

Summary
The in vivo functions of mTORC2, and the signaling mechanisms that control brown adipose tissue (BAT) fuel utilization and activity, are not well understood. Here, by conditionally deleting Rictorin the Myf5-lineage, we provide in vivo evidence that mTORC2 is dispensable for skeletal muscle development and regeneration but essential for BAT growth. Furthermore, deleting RictorinMyf5 precursors shifts BAT metabolism to a more oxidative and less lipogenic state and protects mice from obesity and metabolic disease at thermoneutrality. We additionally find that Rictor is required for brown adipocyte differentiation in vitro, that the mechanismspecifically requires AKT1 hydrophobic motif phosphorylation but is independent of pan-AKT signaling, and is rescued with BMP7. Our findings provide new insights into the signaling circuitry that regulates brown adipocytes and could have important implications for developing therapies aimed at increasing energy expenditure as a means to combat human obesity.
Introduction
Adipose tissue is essential for many biological processes and its dysfunction, for example in obesity, is associated with a growing spectrum of human diseases. Thus, understanding the developmental and metabolic regulation of adipose tissue has broad clinical implications. There are two main classifications of adipose tissue: white adipose tissue (WAT) and brown adipose tissue (BAT). WAT is the major energy storage site in the body and has critical endocrine functions (Gesta et al., 2007) while BAT dissipates energy as heat in a process called nonshivering thermogenesis (Cannon and Nedergaard, 2004). BAT is particularly important in small rodents and newborn humans to defend against cold exposure and its functional relevance in adult humans was only recently appreciated (Harms and Seale, 2013; Nedergaard and Cannon, 2010; Tseng et al., 2010). Brown adipocytes are thermogenic because they express Uncoupling Protein 1 (UCP1), which embeds in the inner mitochondrial membrane and produces heat by uncoupling oxidativemetabolism from ATP production. The energy expending properties of brown adipocytes coupled with the observation that human BAT amount inversely correlates with body fat mass is garnering interest in developing strategies to increase brown adipocyte number and/or activity to treat obesity (Harms and Seale, 2013; Nedergaard and Cannon, 2010; Tseng et al., 2010). However, the mechanisms—and in particular the signaling circuitry—by which BAT regulates its energy supply are poorly understood (Townsend and Tseng, 2014). With the obesity pandemic seemingly out of control, and with a desperate need for novel therapeutics, the importance of elucidating mechanisms controlling adipocyte growth and function cannot be overstated.
Studying the in vivo mechanisms of adipose tissue growth has been challenging because adipocyte origins are poorly understood and consequently few tools are available for genetically targeting adipocyte precursors in vivo (e.g. by Cre-Lox). Lineage tracing studies indicate early mesenchymal precursor cells expressing Myf5give rise to myocytes, brown adipocytes, and a subset of white adipocytes (Sanchez-Gurmaches and Guertin, 2014; Sanchez-Gurmaches et al., 2012; Seale et al., 2008) and several recent studies have used the Myf5-Cre knock-in allele (Tallquist et al., 2000) to study BAT development (Harms et al., 2014; Martinez-Lopez et al., 2013; Ohno et al., 2013; Sanchez-Gurmaches et al., 2012; Schulz et al., 2013). Thus, the multi-fate potential of Myf5precursors provides an opportunity to use genetics to distinguish between signaling mechanisms that are required in vivo for the growth of myocytes versus adipocytes.
The mechanistic target of rapamycin (mTOR) kinase is a master-regulator of growth that functions in two distinct complexes called mTORC1 (defined by the Raptor subunit) and mTORC2 (defined by the Rictor subunit) (Laplante and Sabatini, 2012). While much is known about the inputs, outputs, and regulatory features of mTORC1, mTORC2 regulation and function remains more enigmatic. The best-described biochemical function of mTORC2 is to phosphorylate the hydrophobic motif (HM) of AKT (S473 in AKT1) and the related SGK (S422 in SGK1) kinases (Garcia-Martinez and Alessi, 2008; Sarbassov et al., 2005). AKT has many effectors including GSK3β, FoxO1/3, and mTORC1 (through TSC2 and PRAS40) and most models indicate mTORC2 is an essential upstream regulator of pan-AKT activity (Laplante and Sabatini, 2012). However, the extent to which this is the case in vivo remains unclear because Rictor deficient mouse embryo fibroblasts, which lack mTORC2,have seemingly normal GSK3β phosphorylation, mTORC1 activity, and only partially decreased FoxO1/3 phosphorylation (Guertin et al., 2006; Jacinto et al., 2006; Shiota et al., 2006).
Mice lacking Rictor die around E10.5 (Guertin et al., 2006; Jacinto et al., 2006; Shiota et al., 2006); therefore, mTORC2 function in vivo is mostly being investigated using floxed Rictor alleles. In adipose tissue, two studies using aP2-cre to delete Rictor reported no effect on individual adipocyte size or overall adipose tissue mass (Cybulski et al., 2009; Kumar et al., 2010). One of the studies finds aP2-cre;Rictorfl/fl mice eventually develop mild glucose intolerance and ectopic lipid deposition although a mechanism was not elucidated (Kumar et al., 2010). Notably however, the utility of aP2-cre to target adipocytes has recently been questioned (Lee et al., 2013; Mullican et al., 2013; Wang et al., 2013); therefore, the exact function of mTORC2 in adipose tissue remains unclear. Deleting Rictor in skeletal muscle with Hsa-cre or Mck-cre also has no effect on muscle fiber size or overall muscle mass and only minor effects on insulin-mediated glucose metabolism (Bentzinger et al., 2008; Kumar et al., 2008). These relatively mild phenotypes are somewhat surprising considering the importance of AKT signaling in metabolism; however, in both cases (adipose tissue and muscle) the Cre drivers used target mature cells and thus the in vivo role of mTORC2 in adipose tissue and muscle precursors is unknown.
Here, we take advantage of the fact that Myf5-Cre expresses in precursors of muscle and brown adipocytes to investigate the role of Rictor (i.e. mTORC2) and for comparison Raptor (i.e. mTORC1) in muscle and BAT growth. We report that Raptor is essential in the Myf5 lineage for myogenesis, establishing BAT precursors, and viability. In contrast, Rictor is dispensable for myogenesis and viability, but essential for normal BAT growth. Moreover, Rictor-deficient BAT is more metabolically active, having elevated mitochondrial activity and decreased lipogenesis. Importantly, deleting Rictor in the Myf5-lineage also augments diet-induced thermogenesis, which protects mice from an obesogenic diet at thermoneutrality. We additionally find that Myf5-lineage white adipocytes require Rictor for normal growth in vivo suggesting a broader role for mTORC2 in adipose tissue development. Finally, we show that Rictor is also required in vitro for brown adipocyte differentiation but not for pan-AKT activity, and that this differentiation defect is rescued with BMP7. Collectively, our results provide new insight into the regulation of brown adipocytes and implicate Rictor/mTORC2 as a critical signaling node that balances oxidative and lipogenic metabolic states.
Results
Rictoris dispensable in the Myf5 lineage during embryogenesis
We investigated the role of mTORC1 versus mTORC2 in vivo in fat versus muscle development by generating Myf5-Cre; Raptorfl/fl (RaptorMyf5cKO) and Myf5-Cre; Rictorfl/fl (RictorMyf5cKO) conditional knockout mice. The RictorMyf5cKO mice are born at the expected Mendelian ratio and show no obvious motor or behavioral defects [not shown]. In contrast, RaptorMyf5cKO mice die perinatally. E16.5 RaptorMyf5cKO embryos are smaller due to a muscle development defect that is not apparent in control or RictorMyf5cKO embryos [Figure S1A-D]. Transverse sections through the head and neck of RaptorMyf5cKO embryos reveal an underdeveloped tongue and the absence of the masseter, sternohyoid, hyglossus, supraspinatus, prevertebral, and trapezius muscles, the later deficiency resulting in hind neck body wall fragility during specimen preparation [Figure S1A-D]. Thus, Raptor is essential in the Myf5lineage for viability and muscle development while Rictor is dispensable for both.
