

Abstract
Cholesterol has been shown to promote cell proliferation/migration in many cells; however the mechanism(s) have not yet been fully identified. Here we demonstrate that cholesterol increases Ca2+ entry via the TRPM7 channel, which promoted proliferation of prostate cells by inducing activation of the AKT and/or the ERK pathway. Additionally, cholesterol mediated Ca2+ entry induced calpain activity that showed a decrease in E-cadherin expression, which together could lead to migration of prostate cancer cells. Overexpression of TRPM7 significantly facilitated cholesterol dependent Ca2+ entry, cell proliferation and tumor growth. Whereas, TRPM7 silencing or inhibition of cholesterol synthesis by statin showed a significant decrease in cholesterol-mediated activation of TRPM7, cell proliferation, and migration of prostate cancer cells. Consistent with these results, statin intake was inversely correlated with prostate cancer patients and increase in TRPM7 expression was observed in samples obtained from prostate cancer patients. Altogether, we provide evidence that cholesterol-mediated activation of TRPM7 is important for prostate cancer and have identified that TRPM7 could be essential for initiation and/or progression of prostate cancer.
Keywords: TRPM7, cholesterol, calcium and signal transduction, statin, cell proliferation and migration, prostate cancer
Introduction
Prostate cancer (PCa) is one of the most common malignancies and the second leading cause of cancer-related death in men1-4. Recent studies have shown that cholesterol is an emerging clinically relevant therapeutic target in PCa patients5. Importantly, high circulating cholesterol levels have been shown to increase the risk of overall aggressive PCa5, 6. Consistent with these reports, recent clinical data also showed less aggressive PCa in men taking statins after prostatectomy7. Furthermore, intake of statins also reduced the incidences of PCa treatment failure for patients undergoing radiotherapy8; however, the mechanism as how cholesterol promote PCa is still poorly understood. Early stages of PCa growth depends on androgen which also regulate Ca2+ entry9-11, thus, it is very likely that Ca2+ channels will play an essential role in cellular proliferation and development of PCa12. Additionally, cholesterol has been shown to regulate various ion channels13-15; however the Ca2+ channel(s) involved in cholesterol induced proliferation in prostate cells is not yet identified. Hence, understanding the role of Ca2+ channels that are regulated by cholesterol and induces cell proliferation and/or migration may lead to a better therapeutic target for PCa.
Melastatin-like transient receptor potential (TRPM) subfamilies are a diverse group of voltage-independent Ca2+-permeable cation channels that are expressed in mammalian cells16. One of its member, TRPM7 channels are widely expressed and recently have been shown to be associated with cell survival17, 18. Importantly, TRPM7 has been shown to be required for increased proliferation and migration in several cancers such as breast, pancreatic, gastric, and nasopharyngeal cancers18-20; but its role in PCa has not yet been identified even though TRPM7 has been detected in rat prostate tissues21. TRPM7 is a Mg2+ and Ca2+ permeable ion channel that maintains the cellular Ca2+ and Mg2+ homeostasis22. In addition, Mg2+ is important for various physiological functions, further emphasizing the role of TRPM7 channels in cellular development. Although along with cell survival TRPM7 has been shown to regulate Ca2+ and Mg2+ homeostasis23, the factors that activates and/or regulate TRPM7 expression that can induce cell survival/proliferation has not yet been identified. Importantly, TRPM7 knockout mice are embryonically lethal and targeted disruption of TRPM7 in T cell lineage disrupted thymopoiesis24; further suggesting that these channels are essential for cellular development and abnormal activation of these channels can lead to diseases such as cancer. Our previous studies suggest that TRPM7 is important in prostate cells and maintain cellular Ca2+ and Mg2+ homeostasis. Furthermore, we have shown that alterations in Ca2+ to Mg2+ ratio could be essential for the initiation/progression of PCa25. Here we provide evidence that cholesterol activate TRPM7 channels that initiate Ca2+ entry, which not only facilitateTRPM7 expression, but was also essential for promoting cell proliferation and migration of prostate cancer cells. Finally, inhibition of cholesterol-induced TRPM7 activation by statins or TRPM7 silencing decreased Ca2+ entry, cell proliferation and tumor growth. Consistent with these results, TRPM7 expression was increased in PCa samples and men who used statins showed a decreased incidence of PCa in a retrospective pilot study. Overall, our results indicate that cholesterol-induced activation of TRPM7 increases cytosolic Ca2+ levels, which activates the AKT/ERK pathway and calpain activity that together induces cell proliferation and migration in prostate cancer cells.
Materials and Methods
Cell culture reagents and transfection
Control prostate cell line RWPE1 (CRL 11609), prostate cancer cell line DU145 (HTB-81) and LNCaP (CRL 1740) cells were obtained from the American Type Culture Collection (Manassas, VA). Cells were cultured in their respective medium along with various supplements as suggested by ATCC. Cells were maintained at 37°C with 95% humidified air and 5% CO2 and were passaged as needed. Culture medium was changed twice weekly and cells were maintained in complete media, until reaching 90% confluence. For transfection experiments shRNA plasmid that targets the coding sequence of human TRPM7 were obtained from Origene and TRPM7cDNA construct was used. Cells were transfected with individual shRNA (against TRPM7 or non-target shRNA (Sigma) (50nM) or TRPM7 plasmid (50nM) using Lipofectamine 2000 in Opti-MEM medium as per supplier's instructions (Invitrogen) and assayed after 48 hours. Antibodies that were used in this study are described in the figures. All other reagents used were of molecular biology grade obtained from Sigma chemicals unless mentioned otherwise.
Soft agar colony formation and cell migration assays
Ten thousand cells were grown in a semisolid agar media in 96 well plate for 5 days and later solubilized, lysed and detected by the CyQuant GR dye for soft agar colony formation. The experiment was set up according the manufactures instructions (CytoSelect 96-well cell transformation assay-Cell biolabs, Inc, USA). Images were taken using Nikon E5000 Coolpix prior to solubilizing. For cell migration assays cells were grown on a 12-well plate until 95% confluence and 1 μg/ml of mitomycin C was added to inhibit further proliferation of cells. The cell monolayer was scratched using a 200 μl pipette tip and images were taken using an Olympus CKX41 microscope with QCapture ×64 software (Surrey, Canada) immediately after the scratch, marked as 0 hr and after 24 hours.
Calpain activation assay
Two million cells were grown on 35-mm plates and the cells were treated with 1 μM Cholesterol for 24 hours. The cells were lysed and supernatant was used for the measurement of protein concentration. The cell lysate were diluted using extraction buffer and the activation was measured according to the manufacturer's instructions (Abcam, MA). The samples were analyzed at an excitation of 400 nm and emission at 505 nm using Multiskan spectrum fluorometer (Thermo labsystems) and the colorimetric reading was normalized with the respective total protein concentrations.
Cell Proliferation and Viability assays
The ten thousand cells were plated in 96 well plate and synchronized by stimulating with 10% FBS. Then pulsed with BrdU for 2 h before BrdU incorporation was measured as per manufacturer's instructions (Roche). For viability assays, cells were seeded on 96-well plates at a density of 0.5×105 cells/well. The cultures were grown for 24 hours followed by addition of fresh medium prior to the experiment. Cell viability was measured by using the trypan blue staining method. Cells (5×106 cells/well) were grown under different conditions for 48hrs, trypsinized, stained using equal volume of trypan blue and counted using a light microscope. Cell viability was expressed as a percentage of the control culture.
Calcium Measurements
Cells were incubated with 2 μM fura-2 (Molecular Probes) for 45 min, washed twice with Ca2+ free SES (Standard External Solution, include: 10 mM HEPES, 120 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 10 mM glucose, pH 7.4) buffer. For fluorescence measurements, the fluorescence intensity of Fura-2-loaded control cells was monitored with a CCD camera-based imaging system (Compix) mounted on an Olympus XL70 inverted microscope equipped with an Olympus 40× (1.3 NA) objective. A monochrometer dual wavelength enabled alternative excitation at 340 and 380 nm, whereas the emission fluorescence was monitored at 510 nm with an Okra Imaging camera (Hamamatsu, Japan). The images of multiple cells collected at each excitation wavelength were processed using the C imaging, PCI software (Compix Inc., Cranbery, PA), to provide ratios of Fura-2 fluorescence from excitation at 340 nm to that from excitation at 380 nm (F340/F380). Fluorescence traces shown represent [Ca2+]i values that are averages from at least 30-40 cells and are a representative of results obtained in at least 3-4 individual experiments.
Electrophysiology
For patch clamp experiments, coverslips with cells were transferred to the recording chamber and perfused with an external Ringer's solution of the following composition (mM): NaCl, 145; CsCl, 5; MgCl2, 1; CaCl2, 1; Hepes, 10; Glucose, 10; pH 7.3 (NaOH). Whole cell currents were recorded using an Axopatch 200B (Axon Instruments, Inc.). The patch pipette had resistances between 3 -5 MΩ after filling with the standard intracellular solution that contained the following (mM): cesium methane sulfonate, 150; NaCl, 8; Hepes, 10; EGTA, 10; pH 7.2 (CsOH). With a holding potential 0mV, voltage ramps ranging from -100mV to +100mV and 100ms duration were delivered at 2s intervals after whole cell configuration was formed. Currents were recorded at 2 kHz and digitized at 5–8 kHz. pClamp 10.1 software was used for data acquisition and analysis. Basal leak were subtracted from the final currents and average currents are shown. All experiments were carried out under room temperature.
