

Abstract
Hydrogen peroxide, a reactive oxygen species with unique chemical properties, is produced endogenously in living systems as a destructive oxidant to ward off pathogens or as a finely tuned second messenger in dynamic cellular signaling pathways. In order to understand the complex roles that hydrogen peroxide can play in biological systems, new tools to monitor hydrogen peroxide in its native settings, with high selectivity and sensitivity, are needed. Knowledge of organic synthetic reactivity provides the foundation for the molecular design of selective, functional hydrogen peroxide probes. A palette of fluorescent and luminescent probes that react chemoselectively with hydrogen peroxide has been developed, utilizing a boronate oxidation trigger. These indicators offer a variety of colors and in cellulo characteristics and have been used to examine hydrogen peroxide in a number of experimental setups, including in vitro fluorometry, confocal fluorescence microscopy, and flow cytometry. In this chapter, we provide an overview of the chemical features of these probes and information on their behavior to help researchers select the optimal probe and application.
1. INTRODUCTION
Reactive oxygen species (ROS), including hydrogen peroxide, are products of cellular respiration that can modulate physiology through a variety of homeostatic mechanisms (D’Autréaux & Toledano, 2007; Dickinson & Chang, 2011; Murphy et al., 2011; Rhee, 2006; Stone & Yang, 2006; Veal, Day, & Morgan, 2007; Winterbourn, 2008). On the other hand, aberrant ROS production is implicated in aging (Giorgio, Trinei, Migliaccio, & Peliccil, 2007), as well as illnesses such as Alzheimer’s (Huang & Mucke, 2012; Mattson, 2004), Huntington’s (Xun et al., 2012), Parkinson’s (Exner, Lutz, Haass, & Winklhofer, 2012), and other neurode-generative diseases (Lin & Beal, 2006). In many cases, high levels of ROS may result in lipid peroxidation, protein modifications leading to loss or gain of function, and DNA and RNA damage (Frisard & Ravussin, 2006). As such, the generation of ROS must be tightly regulated, both spatially and temporally, to protect the cell from oxidative stress, while taking advantage of the unique and powerful reactivity of these small molecules. To control ROS levels, organisms have evolved specialized enzymes such as superoxide dismutase and catalase to transform ROS into less harmful species (Zelko, Mariani, & Folz, 2002).
While ROS were long considered molecules that negatively impact human health, emerging studies have since shown that ROS are also essential to maintaining cellular health. In particular, several NADPH oxidases (Noxs) have been identified in many tissue types and are known to play important roles in cellular defense and the modulation of cellular signaling events via the controlled production of hydrogen peroxide (Bedard & Krause, 2007; Lambeth, 2004). Low levels of endogenous hydrogen peroxide can lead to specific protein modifications such as sulfenic acid or disulfide formation upon cysteine oxidation (Nelson et al., 2010; Paulsen & Carroll, 2010). The dual ability of hydrogen peroxide to act as a beneficial or detrimental species in biological systems highlights the challenges of disentangling the complex mechanisms of action of this particular ROS.
Owing to the transient nature of hydrogen peroxide, fluorescent probes, which generally display high sensitivity and can be used to determine spatial and temporal distributions in live specimens, are particularly appealing tools for the detection of ROS and related metabolites. Useful peroxide probes for biological applications must possess the following qualities: (1) compatibility with aqueous media at physiological pH, (2) cell permeability and/or subcellular targetability, (3) high selectivity and sensitivity for hydrogen peroxide over other biologically relevant ROS and reactive species, and (4) spectroscopic properties that are suited for commonly used instrumentation.
To meet this need, our laboratory has developed and continues to expand the toolkit of chemoselective hydrogen peroxide probes based on boronate oxidation, a classic chemical transformation that has its origins in organic synthesis (Kuivila & Armour, 1957; Kuivila & Wiles, 1955). Discovering its compatibility with aqueous environments, our group sought to harness this chemical reaction as a trigger for the selective detection of hydrogen peroxide in biological settings (Chang, Pralle, Isacoff, & Chang, 2004; Lippert, Van de Bittner, & Chang, 2011). This strategy has given rise to a palette of boronate-based fluorescent and luminescent probes that give a turn-on (or ratiometric) response upon reaction with hydrogen peroxide in living systems. In general, these probes are weakly fluorescent in their initial form, and upon oxidation by hydrogen peroxide, they are irreversibly converted to a brighter species. Familiarity with the photophysical properties and the behavior of these probes in biological systems will permit the reader to select and use these probes most effectively in their chosen research endeavors.
The remainder of this chapter is organized as follows. An introduction to fluorescent boronate-based hydrogen peroxide probes is presented in Section 2, summarizing the properties of selected probes and offering a basic guide to choosing a probe for different types of applications. In Section 3, we describe representative experiments using these probes, including fluorometry for in vitro studies, fluorescent microscopy, and flow cytometry. Section 4 outlines methods for inducing the production of endogenous hydrogen peroxide, as well as reagents that can be used to specifically block increases of hydrogen peroxide levels in living cells.
2. FLUORESCENT BORONATE-BASED HYDROGEN PEROXIDE PROBES
2.1. Probe design
The present classes of boronate-based hydrogen peroxide probes rely primarily upon masked fluoresceins and rhodols (Fig. 2.1); the aniline functionality of the latter provides a synthetic handle that can be modified to alter the emission wavelength of the probe (Dickinson, Huynh, & Chang, 2010) or used for targeting the probe to subcellular locations such as the nucleus or mitochondria (Dickinson & Chang, 2008; Dickinson, Tang, Chang, & Chang, 2011). The introduction of alternative fluorophores can also give rise to different emission colors (Miller, Albers, Pralle, Isacoff, & Chang, 2005; Miller, Tulyathan, Isacoff, & Chang, 2007). Additionally, synthetic modification can be performed on the benzene moiety orthogonal to the xanthene with little effect upon the fluorescent properties of the molecule. Tethering a benzyl guanine of benzyl-2-chloro-6-aminopyrimidin-4-amine to PG1 affords the probes SPG1 and SPG2, which react selectively with SNAP-tag fusion proteins to deliver the synthetic hydrogen peroxide probes to selected subcellular localizations (Juillerat et al., 2003; Keppler et al., 2004; Srikun, Albers, Nam, Iavarone, & Chang, 2010). Other variants allow for ratiometric imaging by internal charge transfer (Srikun, Miller, Domaille, & Chang, 2008) or fluorescent resonance energy transfer approaches (Albers, Okreglak, & Chang, 2006) as well as dual-analyte versions on dendrimer scaffolds (Srikun, Albers, & Chang, 2011).
Figure 2.1.

Molecular structures of selected boronate-based hydrogen peroxide probes.
