acidification to pH 2.5 with HCl. The particles were isolated by decanting, washed twice with 1 mL 0.01 N HCl, and lyophilized. Following drying, 20 mg of each of the dried, drug-loaded microparticle samples were added to 40 mL of pH 3, 0.150 M PBS buffer (acidified with HCl) and stirred for 1 h in a Distek dissolution apparatus at 37 °C. The solutions were then neutralized to pH 7.4 by addition of NaOH and stirred for 2 h. Samples were acquired at time points of 1 h during acidic conditions, 1h following neutralization, and 2 h following neutralization. The samples were analyzed for sCT concentration by a Micro BCA protein concentration assay.
3. Results and Discussion
3.1. Hydrogel Synthesis
All hydrogels were successfully prepared by UV-initiated free radical polymerization. IA-based hydrogels were prepared with partial neutralization of acid groups by NaOH, as used by Betancourt et al. (2010). Neutralization allows for incorporation of high levels of IA into the resulting hydrogel by overcoming low solubility (0.083 g/mL) to a level sufficient for creating a homogeneous hydrogel. IA-based gels visibly required longer curing times than MAA-based gels. MAA-based gels had gelled completely within 30 min, whereas IA-based gels required up to 1 h to form a gel.
IA-containing gels also exhibited lower mechanical strength than MAA-containing gels. The IA-based gels were softer and broke into smaller pieces readily upon mild agitation, such as moving the hydrogel film or refilling the wash water. MAA-based gels did not tear nearly as readily and could easily be picked up and moved by hand without tearing. Methyl methacrylate was included as a third monomer in one formulation (P(IA-co-NVP-co-MMA)) at 10 mol% to add additional strength to the IA-based gel. The resulting gel did have improved strength, but was still softer and more easily broken than the MAA-based gels. Nevertheless, the reduced mechanical strength is a bulk property and does not significantly affect microparticle integrity. As a result, it is not expected to be a problem for end-use. Furthermore, the softness suggests higher porosity or greater water imbibition, which would be beneficial for improving bioavailability of proteins by aiding in diffusive protein release from the microparticle carriers.
3.2. Swelling Studies
The fundamental feature of our hydrogel systems is their pH-responsive behavior. At low pH, the carboxylic acid residues in MAA or IA are protonated, allowing hydrogen bonding complexation between these residues and the electronegative oxygen in NVP, yielding a small conformation with small mesh size. At neutral pH, the carboxylic acid residues are deprotonated, losing the hydrogen bonding complexation behavior and becoming anionically charged, thus swelling to a larger conformation with large mesh size due to entropic mixing, osmotic pressure, and coulombic repulsion. As a result, the protein can be imbibed into the hydrogel in neutral conditions, encapsulated by acidification, and released once in neutral conditions again. Without this environmentally-responsive behavior, the hydrogels do not yield protection from proteolytic enzymes and therefore will not work for oral protein delivery. Furthermore, the extent to which swelling occurs is suggestive of the achievable delivery efficiency.
Equilibrium weight swelling experiments were performed to determine the suitability of the synthesized hydrogels. The swelling behavior of P(IA-g-EG) hydrogels was not measured due to low physical stability of the material at the macro-scale. Although the hydrogel remains crosslinked and highly stable as microparticles, the large bulk disks used in weight swelling studies are too prone to breaking, especially when swelled in neutral conditions, to be accurately measured with gravimetry. P(MAA-g-EG) disks were also not studied, as their swelling behavior has been previously very well-characterized (López and Peppas, 2004; Nakamura et al., 2004; Torres-Lugo and Peppas, 1999; Foss et al., 2004).
As shown in Figure 1, during equilibrium swelling studies, the weight swelling ratio, q, defined as the swelled weight divided by the dry weight of a hydrogel disk, is very low for all tested formulations at low pH. All tested formulations exhibit weight swelling ratios ranging from 1.3 to 2.2, indicating that the average pore size remains small in the acidic conditions expected in the stomach, keeping proteins encapsulated inside the hydrogel and preventing proteolytic enzymes from entering and degrading the protein. When swelled at neutral pH, the disks all achieve significantly greater swelling ratios ranging from 13.7 to 23.1, approximately one full order of magnitude greater than at low pH. This result indicates that the mesh size is significantly increased in the neutral conditions expected in the small intestine, such that the protein may diffuse out into the small intestine to cross the epithelial cell layer and enter the bloodstream. All of the tested IA-based gels swelled significantly more (p<0.01) than the MAA-based hydrogel, up to a 69% increase observed with the 45:45:10 P(IA-co-NVP-co-MMA) terpolymer, indicating the potential for the IA-based gels to encapsulate and therefore deliver greater amounts of the protein drug.
Figure 1. Equilibrium Weight Swelling.
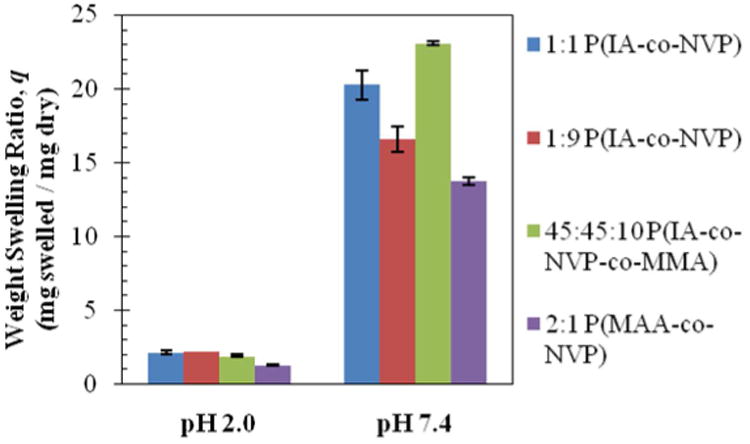
Swelling ratios (weight of swelled disk / weight of dry disk) recorded following 72 h swelling in 0.01 N HCl (pH 2.0) or 150 mM PBS buffer (pH 7.4). Reported as average ± standard deviation.