To confirm that Rictor is dispensable for myogenesis, we purified satellite cells (which express Myf5) from RictorMyf5cKO skeletal muscles, confirmed they are deleted for Rictor [Figure S1E], and show they differentiate ex vivo into myosin heavy chain-positive multinucleate myofibers [Figure S1F-G]. Moreover, deleting Raptor in satellite cells in vivo with Pax7-CreER blocks skeletal muscle repair, while deleting Rictor by the same approach does not prevent muscle regeneration following acute injury [Figure S1H-I]. Thus, Rictor is also dispensable for satellite cell differentiation ex vivo and for adult myogenesis induced by injury.
White adipose tissues develop postpartum in mice, but early brown adipocyte precursor cells (bAPCs) are detectable in E16.5 embryos by Hematoxylin and Eosin (H&E) stain. Qualitatively similar pools of cervical, interscapular, and subscapular bAPCs are detectable in control and RictorMyf5cKO E16.5 embryos [Figure S1A & S1D]. In contrast, interscapular and subscapular bAPCs are absent in E16.5 RaptorMyf5cKO embryos [Figure S1A & S1D]. Notably, a diminished pool of cervical bAPCs is detectable in the RaptorMyf5cKO embryos consistent with our lineage tracing data showing that only about half of the cervical brown adipocytes arise from Myf5-Cre expressing precursors [Figure 1SD] (Sanchez-Gurmaches and Guertin, 2014). Thus, Raptor but not Rictor is also essential in the Myf5-lineage for establishing bAPCs during embryogenesis.
Brown and white adipose tissue growth requires Rictor
Although RictorMyf5cKO mice show no obvious embryonic phenotypes, they tend to weigh less (not significantly) than controls at postnatal day 1 (P1) [Figure S1J], which reaches significance from 6-15 weeks of life [Figure 1A]. Individual tissue analysis indicates that the weight difference results from decreased adipose tissue mass. For example, the interscapular BAT (iBAT) in P1 RictorMyf5cKO neonates weighs about 30% less than normal [Figure S1K] and during the first weeks of life the mutant BAT grows but to a much smaller size, resulting in mutant iBAT and subscapular BAT (sBAT) depots at 6-weeks weighing about 50% less than controls and being darker [Figure 1B]. Adipocytes in the retroperitoneal and anterior subcutaneous WAT depots (rWAT, asWAT) are also derived largely from Myf5-Creexpressing precursors (Sanchez-Gurmaches et al., 2012) and both of these depots also decrease in mass by approximately 50% in the RictorMyf5cKO mice [Figure 1C]. In contrast, the posterior subcutaneous and peri-gonadal WAT depots (psWAT, pgWAT), which are composed ofMyf5-negative lineage adipocytes, do not differ in weight [Figure 1C]. Skeletal muscles (e.g. triceps, quadriceps, and gastrocnemius) and all other lean tissues examined except the kidneys (which are slightly larger) are normal size in the knockout [Figure 1D]. Western analysis for Rictor protein confirms Rictor deletion and reduced AKT-S473 phosphorylation in iBAT and muscle and to a lesser extent in rWAT and asWAT, but not in psWAT, pgWAT or liver [Figure 1E].
Figure 1. Post-natal brown and white adipose tissue growth requires Rictor.

(A) Growth curves (n=13; bars represent mean ± SEM; t-test; *p<0.05, **p<0.01, ***p<0.001).
(B) (Left) BAT massat 6-wks (n=19-21; mean ± SEM; t-test; ***p<0.001) and (Right) representative image.
(C) (Left) Mass of WATs at 6-wks (n=14-16; mean ± SEM; t-test; **p<0.01, ***p<0.001) and (Right) representative image of control and mutant rWAT (6-wks).
(D) Lean tissue massat 6-wks (n=15-19; mean ± SEM; t-test; ***p<0.001).
(E) Westerns of tissue lysates (6-wks).
(F) Average tissue mass (mg) at 6-wks and 6-mos (n= 14-21 for 6-wks; n=7 for 6-mos; mean ± SEM; t-test; ***p<0.001).
See also Figure S1.
From 6 weeks to6 months, the mutant iBAT and sBAT shows no additional growth increase while asWAT and rWAT grow to about half (asWAT) or one third (rWAT) the size of their anatomically matched control tissues [Figure 1F]. In contrast, RictorMyf5cKO psWAT, pgWAT, muscles, and liver grow to their normal size in the same time frame [Figure 1F]. Thus, RictorMyf5cKO mice can grow small BAT tissues in the first weeks of life; however, as RictorMyf5cKO mice age the iBAT and sBAT maintain their weight while asWAT and rWAT grow at a reduced rate. Collectively, these results indicate Rictor is essential in the Myf5lineage for adipose tissue growth but not for skeletal muscle growth.
Brown adipocytes lacking Rictor are smaller
To better define the BAT growth defect we histologically examined iBAT in control and RictorMyf5cKO mice. At E18.5there is no qualitative difference between control and RictorMyf5cKO bAPCs pools [Figure 2A]. In P1 neonates however, lipids begin accumulating in control BAT but not in the RictorMyf5cKO BAT [Figure 2A]. From P1 to 6 monthslipid droplets grow in size in control BAT but remain small in the RictorMyf5cKO BAT [Figure 2A] resulting in smaller cells measured by the increase in nuclei per mm2 [Figure S2A]. Total genomic DNA content is also lower in the RictorMyf5cKO BAT indicating additional hypoplasia [Figure S2B]. In contrast, RictorMyf5cKO skeletal muscle fibers appear histologically identical to control fibers [Figure S2C].
Figure 2. Brown and white adipocytes lacking Rictor are smaller and multilocular.

(A) H&E stains of interscapular BAT (6 wks).
(B) H&E stains of retroperitoneal and anterior subcutaneous WAT.
(C) Representative images of mTFP and mGFP labeled adipocytes. Enlarged images indicated by white box.
See also Figure S2.
Myf5-lineage white adipocytes lacking Rictor are also small and multilocular
Compared to controls, many adipocytes in the RictorMyf5cKO rWAT and as WAT are also smaller and multilocular [Figure 2B] but the pattern is heterogeneous in that several large unilocular white adipocytes are also detectable. The psWAT and pgWAT adipocytes appear unchanged in the knockout [Figure S2C]. The adipocyte precursor poolsin rWAT and asWAT are a mix of Myf5-Crelineage positive and negative precursors (Sanchez-Gurmaches et al., 2012). Therefore, we reasoned that the size heterogeneity in RictorMyf5cKO rWAT and asWAT could reflect a mosaic of Myf5-lineage negative (i.e. undeleted) and Myf5-lineage positive (i.e. Rictor knockout) cells. To test this we incorporated the Rosa26-mTmG reporter (Muzumdar et al., 2007) into control and RictorMyf5cKO mice toirreversibly label Cre-expressing cells and their lineages with membrane-targeted eGFP (mGFP); all other (Creneg) cells and their descendants are labeled with membrane targeted tdTomato fluorescent protein (mTFP). The result is unequivocal; only the small adipocytes are mGFP+ in RictorMyf5cKO rWAT and asWAT while all the large unilocular adipocytes are mTFP+ [Figure 2C]. As expected, in both the control and RictorMyf5cKO mice the iBAT adipocytes are mGFP+ and the psWAT and pgWAT adipocytes are mTFP+[Figure S2D]. We also detect a slight increase in UCP1 staining in the RictorMyf5cKO adipocytes suggesting the cells might have brown-adipocyte like characteristics [Figure S2E] (not shown). These data confirm that the heterogeneous small cell phenotype results from cell-autonomous Rictor deletion in the Myf5-lineage white adipocytes.
Lipogenesis is decreased in Rictor-deficient BAT
We hypothesized that the paucity of lipid and marked color difference between control and RictorMyf5cKO BAT indicates a shift from a lipogenic to oxidative state. To test this we first examined AKT signaling in BAT, which positively regulates lipogenesis. In vivo AKT-T308 phosphorylation is intact in both fasted/re-fed and insulin-stimulatedRictorMyf5cKO BAT despite ablation of pAKT S473 and pAKT T450 (which is also mTORC2 dependent) [Figure 3A & S3A], consistent with the ability of T-loop (T308) and hydrophobic motif (S473) phosphorylation to be regulated independently (Pearce et al., 2010). Surprisingly, phosphorylation of the AKT substrates FoxO1/3, GSK3β, TSC2, PRAS40, and AS160 is normal in RictorMyf5cKO BAT [Figure 3A] indicating Rictor is not essential in BAT for pan-AKT signaling. Rictor loss in BAT also does not affect phosphorylation of the SGK substrate NDRG1 [Figure 3A] indicating mTORC2 is not essential for SGK signaling to NDRG1 in BAT or that a compensatory pathway exists.