A comparison of the spectroscopic properties and applications of commonly used boronate probes reveals a range of excitation and emission wavelengths as well as variations in the fluorescence intensities of these probes (Table 2.1). Background fluorescence, or the brightness of the unreacted probe, varies depending on the structure of the probe and its environment. Boronate probes such as PF1 and PF2, which exist in the “closed” or lactone form, are virtually nonfluorescent before reaction with hydrogen peroxide (Chang et al., 2004; Dickinson, Huynh, & Chang, 2010; Miller et al., 2005). Oxidation of the boronate to the phenol leads to subsequent opening of the lactone ring and tautomerization; the dye in aqueous solution now exists in an equilibrium that lies toward the “open,” fluorescent form (Fig. 2.2). While high background fluorescence should be avoided, probes with low background fluorescence may be desirable over a completely dark or nonfluorescent probe to confirm successful uptake of the dye into cells. Probes such as PG1 (Miller et al., 2007), which is based upon the Tokyo Green scaffold, are weakly fluorescent due to quenching via photoinduced electron transfer (PET) from the electron-donating substituents on the benzene group to the xanthene ring (Urano et al., 2005). Conversion to the phenol and deprotonation to the phenolate results in an anionic molecule, which is electronically less susceptible to PET quenching and therefore more fluorescent.
Table 2.1.
Spectroscopic properties, localization, and example applications of selected boronate-based hydrogen peroxide probes
| Probe | Probe λabs/λem (nm) |
Product λabs/λem (nm) |
Intracellular localization |
Successful applications |
|---|---|---|---|---|
| Peroxyfluor 1 (PF1) | NA/NA | 494/521 | Nontargeted, diffuse |
Exogenous H2O2 detection in cells |
| Peroxyfluor 2 (PF2) | NA/NA | 475/511 | Nontargeted, diffuse |
Detection of H2O2 upon glucose treatment of hematopoietic stem cells in vivo in zebrafish |
| Peroxy green 1 (PG1) |
460/510 | 460/510 | Nontargeted, diffuse |
Detection of H2O2 in EGF signaling in A431 cells |
| Peroxy yellow 1 (PY1) |
494/558 | 519/548 | Nontargeted, diffuse |
Detection of H2O2 in EGF signaling in A431 cells, multiple colors |
| Peroxy orange-1 (PO1) |
507/574 | 540/565 | Nontargeted, diffuse |
Detection of H2O2 and HOCl during immune response in RAW264.7 macrophages; detection of H2O2 in FLT3/STAT5 signaling in leukemia cells |
| Peroxyfluor 6 acetoxymethyl ester (PF6-AM) |
460/530 | 492/517 | Cytosol | Detection of H2O2 in FGF-2 signaling in adult hippocampal progenitor cells, sodium delivery, or angiotensin II stimulation in the medullary thick ascending limb, and during chemotaxis of neutrophils |
| Mitochondria peroxy yellow 1 (MitoPY1) |
489,510/ 540 | 510/528 | Mitochondria | Detection of H2O2 upon paraquat treatment of HeLa cells, S100β, or APP treatment of DS neural progenitors, and sodium delivery in the medullary thick ascending limb |
| Nuclear peroxy emerald 1 (NucPE1) |
468,490/ 530 | 505/530 | Nucleus | Detection of H2O2 in vivo in a long-lived zebrafish model |
Figure 2.2.
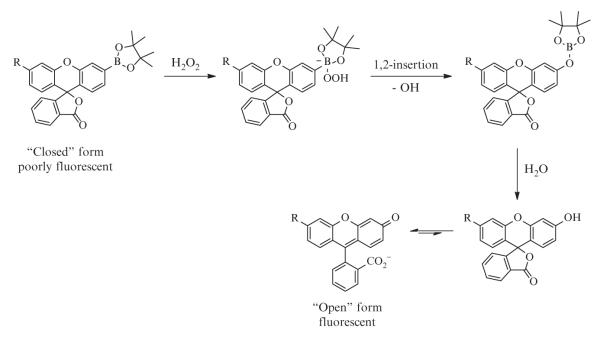
Molecular design of fluorescent boronate-based hydrogen peroxide probes.
2.2. Reactivity
The boronate-based probes are classified as chemodosimeters, which differ from chemosensors in that an irreversible chemical transformation rather than a reversible binding event gives rise to the observed fluorescence increase (Chan, Dodani, & Chang, 2012; Cho & Sessler, 2009; Czarnik, 1994; Du, Hu, Fan, & Peng, 2012; Jun, Roy, & Ahn, 2011; Quang & Kim, 2010). Due to the short lifetimes of ROS in the biological environment, imaging with chemodosimeters is advantageous, particularly when studying systems involving low levels of ROS flux; the accumulation of signal over time can be readily measured via endpoint assays. For real-time detection in intact biological systems, other tools such as genetically encoded hydrogen peroxide sensors of the HyPer family (Belousov et al., 2006; Bilan et al., 2013) and redox-sensitive green fluorescent protein indicators fused to oxidant receptor peroxidase 1 (Albrecht, Barata, Grosshans, Teleman, & Dick, 2011) are available.
In terms of chemical selectivity, incorporation of the boronate functionality into an assortment of dye scaffolds affords a caged fluorophore sensitive to hydrogen peroxide, but not readily oxidized by many other ROS, such as superoxide, hypochlorite, or hydroxyl radical. Selectivity tests have shown negligible turn-on responses of these probes to a wide range of reactive oxygen and nitrogen species (Fig. 2.3B). The boronate oxidation takes advantage of hydrogen peroxide’s specific molecular characteristics, particularly its enhanced nucleophilicity due to the α-effect (Jencks & Carriulo, 1960; Ren & Yamataka, 2007), imparted by adjacent nonbonding orbitals on its oxygen atoms, and the weak O—O bond. Nucleophilic addition of hydrogen peroxide to the boron results in a charged tetrahedral boronate complex, which subsequently undergoes a 1,2-insertion in which the C—B bond migrates to one of the now-electrophilic peroxide oxygens (Fig. 2.2). The resulting borate ester is then hydrolyzed by water to the phenol.
Figure 2.3.

Fluorescence turn-on response and selectivity profile of PO1. (A) Fluorescence emission response of 5 μM PO1 after the addition of 100 μM H2O2. Time points represent 0, 5, 15, 30, 45, and 60 min. (B) Fluorescence emission response of 5 μM PO1 after the addition of a variety of ROS. Bars represent relative responses at 0, 5, 15, 30, 45, and 60 min after addition of each ROS. Data shown are for 200 μM NO and 100 μM for all other ROS. Data were acquired at 25 °C in 20 mM HEPES, pH 7, with excitation at λ = 540 nm and emission was collected between 545 and 750 nm. Reactions were not complete at these time points. Reprinted with permission from Dickinson, Huynh, and Chang (2010). Copyright 2010 American Chemical Society.