In dynamic swelling studies, the difference in swelling behavior between IA and MAA-based gels is even more apparent. Figure 2 shows the results of the dynamic swelling studies, where there are 7 minutes of swelling time between each data point. The IA-based gels all achieved swelling ratios ranging from 11.2 to 15.4 within the 70 min of cumulative swelling time—on the same order of magnitude as the equilibrium swelling ratios. However, the MAA-based gel exhibited nearly imperceptible swelling, achieving a swelling ratio of only 1.5—a full order of magnitude less than its equilibrium weight swelling ratio. Because the residence time in the small intestine is limited—approximately 4 h for both fasted and fed patients (Dressman and Kramer, 2005)—it is important that the microparticles undergo a rapid transition from collapsed to swollen states to maximize the available time in which the encapsulated protein may diffuse out into the small intestine. The significantly improved time-dependent swelling characteristics of the IA-based gels compared to the MAA-based gels should therefore assist in maximizing protein release in the target region by maximizing the time available for diffusive release of the drug.
Figure 2. Dynamic Weight Swelling.
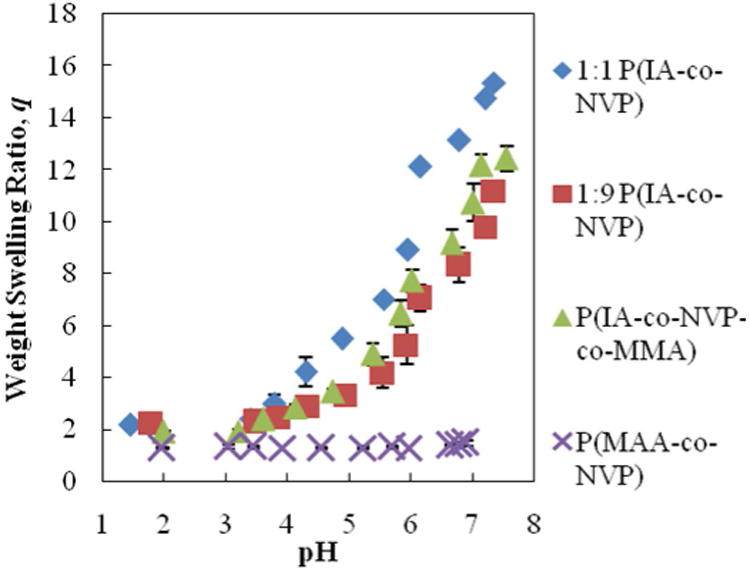
Time-sensitive swelling results as disk transitioned between DMGA buffers in order of increasing pH. Swelling time was 7 minutes between each data point. Reported as average ± standard deviation.
3.3. Cytotoxicity
Having shown an improvement in pH-responsive swelling behavior by utilizing IA as the pH-responsive moiety, we sought to determine whether the polymers were biocompatible and safe for use. Using Caco-2 cells, a proven, effective model of the human small intestine (Sambuy et al., 2005; Pinto, 1983; Hidalgo et al., 1989), a cell viability study was performed in vitro. After culturing cells, cells were exposed to varying microparticle concentrations (0.039 – 5 mg/mL) of all 7 hydrogels, and viability was measured using an MTS assay. The results of this study are shown in Figure 3, with all cell viability values normalized to the positive control. Even at very high concentrations of 5 mg/mL, the microparticles are still well tolerated, achieving over 80% relative viability with nearly every formulation. Only the 1:9 P(IA-co-NVP) shows any indication of toxicity at concentrations of 1.25 and 5.00 mg/mL, achieving only 59% and 74% viability at these concentrations, respectively. However, since 99% viability is observed at 2.50 mg/mL, the potential toxicity at 1.25 mg/mL is likely a random artifact rather than true toxicity. Neverthless, no significant decrease in cell viability was observed with any concentration tested with the other 6 formulations. Therefore, the results indicate a high degree of safety with regard to cytotoxicity, even at high concentrations.
Figure 3. Caco-2 Cell Cytotoxicity.
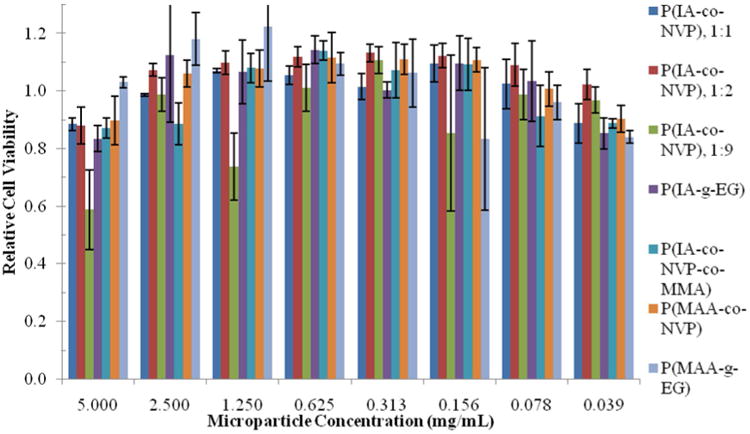
Relative cell viability following 2 h incubation with microparticles, as determined by MTA proliferation assay and normalized to positive control in Caco-2 cell culture. Each bar represents n=5 samples, and is reported as average ± standard deviation.