Figure 3. Rictor-deficient brown adipocytes have a lipid metabolism defect.

(A) Western blots total and phospho-proteins using 6-wk iBAT lysates. Mice were fasted overnight and re-fed for 45mins prior to preparing lysates.
(B-D) qRT-PCR of the indicated genes in P1 (n=6) and 6wk (n=8) iBAT (mean ± SEM; t-test; *p<0.05, **p<0.01)
See also Figure S3.
Next we examined whether deleting Rictor in the Myf5-lineage affects BAT differentiation markers. In P1 neonates, Prdm16, C/ebpα, and C/ebpβ expression do not differ between controls and knockouts while Pparγ and Ucp1 levels slightly decrease [Figure 3B] indicating a possible delay in BAT maturation in the knockouts. However, by 6 weeks Pparγ, Prdm16, and C/ebpα express at control levels while C/ebpβ, Ucp1, and Dio2 express at significantly higher than control levels [Figure 3B]. The mature adipocyte markers Cidea and aP2 are unchanged between control and knockout both at P1 and 6 weeks [Figure 3B]. Consistent with the gene expression data, PPARγ, UCP1, and insulin receptor beta (IRβ) proteins also express at near control levels in RictorMyf5cKO BAT [Figure 3A]. Thus, terminal differentiation per se (i.e. PPARγ, UCP1, and IRβ induction) occurs in vivo in RictorMyf5cKO brown adipocytes.
Next we examined lipogenesis genes. In P1 RictorMyf5cKO BAT, acetyl-coA carboxylase (Acc), fatty acid synthase (Fasn), and fatty acid elongase 6 (Elovl6) decrease expression by 40%, 40%, and 25% respectively [Figure 3C]. By 6 weeks, expression of ATP citrate lyase (Acly) in addition to Acc, Fasn, and Elovl6 is reduced by 90%, 75%, 80%, and 40% respectively [Figure 3C], which we confirmed by Western blot for ACLY and ACC [Figure 3A]. In addition, stearoyl-CoA desaturase (Scd1) decreases expression by 45% in 6 week RictorMyf5cKO BAT [Figure 3C]. The SREBP1c and ChREBP transcription factors regulate lipogenesis gene expression (Czech et al., 2013; Filhoulaud et al., 2013). In both P1 and 6-week RictorMyf5cKO BAT, the mRNA expression of SREBP1c (Srebf1c), which is induced by insulin, and ChREBP (α and β isoforms), which is induced by glucose, is similar [Figure 3D]. However, there is a marked decrease in the amount nuclear SREBP1c (nSREBP1c), the transcriptionally active SREBP1c cleavage product, in Rictor-deficient BAT [Figure 3A] consistent with the decrease in lipogenic gene expression. The levels of insig1, another nSREBP1c target gene and negative regulator of SREBP1c processing, also decreases [Figure 3C]. The mRNA expression of SREBP2 (which regulates cholesterol biosynthesis) slightly decreases in RictorMyf5cKO BAT at 6 weeks, but the SREBP2 target genes HMG-CoA synthase (Hmg-cs) and HMG-CoA reductase (Hmg-cr) express at similar levels in control and knockout BAT [Figure 3D] and nuclear SREBP2 (nSREBP2) accumulates possibly to higher levels in the knockout BAT [Figure 3A]. We find no difference in AMPK or hormone sensitive lipase (HSL) phosphorylation between control and RictorMyf5cKO BAT [Figure 3A & S3A]. Together, these results indicate that despite having seemingly normal AKT signaling, de novo lipogenesis is reduced in RictorMyf5cKO BAT.
Mitochondrial activity is elevated in Rictor-deficient BAT
To further examine the metabolic state of RictorMyf5cKO BAT we examined mitochondrial activity. In P1 neonate RictorMyf5cKO BAT, Pgc1a expresses normally, while expression of mitochondrial transcription factor A (Tfam), which regulates mtDNA replication, and carnitine palmitoyltransferase 1B (Cpt1b), which encodes the rate-limiting enzyme in beta-oxidation, slightly decreases [Figure 4A]. In contrast, Pgc1a, Tfam, and Cpt1bin addition to Ucp1 express at higher levels in the BAT of 6-week old RictorMyf5cKO mice[Figure 4A & 3B] suggesting BAT mitochondrial activity progressively increases or is maintained at a higher level in RictorMyf5cKO mice as they age.
Figure 4. Mitochondrial activity is elevated in Rictor-deficient BAT.
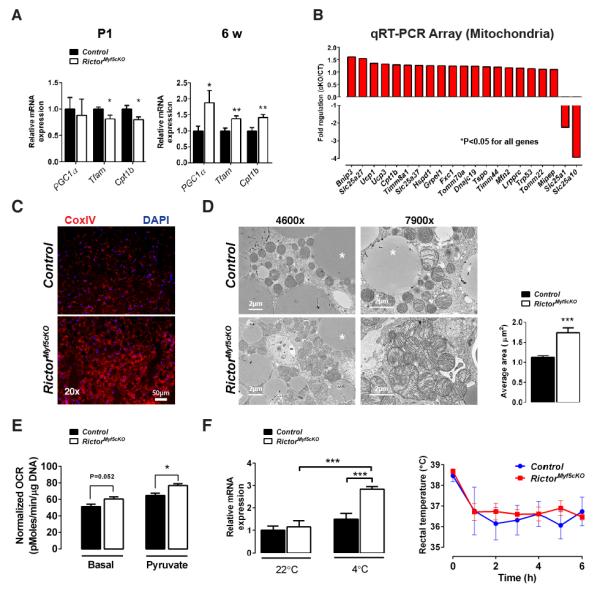
(A) qRT-PCR of mitochondrial genes in P1 (n=6) and 6wk iBAT (n=8) (mean ± SEM; t-test; *p<0.05, **p<0.01)
(B) Differentially expressed genes using mitochondrial qRT-PCR arrays(n=4; t-test; p<0.05)
(C) Representative immunofluorescence images of Cox IV staining in 6wk iBAT (n=3).
(D) (Left) Representative TEM images of 6wk iBAT and (Right) mitochondria size (n=3;mean ± SEM; t-test; ***p<0.001)
(E) Oxygen consumption of iBAT using a Seahorse Flux Analyzer (12 wks, n=5; normalized to DNA content;mean ± SEM; t-test; *p<0.05)
(F) (Left) qRT-PCR of Ucp1 mRNA in iBAT with or without cold exposure (n=3 for 22°C; n=4 for 4°C; mean ± SEM; two-way ANOVA; ***P<0.001) and (Right) rectal temperature in acute cold challenge (n=4; mean ± SEM; t-test; no significant difference).
See also Figure S4.
To explore this in more detail, we used qRT-PCR arrays to broadly measure mitochondrial gene expression in the 6-week old BAT. Using arrays for functional genes involved in mitochondrial molecular transport and biogenesis, we detect increases in several genes indicative of increased mitochondrial activity [Figure 4B]. Furthermore, the mitochondrial citrate and malate transporters Slc25a1 and Slc25a10 respectively—both of which function in fatty acid biosynthesis, the former also being an SREBP1c target gene(Infantino et al., 2007; Mizuarai et al., 2005)—significantly decrease expression in the mutant BAT. Using mitochondrial energy metabolism gene arrays, we found 58 additional genes involved in respiration (OXPHOS) are elevated in RictorMyf5cKO BAT [Figure S4A] suggesting an increase in mitochondrial mass, which we confirmed by CoxIV immunofluorescence [Figure 4C]. Transmission electron microscopy (TEM) reveals individual mitochondria in the mutant BAT are larger and have more disorganized cristae [Figure 4D]. To directly confirm elevated mitochondrial activity, we measured BAT oxygen consumption rate (OCR) in a Seahorse Flux Analyzer and determined that basal and pyruvate-stimulated OCR are elevated by around 18%in RictorMyf5cKO BAT [Figure 4E]. We did not detect a significant increase in overall oxygen consumption when RictorMyf5cKO mice were placed in metabolic cages at 22°C except when normalized for body weight [Figure S4B]. Notably however, mice are under thermal stress at this temperature, which can mask effects on BAT activity (Feldmann et al., 2009).