Membrane preparations and Western blot analyses
Cells were harvested and stored at -80°C. Crude lysates were prepared from RWPE1, DU145, and LNCaP cells as described previously in53 Protein concentrations were determined, using the Bradford reagent (Bio-Rad), and 25-50 ug of proteins were resolved on 3-8% SDS-Tris-acetate gels, transferred to PVDF membranes and probed with respective antibodies. A 1:500 for TRPM7 (Epitomics, CA), 1:1000 for ERK, pERK, E-cadherin (Cell signaling, MA), and 1:1000 for AKT, pAKT, and actin (Santa Cruz, CA) antibodies were used to probe respective proteins. Peroxidase conjugated respective secondary antibodies were used to label the proteins. The proteins were detected by enhanced chemiluminescence detection kit (SuperSignal West Pico; Pierce). Densitometric analysis was performed using image J analysis and results were corrected for protein loading by normalization for β-actin expression as described in53-56.
A pilot hospital-based case-control study
We conducted a pilot case-control study at a community hospital in Grand Forks, ND, USA, to assess the association between statin use and PCa. Cases were men with newly diagnosed, histologically confirmed PCa. Controls were men without clinical cancer who were seen at the same hospital for an annual physical exam. The study was approved by the Institutional Review Boards of the hospital and the university.
Immunohistochemistry and Imaging
Paraffin embedded tissues from age-matched control and adenocarcinoma (Gleason score 3+4) were obtained and 10μm thick cryosections were performed. Hematoxylin and eosin (H&E) staining was performed on the sections using standard procedure (Sigma, St. Louis, MO). For immunolabeling respective samples were permeabilized at room temp with 0.1% TritonX-100 in PBS (pH 7.4), blocked (10% donkey serum and 5% BSA in PBS), and probed overnight with TRPM7 primary antibodies in a hydrated chamber maintained at 4°C. Following incubation with primary antibodies the slides were washed and processed for DAB staining. Images were acquired at 20 or 40X magnifications and total DAB staining from each section was quantified using the image J program.
Statistical Analysis
Mean and standard deviation values were computed for all continuous variables and frequency distributions were calculated for all categorical variables. We compared patients who had statins prescribed in their chart to patients who did not have such prescription on demographic and clinical variables using Wilcoxon signed-rank test for non-normally distributed or t-test for normally distributed continuous variables and chi-square for categorical variables. All statistical tests were two-tailed with p < 0.05 considered to be significant. Statistics were performed using SAS (SAS Institute, Cary, NC; Version 9.3 Users Guide).
Results
Acute cholesterol treatment increases intracellular Ca2+ concentration in prostate cells
Ca2+ plays a vital role in regulating various kinases which are involved in many cellular processes such as proliferation, migration, invasion, motility, gene transcription and apoptosis26, 27 Acute treatment of cholesterol (1μM) enhanced intracellular Ca2+ levels as shown in Figure 1A and 1B. Importantly, cholesterol affects were much more prominent in prostate cancer cells than control RWPE1 cells and significant increase in intracellular Ca2+ levels was observed in cancer cells. Furthermore, cholesterol-mediated increase in Ca2+ influx in DU145 cells was significantly inhibited by 2APB, a known Ca2+ channel inhibitor (Figure 1C, D). To understand the ion channel(s) responsible for cholesterol-mediated increase in Ca2+ entry, current recordings (whole cell) were performed in both control and cancer cells. Importantly, an inward current (that reversed around zero mV) was observed in both control RWPE1 and DU145 cells and application of cholesterol in bath solution showed an increase in inward Ca2+ currents in both control RWPE1 (data not shown) and in DU145 cells (Figure 1E-G), which was similar as observed in MagNuM currents19, 32 To identify the pharmacological properties of these currents, we next studied the effects of 2-APB and Mg-ATP on these cells. Mg-ATP has been shown to inhibit these MagNuM currents, whereas 2-APB has been shown to inhibit TRPM7 function, but potentiates TRPM6 function19, 32, that also shows similar electrophysiological properties. Addition of MgATP or 2APB inhibited the currents (Figure 1H, S1A-H) and addition of cholesterol did not increased the current intensity. Also, the current properties were consistent with previous recording performed in other cells, which have been shown to be linked with TRPM7 channels25, 28-31. Additionally, this cholesterol-mediated enhanced intracellular Ca2+ resulted in an increase in the phosphorylation of AKT and ERK in both RWPE1 and DU145 cells, which was inhibited by either addition of 5mM external EGTA or in the absence of external Ca2+ (Figure 1I-K; S1I), further suggesting that Ca2+ entry upon cholesterol stimulation is needed for their activation.
Figure 1. Acute cholesterol treatment increases intracellular Ca2+ in prostate cells.
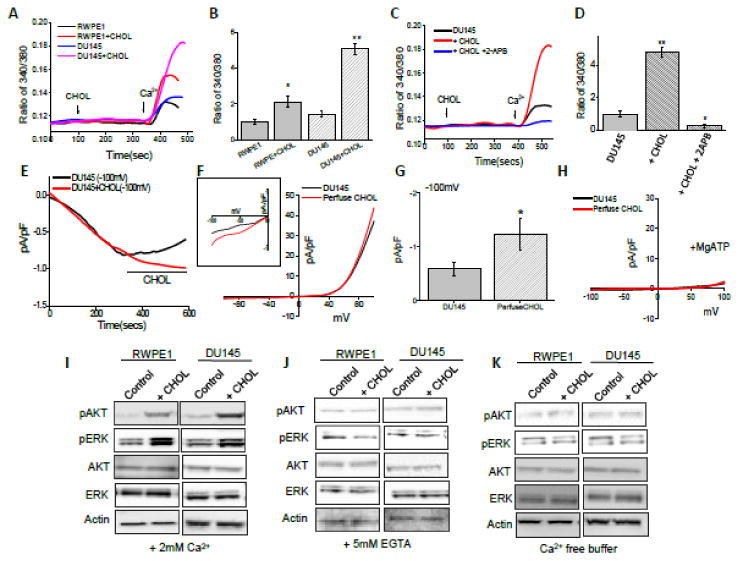
(A) Ca2+ imaging was performed in absence and in the presence of cholesterol (1 μM) in RWPE1 and DU145 cells. Analog plots of the fluorescence ratio (340/380) from an average of 40-60 cells are shown. (B) Quantification (mean ± SD) of fluorescence ratio (340/380). * indicates significance (p<0.05) versus RWPE control. Δ indicates significance (p<0.05) versus DU145 control. (C) Ca2+ imaging was performed in control, in the presence of cholesterol (1 μM) and plus 500 μM 2-APB treatment DU145 cells. Analog plots of the fluorescence ratio (340/380) from an average of 40-60 cells are shown. (D) Quantification (mean ± SD) of fluorescence ratio (340/380). * and ** indicates significance (p<0.05, p<0.01) versus control. (E) Application cholesterol in bath solution slightly facilitated TRPM7-like currents in DU145 cells. Average IV curves under conditions control and cholesterol treatment are shown in (F). Magnification under these conditions is shown as inset. Average (8-10 recordings) current intensity at -100mV under these conditions is shown in (G). (H) Application of MgATP in pipette abolish the TRPM7-like currents. . (I, J, K) Western blot images showing the expression of pAKT, pERK, total AKT, total ERK and loading control β-actin (except, total AKT 1/2/3 (H-136) from Santa Cruz Biotechnology, all antibodies were from Cell Signaling, Inc) in RWPE1 and DU145 cells with treatment of 200μM cholesterol for 15 minutes in presence of calcium, in presence of 5mM calcium chelator, EGTA and in presence of calcium free HBSS buffer, respectively.
Prolonged cholesterol treatment facilitates TRPM7 channel function in prostate cells
Pathological conditions occur mostly due to chronic exposure of cholesterol. Therefore, we examined the effect of prolonged (chronic) cholesterol treatment in prostate cancer cells. We show here that prolonged (24hrs) cholesterol treatment significantly increase TRPM7 activity (Figure 2 A-I). The currents facilitated upon cholesterol treatment were significantly increased in both normal prostate and prostate cancerous cells (in RWPE1, +100 mV was 21.29 ± 1.51 pA/pF, whereas 28.18 ± 1.73 pA/pF was observed with cholesterol treatment) (Figure 2A, B). In DU145 and LNCaP, +100 mV was 34.42 ± 2.04 pA/pF and 31.16 ± 2.86 pA/pF in control, whereas 44.12 ± 3.01 pA/pF and 39.73 ± 3.32 pA/pF was observed in cholesterol treated cells (Figure 2C-I). Importantly, the inward currents were also significantly facilitated in cancerous cells (For DU145 and LNCaP, -100 mV was -0.59 ± 0.13 pA/pF and -0.92 ± 0.18 pA/pF in unstimulated conditions, whereas -1.82 ± 0.25 pA/pF and -1.72 ± 0.12 pA/pF was observed in cholesterol treated cells). However, no significant difference in normal prostate RWPE1 cells was observed upon cholesterol treatment (-100 mV was 0.45 ±0.22 pA/pF in control, whereas 0.75 ± 0.42 pA/pF upon cholesterol treatment) (Figure 2H). Furthermore, extracellular addition of 500 μM of 2-APB, dramatically decreased current amplitude which were able to recover after wash-out of 2APB suggesting that indeed these currents are mediated via TRPM7 (Figure 2I). Additionally, cholesterol-dependent increase in the phosphorylation of AKT and ERK was inhibited in cells treated with 2APB (Figure 2J). Furthermore prolonged cholesterol treatment showed an increase in TRPM7 expression in both RWPE1 and DU145 cells (Figure 2K). Overall, these results suggest that cholesterol treatments potentiates TRPM7 currents and this increase in intracellular Ca2+ levels could also regulate TRPM7 expression via the activation of the ERK pathway.