3.4. Drug Loading/Release
3.4.1. Dependence on Hydrogel Formulation
The ultimate goal of this work is to develop a system enabling oral delivery of high isoelectric point-exhibiting proteins with high bioavailability. The swelling and cytotoxicity studies show that the developed systems exhibit the proper behavior and are biocompatible for use with human cells, but a direct test of delivery capability with high isoelectric point-exhibiting therapeutic proteins was needed. For this test, salmon calcitonin was encapsulated in microparticles of the 7 hydrogel formulations by imbibition and subsequently released by diffusion out of the polymer matrix following swelling in neutral conditions. The observed delivery potential for sCT (mg of sCT released at a certain time per mg of hydrogel used) for each of the hydrogel formulations is shown below in Figure 4 and tabulated in Table 2.
Figure 4. Salmon Calcitonin Release from Hydrogel Microparticles.
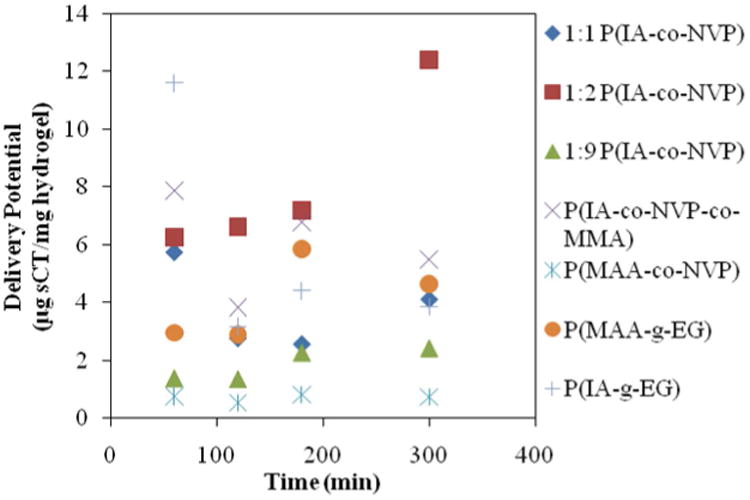
Time point at 60 min is in acidic (pH 3.0) conditions immediately before neutralization with NaOH. All other time points were collected in neutral (pH 7.4) conditions. Delivery potential reported as mg sCT per mg hydrogel, as determined by Micro BCA assay.
Table 2.
Salmon Calcitonin Loading and Release Levels from Various Hydrogel Formulations.
| Hydrogel Formulation | Loading Level (μg sCT/mg hydrogel) | Delivery Potential, t=4 h (μg sCT/mg hydrogel) | Percent Release, t=4 h (%) |
|---|---|---|---|
| 1:2 P(IA-co-NVP) | 45.0 | 12.4 | 27.7 |
| P(IA-co-NVP-co-MMA) | 29.1 | 5.5 | 19.0 |
| P(MAA-g-EG) | 23.4 | 4.6 | 19.8 |
| 1:1 P(IA-co-NVP) | 22.3 | 4.1 | 12.3 |
| P(IA-g-EG) | 38.1 | 3.8 | 10.0 |
| 1:9 P(IA-co-NVP) | 35.9 | 2.4 | 7.4 |
| P(MAA-co-NVP) | 9.8 | 0.7 | 8.0 |
The ideal behavior would be to have zero delivery potential at the 1 h time point, which is taken at the end of the acidic portion of the release, meaning no protein is lost due to premature release in the stomach, and to have very high delivery potential at the 4 h time point that is of similar magnitude as the 24 h time point, indicating fast and complete release in a timeframe and pH similar to what would be observed in the small intestine. None of the systems studied exhibited ideal behavior, with all losing a fraction of encapsulated sCT in the stomach, but by subtracting the amount of sCT released at 1 h from that released at 4 h provides a good indicator of which best approximates ideal behavior. The best candidate is the 1:2 formulation of P(IA-co-NVP), which released 12.4 μg sCT/mg hydrogel after 3 h in neutral conditions— 2.7 times more than the previously studied P(MAA-g-EG) hydrogel, and 16.8 times more than the previously studied P(MAA-co-NVP) hydrogel. Although 6.26 μg sCT/mg hydrogel were released in stomach conditions, the remaining 6.15 μg sCT/mg hydrogel delivered in subsequent small intestinal conditions far exceed the 1.7 μg sCT/mg hydrogel that the P(MAA-g-EG) hydrogel delivered in the same conditions.