While the boronate functionality displays little reactivity toward the majority of biologically relevant oxidizing species, highly reactive oxygen/nitrogen species such as peroxynitrite have been observed to react rapidly with aryl boronates (Sikora, Zielonka, Lopez, Joseph, & Kalyanaraman, 2009). However, we note that peroxynitrite and related species are typically generated at much lower concentrations and exhibit markedly shorter biological half-lives compared to hydrogen peroxide, which can offset differences in in vitro second-order rate constants. In experiments where these species may be added exogenously or produced endogenously in competition with hydrogen peroxide, use of a boronate-based probe for specific detection of hydrogen peroxide must be accompanied by appropriate control experiments (Section 4.3). These controls generally either reduce hydrogen peroxide levels with antioxidants or inhibit its direct enzymatic production to confirm the identity of hydrogen peroxide as the major ROS in a given system of interest.
Finally, the performance of these probes, depending on the fluorophore released, can also be affected by variations in local pH, and therefore it is important to employ buffering systems to maintain a stable pH. The oxidation of boronates to phenols by hydrogen peroxide itself is accelerated at higher pH (Kuivila, 1954), and since the final, deprotected form of the probe is a fluorescein or rhodol derivative, any effect of pH upon the behavior and properties of these fluorophores should be considered. Since fluoresceins are quenched at low pH, appropriate control experiments and adjustments to protocols should be made when imaging with these probes in an acidic environment, such as within the macrophage phagosome.
2.3. Selecting a probe
Choosing an appropriate probe for a particular application is critical, since each probe displays different properties that affect its performance, depending on the system of interest. A number of factors will guide probe selection, including the permeability of the probe or ease with which it diffuses through the cellular membrane or throughout an organism, trappability or extent to which the dye is retained within the cell, color of fluorescence, and targeting of the probe to specific subcellular locations. Trappable probes are effective for flow cytometry experiments, monolayer cell culture, and other experiments requiring media exchange with retention of the dye (Dickinson, Peltier, Stone, Schaffer, & Chang, 2011; Miller, Dickinson, & Chang, 2010; Tsien, 1981). Ester functional groups on the probes are cleaved by intracellular esterases, resulting in a charged molecule that cannot readily diffuse across the plasma membrane. Moreover, most of the boronate-based probes are less cell-permeable upon reaction with hydrogen peroxide, as oxidation of the boronate to a phenol increases the hydrophilicity of the dye. Nontrappable, highly permeable probes such as Peroxy Orange-1 (PO1) and PF2 are more suitable for tissue and in vivo applications than trappable probes (Harris et al., 2013). Additionally, a set of bioluminescent hydrogen peroxide probes has been developed (Van de Bittner, Bertozzi, & Chang, 2013; Van de Bittner, Dubikovskaya, Bertozzi, & Chang, 2010) that can be used in transgenic mice for whole animal experiments.
2.4. Use and storage of probes
To prepare the probes for imaging use, an aliquot of dry probe is dissolved in the designated amount of dry DMSO to give a stock solution of 5–10 mM. This DMSO stock (1–25 μL) is then diluted in 1 mL of an aqueous buffer such as PBS, HEPES, or Tris and mixed thoroughly by pipetting or vortexing to give working concentrations of 1–50 μM. Anhydrous DMSO must be used to dissolve the probe, as high water content may prevent full dissolution of the solid material and interfere with the preparation of a homogenous stock solution.
These probes are most stable in dry, solid form and can be stored for up to 6 months at −20 °C. Solid probe may be dissolved in a small amount of an organic solvent such as chloroform or anhydrous methanol (if soluble) and divided into small portions. The organic solvent can then be removed under reduced pressure in a vacuum desiccator, yielding dried individual aliquots. Once dissolved in anhydrous DMSO, stocks can be stored for 3–4 weeks at −80 °C. Freeze-thaw cycles should be avoided. Any aqueous solutions of boronate-based probes should be freshly prepared and used the same day, as hydrolysis of the boronate reduces permeability and sensitivity of the probe. Furthermore, the probes are sensitive to light and should be protected from photodegradation by storing aliquots and prepared solutions in the dark or covering them with aluminum foil.
3. EXAMPLES OF PROBES AND THEIR USAGE
3.1. PO1 used for fluorometry
3.1.1 Instrumentation and materials
Spectroscopy was performed in 20 mM HEPES buffer, pH 7.4. All solutions were prepared using ultrapure water from a purification system such as Barnstead Nanopure or Millipore. Fluorescence spectra were recorded on a Photon Technology International Quanta Master 4 L-format scanning spectrofluorometer (Lawrenceville, NJ) with an LPS-220B 75-W xenon lamp and power supply, A-1010B lamp housing with integrated igniter, switchable 814 photon-counting/analog photomultiplier detection unit, and MD5020 motor driver. Samples for emission measurements were contained in a quartz cuvette with a path length of 1 cm and width of 0.1 cm (1.5 mL volume, Starna, Atascadero, CA). Absorption spectra were recorded using a Varian Cary 50 spectrophotometer (Walnut Creek, CA).
Probe
PO1 stock solution (5 mM) in anhydrous DMSO.
H2O2. 100 mM aqueous stock made freshly by dissolving 11 μL of 30% H2O2 (Sigma-Aldrich) in 989 μL H2O.
3.1.2 In vitro time course assays
In order to study the response of a probe to 100 μM hydrogen peroxide over time, the dye was freshly diluted to a concentration of 5 μM in 3 mL of buffer in a 15-mL Falcon tube. The dye was mixed by vortexing to ensure a homogeneous solution. The diluted probe was then distributed by adding 1 mL volume to each of two microcentrifuge tubes, one for studying hydrogen peroxide and the other as a negative control. To one tube, 1 μL of 100 mM H2O2 solution was added. The solution was vortexed and rapidly transferred to the quartz cuvette, and absorption and fluorescence emission spectra were collected over time. To the second tube, 1 μL of water was added, and spectra were collected as described above.
For all in vitro fluorometry experiments, a positive and negative control should be done on each day using the same probe aliquot between samples to account for potential variation in environmental factors, aliquot quality, and reagent preparation. Reactions with hydrogen peroxide and other ROS are usually monitored on an hour or longer time scale, with conversion being incomplete after 1 h at room temperature when using 100 μM hydrogen peroxide. Excessive radiation should be avoided to prevent photobleaching of the dye, and excessive heat or very alkaline environments should be avoided due to risk of boronate hydrolysis.
This protocol yields samples with a final DMSO concentration of 0.1%. In experiments where a minimal volume of organic cosolvent is desired, reduced amounts of stock solution may be used or the concentration of the stock solution may be increased. However, care must be taken to ensure that sufficient organic cosolvent is present to prevent the probe from precipitating out of solution upon addition to aqueous buffer. All solutions should appear optically clear and without particulates.
3.1.3 Data processing and analysis
The integrated fluorescence intensity—the area under the fluorescence emission curve—was plotted against time (Fig. 2.3). Fluorescence intensity at a single point, typically the emission maximum, may also be used.