Figure 2. Cholesterol facilitate TRPM7 channel function in prostate cells.
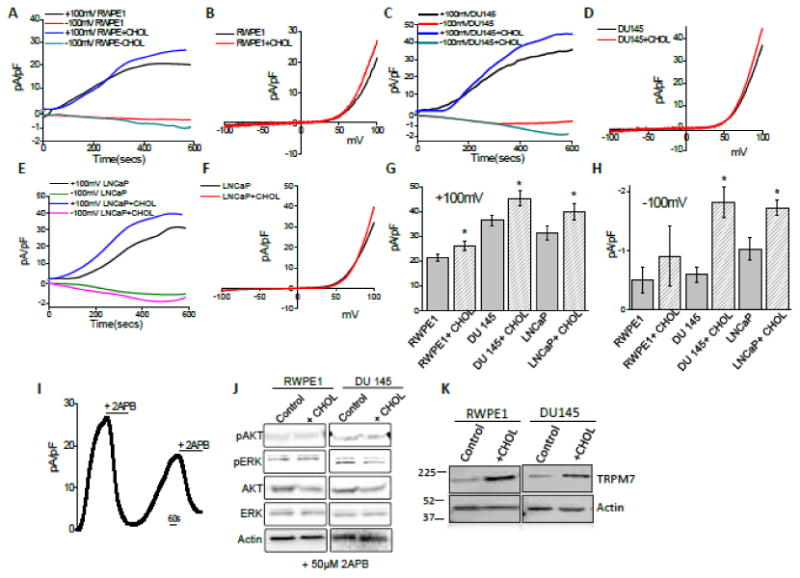
(A) Representative trace showing changes of whole cell currents from RWPE cells that were activated by the depletion of intracellular Mg2+ under various conditions (control and 1 μM cholesterol treatment). Outward currents (top curve) were measured at +100mV; whereas inward currents (bottom curve) was measured at -100mV. Average IV curves (developed from maximum currents) under this condition are shown in (B). (C), (E) Changes of whole cell currents under similar conditions from DU145 and LNCap cells are shown. Outward currents were again measured at +100mV; whereas inward currents were measured at -100mV (bottom line). Respective IV curve of these cells under these conditions are shown in (D) and (F). (G) and (H) average (8-10 recordings) current intensity at +100mV and -100mV under these conditions are shown. * indicates significance (p<0.05) versus untreated cells. (I) Represent outward currents in RWPE1 cells and once the currents reached its peak bath application of 500 μM 2-APB was applied followed by the recovery of the current after washing of 2-APB. (J) Western blot images showing the expression of pAKT, pERK, total AKT (AKT 1/2/3 (H-136), ERK and loading control β-actin in RWPE1 and DU145 cells with treatment of 200μM cholesterol for 15 minutes in presence of 50μM 2APB. (K) 1 μM Cholesterol increased the expression of TRPM7 in both RWPE1 (1.0 ± 0.18 and 1.93 ± 0.09 for control and cholesterol treated RWPE1 cells, respectively; p<0.01, N= 4) and DU145 (1.0 ± 0.15 and 2.26 ± 0.35 for control and cholesterol treated DU145 cells, respectively; p<0.05: N= 4) cells.
TRPM7 activation by cholesterol promotes cell proliferation in prostate cells
Cholesterol is an essential membrane component of animal cells, which makes up about one-third of the lipid content of the plasma membrane5. Therefore effect of prolong cholesterol treatment on TRPM7 expression along with other cellular functions were studied in prostate cells. Prolonged cholesterol treatment also increased cell viability in both RWPE1 and DU145 cells (Figure 3A). Consistent with this observation cellular proliferation was also increased as defined by using BrdU incorporation assay in RWPE1, androgen independent DU145, and androgen dependent LNCaP cells (Figure 3B), which was again significantly higher in prostate cancer cells. To further understand the significance of cholesterol, cells were treated with 1μM cholesterol and cell migration was studied after 24 hours. As shown in Figure 3C and Figure S1J, increased migration of RWPE-1, DU145 and LNCaP cells was observed in cholesterol treated cells, which was inhibited in the presence of 2APB (data not shown). Increased migration of cells under cholesterol treatment might have resulted from increased activity of calpain that are also Ca2+ dependent33, 34. Thus, calpain activity was measured, which showed a significant increase in calpain activity in cells incubated with cholesterol (Figure 3D). Calpain-dependent proteolysis of E-cadherin is known to be associated with prostate cancer35, hence we next studied the cholesterol mediated E–cadherin expression in RWPE1 and DU-145 cells which was down-regulated in the presence of cholesterol (Figure 3E). Since, cholesterol treatment increases ERK phosphorylation, and showed increase in TRPM7 expression, we thus studied whether TRPM7 expression is dependent on Ca2+-induced activation of ERK. Importantly, cells treated with ERK inhibitor PD98059 (10μM for 24 hours) decreased TRPM7 expression in both cells (Figure 3F). Importantly, 2APB also reduced TRPM7 expression when cells were incubated with 50μM 2APB for 24 hours (Figure 3G), suggesting that expression of TRPM7 is dependent on Ca2+ -dependent activation of ERK.
Figure 3. Cholesterol-mediated activation of TRPM7 regulates cellular function in prostate cells.
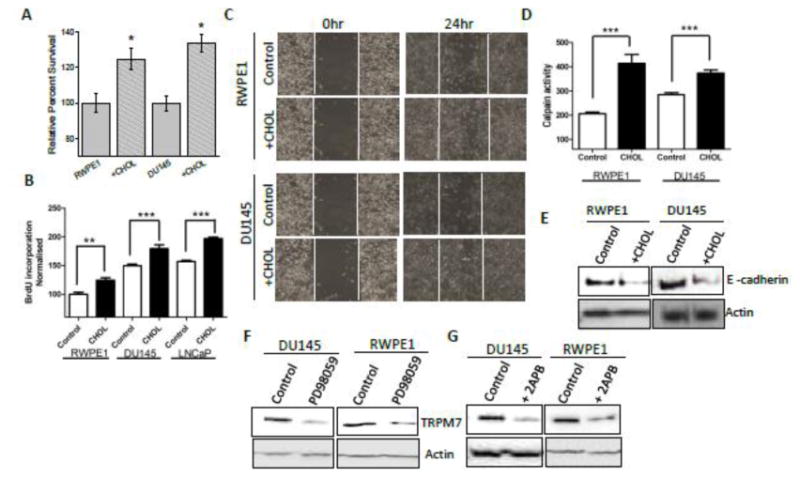
Cell viability under cholesterol treated conditions in RWPE1 and DU145 cells are shown in (A). * indicates values that are significantly different from untreated cells p<0.05. (B) Bar diagram showing the relative absorbance at 450nm of RWPE1, DU145 and LNCaP cells after BrDU incorporation. Each bar gives the mean ± SEM of 4 separate experiments. **, p<0.01, ***, p<0.001. (C) Images showing the wound-healing assay for cellular migration of RWPE11 cells and DU-145 cells treated with 1 μM cholesterol. Images were taken after the wound scratch (0 hr) and after 24 hours. The pictures are representative of 4 separate experiments. (D) Calpain activity measured using calpain activity kit from Abcam, in RWPE1 and DU145 cells and after treatment with 1 μM cholesterol for 24 hours (marked as none treated as control and Chol 1 μM for cells treated with 1μM cholesterol for 24 hours). Each bar gives the mean ± SEM (N=4, ***, p<0.001. (F) 1 μM Cholesterol treatment for 24hr decreases the expression of E cadherin (Cell Signaling Technology) in both RWPE1 (1.0 ± 0.23 and 0.63 ± 0.18 for control and cholesterol treated RWPE1 cells, respectively; p<0.05, N= 4) and DU145 (1.0 ± 0.05 and 0.46 ± 0.21 for control and cholesterol treated DU145 cells, respectively; p<0.05: N= 4). (G) 10 μM PD98059 (ERK inhibitor) treatment for 24 hours decreased the expression of TRPM7 in both DU145 (0.98± 0.17 and 0.40 ± 0.11 for control and PD98059 treated DU145 cells, respectively; p<0.05, N= 3) and RWPE1 (1.2 ± 0.27 and 0.51 ± 0.11 for control and PD98059 treated RWPE1 cells, respectively; p<0.05, N= 4) cells. (H) 50μM 2APB treatment for 24 hours decreased the expression of TRPM7 in both DU145 (1.0 ± 0.07 and 0.60 ± 0.03 for control and treated DU145 cells, respectively; p<0.01, N= 3) and RWPE1 (“1.3± 0.27 and 0.40 ± 0.051 for control and treated RWPE1 cells, respectively; p<0.05: N= 4) cells.