Interestingly, we also detect an approximate 2-fold increase in basal glucose uptake in RictorMyf5cKO BAT measured by 18 FDG-PET CT scanning [Figure S4C] and an increase in lipoprotein lipase (Lpl) expression [Figure 3C] suggesting that RictorMyf5cKO BAT may consume more nutrients than age matched control BAT. Small metabolite profiling reveals that RictorMyf5cKO BAT also has elevated levels of inosine monophosphate (IMP)[Figure S4D], a deamination product of adenosine monophosphate (AMP), the accumulation of which suggests increased uncoupling (Balcke et al., 2011). In an acute cold challenge Rictor-deficient BAT also induces Ucp1 expression significantly more than control BAT and the mutants have no difficulty maintaining body temperature although body temperature regulation in an acute cold challenge is largely a function of muscle [Figure 4F]. Finally, we see no compensatory “browning” in the psWAT as would be expected if RictorMyf5cKO BAT were dysfunctional [Figure S2C-D] (Schulz et al., 2013). These results are consistent with Rictor loss in BAT shifting metabolism to a more oxidative and less lipogenic state.
Brown preadipocytesrequire Rictor to differentiate in vitro
To examine if brown adipocyte differentiation also requires Rictor in vitro, we generated brown adipocyte precursor cells (bAPCs)harboring an inducible knockout system (i.e. RictoriKO) in which Rictor deletion is triggered by 4-hydroxytamoxifen (4-OHT)[Figure S5A]. Compared to isogenic controls, inducibly deleting Rictor rapidly and robustly depletes Rictor protein and AKT-S473 phosphorylation, and consistent with our in vivo data, leaves AKT-T308 phosphorylation intact [Figure S5B]. Also consistent with the in vivo results, both basal and insulin-stimulated phosphorylation of FoxO1/3, GSK3β, TSC2, and PRAS40are normal in RictoriKO bAPCs [Figure 5A]. S6K1 phosphorylation is also unaffected [Figure 5A]. Contrary to the in vivo results, acute Rictor loss in vitro decreases NDRG1 phosphorylation [Figure 5A]. This indicates Rictor is required in cultured bAPCs for SGK activity to NDRG1 but not for pan-AKT or mTORC1 activity.
Figure 5. Rictor is required for brown adipocyte differentiation in vitro.

(A) Western immunoblots using control and RictoriKO brown preadipocyte lysates. Cells were serum deprived 3 hours then stimulated with 0, 5, 25, 120, or 600nM insulin for 15 minutes prior to lysis.
(B) Oil Red O staining after differentiation.
(C) Western immunoblots using lysates from the indicated days of differentiation.
(D) qRT-PCR for differentiation-related genes (n=3; mean ± SEM; t-test; *p<0.05, ***p<0.001).
(E) Same as (C).
(F) Western immunoblots of cell lysates collected at day 10 of differentiation. M=mock; V=empty vector; γ2=recombinant PPARγ2. The γ1 and γ2 isoforms are indicated.
(G) Oil Red O staining of cells in (F).
See also Figure S5.
To our surprise, RictoriKO bAPCs are completely incapable of synthesizing lipid droplets when induced to differentiate [Figure 5B]. This is surprising because RictoriKO cells maintain normal levels of pAKT-T308, pGSK3β-S9, and pS6K1-T389 (i.e. PDK1, AKT, and mTORC1 activity respectively) throughout the differentiation protocol[Figure 5C]. The differentiation block occurs early as RictoriKO bAPCs fail to induce C/ebpα, Pparγ, Prdm16, Pgc1α, Srebf1c, Ucp1, and Glut4 [Figure 5D & S5C]. The expression of C/ebpδ and C/ebpβ on the other hand induces normally and slightly higher respectively in the RictoriKO bAPCs at differentiation day 6 [Figure 5D]. Consistent with the gene expression data, PPARγ, IRβ, UCP1, nSREBP1c, ACC, and ACLY levels fail to increase during differentiation in RictoriKO bAPCs [Figure 5Eh]. Notably, 4-OHT or CreER activation alone (i.e. in the absence of Rictor floxed alleles) has no effect on differentiation [not shown]. Moreover, bAPCs prepared from P1 RictorMyf5cKO neonates also fail to differentiate indicating that the ex vivo differentiation block is not unique to using the inducible knockout-system [Figure S5D]. Importantly, expressing recombinant PPARγ in RictoriKO bAPCs rescues IRβ, UCP1 and nSREBP1c expression [Figure 5F] and lipid droplet production [Figure 5G] indicating Rictor promotes differentiation at least in part by facilitating PPARγ induction.
Insulin receptor substrate 1 (Irs1) and Irs3 knockout bAPCs also fail to induce PPARγex vivo (Fasshauer et al., 2001). It was later shown that Irs1/3 KO bAPCs are unable to differentiate because they express high levels of Pref-1, Wnt10a, and Necdin, which encode adipogenesis inhibitors (Tseng et al., 2005). In contrast, Rictor-deficient bAPCs express normal levels of Pref-1, Wnt10a, and Necdinin culture, and during differentiation Necdinand Pref-1 increase but only late in the differentiation protocol [Figure S5C]. Thus, the mechanism by which deleting Rictor inhibits brown adipocyte differentiation differs from that of deleting Irs1/3.
AKT1 functions downstream of Rictor in brown adipocyte differentiation
To further explore the mechanism by which Rictor regulates differentiation, we next asked whether an AKT or SGK pathway is required downstream of Rictor. To this end, we generated RictoriKO bAPCs that express HA-SGK1, HA-AKT1, HA-AKT2, or their phosphomimetic counterparts HA-SGK-S422D, HA-AKT1-S473D, HA-AKT2-S474D in which a phosphomimetic residue was placed at the mTORC2 hydrophobic motif site, confirmed they were functional [Figure S6A], and asked whether any of these constructs rescue differentiation. Only HA-AKT1-S473D efficiently rescueslipid biosynthesis [Figure 6A]. HA-AKT1-S473D-expressing RictoriKO bAPCs also induce PPARγ and restore IRβ, UCP1, nSREBP1c, ACLY, and ACC expression [Figure 6B]. Thus, Rictor promotes differentiation as part of mTORC2 through an AKT pathway.
Figure 6. Recombinant AKT1-S473D or BMP7 supplementation rescue differentiation in the absence of Rictor.

(A) Oil Red O staining of differentiated control (vehicle) and RictoriKO cells (4-OHT) cells stably expressing the indicated constructs.
(B) Western immunoblots corresponding to (A).
(C) Oil Red O staining of differentiated Akt1 and Akt2 conditional knockout and control bAPCs.
(D) Western immunoblots corresponding to (C).
(E) Oil Red O staining of differentiated control and RictoriKO cells in the presence or absence of BMP7 (3.2nM added day 1 during differentiation).
(F) Western immunoblots of corresponding to (E).
(G) Model summarizing the role of mTORC2 in vitro in brown adipocyte differentiation.
See also Figure S6.
Our rescue experiments point toAKT1 as the isoform driving bAPC differentiation in vitro. Consistently, AKT1 is highly expressed in undifferentiated precursors and decreases expression during differentiation while AKT2 expression increases during differentiation [Figure S6B]. To further examine the role of AKT1 and AKT2 in bAPC differentiation we generated bAPC lines that specifically lack either Akt1 or Akt2 and determined their in vitro differentiation capacity. Consistent with AKT1 but not AKT2 being required for differentiation, Akt1-deficient bAPCs cannot efficiently synthesize lipid droplets [Figure 6C] or up-regulate PPARγ, IRβ,or UCP1 when induced to differentiate[Figure 6D]. In contrast, Akt2-deficient bAPCs induce PPARγ, IRβ, and UCP1 normally [Figure 6C-D] indicating that AKT1 is indeed the isoform required downstream of Rictor/mTORC2 for brown adipocyte differentiation. Interestingly, we noticed in our in vitro differentiation assays that while the Akt2-deficient cells differentiate, they fail to induce nSREBP1C, that ACLY and ACC express at low levels and that lipid droplet content is reduced [Figure 6C-D]. This suggests that while AKT2 is not essential for differentiation it is important downstream of Rictor/mTORC2 for lipid metabolism. Indeed, when we immunoprecipitate AKT1 or AKT2 from undifferentiated bAPCs most of the AKT phosphorylation is on AKT1 while in vivo the bulk of AKT phosphorylation shifts to AKT2 [Figure S6C-D]. Thus, while the inability of RictoriKO bAPCs to differentiate in culture reflects an AKT1 deficiency, the in vivo metabolic phenotype appears to reflect an AKT2 deficiency.