The large, significant (p<0.01) increase in delivery potential observed with the 1:2 molar ratio of IA to NVP compared to the other formulations shows that the 1:2 ratio optimizes swelling and binding behavior to best accommodate oral delivery of sCT, potentially due to the distribution of anions through the hydrogel. The anionic groups in the polymer backbone are useful in achieving high drug loading by taking advantage of the same coulombic interactions that are best avoided during release and by improving hydrogel swelling via anionic repulsion for greater loading and release. The important distinction between IA and MAA is that IA has two potential anions (–COO−) present per molecule while MAA possesses only one. Therefore, by having 1 in 3 monomer subunits ionizable (IA) in the 1:2 P(IA-co-NVP) hydrogel versus 2 in 3 ionizable subunits (MAA) in the 2:1 P(MAA-co-NVP) hydrogel, the overall number of anions could be similar between the two gels. However, with IA, the 2 anions are localized on a single subunit rather than spread out among 2 different monomers, which results in a greater average distance of approach for the sCT from the anions during release. Because coulombic forces decrease by the square of the distance between ions, any increase in approach distance will have a significant effect in assisting diffusive release. Therefore, the localization of anions to a single monomer is likely to be a contributing factor in the improved delivery potential observed with the 1:2 P(IA-co-NVP) hydrogel formulation. Coupled with faster and greater overall swelling response, which further increase the average approach distance and maximize time available for diffusive release, the 1:2 molar ratio experimentally proves to best take advantage of these multiple factors to achieve improved delivery potential.
3.4.2. Dependence on Ionic Strength of Loading Solution
In addition to using different materials based on IA instead of MAA, there may be delivery improvements available from a procedural standpoint. Unfortunately, the release environment is set entirely by the small intestine and is not easily alterable. Therefore, only variables involved in the loading are easily utilized for improving bioavailability of high isoelectric point-exhibiting proteins.
One of the ideal variables available for study is the ionic strength of the loading solution. The ionic strength can affect the loading of the drug into the hydrogel in two ways. First, ionic strength strongly affects the swelling behavior of the pH-responsive hydrogel as described by Brannon-Peppas and Peppas (1991), with higher ionic strength resulting in decreased swelling and lower ionic strength resulting in increased swelling. Therefore, by reducing the ionic strength of the loading solution, the hydrogel will swell to a greater degree, allowing it to imbibe a greater amount of the protein. With more drug loaded, the driving force for diffusive release is increased, resulting in greater delivery capability. Second, the ionic strength affects the degree to which coulombic interactions take place, as described by the Debye length. The Debye length is the effective distance over which an ion's charge is offset by the charges of ions present in the surrounding medium. The Debye length can be calculated as
where I is the ionic strength (mol/m3), ε is the permittivity of the medium, kB is the Boltzmann constant, T is the absolute temperature, NA is Avogadro's number, and e is the elementary charge. The important relationship is that the Debye length is inversely proportional to the square root of the ionic strength. Therefore, by decreasing the ionic strength of the loading solution, in addition to increasing swelling, the distance over which ionic interactions are expected to occur increases, meaning there is a greater likelihood of coulombic binding. Although we hope to avoid such interactions during drug release, they can be used beneficially during loading by encouraging binding to the interior of the microparticles, thereby increasing the driving force for diffusive release.
To test these hypotheses, an experiment involving loading and releasing sCT in four loading solutions of different ionic strength was performed in three different trials. The results are shown below in Figures 5 through 7 and Tables 3 through 5.
Figure 5. Salmon Calcitonin Release--Ionic Strength Trial 1.
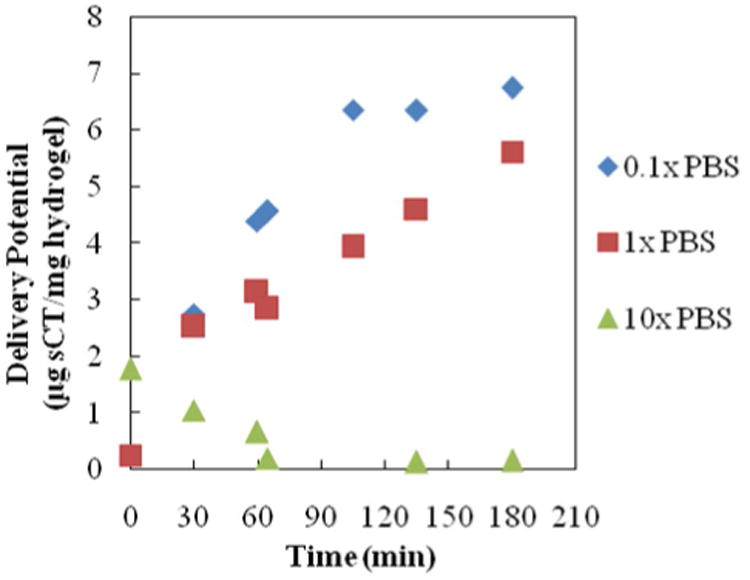
All profiles are for release of salmon calcitonin following loading into 1:1 P(IA-co-NVP) microparticles in 0.1×, 1×, or 10× concentrated PBS buffer. Time points from 0 – 60 min are in acidic conditions (pH 3.0) and time points from 65 – 180 minutes are in neutral conditions (pH 7.4).
Figure 7. Salmon Calcitonin Release--Ionic Strength Trial 3.