Cholesterol mediated increase in Ca2+ entry and cell proliferation of prostate cells is dependent on TRPM7 expression
To further establish that effect cholesterol in cell proliferation and migration was dependent on TRPM7 expression, we silenced TRPM7 and evaluated the effect of cholesterol on prostate cells. As shown in Figure 4A and in supplementary figure 2, expression of shTRPM7 (shTRPM7), but not the non-targeting shRNA, in prostate cells showed a decrease in TRPM7 expression. Control actin levels were however not changed under these conditions. Importantly, cholesterol induced Ca2+ influx was also inhibited in both control RWPE1 and prostate cancer DU145 cells that was expressing shTRPM7 (Figure 4B-D). Consistent with these results, TRPM7 currents were also significantly inhibited in both control and prostate cancer cells (DU145 and LNCaP) that expresses shTRPM7 (Figure 4E-J and S2B, D, E). Additionally, cholesterol treatment showed no increase in TRPM7 currents in cells expressing shTRPM7 (Figure 4E-J); further showing that cholesterol activates TRPM7 currents. More importantly, consistent with these results, cholesterol induced increase in cell survival and cell proliferation of control or prostate cancer cells was also inhibited in cells treated with shTRPM7 (Figure 5A, B; S2G). Additionally, cholesterol-dependent increase in the phosphorylation of AKT and ERK was inhibited in cells expressing shTRPM7 (Figure 5C and 5D). In contrast control shRNA expressing DU145 cells showed a significant increase in AKT and ERK phosphorylation (5C, D). Similar data was also observed with RWPE1, where control shRNA showed an increase in cholesterol-mediated activation of AKT and ERK (data not shown), whereas cells expressing TRPM7shRNA had no increase in AKT or ERK phosphorylation (5C). Calpain activity upon cholesterol activation was also inhibited in prostate cancer DU145 cells that was expressing shTRPM7 (Figure 5E). Finally, the tumor growth was also reduced in shTRPM7 cells, studied using soft agar colony formation assays in both DU145 and LNCaP cells (Figure 5F, G, S2G, H). These results strongly suggest that TRPM7 expression in prostate cancerous cells play an important role in cholesterol-mediated increase in cytosolic Ca2+ that is essential for increase in cell proliferation/survival and migration, therefore may be critical for the initiation and or progression of PCa.
Figure 4. Knockdown of TRPM7 channel abolished Ca2+ signal induced by cholesterol in prostate cells.
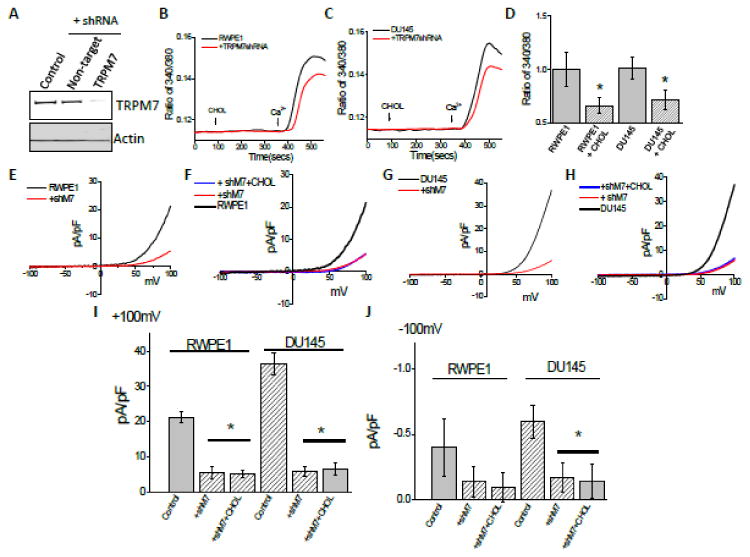
(A) Representative blots indicating DU145 cells expressing shRNA targeting TRPM7 or control non-targeting shRNA. Control represent similar conditions without plasm id (1.0 ± 0.03, 0.89 ± 0.16 and 0.38 ± 0.17 for control, non-targeting and TRPM7shRNA cells, respectively; p <0.05, N= 4). Cell lysates from DU145 cells were resolved on NuPAGE 3-8% Tris-Acetate gels and analyzed by western blotting using TRPM7 antibody (Epitomics, CA). β-actin was used as loading control. (B) Ca2+ imaging was performed in the presence of cholesterol (1 μM) in control RWPE cells and cells transfected with shRNA targeting TRPM7. Analog plots of the fluorescence ratio (340/380) from an average of 40-60 cells are shown. (C) Changes of Ca2+ influx under similar conditions from DU145 cells are shown. (D) Quantification (mean ± SD) of fluorescence ratio (340/380). * indicates significance (p<0.05) versus control. In RWPE cells transfected with shRNA targeting TRPM7 and Cholesterol pretreatment for 24 hours affect TRPM7-like currents, which average IV curves (developed from maximum currents) under various conditions are shown in (E) and (F). (G), (H) Changes of whole cell current under similar conditions from DU145 cells are shown. (I), (J) Average (8-10 recordings) current intensity at +100mV and -100mV under these conditions is shown. * indicates significance (p<0.05) versus untreated cells.
Figure 5. Knockout TRPM7 channel resulted in cholesterol induce function in prostate cells.
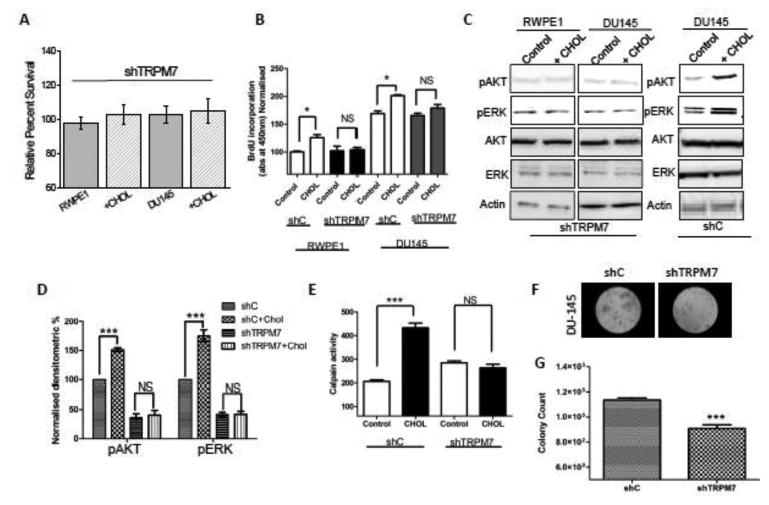
Cell viability under cholesterol treated conditions in RWPE1 and DU145 cells are shown in (A). * indicates values that are significantly different from untreated cells p<0.05. (B). Bar diagram showing the relative absorbance at 450nm of RWPE1 and DU145 (shRNA control non-targeting marked as shC and TRPM7 knockdown cells marked as shTRPM7) cells after BrDU incorporation. Each bar gives the mean ± SEM of 4 separate experiments. * indicates significance p<0.05. (C) Western blot images showing the expression of pAKT, pERK, total AKT (AKT 1/2/3 (H-136), ERK and loading control β-actin in shTRPM7 (TRPM7 knockdown) RWPE1 and DU145 cells with treatment of 200μM cholesterol for 15 minutes. Panel on the right shows stimulation of DU145 cells overexpressing control shRNA (shC). (D) Bar diagram representing the densitometry reading showing the activity of phospho form of AKT and ERK, in shC and shTRPM7 in DU145 cells. Each bar represents percentage of respective pAKT or pERK normalized with the total AKT or ERK expression of the respective samples. Each bar gives the mean ± SEM (N=4, ***, p<0.001, NS= non significance). (E) Calpain activity measured using calpain activity kit from Abcam, in DU145 (shRNA control marked as shC and TRPM7 knockdown cells marked as shTRPM7) cells and after treatment with 1 μM cholesterol for 24 hours (marked as none treated as control and Chol 1 μM for cells treated with 1 μM cholesterol for 24 hours). Each bar gives the mean ± SEM (N=4, ***, p<0.001 (F). Images representing the soft agar colony tumor growth in DU145 cells and TRPM7 knockdown cells. Bar diagram represents the relative fluorescence reading at 485/525 nm filters, of control and TRPM7 knockdown DU145 cells after agar media being solubilized, lysed and detected by the patented CyQuant® GR Dye in a fluorescence plate reader.