BMP7 rescues brown adipocyte differentiation in the absence of Rictor
In vitro RictoriKO bAPCs cannot differentiate (i.e. induce PPARγ and UCP1), but in vivo PPARγ and UCP1 positive Rictor-deficient BAT develops. One possible explanation for this paradoxis that in vivo there are developmental signals present that are missing from the artificial in vitro differentiation assay. The signals that drive brown adipocyte differentiation in vivo are poorly understood. One proposed inducer of brown adipocyte differentiation is the transforming growth factor-β superfamily member BMP7(Schulz and Tseng, 2013; Tseng et al., 2008). When given to control or RictoriKO bAPCs, BMP7 does not induce AKT phosphorylation [Figure S6E]. However, when supplemented into the differentiation cocktail BMP7 restores to RictoriKO bAPCs their ability to synthesizelipid droplets [Figure 6E] and express PPARγ, IRβ, UCP1 and to a lesser extent nSREBP1c, ACLY, and ACC [Figure 6F] This is consistent with the in vitro differentiation assay lacking signaling molecules present in vivo and suggests BMP7 and mTORC2-AKT1signaling converge during brown adipocyte differentiation. A model depicting the role mTORC2-AKT signaling in vitro in brown adipocyte differentiation is shown in Figure 6G.
RictorMyf5cKO mice are less susceptible to obesity and metabolic disease at thermoneutrality
The higher metabolic activity of Rictor-deficient BAT led us to wonder whether RictorMyf5cKO mice are resistant to obesity. Chronic consumption of high fat diet (HFD) triggers a phenomenon in mice called diet-induced thermogenesis, which requires UCP1, and counteracts obesity (Cannon and Nedergaard, 2010; Feldmann et al., 2009). Because BAT activity is masked by chronic thermal stress at 22° Cwe conducted the following studies at thermoneutrality (30°C for mice), which exempts mice from thermal stress (Feldmann et al., 2009). When eating a normal chow diet, control and RictorMyf5cKO mice gain equal weight [Figure 7A] and consume the same total energy [Figure 7B] over 12-weeks. In contrast, when eating a HFD control mice gain 14.67±1.05 grams while RictorMyf5cKO mice gain 10.57±1.18 grams [Figure 7A] despite both groups consuming the same energy [Figure 7B]. Thus, controls gain 64% more weight when eating HFD versus chow compared to RictorMyf5cKO mice. This suggests RictorMyf5cKO mice living at thermoneutrality and eating a HFD are less metabolically efficient than controls, which is indeed the case [Figure 7C].
Figure 7. RictorMyf5cKO mice exempt from thermal stress and consuming a high fat diet are resistant to obesity and metabolic disease.

(A)Weight gain of control and RictorMyf5cKO mice during 12-weeks of normal chow (chow) or high fat diet (HFD) (n=8 control and n=12 for KO in chow; n=10 for both genotypes on HFD;mean ± SEM; t-test; *p<0.05). Control mice initially weighed 21.63±0.812g in the chow group and 21.24±0.621 in the HFD group; The RictorMyf5cKO mice initially weighed 19.42±0.305g in the chow group and 19.32±0.348 in the HFD group.
(B) Total energy intake (MJ) during the feeding regimen in (A). Control mice consumed 3.75±0.56g of chow and 2.81±0.12g of HFD; RictorMyf5cKO mice consumed 3.85±0.24g of chow and 2.95±0.35g of HFD.
(C) Metabolic efficiency determined as the amount of body weight increase (g) per MJ food consumed (n=8 control and n=12 KO on chow; n=10 for both genotypes on HFD; mean ± SEM; two-way ANOVA; *p<0.05, ***p<0.001).
(D) Mass (mg) of the indicated tissues collected from control and KO mice after 12 weeks on chow or HFD. (n=8 control and n=12 KO on chow; n=10 for both genotypes on HFD; mean ± SEM; two-way ANOVA; *p<0.05, ***p<0.001).
(E-F) H&E staining of iBAT and pgWAT and Oil red O staining of livers after 12-weeks of eating chow (E) or high fat diet (F).
(G) qRT-PCR of the indicated brown and white fat genes in iBAT from chow or HFD mice (n=8 control and n=12 KO on chow; n=10 for both genotypes on HFD; mean ± SEM; two-way ANOVA; *p<0.05, **p<0.01, ***p<0.001; # indicates significant difference over the control chow group).
(H) qRT-PCR of the indicated metabolic genes in iBAT from chow or HFD mice (n=8 control and n=12 KO in chow; n=10 for both genotypes in HFD;mean ± SEM; two-way ANOVA; *p<0.05, **p<0.01, ***p<0.001; # indicates significant difference over the control chow group).
(I)Western immunoblotsofiBAT lysates.
See also Figure S7.
The resistance to weight gain in the HFD-fed RictorMyf5cKO cohort is partly due to reduced growth of adipose tissue. For example, the pgWAT gains significantly less mass in the HFD-fed RictorMyf5cKO cohort than in HFD-fed controls [Figure 7D]. Liver and heart also grow larger in controlseating HFD compared tochow, while liver and heart grow to the same mass in the RictorMyf5cKO cohorts regardless of diet [Figure 7D & S7A]. Diet has no effect on other lean tissues in either the controls or RictorMyf5cKO cohorts [Figure 7D & S7A]. That pgWAT grows less in HFD-fed RictorMyf5cKO mice compared to HFD-fed controls indicates systemic protection against obesity is occurring because Myf5-Cre does not target pgWAT [Figure 1F & S2C]. The reduction in pgWAT mass is due in part to smaller adipocyte size [Figure 7E]; the livers of RictorMyf5cKO mice also resist hepatic steatosis [Figure 7E]; and the HFD-fed RictorMyf5cKO mice perform better in a glucose tolerance test [Figure S7B].
In chow-fed cohortshistology reveals that control BAT adopts a more “white adipocyte-like” appearance [Figure 7E]. In contrast, the BAT in chow-fed RictorMyf5cKO mice resists the whitening effects of living at thermoneutrality and maintains a more “brown-adipocyte-like” appearance [Figure 7E]. The resistance of RictorMyf5cKO BAT to whitening is reflected in the gene expression signature; for example, when normalized to BAT gene expression at 22°C, the shift to thermoneutrality decreases the expression of BAT-selective genes (Prdm16, Sgk2, cideb, cyp2b10) and increases the expression of WAT-selective genes (Dpt1, Retn, Trim14, Nnmt) to a greater extent in control BAT than in RictorMyf5cKO BAT, which maintains a more BAT-like identity. [Figure S7C].
In HFD-fed cohortshistology reveals a large number of multi-locular adipocytes in control BAT [Figure 7F] that are not apparent in chow-fed controls [Figure 7E] suggesting diet-induced thermogenesis. This is reflected in the gene expression data as Prdm16 increasesin control BAT in HFD-fed mice compared to chow-fed [Figure 7G], while the WAT-specific genes Retn, Trim14, and Nnmt decrease [Figure 7G]. Histology also reveals that RictorMyf5cKO BAT is evenmore“brown-like” in the HFD-fed cohort exhibiting auniform abundance of small lipid droplets [Figure 7F] and a stronger BAT gene signature(i.e. elevated Prdm16, Sgk2, Cideb, Cyp2b10; decreased Dpt1, Retn, Trim14, Nnmt) [Figure 7G]. Consistently, BAT functional genes (Ucp1, Pgc1α, Cpt1β, Dio2) induce to a greater extent in HFD-fed RictorMyf5cKO mice [Figure 7H], which also maintainlow Acly, Acc, and Fasnexpression [Figure S7D]. Importantly, UCP1 protein level is higher in the BAT of RictorMyf5cKO mice eating HFD [Figure 7I]. Notably, after 20 weeks of eating a HFD the control BAT reverts to a more “white-adipocyte-like” histology; however, BAT character is preserved in RictorMyf5cKO mice [Figure S7E]. Collectively, these results suggest that inhibiting mTORC2 in BAT increases diet-induced thermogenesis and consequently, RictorMyf5cKO mice living without thermal stress and consuming an obesogenic diet are less susceptible to developing obesity and metabolic disease.