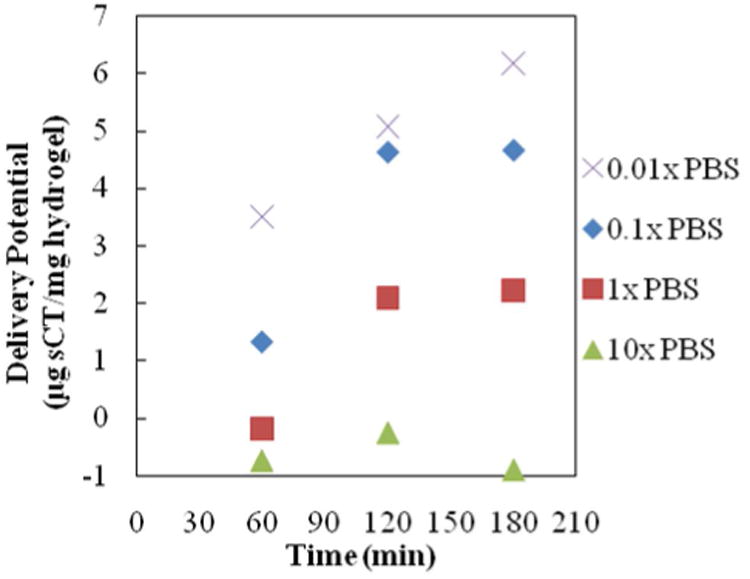
All profiles are for release of salmon calcitonin following loading into 1:1 P(IA-co-NVP) microparticles in 0.01×, 0.1×, 1×, or 10× concentrated PBS buffer. Time point at 60 min is in acidic conditions (pH 3.0), and time points at 120 and 180 minutes are in neutral conditions (pH 7.4).
Table 3.
Salmon Calcitonin Loading and Release Levels, Ionic Strength Trial 1.
| Loading Solution | Loading Level (μg sCT/mg hydrogel) | Delivery Potential, t=180 min (μg sCT/mg hydrogel) | Percent Release, t=180 min (%) |
|---|---|---|---|
| 0.1× PBS (15mM) | 10.7 | 6.74 | 63.0 |
| 1× PBS (150 mM) | 23.5 | 5.62 | 23.9 |
| 10× PBS (1500 mM) | 7.4 | 0.16 | 2.1 |
Table 5.
Salmon Calcitonin Loading and Release Levels—Ionic Strength Trial 3.
| Loading Solution | Loading Level (μg sCT/mg hydrogel) | Delivery Potential, t=180 min (μg sCT/mg hydrogel) | Percent Release, t=180 min (%) |
|---|---|---|---|
| 0.01× PBS (1.5 mM) | 57.8 | 6.18 | 10.7 |
| 0.1× PBS (15 mM) | 54.8 | 4.67 | 8.5 |
| 1× PBS (150 mM) | 49.6 | 2.23 | 4.5 |
| 10× PBS (1500 mM) | 47.3 | -0.90 | -1.9 |
In Trial 1 (Figure 5, Table 3), it is observed that lower ionic strength loading solution (0.1× PBS) results in an overall improvement in delivery potential compared to the previously used standard (1×) PBS solution. The loading level is 54% lower in the 0.1× PBS buffer compared to the 1× PBS buffer, contrary to expectations, but the percent release is 164% greater, yielding 20% greater overall delivery potential. This result is preferable, because it means that less hydrogel is required to deliver a therapeutic dose of drug and that less of the drug is being wasted by remaining in the hydrogel—both of which are benefits that will help decrease cost of an oral drug formulation.
In Trial 2 (Figure 6, Table 4), the experiment was extended to include an even lower ionic strength loading solution (0.0 1× PBS). The results show that the further reduction provides even greater benefits to the delivery potential. Within three hours of release (2 h at neutral pH), the 0.01×-PBS-loaded sample delivered 48.4 μg sCT/mg hydrogel, compared to the 0.1×-PBS-loaded sample delivering 16.1 μg sCT/mg (a 3.0-fold improvement) and the 1×-PBS-loaded sample delivering only 0.6 μg sCT/mg (an 83-fold improvement). Percent release also increased with decreasing ionic strength in the loading solution. Again, lower ionic strength loading solutions yielded greater percent release and greater overall delivery, which results in a smaller pill for the user at cheaper cost due to less wasted drug.
Figure 6. Salmon Calcitonin Release--Ionic Strength Trial 2.
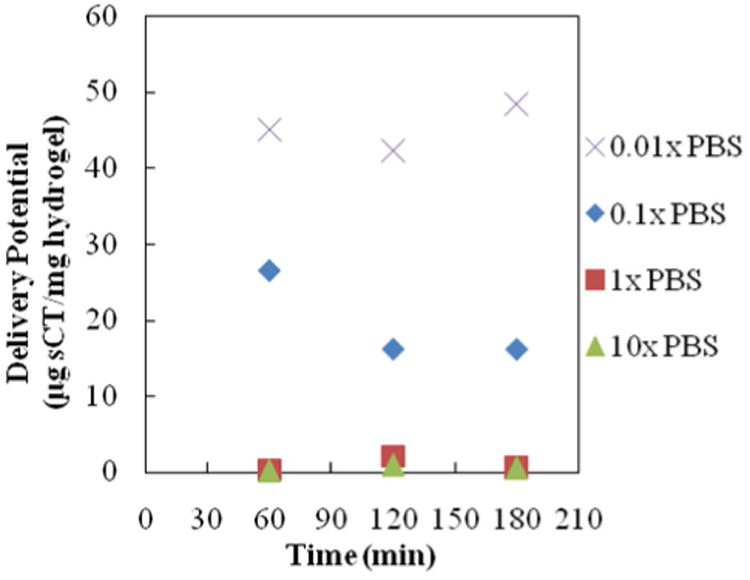
All profiles are for release of salmon calcitonin following loading into 1:1 P(IA-co-NVP) microparticles in 0.01×, 0.1×, 1×, or 10× concentrated PBS buffer. Time point at 60 min is in acidic conditions (pH 3.0), and time points at 120 and 180 minutes are in neutral conditions (pH 7.4).
Table 4.