Overexpression of TRPM7 enhances cholesterol-mediated effects in prostate cancer cells
To understand the significance of TRPM7 in cholesterol-mediated activation, cells where transfected with TRPM7. Western blot images confirm the overexpression of TRPM7 in DU145 cells (Figure 6A) and LNCaP cells (Figure S2A). Furthermore, overexpression of TRPM7 showed a significant increase in MagNuM currents in both DU145 cells and LNCaP cells (Figure 6B-E and Figure S2B). Additionally, cholesterol treatment showed a further increase in TRPM7 currents in cells overexpressing TRPM7 (Figure 6C-E and Figure S.2B). TRPM7 overexpression also enhanced cholesterol induced cell proliferation of prostate cancer cells (Figure 6F and Figure S2F). Overexpression of TRPM7 in both DU145 cells and LNCaP also resulted in an increase tumor growth, studied using soft agar colony formation assay (Figure 6G and Figure S2G, H). Finally, cholesterol levels were found to be significantly increased in prostate cancer cells (DU145, and LNCaP), when compared with control RWPE1 cells (Figure S2I), further suggesting that cholesterol mediated activation of TRPM7 cells is perhaps critical for cancer cell growth. Consistent with these results increased TRPM7 expression was observed in samples obtained from adenocarcinoma patients when compared with age-matched control samples (Figure 6H). These results further suggest that TRPM7 expression could lead to prostate cancer.
Figure 6. Overexpression of TRPM7 resulted in enhanced effect in cholesterol induce fuction.
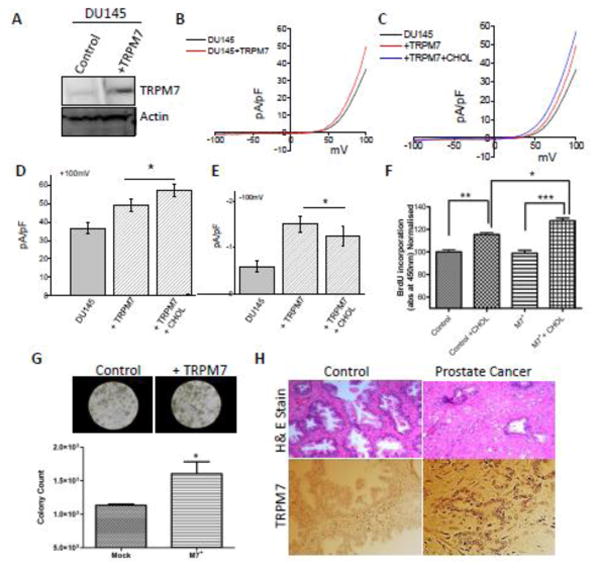
(A) Representative blots indicating DU145 cells overexpressing TRPM7 or control non-targeting shRNA. Cell lysates from DU145 cells were resolved on NuPAGE 3-8% Tris-Acetate gels and analyzed by western blotting using TRPM7 antibody (1.0 ± 0.09 and 2.85 ± 0.42 for control and DU145 TRPM7 overexpressed cells marked as +M7, respectively; p<0.05: N= 4) β-actin was used as loading control. In DU145 cells transfected with overexpress TRPM7 and cholesterol pretreatment for 24 hours affect TRPM7-like currents, which average IV curves (developed from maximum currents) under various conditions are shown in (B) and (C). (D), (E) Average (8-10 recordings) current intensity at +100mV and -100mV under these conditions is shown. * indicates significance (p<0.05) versus untreated cells. (F) Bar diagram showing the relative absorbance at 450nm of DU145 (shRNA control marked as shC and TRPM7 overexpressed cells marked as +M7) with and without 1 μM cholesterol treatment for 24 hours and 2 hours of BrDU incorporation. Each bar gives the mean ± SEM of 4 separate experiments. * indicates significance *, p<0.05, ** p<0.01 and *** p<0.001. (G) Images representing the soft agar colony tumor growth in control DU145 cells and TRPM7 overexpressing cells. Bar diagram represents the relative fluorescence reading at 485/525 nm filters, of control and TRPM7 overexpressing DU145 cells after agar media being solubilized, lysed and detected by the patented CyQuant® GR Dye in a fluorescence plate reader. (H) Expression of TRPM7 in control and prostate cancer tissues.
Statins attenuates cholesterol mediated activation of TRPM7 channel in prostate cells
Statins inhibit the mevalonate pathway, thereby preventing the synthesis of cholesterol36. Use of statin has been shown to relate to PCa outcomes37, but the mechanism(s) is not known. We thus used two statin drugs (simvastatin and mevastatin) to examine if they block cholesterol-mediated activation of TRPM7 that induces cell proliferation in prostate cells. Statin treatment inhibited Ca2+ influx in a dose dependent manner. Pretreatment with simvastatin and mevastatin for 24 hours inhibited Ca2+ influx induced by cholesterol in both normal prostate and cancerous cells (Figure 7A-D). Cancerous prostate cells (DU145) were however more sensitive to statin treatment than normal RWPE1 prostate cells. Importantly in membrane current recording (TRPM7 function), pretreatment with simvastatin for 24 hours inhibited TRPM7 currents only in cancerous prostate cells, but not in control cells (Figure 7E-H). Also mevastatin decreased TRPM7 currents more than simvastatin indicating that different statins can alter TRPM7 function differently. Consistent with these results addition of external cholesterol restored statin-mediated decrease of TRPM7 currents in prostate cells (Figure 7I-K). Importantly, Ca2+ influx was not further decreased upon statin treatment in TRPM7 silenced cells (Figure S3A-D). Similar results were also observed with TRPM7 currents and no significant decrease was observed in TRPM7 currents in cells expressing shTRPM7 group and shTRPM7 treated with statins (Figure S3E-H), suggesting that the effects observed above were due to TRPM7. To understand as how statin treatment is able to inhibit cholesterol mediated increase in Ca2+ influx, we assayed TRPM7 expression in these cells. Importantly, pretreatment with statins decreased TRPM7 protein expression in both normal and prostate cancerous cells (Figure 8A). These results again suggest that expression of TRPM7 was dependent on cholesterol-mediated Ca2+ influx via TRPM7. Overall, results presented thus far indicate that TRPM7 channel is involved in cholesterol mediated activation of Ca2+ influx which can be reversed by pretreatment of statins.
Figure 7. Statins inhibit TRPM7 channel function in prostate cells.
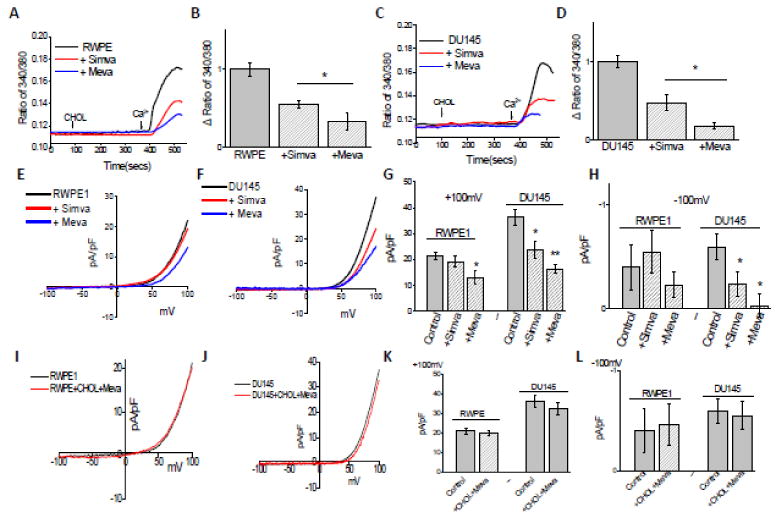
(A) Ca2+ imaging was performed in the presence of cholesterol (1 μM) in control and statins treated for 24Hour in RWPE1 cells. Analog plots of the fluorescence ratio (340/380) from an average of 40-60 cells are shown. (B) Quantification (mean ± SD) of fluorescence ratio (340/380). * indicates significance (p<0.05) versus control. (C), (D) Changes of Ca2+ influx under similar conditions from DU145 cells are shown. Statins pretreatment for 24 hours inhibited TRPM7-like currents. Average IV curves (developed from maximum currents) under various conditions (control, 50μM mevastatin treatment and 10μM simvastatin treatment) are shown in (E). IV curve of whole cell recording under similar conditions from DU145 cells are shown in (F). (G), (H) Average (8-10 recordings) current intensity at +100mV and -100mV under these conditions is shown. * indicates significance (p<0.05) versus untreated cells. Respectively IV curves of cells treated cholesterol and statins same time in RWPE and DU145 cells are show in (I) and (J). (K), (L) Average (8-10 recordings) current intensity at +100mV and -100mV under these conditions is shown.
Figure 8. Statin inhibits the cholesterol mediated cellular function in prostate cells.