3.2. Trappable PF6-AM for use in flow cytometry experiments
3.2.1 Instrumentation and materials
Instrumentation
We performed flow cytometry experiments on either a Beckman-Coulter EPICS XV-MCL flow cytometer or a LSR Fortessa cell analyzer (BD Biosciences) equipped with a 488-nm laser. The data were analyzed with FlowJo.
Media
(1) Dulbecco’s modified Eagle medium (DMEM, Mediatech) with 10% heat-deactivated fetal bovine serum (FBS, HyClone) and 1% penicillin–streptomycin (Mediatech); (2) washing media: Dulbecco’s phosphate-buffered saline (DPBS, Invitrogen), DMEM containing no dye, serum, or antibiotics.
Probe
5 mM PF6-AM stock solution in anhydrous DMSO made the day of the experiment.
Exogenous H2O2
100 mM aqueous stock made by dissolving 11 μL of 30% H2O2 (Sigma-Aldrich) in 989 μL H2O.
Cell culture supplies
Standard supplies for mammalian cell culture.
3.2.2 Cell culture, dye loading, stimulation, and flow cytometry
Culture HeLa cells in DMEM with high glucose, GlutaMAX™ (Invitrogen, Carlsbad, CA), and 10% FBS in an incubator at 37 °C, 5% CO2. Passage the cells every 3–4 days at nearly 100% confluence. We typically assay cells with passage numbers of 4–20, both to ensure a stable, exponentially growing culture and to avoid cumulative changes at high passage numbers.
Two days before imaging
Passage the cells and plate in 35-mm tissue culture plates. At the time of the experiment, the confluence level was ca. 70%.
Approximately 1 h before assay
Wash the cells twice with DPBS, detach with 200 μL 0.05% trypsin (Invitrogen), quench the trypsin with 1 mL DMEM with 10% FBS, and collect the cells by centrifugation. Redissolve the cell pellets in 1 mL of 5 μM PF6-AM (from 5 mM stock solution in DMSO) in DPBS with calcium chloride and magnesium chloride and incubate the cell solution for 20 min in an incubator at 37 °C, 5% CO2. Divide the cell solution into two portions, one for treatment conditions and one for control conditions. Treat one cell suspension with 100–300 μM H2O2 and the other cell suspension with an equal volume of water. Incubate the cell solutions for 40 min in an incubator at 37 °C, 5% CO2. Analyze each cell solution by flow cytometry using excitation by a 488-nm laser and collection by a 525-mm band pass filter. Collect data for at least 10,000 cells for each condition using identical instrument settings.
3.2.3 Data processing and results
In order to exclude dead cells and debris from the final quantification, the forward scatter (FS) and side scatter (SS) results should be used to gate only on the live cell population in each dataset. It is critical to use an identical gate for each condition, and note any significant changes in cell morphology as indicated by FS/SS results upon any condition as these changes may indicate toxicity issues. Using the gate on the live cell population, a histogram should be generated for PF6-AM intensity. The average fluorescence intensity of each population can then be quantified and compared between conditions. This experiment can be performed in multiple biological replicates to generate statistics for the average fluorescence intensity of each condition. When detecting endogenous H2O2, control experiments coadministering antioxidants, inhibitors, or RNA interference (RNAi; Section 4.3) are particularly important in identifying the source of fluorescence turn-on.
3.3. Targeted MitoPY1 for fluorescence microscopy
3.3.1 Instrumentation and materials
Confocal microscope
Fluorescence microscopy studies were performed with a LSM 710 laser scanning confocal microscope (Carl Zeiss) with a 63 × oil objective lens, with Zen 2009 software (Carl Zeiss). MitoPY1 was excited using a 488-nm Ar laser, and emission was collected using a META detector between 527 and 580 nm. Hoechst 33342 was excited with a 405-nm diode laser, and emission was collected using a META detector between 450 and 500 nm. The cells were imaged at 37 °C throughout the course of the experiment. Data processing and analysis were conducted using ImageJ (National Institute of Health) or Zen 2009 software (Carl Zeiss).
Probe
MitoPY1 stock solution (5 mM) in anhydrous DMSO.
Mitochrondria marker
MitoTracker Deep Red (Invitrogen) stock solution (50 μM) in DMSO.
Nuclear stain
Hoechst 33342 (Sigma-Aldrich) stock solution (1 mM) in DMSO.
Exogenous H2O2
100 mM aqueous stock made by dissolving 11 μL of 30% H2O2 (Sigma-Aldrich) in 989 μL H2O.
3.3.2 Cell culture
HeLa cells were maintained and passaged using standard protocols. For imaging by fluorescence microscopy, cells were plated at 70–90% confluency in 0.5 mL DMEM supplemented with 10% FBS and 2 mM glutamine without phenol red in glass 4-well chamber slides or in a 24-well plate with 18-mm glass coverslips 1–2 days prior to imaging. HeLa cells will grow on uncoated glass. An appropriate coating should be used for plating other adherent cell lines to facilitate growth on glass.
3.3.3 Dye loading, imaging, and data analysis
A dry aliquot of MitoPY1 was allowed to reach room temperature, and then it was diluted in an appropriate volume of anhydrous DMSO to yield a 5 mM stock solution. An aqueous solution of 5 μM MitoPY1 in DPBS was prepared by adding 2 μL of the stock solution to 2 mL warm DPBS and mixing thoroughly by vortexing. The cell media was exchanged for the MitoPY1 solution and the cells incubated for 45 min at 37 °C. The probe solution was then removed and buffer replaced with warm DPBS containing 1 μM Hoechst and 25 nM MitoTracker Deep Red. At this point, H2O (control) or 100 μM H2O2 was added. The HeLa cells were then incubated for an additional 60 min at 37 °C before imaging.
Image analysis was performed in ImageJ or Zen 2009 software. Z-stacks and maximum intensity projections should be acquired for cells with variation in topology to ensure reproducible quantification of fluorescence. As seen in Fig. 2.4, hydrogen peroxide triggers a robust mitochondrial fluorescent enhancement in the MitoPY1 channel, without change in the MitoTracker Deep Red channel. Only images acquired under the same parameters can be compared during analysis, and analysis should be performed using a standardized method. This type of experiment allows for colocalization studies using a targeted fluorescent hydrogen peroxide probe (Dickinson, Lin, & Chang, in press).
Figure 2.4.