DISCUSSION
While transcriptional regulation of BAT development has been extensively described (Kajimura et al., 2010), less is known about the signaling mechanisms that regulate BAT. The control of brown fat fuel utilization is also incompletely understood (Townsend and Tseng, 2014). Previous studies reported that conditionally deleting Rictor in white and brown adipose tissue or skeletal muscle has no affect on WAT or BAT mass or individual adipocyte or myocyte size (Bentzinger et al., 2008; Cybulski et al., 2009; Kumar et al., 2008; Kumar et al., 2010). However, these studies used Cre drivers that reportedly delete Rictor in mature cells, which led us to hypothesize that Rictor/mTORC2 may be more important for brown/white adipose tissue and/or muscle development. By conditionally deleting Rictor in Myf5 precursors we discovered that Rictor is not essential in vivo for muscle development or regeneration. In contrast, Myf5-lineage brown and white adipocytes lacking Rictor are reduced in size. Furthermore, Rictor-deficient BAT undergoes a metabolic shift to a more oxidative and less lipogenic metabolic despite having seemingly normal pan-AKT signaling. Importantly, atthermoneutrality this protects mice against an obesogenic diet. These findings implicate Rictor/mTORC2 as an essential signaling node in BAT that regulates the balance between fatty acid oxidation and storage. These findings could have important implications for understanding the signaling mechanisms that regulate fuel usage and metabolic activity in human BAT.
We also report that in vitro brown adipocyte differentiation requires Rictor/mTORC2. Mechanistically, Rictor/m TORC2 promotes Pparγ induction through AKT1 independently of pan-AKT signaling and mTORC1 activity. In vivo however, brown adipocytes differentiate in RictorMyf5cKO mice despite lacking Rictor expression. We hypothesize that this paradox indicates that the artificial in vitro culture conditions lack important signals present in vivo that overcome this deficiency. Supporting this notion, supplementing the differentiation assay with BMP7, a proposed in vivo inducer of brown adipocyte differentiation and thermogenesis (Schulz and Tseng, 2013; Tseng et al., 2008), rescues differentiation in the absence of Rictor. Notably, we do detect lowpparγexpression in RictorMyf5cKO P1 BAT, which may reflect the role of Rictor/mTORC2 in early brown adipocyte differentiation and explain the mutant BAT hypoplasia. Exactly how Rictor/mTORC2 and BMP7 signaling might converge on PPARγ is not yet clear. We also show that during brown adipocyte differentiation, the major AKT isoform switches from AKT1 to AKT2; thus, while Rictor/mTORC2 may regulate differentiation through an AKT1 pathway that can be bypassed in vivo, its role in BAT metabolism is likely mediated through an AKT2 pathway that cannot be compensated for. Consistent with this idea, whole body Akt2 knockout mice among many other phenotypes have smaller BATs (Cho et al., 2001; Garofalo et al., 2003).
Why does deleting Rictor in BAT cause a metabolic shift? One possibility is that forkhead box O (FOXO) transcription factors are more active in Rictor-deficient brown adipocytes. FOXOs are regulated by multiple signals and function as cellular homeostasis regulators under stressful conditions (Eijkelenboom and Burgering, 2013). FoxO1 and FoxO3 are AKT substrates that are partially dephosphorylated in some Rictor-deficient cells (Guertin et al., 2009; Guertin et al., 2006; Hagiwara et al., 2012; Jacinto et al., 2006; Yuan et al., 2012). When dephosphorylated, FoxO1/3 translocate to the nucleus where they affect metabolism, survival, and cell cycle genes and the activity of transcriptional regulators (including PPARγ and C/EBPα) (Eijkelenboom and Burgering, 2013). However, FoxO1/3 phosphorylation is not affected in Rictor-deficient BAT; thus, if the metabolic shift is driven by FoxO1/3, it may be through an alterative mechanism such as acetylation (Banks et al., 2011; Masui et al., 2013). Another possibility is that FoxC2 mediates the metabolic shift (Cederberg et al., 2001; Yao et al., 2013); however, we do not observe any change in FoxC2 expression in Rictor-deficient preadipocytes (not shown), nor do we see effects on the FoxC2 targets C/ebpβ or Wnt10b during differentiation (Gerin et al., 2009). The shift could also bemediated through unidentified AKT substrates thatuniquelyrequirehydrophobic motif phosphorylation. This is an important ongoing area of investigation.
Consistent with the Myf5-lineage giving rise to a subset of white adipocytes, we also uncovered an essential role for Rictor/mTORC2 in vivo in white adipocyte growth. This confirms our previous discovery that some white adipocytes arise from Myf5-Cre expressing precursors (Sanchez-Gurmaches and Guertin, 2014; Sanchez-Gurmaches et al., 2012). However, because in the RictorMyf5cKO mice the Rictor-deficient white adipocytes are interspersed heterogeneously with non-deleted adipocytes within the same depot, we could not perform the appropriate whole-tissue biochemical studies using Rictor-deficient WAT. We did however determine that RictoriKO white adipocyte precursors purified from the stromal vascular fraction of psWAT (which are not Myf5-lineage derived) are also defective at differentiating in vitro (not shown) indicating Rictor also has a cell autonomous role in white adipocyte differentiation that is not dependent upon being Myf5-lineage derived. To determine the in vivo relevance of these findings we will need to identify Cre drivers that express uniformly and specifically in white adipocyte precursors; however, the origins of adipocytes are just beginning to be revealed and appropriate tools are not yet available for this line of investigation.
Is Rictor/mTORC2 a master regulator of lipid metabolism? Recent studies of liver collectively report that deleting hepatic Rictor results in a complex phenotype including increased gluconeogenesis, decreased glycolysis, and impaired lipogenesis (Hagiwara et al., 2012; Lamming et al., 2012; Yuan et al., 2012). Two studies find that hepatic Rictor loss also decreases SREBP1c activity; however, one study suggests AKT2 mediates this function (Hagiwara et al., 2012) while the other proposes an AKT-independent pathway (Yuan et al., 2012). These two studies are also inconsistent with respect to how Rictor loss affects AKT signaling and thus the role of hepatic Rictor/mTORC2 is currently controversial. Nevertheless, the glucose uptake and glycolysis defect is reportedly independent of the lipogenesis defect because restoring glucose flux in Rictor-KO hepatocytes did not rescue lipogenesis (Hagiwara et al., 2012). This study also reports that fatty acid oxidation genes are elevated in Rictor-deficient hepatocytes (Hagiwara et al., 2012). Thus, Rictor/mTORC2 may have a broad role in establishing a pro-lipogenic metabolic state. Going forward it is important to determine if Rictor/mTORC2 regulates de novo lipogenesis and beta-oxidation by a common or coordinate set of mechanisms, or whether one metabolic deficiency is indirectly driving the other. Notably, we detect a decrease in lipogenesis gene expression in P1 BAT lacking Rictor, but the increase in fatty acid oxidation gene expression we first detect in 6-week mutant BAT. Thus, mitochondrial activity may progressively increase in the Rictor-deficient BAT and be secondary to a lipogenesis defect. Regardless, our findings support the idea that targeting lipogenesis and/or beta-oxidation pathways in adipocytes could be one approach to treating obesity and diabetes.