Salmon Calcitonin Loading and Release Levels, Ionic Strength Trial 2.
| Loading Solution | Loading Level (μg sCT/mg hydrogel) | Delivery Potential, t=180 min (μg sCT/mg hydrogel) | Percent Release, t=180 min (%) |
|---|---|---|---|
| 0.01× PBS (1.5 mM) | 105.6 | 48.4 | 45.8 |
| 0.1× PBS (15 mM) | 171.0 | 16.1 | 9.4 |
| 1× PBS (150 mM) | 12.2 | 0.6 | 4.8 |
| 10× PBS (1500mM) | 53.4 | 0.6 | 1.1 |
Finally, in Trial 3 (Figure 7, Table 5), similar, but less pronounced behavior is observed. The 0.01×-PBS-loaded sample releases 6.18 mg sCT/mg hydrogel within 2 h at neutral pH, compared to the 0.1×-PBS-loaded sample delivering 4.67 μg sCT/mg (a 1.3-fold improvement) and the 1×-PBS-loaded sample delivering only 2.23 μg sCT/mg (a 2.8-fold improvement). Additionally, the percent release increases with decreasing ionic strength of the loading solution. Once again, this data collectively shows that a small procedural change using a reduced ionic strength loading solution yields a cheaper, better delivery system that requires less hydrogel and wastes less of the drug.
Unfortunately the degree of improvement achieved by utilizing a lower ionic strength loading solution is not consistent across all three trials, ranging from a 2.8-fold improvement to an 83-fold improvement by moving to the 0.01× PBS loading solution from the 1×. However, the general trend is consistent across all three trials: that a reduced ionic strength loading solution yields higher delivery potential and a higher percentage of encapsulated drug being released, which holds true from the 0.01× solution to the 10× solution. Of course, this trend is only necessarily true for salmon calcitonin as tested here, not other proteins with different sizes and shapes. Nevertheless, given the core principles behind the improvement (altering swelling and coulombic binding properties), the trend is expected to extend to all high isoelectric point-exhibiting proteins. Although not tested here, using a lower ionic strength loading solution may yield improvement for low isoelectric point-exhibiting proteins as well. Although the Debye length changes are then unfavorable (encouraging repulsion from the microparticles rather than binding for loading), the improvement in swelling should be more beneficial than the slight increase in ionic repulsion will be detrimental.
3.5. Pharmacological Feasibility
The normal recommended dosage of salmon calcitonin for a patient treating osteoporosis is 50-100 IU/day by injection, equating to 8.3-16.7 μg/day. To deliver an equivalent amount of protein by the oral route, a greater amount will need to be delivered, which can be estimated by a simple calculation.
To ensure a conservative determination of feasibility, the required dose used in the calculation will be the high end value of 16.7 μg/day. Aside from limited delivery potential from the microparticle carriers, loss of bioavailability resulting from intestinal enzymatic degradation and low intestinal absorption must also be accounted for. Data from Youn et al. (2006) showed 0.05% of unmodified sCT was transported across a Caco-2 monolayer in in vitro conditions over 60 min. Assuming a constant permeability (as is observed during those 60 min), this means 0.15% of released sCT can be expected to be transported across the intestinal epithelium into the bloodstream over the 3 h average residence time. The actual percentage will likely be significantly higher than this, as Caco-2 monolayers are reported to exhibit less permeable tight junctions than are observed in vivo (Artursson, 1991; Lennernas et al., 1996; Dantzig and Bergin, 1990). Correlations between Caco-2 permeability and in vivo absorption (Artursson and Karlsson, 1991; Yee, 1997) suggest that the apparent permeability of 1.5 × 10−7/cm2 reported by Youn et al. (2006) lies in the range of poorly absorbed compounds (0-20% uptake); indeed, compounds with similar permeability in Caco-2 cultures such as doxorubicin (1.6 × 10−7/cm2) or 1-deamino-8-D-arginine-vasopressin (1.3 × 10−7/cm2) show 5% and 1% absorption respectively, so the actual absorption could be expected to be somewhere between 1 and 5%. Furthermore, the presence of P(MAA-g-EG) microparticles has been shown to inhibit enzymatic degradation following intestinal release and enhance permeability by reversible opening of the tight junctions in the intestinal epithelium, which would further enhance bioavailability and intestinal absorption (Madsen and Peppas, 1999; Kavimandan et al., 2006; Kavimandan and Peppas, 2008; Ichikawa and Peppas, 2003). However, for the purpose of maintaining a conservative estimate of feasibility, the very low 0.15% absorption for Caco-2 cultures over 3 h will be used.
With the selection of a high dosage and a low level of absorption, the approximate amount of polymer microparticles needed for a daily dose given 48.2 or 6.2 μg sCT/mg hydrogel delivery potentials (results from trials 3 and 2, respectively) are
Given a density of 0.66 g/mL for dry P(IA-co-NVP) microparticles, 230 mg will easily fit within a size 2 gel capsule (15.3 mm in length), meaning a daily dose may be given with one pill that is smaller than a standard Tylenol gel capsule (19 mm in length) per day. The larger estimate of 1796 mg of hydrogel will require multiple gel capsules, such as three size 00 (20.2 mm) or four size 0 (18.4 mm) capsules. Therefore, depending strongly on the actual absorption in the small intestine and the prescribed dosage, our results show that a normal regimen of salmon calcitonin may be received with our system with the improved loading procedure and delivery material described herein, likely with no more than one or two pills taken per day. Better intestinal absorption well beyond 0.15% absorption is needed to make the cost reasonable, but improved absorption is expected to result from greater in vivo permeability compared to Caco-2 cultures as described above and from the reversible opening of tight junctions by the polymer microparticles.