Confocal fluorescence images of exogenous H2O2 added to live HeLa cells with mitochondrial-targeted H2O2 probe MitoPY1. HeLa cells were loaded with 10 μM MitoPY1 in DPBS for 45 min at 37 °C. The media was then exchanged for fresh DPBS containing 25 nM Mitotracker Deep Red and 1 μM Hoechst. After the addition of either H2O (control) or 100 μM H2O2, the cells were incubated for 60 min at 37 °C. Control cells were then imaged with MitoPY1 (A), Mitotracker Deep Red (B), overlay of MitoPY1 (green), Mitotracker Deep Red (red), and Hoechst (blue) (C), or overlay of bright field and Hoechst (blue) (D) with 40 μm scale bar. H2O2-treated cells were imaged with MitoPY1 (E), Mitotracker Deep Red (F), overlay of MitoPY1 (green), Mitotracker Deep Red (red), and Hoechst (blue) (G), or overlay of bright field and Hoechst (blue) (H) with 40 μm scale bar. The region of H2O2-treated cells denoted in (E) is enlarged, showing MitoPY1 (I), Mitotracker Deep Red (J), overlay of MitoPY1 (green), Mitotracker Deep Red (red), and Hoechst (blue) (K), or overlay of bright field and Hoechst (blue) (L) with 10 μm scale bar. Reprinted with permission from Dickinson, Lin, and Chang (2013). Copyright 2013 Nature Publishing Group.
4. IMAGING ENDOGENOUS H2O2 USING FLUORESCENT PROBES
In this section, we provide examples to illustrate how H2O2 fluorescent probes allow for imaging endogenous H2O2 in two representative experimental systems. To demonstrate the simultaneous detection of two separate classes of ROS, dual-imaging experiments with macrophages were carried out using PO1, a red-shifted probe, and aminophenyl fluorescein (APF), a green probe that detects highly reactive oxygen species (hROS; Setsukinai, Urano, Kakinuma, Majima, & Nagano, 2003). The growth factor stimulation experiment was performed with adult hippocampal neural progenitor (AHP) cells using a cytosolic trappable probe, PF6-AM, to demonstrate the detection of H2O2 in a physiologically relevant cell line. As sample preparation is highly dependent on the specific biological questions being addressed, we focus on the methods most applicable to imaging H2O2 in a variety of systems. This section concludes with a discussion of control experiments to verify that the observed fluorescent response from the probes is hydrogen peroxide-dependent.
4.1. Imaging hydrogen peroxide and hROS simultaneously in macrophages
4.1.1 Materials and instrumentation
Media
(1) DMEM (Mediatech) with 10% heat-deactivated FBS (HyClone) and GlutaMAX™ (Invitrogen); (2) washing media: DPBS (Invitrogen), DMEM containing no dye, serum, or antibiotics.
Probes
5 mM PO1 and 5 mM APF (Invitrogen, A36003) stock solutions in anhydrous DMSO made the day of the experiment.
Exogenous H2O2 and HOCl
100 mM aqueous stocks made by dissolving 30% H2O2 (Sigma-Aldrich) or HOCl in H2O.
Cell culture supplies
Standard supplies for mammalian cell culture.
Instrumentation
We performed fluorescence imaging experiments with a LSM 510 or LSM 710 laser scanning confocal microscope (Carl Zeiss).
4.1.2 Cell culture, dye loading, stimulation, and imaging
RAW264.7 macrophages were cultured in DMEM containing high glucose with GlutaMAX™ and supplemented with 10% FBS. The cells were passaged every 3–4 days at nearly 100% confluence. We typically assay cells with passage numbers of 4–20, both to ensure a stable, exponentially growing culture and to avoid cumulative changes at high passage numbers.
Two days before imaging
The cells were passaged and plated in culture plates suitable for imaging. At the time of the experiment, the confluence level was ca. 70%.
Approximately 1 h before imaging
The culturing media of the RAW264.7 macrophages was replaced with DPBS containing 5 μM PO1 and 5 μM APF and the cells incubated for 20 min at 37 °C. At least one well of cells for each experimental condition was prepared to be imaged simultaneously. After the 20 min incubation, the cells were treated with either experimental or control conditions. For example, one well was treated with H2O as a control, one well with 100 μM H2O2, one well with 100 μM HOCl, and one well with 1 μg/mL phorbal myristate acetate (PMA), a small molecule activator of protein kinase C. The cells were incubated at 37 °C for another 20 min.
Immediately before imaging
The dye-containing media and stimulants were removed from culture dishes. The cells were washed twice with 2 mL of prewarmed DPBS and bathed in 2 mL of dye- and serum-free DMEM for use during imaging.
4.1.3 Imaging and results
Excitation of PO1- and APF-loaded cells at 488 and 543 nm was carried out with Ar and HeNe lasers, respectively, and emission was collected using a META detector between 495–538 and 548–602 nm, respectively, using sequential scans. Image analysis was performed in ImageJ. Additional types of microscopes can be used, but the settings will be specific to the given microscope. In general, care must be taken to find the optimal settings that permit detection of both the PO1 and APF channels independently from one another.
In order to compare the fluorescence intensities before and after addition of exogenous H2O2, the images must be acquired and analyzed with identical parameters. As seen in Fig. 2.5, the addition of H2O2 resulted in an increase in fluorescent intensity in the PO1 channel, while the addition of HOCl, a hROS detected by APF, was detected only in the APF channel. Activation of the macrophages by PMA, which leads to a robust respiratory burst response and endogenous production of a variety of ROS, including H2O2 and HOCl, resulted in punctate staining in both the PO1 and APF channels. Experiments such as these permit the measurement of multiple classes of endogenously produced ROS simultaneously in live specimens.
Figure 2.5.
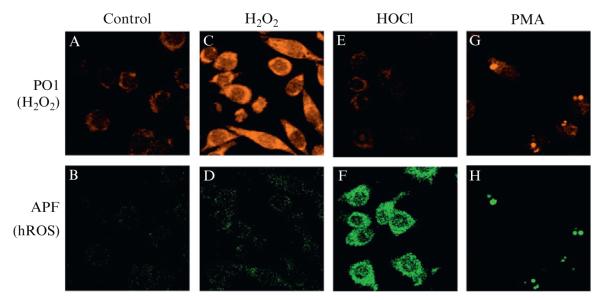
Confocal fluorescence images of PMA-induced ROS production in live RAW264.7 macrophages with PO1 and APF simultaneously. Macrophages incubated with 5 μM PO1 and 5 μM APF for 40 min at 37 °C and imaged for PO1 (A) and APF (B). Macrophages incubated with 5 μM PO1 and 5 μM APF for 40 min at 37 °C with 50 μM H2O2 added for the final 20 min and imaged for PO1 (C) and APF (D). Macrophages incubated with 5 μM PO1 and 5 μM APF for 40 min at 37 °C with 100 μM HOCl added for the final 20 min and imaged for PO1 (E) and APF (F). Macrophages incubated with 5 μM PO1 and 5 μM APF for 40 min at 37 °C with 1 μg/mL PMA added for the final 20 min and imaged for PO1 (G) and APF (H). Reprinted with permission from Dickinson, Huynh, and Chang (2010). Copyright 2010 American Chemical Society.
4.2. Growth factor stimulation
4.2.1 Materials and instrumentation
Media
(1) DMEM (Mediatech) with 10% heat-deactivated FBS (HyClone) and GlutaMAX™ (Invitrogen); (2) washing media: DPBS (Invitrogen), DMEM containing no dye, serum, or antibiotics.