One prediction is that increasing BAT energy expenditure could have anti-obesity therapeutic potential (Tseng et al., 2010). To achieve this goal, a deeper understanding of how BAT utilizes fuel is required (Townsend and Tseng, 2014). An important finding in our study is that RictorMyf5cKO mice living at thermoneutrality, when challenged with an obesogenic diet, induce higher levels of UCP1and are more resistant to developing obesity and metabolic disease compared to HFD-fed controls. This suggests that inhibiting mTORC2 in BAT augments diet-induced thermogenesis (Cannon and Nedergaard, 2010; Feldmann et al., 2009)although we cannot yet rule out that Rictor loss in other Myf5-lineage tissues might also contribute to this phenotype. It is currently being debated as to whether humans have classic brown adipocytes or a potential third class of adipocyte called a brite/beige adipocyte (Nedergaard and Cannon, 2013). Recent work indicates that in the neck, deep fat is similar to rodent BAT and expresses high levels of UCP1 while more super ficial fat expresses lower UCP1 levels and has more brite/beige characteristics(Cypess et al., 2013). Notably, humans typically adjust temperature to be around thermoneutrality (Cannon and Nedergaard, 2010) and the BAT of mice living at thermoneutrality appears more “white-fat like”, or perhaps more “brite/beige-fat” like [Figure 7]. Thus, it seems likely that humans possess classic brown fat and that studies of brown fat in mice will provide important insights into human BAT regulation. Continued elucidation of mTORC2 pathways in BAT bioenergetics could therefore lead to novel anti-obesity therapies that target cellular energy expenditure.
EXPERIMENTAL PROCEDURES
Gene Expression
Total RNA was isolated from using Qiazol (Invitrogen) and an RNeasy kit (Invitrogen).Equal amounts of RNA were retro-transcribed to cDNA using a High capacity cDNA reverse transcription kit (Applied Biosystems). Tbp expression was used as a normalization gene. A different set of iBAT samples was used in RT-PCR arrays (Qiagen) according to manufacturer’s instruction. See also Supplemental Experimental Procedures.
In vitro differentiation
Primary brown adipocyte precursors (bAPC) cells were isolated from P1 neonates and immortalized with pBabe-SV40 Large T antigen. To induce Rictor deletion ubc-creERT2;Rictorfl/fl cells were treated on three consecutive days with 1μM 4-OHT. bAPCs were seeded at 4×104 cells/ml and allowed reach confluence over 3 days in mediumcontaining 20nM insulin, 1nM T3 (differentiation medium). On day 4, cells were induced with 20nM insulin, 1nM T3, 0.125mM indomethacin, 2μg/mL dexamethasone and 0.5mM 3-isobutyl-1-methylxanthine. Two days later the induction medium was replaced with fresh differentiation medium and changed every two days until day 10. See also Supplemental Experimental Procedures.
Metabolic studies
For thermoneutrality studies, 6 week-old male mice were transferred to 30°C. At 7 weeks of age, mice were fed chow (Prolab Isopro RMH3000, LabDiet) or high fat diet (45% calories from fat; ResearchDiet # D12451). Body weight and food intake were accessed weekly for 12 weeks. Glucose tolerance tests were performed at the 11th week. Overnight fasted animals were subjected to GTT by i.p. injecting glucose at 2g/Kg of body weight and blood glucose levels were measured with a commercially available glucose meter. A small group (n=4) of mice were kept for 20 weeks on HFD for morphological studies.
Statistics
Unless otherwise stated, the results are described as mean ± SEM. Two-way ANOVA was performed where indicated. For most experiments Student’s t-test was used to determine statistical significance: *indicates P<0.05; **, P<0.01; ***, P<0.001.
Supplementary Material
HIGHLIGHTS.
Brown and white adipocyte growth requires mTORC2
mTORC2 promotes lipogenesis and suppresses β-oxidation in brown fat
Brown preadipocytes also require mTORC2 to differentiate in vitro
Inhibiting mTORC2 in BAT enhances diet-induced thermogenesis
ACKNOWLEDGEMENTS
This work was supported by NIH (R00CA129613 &R01DK094004), American Diabetes Association (ADA113BS-066), Pew Charitable Trusts and Charles Hood Foundationgrants to D.A.G. We thank Yuefeng Tang, Xiaohao Yao, and Christine Powersfor technical assistance, Morris Birnbaum for Aktfloxed mice, and Marcus Cooperfor the PPARγ2 construct. Metabolic cage studies were performed in the UMass Mouse Phenotype Center (DK09300).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
See also Supplemental Experimental Procedures
AUTHOR CONTRIBUTIONS
C-M.H. and D.A.G. designed the projectand wrote and edited the manuscript. C-M.H. performed most experiments and D.A.G. assisted in analysis and interpretation. C.M.C., J.S-G., and H.L.assisted with rescue, lineage tracing, and mouseexperiments. C.B.C. performed metabolite profiling. S.H. and A.J.W. assisted with muscle experiments.
The authors declare no conflict of interests.
REFERENCES
- Balcke GU, Kolle SN, Kamp H, Bethan B, Looser R, Wagner S, Landsiedel R, van Ravenzwaay B. Linking energy metabolism to dysfunctions in mitochondrial respiration--a metabolomics in vitro approach. Toxicology letters. 2011;203:200–209. doi: 10.1016/j.toxlet.2011.03.013. [DOI] [PubMed] [Google Scholar]
- Banks AS, Kim-Muller JY, Mastracci TL, Kofler NM, Qiang L, Haeusler RA, Jurczak MJ, Laznik D, Heinrich G, Samuel VT, Shulman GI, Papaioannou VE, Accili D. Dissociation of the glucose and lipid regulatory functions of FoxO1 by targeted knockin of acetylation-defective alleles in mice. Cell metabolism. 2011;14:587–597. doi: 10.1016/j.cmet.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzinger CF, Romanino K, Cloetta D, Lin S, Mascarenhas JB, Oliveri F, Xia J, Casanova E, Costa CF, Brink M, Zorzato F, Hall MN, Ruegg MA. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell metabolism. 2008;8:411–424. doi: 10.1016/j.cmet.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiological reviews. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J. Metabolic consequences of the presence or absence of the thermogenic capacity of brown adipose tissue in mice (and probably in humans) Int J Obes (Lond) 2010;34(Suppl 1):S7–16. doi: 10.1038/ijo.2010.177. [DOI] [PubMed] [Google Scholar]
- Cederberg A, Gronning LM, Ahren B, Tasken K, Carlsson P, Enerback S. FOXC2 is a winged helix gene that counteracts obesity, hypertriglyceridemia, and diet-induced insulin resistance. Cell. 2001;106:563–573. doi: 10.1016/s0092-8674(01)00474-3. [DOI] [PubMed] [Google Scholar]
- Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, 3rd, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- Cybulski N, Polak P, Auwerx J, Ruegg MA, Hall MN. mTOR complex 2 in adipose tissue negatively controls whole-body growth. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9902–9907. doi: 10.1073/pnas.0811321106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cypess AM, White AP, Vernochet C, Schulz TJ, Xue R, Sass CA, Huang TL, Roberts-Toler C, Weiner LS, Sze C, Chacko AT, Deschamps LN, Herder LM, Truchan N, Glasgow AL, Holman AR, Gavrila A, Hasselgren PO, Mori MA, Molla M, Tseng YH. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nature medicine. 2013;19:635–639. doi: 10.1038/nm.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech MP, Tencerova M, Pedersen DJ, Aouadi M. Insulin signalling mechanisms for triacylglycerol storage. Diabetologia. 2013 doi: 10.1007/s00125-013-2869-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nature reviews. Molecular cell biology. 2013;14:83–97. doi: 10.1038/nrm3507. [DOI] [PubMed] [Google Scholar]