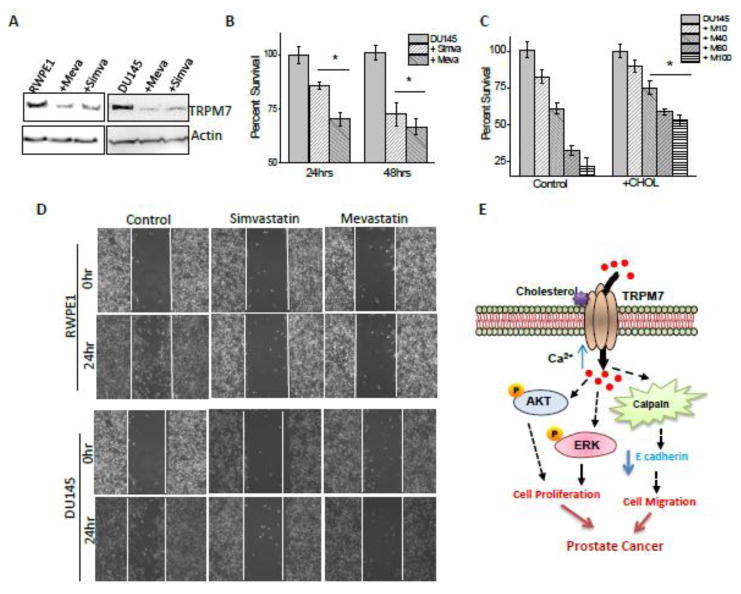
(A) Representative blots indicating expression of TRPM7 in RWPE1 (1.0 ± 0.05, 0.45 ± 0.23 and 0.63 ± 0.11 for control, mevastatin, and simvastatin treated RWPE1 cells, respectively; p<0.05, N= 4) and DU145 cells (1.0 ± 0.06, 0.17 ± 0.38 and 0.35 ± 0.27 for control, mevastatin, and simvastatin treated DU145 cells, respectively; p<0.05, N= 4) cells. Cell lysates from cells were resolved on NuPAGE 3-8% Tris-Acetate gels and analyzed by western blotting using TRPM7 antibody. MTT assays under simvastatin and mevastatin treated conditions in DU145 cells are shown in (B). * indicates values that are significantly different from control p<0.05. (C), Cholesterol attenuated effect of inhibition by statin in DU145 cells. (D) Images showing the wound-healing assay for cellular migration of RWPE-1 and DU145 cells treated with 10μM simvastatin or 10 μM mevastatin. Images were taken after the wound scratch (0 hr) and after 24 hours. The pictures are representative of 4 separate experiments. (E) Schematic picture of proposed model. Acute extracellular cholesterol activates the TRPM7 resulting in an increase cellular calcium which results in increased AKT and ERK phosphorylation. Prolong cholesterol treatment via ERK/AKT pathway increases the TRPM7 expression, thereby, increasing the intracellular calcium concentration that increases the calpain activity which in turn reduce E cadherin expression. Hence, resulting in an increase in cell proliferation, migration, and cancer progression.
Statin inhibits cholesterol mediated increase in cell proliferation and migration in prostate cancer cells
The results presented above suggest that cholesterol increases Ca2+ entry via TRPM7 channels, which is critical for cell proliferation and or cancer progression, but can be inhibited by statins. Hence we studied proliferation and migration of cancer cells in the presence of statins. Importantly, a dominant decrease in cell survival was observed in prostate cancerous cells in a time dependent manner. Additionally, mevastatin treatment for 24 hours, showed a significant decrease in cell proliferation when compared with simvastatin (Figure 8B, C). These results are consistent as mevastatin decreased TRPM7 currents more than simvastatin. To establish if cholesterol treatment can revert the effects of mevastatin we treated DU145 cells simultaneously with cholesterol along with varying doses of mevastatin. As indicated in Figure 8C, mevastatin showed a dose dependent decrease in cell survival and addition of cholesterol partially rescued cell proliferation in cancer cells. Importantly, cholesterol was much efficient at higher statin concentration. Consistent with these results, a dominant inhibition in cell migration was also observed when prostate cancer cells were treated with simvastatin and mevastatin for 24hours (Figure 8D). Overall, these results suggest that statin treatment inhibits TRPM7 expression and function thereby inhibiting cancer cell proliferation and migration.
Statin use was inversely associated with PCa
To finally establish the link between statin use and PCa, records from PCa and age-matched control patients was used. The mean age was not significantly different in both the group (Table 1). Importantly, patients that used tobacco (1 pack per day) had a significantly higher prevalence of PCa (20% vs. 8%, respectively for control; p=0.04). This is consistent as cigarette smoking has been shown to increase cholesterol levels38, 39. Median PSA was also lower in PCa patients that took statins; however no difference in the Gleason score was observed (data not shown). Importantly, use of statin showed an inverse correlation, where statin users had significantly lower prevalence of newly diagnosed prostate cancer (78% had no cancer that used statins versus 22% that had cancer and did not used statins) (Table 1). Taken together, these results suggest that statin treatment could inhibit prostate cancer cell proliferation and migration, which was dependent on TRPM7 function as they can inhibit TRPM7 function.
Table 1. Demographic, clinical characteristics and prostate cancer status among statin users and none users.
| Variables | Statin use | No statin use | p-value | ||
|---|---|---|---|---|---|
| n | % | n | % | ||
| Total (n=121) | 66 | 55 | 55 | 45 | |
| Age, mean ± SD | 69 ± 7 | 67 ± 8 | .11 | ||
| Smoking status | .006 | ||||
| Current | 12 | 19 | 7 | 15 | |
| Former | 36 | 56 | 15 | 31 | |
| Never | 16 | 25 | 26 | 54 | |
| Prostate-Specific Antigen, ng/ml | 3.6 (0.1-22.5) | 5.7 (0.3-748.1) | .003 | ||
| Gleason scores | .77 | ||||
| Low grade (< 7) | 20 | 57 | 26 | 60 | |
| High grade (≥ 7) | 15 | 43 | 26 | 40 | |
| Serum albumin, g/dL | 3.86 ± 0.4 | 3.99 ± 0.3 | .16 | ||
| Corrected serum calcium, mg/dl* | 9.52 ± 0.5 | 9.39 ± 0.42 | .25 | ||
| Type of statin | |||||
| Hydrophobic | 60 | 91 | - | ||
| Hydrophilic | 6 | 9 | - | ||
| Cancer status | .004 | ||||
| No cancer (n=43) | 31 | 47 | 12 | 22 | |
| Newly diagnosed prostate cancer (n=78) | 35 | 53 | 43 | 78 | |
Hydrophobic: atorvastatin, lovastatin, and simvastatin.
Hydrophilic: rosuvastatin, and pravastatin.
Based on 44 and 26 patients for statin users and none users respectively
Discussion
TRPM7 channels are widely expressed in cells including prostate tissues21. We recently reported that TRPM7 channels were expressed in both normal (RWPE1) and prostate cancerous cells (DU145, PC3) and alterations in the Ca2+/Mg2+ ratio facilitated cell proliferation in cancer cells12. Similarly TRPM7 has been shown to be critical for various other cancers and together our results suggest that TRPM7 can be critical for PCa initiation and/or progression. However the factors that can increase TRPM7 expression and function are not known. In the present study, we found that acute and prolonged cholesterol treatment induces Ca2+ influx in prostate cells that were dependent on TRPM7 channels. Moreover, this cholesterol-mediated increase in Ca2+ influx was also essential for regulating TRPM7 expression which could be a feed forward mechanism that can be activated in prostate cancer cells. Cholesterol is a key component of the plasma membrane and has been shown to regulate the function of various ion channels in the membrane14, 40; however it is unknown if cholesterol can regulate TRPM7 channel. Furthermore, the consequence of this activation of TRPM7 channel is not clear in diseases such as PCa. A recent report has shown that cholesterol can regulate Ca2+ entry via nonspecific Ca2+ channels41. Our data specifically showed that acute and prolong cholesterol treatment activates TRPM7 channels thereby increasing cytosolic Ca2+ levels that not only increase TRPM7 expression, but also promote cell proliferation in prostate cancer cells. TRPM7 has been implicated in the control of cellular proliferation and viability by transporting metal ions to other cells29, which is consistent with the results presented here. Additionally, inhibition of TRPM7 channel activity has been shown to decrease proliferation of breast cancerous cells and hypopharyngeal squamous cell carcinoma cells42, 43, further suggesting that it can have a role in PCa. Importantly higher cholesterol levels as well as cholesterol-mediated increase in TRPM7 function was observed only in cancerous cells. Similarly, cholesterol can also activate other ion channels and more research is needed to fully identify the role of cholesterol in activating Ca2+ channels. In addition, cholesterol is important for lipid rafts that have been previously shown to be important for Ca2+ entry40, 55 thus, there may be additional Ca2+ entry channels that can also contribute towards this process.
To further understand the role of prolonged cholesterol in PCa, we found that cholesterol mediated increase in Ca2+ entry facilitated cell migration in prostate cancerous cells. This increase in migration was mainly due to the activation of Ca2+-activated calpains, which inhibits E cadherins expression that could result in the loss of tight junction's thus inducing cell migration. Epidemiologic studies indicated that cholesterol increases the risk of PCa and cholesterol is shown to regulate proliferation and migration in PCa5, 44, 45. Our studies provide further evidence that this increase of cell proliferation, due to cholesterol dependent increase in Ca2+ influx via TRPM7, was dependent on AKT and ERK phosphorylation, which is consistent with previous studies36, 46. Previous studies have suggested the role of cholesterol in PCa5, 44, where patients diagnosed with hyper-cholesterolemia had higher likelihood for PCa44. In agreement with these reports, our result also implicate that cholesterol plays a critical role in prostate cancer cells migration and proliferation, which was again dependent on the activation of TRPM7. Thus, our studies not only compliment these previous studies but also provide the mechanism as to how cholesterol induces PCa.