4. Conclusions
In this paper, a potential carrier enabling oral delivery of high isoelectric point-exhibiting therapeutic proteins has been studied. Although bioavailability was limited by unfavorable coulombic interactions in previously-studied pH-responsive P(MAA-g-EG) or P(MAA-co-NVP) hydrogels, use of itaconic acid-based hydrogels copolymerized with N-vinylpyrrolidone show improved material properties for oral delivery of high isoelectric point proteins, including greater overall swelling (up to 68% improvement over the tested P(MAA-co-NVP) hydrogel) and vastly improved time-limited response to pH shifts (up to 10.4 times higher weight swelling ratio in same 70 minute time frame) that will collectively aid in imbibing and releasing therapeutic proteins within the time frame during which the microparticles will be in the target area of the small intestine. When tested for delivery of salmon calcitonin, the 1:2 molar ratio of itaconic acid to N-vinylpyrrolidone in the monomer feed produced the best combination of distribution of ionic charge throughout the hydrogel network and swelling properties, achieving 12.4 μg sCT/mg hydrogel delivered in neutral conditions in 4 h—a 2.7-fold improvement over the tested P(MAA-g-EG) hydrogel. Additional improvement was shown using a procedural change utilizing a lower ionic strength loading solution during drug encapsulation. By using a 1.50 mM PBS buffer instead of the traditional 150 mM PBS buffer, a dramatic increase in delivery potential is achieved; results indicated an improvement ranging from 2.8- to 83-fold in sCT delivery per unit hydrogel and a 2.6- to 9.6-fold improvement in percent of the encapsulated drug that was released. The general trend of lower ionic strength yielding improved delivery and greater degree of release was consistent across all three trials and for all four ionic strength loading solutions tested. With the observed delivery potential and a very conservative estimate of transport across the intestinal epithelium, it is shown that the delivery levels achieved via combination of the new material and new loading procedure are sufficient for effective daily dosing of sCT in a regularly-sized pill form. The tested system therefore holds promise as a potential delivery vehicle for enabling delivery of high isoelectric point-exhibiting therapeutic proteins via the strongly preferred oral pathway.
Acknowledgments
This work is funded by the National Institutes of Health, grant R01-EB000246-19, and by the Fletcher S. Pratt Foundation. The authors would also like to acknowledge David Tai for assistance with laboratory work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Artursson P. Cell cultures as models for drug absorption across the intestinal mucosa. Crit Rev Therap Drug Carrier Systems. 1991;8:305–330. [PubMed] [Google Scholar]
- 2.Artursson P, Karlsson J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Comm. 1991;175:880–885. doi: 10.1016/0006-291x(91)91647-u. [DOI] [PubMed] [Google Scholar]
- 3.Betancourt T, Pardo J, Soo K, Peppas NA. Characterization of pH-responsive hydrogels of poly(itaconic acid-g-ethylene glycol) prepared by UV-initiated free radical polymerization as biomaterials for oral delivery of bioactive agents. J Biomed Mater Res. 2010;93A:175–188. doi: 10.1002/jbm.a.32510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brannon-Peppas L, Peppas NA. Equilibrium swelling behavior of pH-sensitive hydrogels. Chem Eng Sci. 1991;46:715–722. [Google Scholar]
- 5.Carr DA, Gómez-Burgaz M, Boudes MC, Peppas NA. Complexation Hydrogels for the Oral Delivery of Growth Hormone and Salmon Calcitonin. Ind Eng Chem Res. 2010;49:11991–11995. doi: 10.1021/ie1008025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carr DA, Peppas NA. Molecular Structure of Physiologically-Responsive Hydrogels Controls Diffusive Behavior. Macromol Biosci. 2009;9:497–505. doi: 10.1002/mabi.200800235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carr DA, Peppas NA. Assessment of poly(methacrylic acid-co-N-vinyl pyrrolidone) as a carrier for the oral delivery of therapeutic proteins using Caco-2 and HT29-MTX cell lines. J Biomed Mater Res. 2010;92A:504–512. doi: 10.1002/jbm.a.32395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dantzig AH, Bergin L. Uptake of the cephalosporin, cephalexin, by a dipeptide transport carrier in the human intestinal cell line, Caco-2. Biochim Biophys Acta. 1990;1027:211–217. doi: 10.1016/0005-2736(90)90309-c. [DOI] [PubMed] [Google Scholar]
- 9.Dimitrov DS. Therapeutic Proteins. Methods Mol Biol. 2012;899:1–26. doi: 10.1007/978-1-61779-921-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dressman J, Kramer J. Pharmaceutical Dissolution Testing. Taylor & Francis; Boca Raton, FL: 2005. [Google Scholar]
- 11.Foss AC, Goto T, Morishita M, Peppas NA. Development of acrylic-based copolymers for oral insulin delivery. Eur J Pharm Biopharm. 2004;57:163–169. doi: 10.1016/S0939-6411(03)00145-0. [DOI] [PubMed] [Google Scholar]