Probes
5 mM PF6-AM stock solution in anhydrous DMSO made the day of the experiment.
Exogenous H2O2
100 mM aqueous stock made by dissolving 11 μL of 30% H2O2 (Sigma-Aldrich) in 989 μL H2O.
Cell culture supplies
Standard supplies for mammalian cell culture.
Instrumentation
We performed fluorescence imaging experiments with a LSM510 or LSM 710 laser scanning confocal microscope (Zeiss).
4.2.2 Cell culture, dye loading, stimulation, and imaging
AHP cells were isolated from the hippocampi of 6-week-old female Fisher 344 rats as described (Palmer, Markakis, Willhoite, Safar, & Gage, 1999). Cells were cultured on tissue culture polystyrene coated with poly-ornithine and 5 μg/mL of laminin (Invitrogen) and grown in (DMEM)/F-12 (1:1) high-glucose medium (Invitrogen) containing N-2 supplement (Invitrogen) and 20 ng/mL recombinant human FGF-2 (Peprotech). Cells for H2O2 production assays were grown in home-made incubators ((2006). Nature Protocols, 1, 2088) under an atmosphere of 2% O2/5% CO2/94% N2.
The cells were passaged every 2–3 days at ca. 80% confluence. We typically assay cells with passage numbers of 10–30, both to ensure a stable, exponentially growing culture and to avoid cumulative changes at high passage numbers.
Two days before imaging
The cells were passaged and plated in culture plates suitable for imaging. At the time of the experiment, the confluence level was ca. 50%.
12 h before imaging
The culturing media was replaced with (DMEM)/F-12 (1:1) high-glucose medium containing N-2 supplement without FGF-2.
Approximately 1 h before imaging
The culturing media of the AHPs was replaced with (DMEM)/F-12 (1:1) high-glucose medium with N-2 supplement and 5 μM PF6-AM. Cells were incubated for 20 min at 37 °C. At least one well of cells was prepared for each experimental condition to be imaged simultaneously. After the 30 min incubation, the cells were treated with either experimental or control conditions. For example, one well was treated with H2O as a control and one well with 20 ng/mL FGF-2. The cells were incubated at 37 °C for 30 min, then imaged.
Immediately before imaging
The dye-containing media and stimulants were removed from the culture dishes. The cells were washed twice with 2 mL of prewarmed (DMEM)/F-12 (1:1) high-glucose medium and bathed in 2 mL of dye- and serum-free (DMEM)/F-12 (1:1) high-glucose medium for use during imaging.
4.2.3 Imaging and results
Confocal fluorescence imaging studies were performed with a Zeiss LSM510 NLO Axiovert 200 laser scanning microscope and a 40× Achroplan IR water-immersion objective lens. Excitation of PF6-AM-loaded cells at 488 nm was carried out with an Ar laser and emission was collected using a META detector between 501 and 554 nm. Image analysis was performed in ImageJ. Additional types of microscopes can be used, but the settings should be specific to the given microscope.
In order to compare the fluorescence intensities before and after addition of FGF-2, the images must be acquired and analyzed with identical parameters. As seen in Fig. 2.6, the addition of FGF-2 results in an increase in fluorescent intensity. This increase can be attenuated through the use of inhibitors, antioxidants, or genetic manipulation of the molecular source (Fig. 2.6) as outlined in Section 4.3.
Figure 2.6.
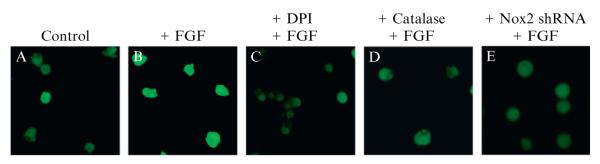
FGF-2 stimulated H2O2 production in AHPs. After FGF-2 starvation, AHPs were loaded with 5 μM PF6-AM for 30 min, stimulated with H2O carrier (A) or 20 ng/mL FGF-2 (B), and then imaged. For DPI treatment, cells were preincubated in media containing 5 μM DPI before FGF-2 stimulation (C). For catalase addition, AHPs were transfected with either Catalase or control vector prior to FGF-2 stimulation (D). For Nox2 shRNA addition, AHPs were transfected with either Nox2-shRNA or control vector prior to treatment with FGF-2 (E).
4.3. Controls utilizing inhibitors of H2O2
In order to confirm that signal differences detected from a particular boronate probe are indeed due to endogenous H2O2 production, it is critical to perform a variety of additional control experiments. The specific controls will depend upon the cell or tissue type under examination and the purported source of the H2O2 signal. In this section, we list selected control experimental conditions that may be useful to validate the H2O2 signal and identify its source in a particular biological system.
The production of H2O2 may be suppressed via chemical means, enzyme scavenging, or gene silencing. Diphenylene iodonium (DPI) is a general flavin inhibitor that inhibits the most common intracellular sources of H2O2, including the mitochondrial respiratory chain and Noxs. N-acetylcysteine (NAC) is a thiol-based general antioxidant that increases the redox capacity of the cell. Catalase is a highly reactive and specific H2O2 scavenger enzyme. After validating the endogenous H2O2 production by the aforementioned pharmacological and molecular approaches, genetic manipulation of the putative source using RNAi will provide further verification of the cellular machinery responsible for the observed H2O2.
DPI
Incubate the cells or tissue samples with 5–10 μM DPI chloride (Sigma-Aldrich, D2926) from a 5 mM stock in DMSO for 20–40 min prior to stimulation. The DPI can be administered simultaneously with fluorescent probes. For systems sensitive to chloride, a DPI sulfate salt is also available.
NAC
Incubate cells with 1–10 mM NAC from a 1 M stock for 40 min prior to stimulation. The NAC can be administered simultaneously with the fluorescent probes.
Catalase
Catalase can be expressed from a pCMV6 mammalian expression vector (Origene) and the empty vector, without the catalase gene, can be utilized as a control. The vectors should be delivered to the cell or tissue type under study via the DNA delivery system commonly used for that system. After transfection, the cells should be allowed to recover and produce catalase for at least 24 h prior to analysis.
RNAi
Short-hairpin RNA (shRNA) constructs or small-interfering RNA (siRNA) directed against the putative source of H2O2 can be designed by OligoEngine or other design tools, along with relevant control vectors. Each siRNA oligomer or shRNA expression vector should be validated using Western Blot to confirm the transfection has sufficiently knocked down the target protein.
Performing a series of control experiments can allow for the elucidation of H2O2 generation pathways. In the previous section, we discussed the use of PF6-AM as an imaging agent to detect endogenous H2O2 production via FGF-2 stimulation in AHPs. AHPs can be transfected with vectors producing shRNA directed against either Nox2, the source of FGF-2 stimulated H2O2 production, or Nox3, a related isoform of the Nox proteins. After transfection, the cells were allowed to recover for 24 h and then imaged for H2O2 production.