Epidemiologic data also suggest that cholesterol increases the risk of PCa47-50. Statins are used clinically to reduce LDL levels and they inhibit the rate-limiting step in cholesterol synthesis, but their role in PCa is not fully defined. We found that statins decrease viability and migration of prostate cancer cells. More importantly, addition of cholesterol partially recovered cell viability that was inhibited by statin treatment. To understand the mechanism we show here that statins reduced Ca2+ influx that was induced by cholesterol, by inhibiting TRPM7 currents. Furthermore, statins decreased TRPM7 expression, which could be important for the inhibition of cell migration and cell proliferation critical for PCa. Evaluating the clinically relevant factors that affect PCa would not only provide a better understanding on the mechanism that could alter PCa progression, but could also identify new drug targets for PCa. Our results further show that TRPM7 expression was increased in samples obtained from PCa patients and individuals that used statin had a significant decrease in the likelihood of developing PCa. However, these findings should be interpreted with caution since this is an observational study using a small sample size. Additionally, although we did not separate the risk reduction with regard to PSA or Gleason score, it has been previously shown that statin use is greatest for decreasing the risk of clinically more aggressive disease (Gleason score <7)7. Together these results suggest that statins not only inhibit cholesterol levels51, 52, but can also inhibit PCa, by inhibiting TRPM7 function. Although other mechanisms whereby statins may inhibit PCa are also possible, our results strongly suggest that statin treatment inhibits TRPM7 expression and function thereby inhibiting cell proliferation and migration in PCa.
To further define the role of TRPM7 in cholesterol mediated induction of prostate cell proliferation, we reciprocally silenced and overexpressed TRPM7 channels. Importantly in TRPM7 silenced cells statins were unable to further reduce Ca2+ influx and TRPM7 currents. Although at present we cannot limit that cholesterol dependent effect were exclusively due to TRPM7, silencing of TRPM7 was able to inhibit cholesterol dependent increase in Ca2+ entry as well as cell proliferation. Consistent with this overexpression of TRPM7 showed a significant increase in cell proliferation, migration and tumor growth. Additionally, TRPM7 expression was increased in PCa samples. Together these results establish that TRPM7 channels are involved in PCa. In addition, cholesterol is an emerging clinically relevant therapeutic target in PCa 5 and our studies gives a new dimension into the role of non-selective transient receptor potential cation channels which can be inhibited to prevent PCa (Figure 7H). However prospective epidemiologic studies are needed to assess the function of TRPM7 in wet tissue and whether statin use impact serum Ca2+ and Mg2+ that can activate TRPM7.
Supplementary Material
Highlights.
Expression and function of TRPM7 is increased in prostate cancer cells
Cholesterol activate TRPM7 to induce cell proliferation/migration in prostate cells
Ca2+ via TRPM7 activate the ERK/AKT pathway to induce cell proliferation
Statins inhibit cholesterol-dependent activation of TRPM7 and decreases cancer
Acknowledgments
We thank Drs. Don Sens and Xudong Zhou for providing the human samples and for insightful advice discussions and interpretations of the data. This work was supported by NIH grants R01DE017102 and 1R03AI097532 awarded to BBS.
Footnotes
Author contributions: Y.S., P.S., A.V., and S.D. performed experiments, analyzed data and wrote the paper; A.S. and B.S. developed experimental tools, designed experiments, analyzed data and wrote the paper. All authors discussed the results and implications, and commented on the manuscript at all stages.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Center MM, Jemal A, Lortet-Tieulent J, et al. International variation in prostate cancer incidence and mortality rates. Eur Urol. 2012;61:1079–1092. doi: 10.1016/j.eururo.2012.02.054. [DOI] [PubMed] [Google Scholar]
- 2.Flourakis M, Prevarskaya N. Insights into Ca2+ homeostasis of advanced prostate cancer cells. Biochim Biophys Acta. 2009;1793:1105–1109. doi: 10.1016/j.bbamcr.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 3.Prevarskaya N, Skryma R, Shuba Y. Ca2+ homeostasis in apoptotic resistance of prostate cancer cells. Biochem Biophys Res Commun. 2004;322:1326–1335. doi: 10.1016/j.bbrc.2004.08.037. [DOI] [PubMed] [Google Scholar]
- 4.Wissenbach U, Niemeyer B, Himmerkus N, Fixemer T, Bonkhoff H, Flockerzi V. TRPV6 and prostate cancer: cancer growth beyond the prostate correlates with increased TRPV6 Ca2+ channel expression. Biochem Biophys Res Commun. 2004;322:1359–1363. doi: 10.1016/j.bbrc.2004.08.042. [DOI] [PubMed] [Google Scholar]
- 5.Pelton K, Freeman MR, Solomon KR. Cholesterol and prostate cancer. Current opinion in pharmacology. 2012;12:751–759. doi: 10.1016/j.coph.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Twiddy AL, Leon CG, Wasan KM. Cholesterol as a potential target for castration-resistant prostate cancer. Pharmaceutical research. 2011;28:423–437. doi: 10.1007/s11095-010-0210-y. [DOI] [PubMed] [Google Scholar]
- 7.Loeb S, Kan D, Helfand BT, Nadler RB, Catalona WJ. Is statin use associated with prostate cancer aggressiveness? BJU Int. 2010;105:1222–1225. doi: 10.1111/j.1464-410X.2009.09007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gutt R, Tonlaar N, Kunnavakkam R, Karrison T, Weichselbaum RR, Liauw SL. Statin use and risk of prostate cancer recurrence in men treated with radiation therapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:2653–2659. doi: 10.1200/JCO.2009.27.3003. [DOI] [PubMed] [Google Scholar]
- 9.Chen R, Zeng X, Zhang R, et al. Ca1.3 channel alpha protein is overexpressed and modulates androgen receptor transactivation in prostate cancers. Urologic oncology. 2013 doi: 10.1016/j.urolonc.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 10.Datta M, Schwartz GG. Calcium and vitamin D supplementation during androgen deprivation therapy for prostate cancer: a critical review. The oncologist. 2012;17:1171–1179. doi: 10.1634/theoncologist.2012-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Racioppi L. CaMKK2: a novel target for shaping the androgen-regulated tumor ecosystem. Trends in molecular medicine. 2013;19:83–88. doi: 10.1016/j.molmed.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun Y, Selvaraj S, Varma A, Derry S, Sahmoun AE, Singh BB. Increase in serum Ca2+/Mg2+ ratio promote proliferation of prostate cancer cells by activating TRPM7 channel. J Biol Chem. 2012 doi: 10.1074/jbc.M112.393918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee SY, Choi HK, Kim ST, et al. Cholesterol inhibits M-type K+ channels via protein kinase C-dependent phosphorylation in sympathetic neurons. J Biol Chem. 2010;285:10939–10950. doi: 10.1074/jbc.M109.048868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chun YS, Shin S, Kim Y, et al. Cholesterol modulates ion channels via down-regulation of phosphatidylinositol 4,5-bisphosphate. J Neurochem. 2010;112:1286–1294. doi: 10.1111/j.1471-4159.2009.06545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dopico AM, Bukiya AN, Singh AK. Large conductance, calcium- and voltage-gated potassium (BK) channels: regulation by cholesterol. Pharmacol Ther. 2012;135:133–150. doi: 10.1016/j.pharmthera.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Runnels LW, Yue L, Clapham DE. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science. 2001;291:1043–1047. doi: 10.1126/science.1058519. [DOI] [PubMed] [Google Scholar]
- 17.Runnels LW. TRPM6 and TRPM7: A Mul-TRP-PLIK-cation of channel functions. Current pharmaceutical biotechnology. 2011;12:42–53. doi: 10.2174/138920111793937880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lam DH, Grant CE, Hill CE. Differential expression of TRPM7 in rat hepatoma and embryonic and adult hepatocytes. Can J Physiol Pharmacol. 2012;90:435–444. doi: 10.1139/y11-136. [DOI] [PubMed] [Google Scholar]
- 19.Mishra R, Rao V, Ta R, Shobeiri N, Hill CE. Mg2+- and MgATP-inhibited and Ca2+/calmodulin-sensitive TRPM7-like current in hepatoma and hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2009;297:G687–694. doi: 10.1152/ajpgi.90683.2008. [DOI] [PubMed] [Google Scholar]
- 20.Guilbert A, Gautier M, Dhennin-Duthille I, Haren N, Sevestre H, Ouadid-Ahidouch H. Evidence that TRPM7 is required for breast cancer cell proliferation. Am J Physiol Cell Physiol. 2009;297:C493–502. doi: 10.1152/ajpcell.00624.2008. [DOI] [PubMed] [Google Scholar]