- 12.Foss AC, Peppas NA. Investigation of the cytotoxicity and insulin transport of acrylic-based copolymer protein delivery systems in contact with caco-2 cultures. Eur J Pharm Biopharm. 2004;57:447–455. doi: 10.1016/j.ejpb.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 13.Gupta S, Jain A, Chakraborty M, Sahni JK, Ali J, Dang S. Oral delivery of therapeutic proteins and peptides: a review on recent developments. Drug Deliv. 2013;20:237–246. doi: 10.3109/10717544.2013.819611. [DOI] [PubMed] [Google Scholar]
- 14.Hidalgo IJ, Raub TJ, Borchardt RT. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology. 1989;96:736–749. [PubMed] [Google Scholar]
- 15.Ichikawa H, Peppas NA. Novel complexation hydrogels for oral peptide delivery: in vitro evaluation of their cytocompatibility and insulin-transport enhancing effects using Caco-2 cell monolayers. J Biomed Mater Res. 2003;67A:609–617. doi: 10.1002/jbm.a.10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamei N, Morishita M, Chiba H, Kavimandan NJ, Peppas NA, Takayama K. Complexation hydrogels for intestinal delivery of interferon β and calcitonin. J Control Release. 2009;134:98–102. doi: 10.1016/j.jconrel.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kavimandan NJ, Losi E, Peppas NA. Novel delivery system based on complexation hydrogels as delivery vehicles for insulin-transferrin conjugates. Biomater. 2006;27:3846–3854. doi: 10.1016/j.biomaterials.2006.02.026. [DOI] [PubMed] [Google Scholar]
- 18.Kavimandan NJ, Peppas NA. Confocal Microscopic Analysis of Transport Mechanisms of Insulin Across the Cell Monolayer. Int J Pharm. 2008;354:143–148. doi: 10.1016/j.ijpharm.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 20.Lennernas H, Palm K, Fagerholm U, Artursson P. Comparison between active and passive drug transport in human intestinal epithelial (Caco-2) cells in vitro and human jejunum in vivo. Int J Pharm. 1996;127:103–107. [Google Scholar]
- 21.López JE, Peppas NA. Effect of Poly (Ethylene Glycol) Molecular Weight and Microparticle Size on Oral Insulin Delivery from P(MAA-g-EG) Microparticles. Drug Dev Ind Pharm. 2004;30:497–504. doi: 10.1081/ddc-120037480. [DOI] [PubMed] [Google Scholar]
- 22.Lowman AM, Morishita M, Kajita M, Nagai T, Peppas NA. Oral Delivery of Insulin Using pH-Responsive Complexation Gels. J Pharm Sci. 1999;88:933–937. doi: 10.1021/js980337n. [DOI] [PubMed] [Google Scholar]
- 23.Madsen F, Peppas NA. Complexation graft copolymer networks: swelling properties, calcium binding and proteolytic enzyme inhibition. Biomater. 1999;20:1701–1708. doi: 10.1016/s0142-9612(99)00071-x. [DOI] [PubMed] [Google Scholar]
- 24.Morishita M, Peppas NA. Is the oral route possible for peptide and protein drug delivery? Drug Discov Today. 2006;11:905–910. doi: 10.1016/j.drudis.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 25.Mullard A. 2012 FDA drug approvals. Nat Rev Drug Discov. 2013;12:87–90. doi: 10.1038/nrd3946. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura K, Murray RJ, Joseph JI, Peppas NA, Morishita M, Lowman AM. Oral insulin delivery using P(MAA-g-EG) hydrogels: effects of network morphology on insulin delivery characteristics. J Control Release. 2004;95:589–599. doi: 10.1016/j.jconrel.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 27.Peyrot M, Rubin RR, Kruger DF, Travis LB. Correlates of insulin injection omission. Diabetes Care. 2010;33:240–245. doi: 10.2337/dc09-1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinto M. Enterocyte-like differentiation and polarization of the human colon carcinoma cell line Caco-2 in culture. Biol Cell. 1983;47:323–330. [Google Scholar]
- 29.Renukuntla J, Vadlapudi AD, Patel A, Boddu SHS, Mitra AK. Approaches for enhancing oral bioavailability of peptides and proteins. Int J Pharm. 2013;447:75–93. doi: 10.1016/j.ijpharm.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salama NN, Eddington ND, Fasano A. Tight junction modulation and its relationship to drug delivery. Adv Drug Deliv Rev. 2006;58:15–28. doi: 10.1016/j.addr.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Sambuy Y, De Angelis I, Ranaldi G, Scarino ML, Stammati A, Zucco F. The Caco-2 cell line as a model of the intestinal barrier: influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol Toxicol. 2005;21:1–26. doi: 10.1007/s10565-005-0085-6. [DOI] [PubMed] [Google Scholar]
- 32.Torres-Lugo M, Peppas NA. Molecular Design and in Vitro Studies of Novel pH-Sensitive Hydrogels for the Oral Delivery of Calcitonin. Macromol. 1999;32:6646–6651. [Google Scholar]
- 33.U.S. Food and Drug Administration. FY 2011 Innovative Drug Approvals. 2012:1–28. http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/UCM278358.pdf.
- 34.Yee S. In Vitro Permeability Across Caco-2 Cells (Colonic) Can Predict In Vivo (Small Intestinal) Absorption in Man—Fact or Myth. Pharm Res. 1997;14:763–766. doi: 10.1023/a:1012102522787. [DOI] [PubMed] [Google Scholar]
- 35.Youn YS, Jung JY, Oh SH, Yoo SD, Lee KC. Improved intestinal delivery of salmon calcitonin by Lys18-amine specific PEGylation: Stability, permeability, pharmacokinetic behavior and in vivo hypocalcemic efficacy. J Control Release. 2006;114:334–342. doi: 10.1016/j.jconrel.2006.06.007. [DOI] [PubMed] [Google Scholar]