- Fasshauer M, Klein J, Kriauciunas KM, Ueki K, Benito M, Kahn CR. Essential role of insulin receptor substrate 1 in differentiation of brown adipocytes. Molecular and cellular biology. 2001;21:319–329. doi: 10.1128/MCB.21.1.319-329.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann HM, Golozoubova V, Cannon B, Nedergaard J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell metabolism. 2009;9:203–209. doi: 10.1016/j.cmet.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Filhoulaud G, Guilmeau S, Dentin R, Girard J, Postic C. Novel insights into ChREBP regulation and function. Trends in endocrinology and metabolism: TEM. 2013;24:257–268. doi: 10.1016/j.tem.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) The Biochemical journal. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, Coskran T, Black SC, Brees DJ, Wicks JR, McNeish JD, Coleman KG. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. 2003;112:197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerin I, Bommer GT, Lidell ME, Cederberg A, Enerback S, Macdougald OA. On the role of FOX transcription factors in adipocyte differentiation and insulin-stimulated glucose uptake. The Journal of biological chemistry. 2009;284:10755–10763. doi: 10.1074/jbc.M809115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell. 2007;131:242–256. doi: 10.1016/j.cell.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Stevens DM, Saitoh M, Kinkel S, Crosby K, Sheen JH, Mullholland DJ, Magnuson MA, Wu H, Sabatini DM. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer cell. 2009;15:148–159. doi: 10.1016/j.ccr.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Developmental cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, Terracciano L, Heim MH, Ruegg MA, Hall MN. Hepatic mTORC2 Activates Glycolysis and Lipogenesis through Akt, Glucokinase, and SREBP1c. Cell metabolism. 2012;15:725–738. doi: 10.1016/j.cmet.2012.03.015. [DOI] [PubMed] [Google Scholar]
- Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nature medicine. 2013;19:1252–1263. doi: 10.1038/nm.3361. [DOI] [PubMed] [Google Scholar]
- Harms MJ, Ishibashi J, Wang W, Lim HW, Goyama S, Sato T, Kurokawa M, Won KJ, Seale P. Prdm16 is required for the maintenance of brown adipocyte identity and function in adult mice. Cell metabolism. 2014;19:593–604. doi: 10.1016/j.cmet.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infantino V, Iacobazzi V, De Santis F, Mastrapasqua M, Palmieri F. Transcription of the mitochondrial citrate carrier gene: role of SREBP-1, upregulation by insulin and downregulation by PUFA. Biochemical and biophysical research communications. 2007;356:249–254. doi: 10.1016/j.bbrc.2007.02.114. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Kajimura S, Seale P, Spiegelman BM. Transcriptional control of brown fat development. Cell metabolism. 2010;11:257–262. doi: 10.1016/j.cmet.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Harris TE, Keller SR, Choi KM, Magnuson MA, Lawrence JC., Jr. Muscle-specific deletion of rictor impairs insulin-stimulated glucose transport and enhances Basal glycogen synthase activity. Molecular and cellular biology. 2008;28:61–70. doi: 10.1128/MCB.01405-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Lawrence JC, Jr., Jung DY, Ko HJ, Keller SR, Kim JK, Magnuson MA, Harris TE. Fat cell-specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes. 2010;59:1397–1406. doi: 10.2337/db09-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, Guertin DA, Sabatini DM, Baur JA. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638–1643. doi: 10.1126/science.1215135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR Signaling in Growth Control and Disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Russell SJ, Ussar S, Boucher J, Vernochet C, Mori MA, Smyth G, Rourk M, Cederquist C, Rosen ED, Kahn BB, Kahn CR. Lessons on conditional gene targeting in mouse adipose tissue. Diabetes. 2013;62:864–874. doi: 10.2337/db12-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Lopez N, Athonvarangkul D, Sahu S, Coletto L, Zong H, Bastie CC, Pessin JE, Schwartz GJ, Singh R. Autophagy in Myf5+ progenitors regulates energy and glucose homeostasis through control of brown fat and skeletal muscle development. EMBO reports. 2013;14:795–803. doi: 10.1038/embor.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, Iwanami A, Liu F, Villa GR, Gu Y, Campos C, Zhu S, Yang H, Yong WH, Cloughesy TF, Mellinghoff IK, Cavenee WK, Shaw RJ, Mischel PS. mTOR Complex 2 Controls Glycolytic Metabolism in Glioblastoma through FoxO Acetylation and Upregulation of c-Myc. Cell metabolism. 2013 doi: 10.1016/j.cmet.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuarai S, Miki S, Araki H, Takahashi K, Kotani H. Identification of dicarboxylate carrier Slc25a10 as malate transporter in de novo fatty acid synthesis. The Journal of biological chemistry. 2005;280:32434–32441. doi: 10.1074/jbc.M503152200. [DOI] [PubMed] [Google Scholar]
- Mullican SE, Tomaru T, Gaddis CA, Peed LC, Sundaram A, Lazar MA. A novel adipose-specific gene deletion model demonstrates potential pitfalls of existing methods. Mol Endocrinol. 2013;27:127–134. doi: 10.1210/me.2012-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Nedergaard J, Cannon B. The changed metabolic world with human brown adipose tissue: therapeutic visions. Cell metabolism. 2010;11:268–272. doi: 10.1016/j.cmet.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Nedergaard J, Cannon B. How brown is brown fat? It depends where you look. Nature medicine. 2013;19:540–541. doi: 10.1038/nm.3187. [DOI] [PubMed] [Google Scholar]
- Ohno H, Shinoda K, Ohyama K, Sharp LZ, Kajimura S. EHMT1 controls brown adipose cell fate and thermogenesis through the PRDM16 complex. Nature. 2013;504:163–167. doi: 10.1038/nature12652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nature reviews. Molecular cell biology. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- Sanchez-Gurmaches J, Guertin DA. Adipocytes arise from multiple lineages that are heterogeneously and dynamically distributed. Nature communications. 2014 doi: 10.1038/ncomms5099. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Gurmaches J, Hung CM, Sparks CA, Tang Y, Li H, Guertin DA. PTEN loss in the Myf5 lineage redistributes body fat and reveals subsets of white adipocytes that arise from Myf5 precursors. Cell metabolism. 2012;16:348–362. doi: 10.1016/j.cmet.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Schulz TJ, Huang P, Huang TL, Xue R, McDougall LE, Townsend KL, Cypess AM, Mishina Y, Gussoni E, Tseng YH. Brown-fat paucity due to impaired BMP signalling induces compensatory browning of white fat. Nature. 2013;495:379–383. doi: 10.1038/nature11943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TJ, Tseng YH. Brown adipose tissue: development, metabolism and beyond. The Biochemical journal. 2013;453:167–178. doi: 10.1042/BJ20130457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scime A, Devarakonda S, Conroe HM, Erdjument-Bromage H, Tempst P, Rudnicki MA, Beier DR, Spiegelman BM. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota C, Woo JT, Lindner J, Shelton KD, Magnuson MA. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Developmental cell. 2006;11:583–589. doi: 10.1016/j.devcel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Tallquist MD, Weismann KE, Hellstrom M, Soriano P. Early myotome specification regulates PDGFA expression and axial skeleton development. Development. 2000;127:5059–5070. doi: 10.1242/dev.127.23.5059. [DOI] [PubMed] [Google Scholar]
- Townsend KL, Tseng YH. Brown fat fuel utilization and thermogenesis. Trends in endocrinology and metabolism: TEM. 2014;25:168–177. doi: 10.1016/j.tem.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng YH, Butte AJ, Kokkotou E, Yechoor VK, Taniguchi CM, Kriauciunas KM, Cypess AM, Niinobe M, Yoshikawa K, Patti ME, Kahn CR. Prediction of preadipocyte differentiation by gene expression reveals role of insulin receptor substrates and necdin. Nature cell biology. 2005;7:601–611. doi: 10.1038/ncb1259. [DOI] [PubMed] [Google Scholar]
- Tseng YH, Cypess AM, Kahn CR. Cellular bioenergetics as a target for obesity therapy. Nature reviews. Drug discovery. 2010;9:465–482. doi: 10.1038/nrd3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng YH, Kokkotou E, Schulz TJ, Huang TL, Winnay JN, Taniguchi CM, Tran TT, Suzuki R, Espinoza DO, Yamamoto Y, Ahrens MJ, Dudley AT, Norris AW, Kulkarni RN, Kahn CR. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature. 2008;454:1000–1004. doi: 10.1038/nature07221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Mullican SE, DiSpirito JR, Peed LC, Lazar MA. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARgamma. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:18656–18661. doi: 10.1073/pnas.1314863110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Suraokar M, Darnay BG, Hollier BG, Shaiken TE, Asano T, Chen CH, Chang BH, Lu Y, Mills GB, Sarbassov D, Mani SA, Abbruzzese JL, Reddy SA. BSTA promotes mTORC2-mediated phosphorylation of Akt1 to suppress expression of FoxC2 and stimulate adipocyte differentiation. Science signaling. 2013;6:ra2. doi: 10.1126/scisignal.2003295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Pino E, Wu L, Kacergis M, Soukas AA. Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. The Journal of biological chemistry. 2012;287:29579–29588. doi: 10.1074/jbc.M112.386854. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.