In order to confirm that the changes in signal from PF6-AM in the AHPs are due to intracellular H2O2 production and to determine the specific molecular source, a variety of control imaging experiments were performed. Stimulation of PF6-AM-loaded AHPs with 20 ng/mL FGF-2 results in an increase in PF6-AM fluorescence intensity (Fig. 2.6A and B). However, pretreatment with 5 μM DPI, expression of catalase, or expression of Nox2-directed shRNA all abolish the signal increase compared to relevant control experiments (Fig. 2.6C–E). This combination of experiments confirms that the changes in PF6-AM signal are due to (1)intracellular enzymatic activity (validated by DPI inhibition), (2) H2O2 (validated by catalase expression), and (3) Nox2-dervied ROS (validated by Nox2 shRNA knockdown).
5. CONCLUSIONS
Installation of the boronate functionality on a variety of fluorescent scaffolds has provided access to a diverse selection of fluorescent hydrogen peroxide probes with a range of spectroscopic properties and in cellulo characteristics. Chemical principles have allowed for the design of molecules with traits tailored for different types of biological applications, including in vitro assays and fluorescence microscopy of monolayer cell culture which can elucidate the spatial and temporal distribution of hydrogen peroxide in cells. The combination of these chemoselective tools with existing and newly developed methods for the study of hydrogen peroxide in living systems enables further exploration of novel biological systems and the discovery and elucidation of the essential roles for hydrogen peroxide in a variety of signaling and stress pathways.
ACKNOWLEDGMENTS
We thank the University of California, Berkeley, the Packard Foundation, and the National Institute of General Medical Sciences (NIH GM 79465), as well as Amgen, Astra Zeneca, and Novartis for funding our work on redox imaging probes. B. C. D. is a fellow of the Jane Coffin Childs Memorial Fund for Medical Research. C. J. C. is an investigator with the Howard Hughes Medical Institute.
REFERENCES
- Albers AE, Okreglak VS, Chang CJ. A FRET-based approach to ratiometric fluorescence detection of hydrogen peroxide. Journal of the American Chemical Society. 2006;128:9640–9641. doi: 10.1021/ja063308k. [DOI] [PubMed] [Google Scholar]
- Albrecht SC, Barata AG, Grosshans J, Teleman AA, Dick TP. In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metabolism. 2011;14:819–829. doi: 10.1016/j.cmet.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause K-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiological Reviews. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Belousov VV, Fradkov AF, Lukyanov KA, Staroverov DB, Shakhbazov KS, Terskikh AV, et al. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nature Methods. 2006;3:281–286. doi: 10.1038/nmeth866. [DOI] [PubMed] [Google Scholar]
- Bilan DS, Pase L, Joosen L, Gorokhovatsky AY, Ermakova YG, Gadella TWJ, et al. HyPer-3: A genetically encoded H2O2 probe with improved performance for ratiometric and fluorescence lifetime imaging. ACS Chemical Biology. 2013;8:535–542. doi: 10.1021/cb300625g. [DOI] [PubMed] [Google Scholar]
- Chan J, Dodani SC, Chang CJ. Reaction-based small-molecule fluorescent probes for chemoselective bioimaging. Nature Chemistry. 2012;4:973–984. doi: 10.1038/nchem.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MCY, Pralle A, Isacoff EY, Chang CJ. A selective, cell-permeable optical probe for hydrogen peroxide in living cells. Journal of the American Chemical Society. 2004;126:15392–15393. doi: 10.1021/ja0441716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho DG, Sessler JL. Modern reaction-based indicator systems. Chemical Society Reviews. 2009;38:1647–1662. doi: 10.1039/b804436h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarnik AW. Chemical communication in water using fluorescent chemosensors. Accounts of Chemical Research. 1994;27:302–308. [Google Scholar]
- D’Autréaux B, Toledano MB. ROS as signaling molecules: Mechanisms that generate specificity in ROS homeostasis. Nature Reviews Molecular Cell Biology. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- Dickinson BC, Chang CJ. A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. Journal of the American Chemical Society. 2008;130:9638–9639. doi: 10.1021/ja802355u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nature Chemical Biology. 2011;7:504–511. doi: 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson BC, Huynh C, Chang CJ. A palette of fluorescent probes with varying emission colors for imaging hydrogen peroxide signaling in living cells. Journal of the American Chemical Society. 2010;132:5906–5915. doi: 10.1021/ja1014103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson BC, Lin VS, Chang CJ. Preparation and use of MitoPY1, a synthetic fluorophore for imaging hydrogen peroxide in mitochondria of live biological specimens. Nature Protocols. doi: 10.1038/nprot.2013.064. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson BC, Peltier J, Stone D, Schaffer DV, Chang CJ. Nox2 redox signaling maintains essential cell populations in the brain. Nature Chemical Biology. 2011;7:106–112. doi: 10.1038/nchembio.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson BC, Tang Y, Chang Z, Chang CJ. A nuclear-localized fluorescent hydrogen peroxide probe for monitoring sirtuin-mediated oxidative stress responses in vivo. Chemical Biology. 2011;18:943–948. doi: 10.1016/j.chembiol.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Hu M, Fan J, Peng X. Fluorescent chemodosimeters using ‘mild’ chemical events for the detection of small anions and cations in biological and environmental media. Chemical Society Reviews. 2012;41:4511–4535. doi: 10.1039/c2cs00004k. [DOI] [PubMed] [Google Scholar]
- Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson’s disease: Molecular mechanisms and pathophysiological consequences. EMBO Journal. 2012;31:3038–3062. doi: 10.1038/emboj.2012.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisard M, Ravussin E. Energy metabolism and oxidative stress: Impact on the metabolic syndrome and the ageing process. Endocrine. 2006;29:27–32. doi: 10.1385/ENDO:29:1:27. [DOI] [PubMed] [Google Scholar]
- Giorgio M, Trinei M, Migliaccio E, Peliccil PG. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nature Reviews Molecular Cell Biology. 2007;8:722–728. doi: 10.1038/nrm2240. [DOI] [PubMed] [Google Scholar]
- Harris JM, Esain V, Frechette GM, Harris LJ, Cox AG, Cortes M, et al. Glucose Metabolism Impacts the Spatiotemporal Onset and Magnitude of HSC Induction in Vivo. Blood. 2013;121:2483–2493. doi: 10.1182/blood-2012-12-471201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks WP, Carriulo JC. Reactivity of nucleophilic reagents towards esters. Journal of the American Chemical Society. 1960;82:1778–1786. [Google Scholar]