- 21.Wang HP, Pu XY, Wang XH. Distribution profiles of transient receptor potential melastatinrelated and vanilloid-related channels in prostatic tissue in rat. Asian J Androl. 2007;9:634–640. doi: 10.1111/j.1745-7262.2007.00291.x. [DOI] [PubMed] [Google Scholar]
- 22.Paravicini TM, Chubanov V, Gudermann T. TRPM7: a unique channel involved in magnesium homeostasis. Int J Biochem Cell Biol. 2012;44:1381–1384. doi: 10.1016/j.biocel.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 23.Chen HC, Su LT, Gonzalez-Pagan O, Overton JD, Runnels LW. A key role for Mg(2+) in TRPM7's control of ROS levels during cell stress. Biochem J. 2012;445:441–448. doi: 10.1042/BJ20120248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin J, Desai BN, Navarro B, Donovan A, Andrews NC, Clapham DE. Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science. 2008;322:756–760. doi: 10.1126/science.1163493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun Y, Selvaraj S, Varma A, Derry S, Sahmoun AE, Singh BB. Increase in serum Ca2+/Mg2+ ratio promotes proliferation of prostate cancer cells by activating TRPM7 channels. J Biol Chem. 2013;288:255–263. doi: 10.1074/jbc.M112.393918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nature reviews Molecular cell biology. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 27.Braun A, Vogtle T, Varga-Szabo D, Nieswandt B. STIM and Orai in hemostasis and thrombosis. Frontiers in bioscience. 2011;16:2144–2160. doi: 10.2741/3844. [DOI] [PubMed] [Google Scholar]
- 28.Jiang X, Newell EW, Schlichter LC. Regulation of a TRPM7-like current in rat brain microglia. J Biol Chem. 2003;278:42867–42876. doi: 10.1074/jbc.M304487200. [DOI] [PubMed] [Google Scholar]
- 29.Nadler MJ, Hermosura MC, Inabe K, et al. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature. 2001;411:590–595. doi: 10.1038/35079092. [DOI] [PubMed] [Google Scholar]
- 30.Runnels LW, Yue L, Clapham DE. The TRPM7 channel is inactivated by PIP(2) hydrolysis. Nat Cell Biol. 2002;4:329–336. doi: 10.1038/ncb781. [DOI] [PubMed] [Google Scholar]
- 31.Voets T, Nilius B, Hoefs S, et al. TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. J Biol Chem. 2004;279:19–25. doi: 10.1074/jbc.M311201200. [DOI] [PubMed] [Google Scholar]
- 32.Li M, Du J, Jiang J, et al. Molecular determinants of Mg2+ and Ca2+ permeability and pH sensitivity in TRPM6 and TRPM7. J Biol Chem. 2007;282:25817–25830. doi: 10.1074/jbc.M608972200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franco SJ, Huttenlocher A. Regulating cell migration: calpains make the cut. J Cell Sci. 2005;118:3829–3838. doi: 10.1242/jcs.02562. [DOI] [PubMed] [Google Scholar]
- 34.Sukumaran P, Lof C, Pulli I, Kemppainen K, Viitanen T, Tornquist K. Significance of the transient receptor potential canonical 2 (TRPC2) channel in the regulation of rat thyroid FRTL-5 cell proliferation, migration, adhesion and invasion. Molecular and cellular endocrinology. 2013;374:10–21. doi: 10.1016/j.mce.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 35.Rios-Doria J, Day KC, Kuefer R, et al. The role of calpain in the proteolytic cleavage of E-cadherin in prostate and mammary epithelial cells. J Biol Chem. 2003;278:1372–1379. doi: 10.1074/jbc.M208772200. [DOI] [PubMed] [Google Scholar]
- 36.Roy M, Kung HJ, Ghosh PM. Statins and prostate cancer: role of cholesterol inhibition vs. prevention of small GTP-binding proteins. American journal of cancer research. 2011;1:542–561. [PMC free article] [PubMed] [Google Scholar]
- 37.Geybels MS, Wright JL, Holt SK, Kolb S, Feng Z, Stanford JL. Statin Use in Relation to Prostate Cancer Outcomes in a Population-based Patient Cohort Study. Prostate. 2013 doi: 10.1002/pros.22671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graham I, Cooney MT, Bradley D, Dudina A, Reiner Z. Dyslipidemias in the prevention of cardiovascular disease: risks and causality. Current cardiology reports. 2012;14:709–720. doi: 10.1007/s11886-012-0313-7. [DOI] [PubMed] [Google Scholar]
- 39.Critchley JA, Capewell S. WITHDRAWN: Smoking cessation for the secondary prevention of coronary heart disease. Cochrane database of systematic reviews. 2012;2:CD003041. doi: 10.1002/14651858.CD003041.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pani B, Ong HL, Liu X, Rauser K, Ambudkar IS, Singh BB. Lipid rafts determine clustering of STIM1 in endoplasmic reticulum-plasma membrane junctions and regulation of store-operated Ca2+ entry (SOCE) J Biol Chem. 2008;283:17333–17340. doi: 10.1074/jbc.M800107200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kannan KB, Barlos D, Hauser CJ. Free cholesterol alters lipid raft structure and function regulating neutrophil Ca2+ entry and respiratory burst: correlations with calcium channel raft trafficking. J Immunol. 2007;178:5253–5261. doi: 10.4049/jimmunol.178.8.5253. [DOI] [PubMed] [Google Scholar]
- 42.Dou Y, Li Y, Chen J, et al. Inhibition of cancer cell proliferation by midazolam by targeting transient receptor potential melastatin 7. Oncology letters. 2013;5:1010–1016. doi: 10.3892/ol.2013.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim BJ. Involvement of melastatin type transient receptor potential 7 channels in ginsenoside Rd-induced apoptosis in gastric and breast cancer cells. Journal of ginseng research. 2013;37:201–209. doi: 10.5142/jgr.2013.37.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Solomon KR, Freeman MR. The complex interplay between cholesterol and prostate malignancy. The Urologic clinics of North America. 2011;38:243–259. doi: 10.1016/j.ucl.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kitahara CM, Berrington de Gonzalez A, Freedman ND, et al. Total cholesterol and cancer risk in a large prospective study in Korea. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:1592–1598. doi: 10.1200/JCO.2010.31.5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown M, Hart C, Tawadros T, et al. The differential effects of statins on the metastatic behaviour of prostate cancer. Br J Cancer. 2012;106:1689–1696. doi: 10.1038/bjc.2012.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Platz EA, Leitzmann MF, Visvanathan K, et al. Statin drugs and risk of advanced prostate cancer. Journal of the National Cancer Institute. 2006;98:1819–1825. doi: 10.1093/jnci/djj499. [DOI] [PubMed] [Google Scholar]
- 48.Murtola TJ, Tammela TL, Lahtela J, Auvinen A. Cholesterol-lowering drugs and prostate cancer risk: a population-based case-control study. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2007;16:2226–2232. doi: 10.1158/1055-9965.EPI-07-0599. [DOI] [PubMed] [Google Scholar]
- 49.Platz EA, Till C, Goodman PJ, et al. Men with low serum cholesterol have a lower risk of high-grade prostate cancer in the placebo arm of the prostate cancer prevention trial. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2009;18:2807–2813. doi: 10.1158/1055-9965.EPI-09-0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khosropanah I, Falahatkar S, Farhat B, et al. Assessment of atorvastatin effectiveness on serum PSA level in hypercholesterolemic males. Acta medica Iranica. 2011;49:789–794. [PubMed] [Google Scholar]
- 51.Rauthan M, Ranji P, Aguilera Pradenas N, Pitot C, Pilon M. The mitochondrial unfolded protein response activator ATFS-1 protects cells from inhibition of the mevalonate pathway. Proc Natl Acad Sci U S A. 2013;110:5981–5986. doi: 10.1073/pnas.1218778110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garcia-Ruiz C, Morales A, Fernandez-Checa JC. Statins and protein prenylation in cancer cell biology and therapy. Anti-cancer agents in medicinal chemistry. 2012;12:303–315. doi: 10.2174/187152012800228715. [DOI] [PubMed] [Google Scholar]
- 53.Singh BB, Lockwich TP, Bandyopadhyay BC, et al. VAMP2-dependent exocytosis regulates plasma membrane insertion of TRPC3 channels and contributes to agonist-stimulated Ca2+ influx. Mol Cell. 2004;15:635–646. doi: 10.1016/j.molcel.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 54.Pani B, Cornatzer E, Cornatzer W, et al. Up-regulation of transient receptor potential canonical 1 (TRPC1) following sarco(endo)plasmic reticulum Ca2+ ATPase 2 gene silencing promotes cell survival: a potential role for TRPC1 in Darier's disease. Mol Biol Cell. 2006;17:4446–4458. doi: 10.1091/mbc.E06-03-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pani B, Ong HL, Brazer SC, et al. Activation of TRPC1 by STIM1 in ER-PM microdomains involves release of the channel from its scaffold caveolin-1. Proc Natl Acad Sci U S A. 2009;106:20087–20092. doi: 10.1073/pnas.0905002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Selvaraj S, Watt JA, Singh BB. TRPC1 inhibits apoptotic cell degeneration induced by dopaminergic neurotoxin MPTP/MPP(+) Cell Calcium. 2009;46:209–218. doi: 10.1016/j.ceca.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.