- Juillerat A, Gronemeyer T, Keppler A, Gendreizig S, Pick H, Vogel H, et al. Directed evolution of O6-alkylguanine-DNA alkyltransferase for efficient labeling of fusion proteins with small molecules in vivo. Chemical Biology. 2003;10:313–317. doi: 10.1016/s1074-5521(03)00068-1. [DOI] [PubMed] [Google Scholar]
- Jun ME, Roy B, Ahn KH. Turn-on’ fluorescent sensing with ‘reactive’ probes. Chemical Communications. 2011;47:7583–7601. doi: 10.1039/c1cc00014d. [DOI] [PubMed] [Google Scholar]
- Keppler A, Kindermann M, Gendreizig S, Pick H, Vogel H, Johnsson K. Labeling of fusion proteins of O6-alkylguanine-DNA alkyltransferase with small molecules in vivo and in vitro. Methods. 2004;32:437–444. doi: 10.1016/j.ymeth.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Kuivila HG. Electrophilic displacement reactions: Kinetics of the reaction between hydrogen peroxide and benzeneboronic acid. Journal of the American Chemical Society. 1954;76:870–874. [Google Scholar]
- Kuivila HG, Armour AG. Electrophilic displacement reactions: Effects of substituents on rates of reactions between hydrogen peroxide and benzeneboronic acid. Journal of the American Chemical Society. 1957;79:5659–5662. [Google Scholar]
- Kuivila HG, Wiles RA. Electrophilic displacement reactions: Catalysis by chelating agents in the reaction between hydrogen peroxide and benzeneboronic acid. Journal of the American Chemical Society. 1955;77:4830–4834. [Google Scholar]
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nature Reviews Immunology. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neuro-degenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Lippert AR, Van de Bittner GC, Chang CJ. Boronate oxidation as a bio-orthogonal reaction approach for studying the chemistry of hydrogen peroxide in living systems. Accounts of Chemical Research. 2011;44:793–804. doi: 10.1021/ar200126t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EW, Albers AE, Pralle A, Isacoff EY, Chang CJ. Boronate-based fluorescent probes for imaging cellular hydrogen peroxide. Journal of the American Chemical Society. 2005;127:16652–16659. doi: 10.1021/ja054474f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EW, Dickinson BC, Chang CJ. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15681–15686. doi: 10.1073/pnas.1005776107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EW, Tulyathan O, Isacoff EY, Chang CJ. Molecular imaging of hydrogen peroxide produced for cell signaling. Nature Chemical Biology. 2007;3:263–267. doi: 10.1038/nchembio871. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Holmgren A, Larsson N-G, Halliwell B, Chang CJ, Kalyanaraman B, et al. Unraveling the biological roles of reactive oxygen species. Cell Metabolism. 2011;13:361–366. doi: 10.1016/j.cmet.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson KJ, Klomsiri C, Codreanu SG, Soito L, Liebler DC, Rogers LC, et al. Use of dimedone-based chemical probes for sulfenic acid detection: Methods to visualize and identify labeled proteins. Methods in Enzymology. 2010;473:95–115. doi: 10.1016/S0076-6879(10)73004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer TD, Markakis EA, Willhoite AR, Safar F, Gage FH. Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. Journal of Neuroscience. 1999;19:8487–8497. doi: 10.1523/JNEUROSCI.19-19-08487.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen CE, Carroll KS. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chemical Biology. 2010;5:47–62. doi: 10.1021/cb900258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang DT, Kim JS. Fluoro- and chromogenic chemodosimeters for heavy metal ion detection in solution and biospecimens. Chemical Reviews. 2010;110:6280–6301. doi: 10.1021/cr100154p. [DOI] [PubMed] [Google Scholar]
- Ren Y, Yamataka H. The α-effect in gas-phase SN2 reactions: Existence and the origin of the effect. Journal of Organic Chemistry. 2007;72:5660–5667. doi: 10.1021/jo070650m. [DOI] [PubMed] [Google Scholar]
- Rhee SG. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- Setsukinai K, Urano Y, Kakinuma K, Majima HJ, Nagano T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. Journal of Biological Chemistry. 2003;278:3170–3175. doi: 10.1074/jbc.M209264200. [DOI] [PubMed] [Google Scholar]
- Sikora A, Zielonka J, Lopez M, Joseph J, Kalyanaraman B. Direct oxidation of boronates by peroxynitrite: Mechanism and implications in fluorescence imaging of peroxynitrite. Free Radical Biology & Medicine. 2009;47:1401–1407. doi: 10.1016/j.freeradbiomed.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikun D, Albers AE, Chang CJ. A dendrimer-based platform for simultaneous dual fluorescence imaging of hydrogen peroxide and pH gradients produced in living cells. Chemical Science. 2011;2:1156–1165. [Google Scholar]
- Srikun D, Albers AE, Nam CI, Iavarone AT, Chang CJ. Organelletargetable fluorescent probes for imaging hydrogen peroxide in living cells via SNAP-tag protein labeling. Journal of the American Chemical Society. 2010;132:4455–4465. doi: 10.1021/ja100117u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikun D, Miller EW, Domaille DW, Chang CJ. An ICT-Based approach to ratiometric fluorescence imaging of hydrogen peroxide produced in living cells. Journal of the American Chemical Society. 2008;130:4596–4597. doi: 10.1021/ja711480f. [DOI] [PubMed] [Google Scholar]
- Stone JR, Yang S. Hydrogen peroxide: A signaling messenger. Antioxid Redox Signaling. 2006;8:243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- Tsien RY. A non-disruptive technique for loading calcium buffers and indicators into cells. Nature. 1981;290:527–528. doi: 10.1038/290527a0. [DOI] [PubMed] [Google Scholar]
- Urano Y, Kamiya M, Kanda K, Ueno T, Hirose K, Nagano T. Evolution of fluorescein as a platform for finely tunable fluorescence probes. Journal of the American Chemical Society. 2005;127:4888–4894. doi: 10.1021/ja043919h. [DOI] [PubMed] [Google Scholar]
- Van de Bittner GC, Bertozzi CR, Chang CJ. A strategy for dual-analyte luciferin imaging: In vivo bioluminescence detection of hydrogen peroxide and caspase activity in a murine model of acute inflammation. Journal of the American Chemical Society. 2013;135:1783–1795. doi: 10.1021/ja309078t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Bittner GC, Dubikovskaya EA, Bertozzi CR, Chang CJ. In vivo imaging of hydrogen peroxide production in a murine tumor model with achemoselective bioluminescent reporter. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21316–21321. doi: 10.1073/pnas.1012864107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Molecular Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nature Chemical Biology. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- Xun Z, Rivera-Sánchez S, Ayala-Peña S, Lim J, Budworth H, Skoda EM, et al. Targeting of XJB-5-131 to mitochondria suppresses oxidative DNA damage and motor decline in a mouse model of Huntington’s Disease. Cell Reports. 2012;2:1137–1142. doi: 10.1016/j.celrep.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radical Biology & Medicine. 2002;33:337–349. doi: 10.1016/s0891-5849(02)00905-x. [DOI] [PubMed] [Google Scholar]