

Abstract
Introduction
Sedentarism, also termed physical inactivity, is an independent risk factor for cardiovascular diseases. Mechanisms thought to be involved include insulin resistance, dyslipidemia, hypertension, and increased inflammation. It is unknown whether changes in vascular and endothelial function also contribute to this excess risk. We hypothesized that short-term exposure to inactivity would lead to endothelial dysfunction, arterial stiffening and increased vascular inflammation.
Methods
Five healthy subjects (4 males and 1 female) underwent 5 days of bed rest (BR) to simulate inactivity. Measurements of vascular function [flow-mediated vasodilation (FMD) to evaluate endothelial function; applanation tonometry to assess arterial resistance], inflammation and metabolism were made before BR, daily during BR and after 2 recovery days. Subjects maintained an isocaloric diet throughout.
Results
Bed rest led to significant decreases in brachial artery and femoral artery FMD [Brachial: 11 ± 3% pre-BR vs. 9 ± 2% end-BR, P=0.04; Femoral: 4 ± 1% vs. 2 ± 1%, P=0.04]. The central augmentation index increased with BR [−4 ± 9% vs. 5 ± 11%, P=0.03]. Diastolic blood pressure (DBP) increased [58 ± 7 mmHg vs. 62 ± 7 mmHg, P=0.02], while neither systolic blood pressure nor heart rate changed. 15-HETE, an arachidonic acid metabolite, increased but the other inflammatory and metabolic biomarkers were unchanged.
Conclusions
Our findings show that acute exposure to sedentarism results in decreased endothelial function, arterial stiffening, increased DBP, and an increase in 15-HETE. We speculate that inactivity promotes a vascular “deconditioning” state characterized by impaired endothelial function, leading to arterial stiffness and increased arterial tone. Although physiologically significant, the underlying mechanisms and clinical relevance of these findings need to be further explored.
INTRODUCTION
Sedentarism, also known as habitual physical inactivity, is proposed to be an important and independent contributor to atherosclerosis and cardiovascular disease,1, 2 as well as to other chronic conditions.3 Sedentary behavior, by most definitions, refers to activities that do not increase energy expenditure substantially above the resting level, such as sleeping, sitting, lying down, and watching television and other variants of screen-based entertainment.4 In 2007, less than half of US adults met the recommended physical activity standards.5 Since prevalence of sedentary living continues to rise,6 understanding the physiologic effects of physical inactivity and how it contributes to increased cardiovascular risk is crucial.
Bed rest (BR) has previously been employed as a model to study the effects of sedentarism. Prior BR studies demonstrated that prolonged inactivity leads to reductions in conduit artery diameter,7 decreased reactive hyperemia (RH),8 development of insulin resistance,9 type 2 diabetes,10 up-regulation of the renin-angiotensin axis11 and possibly, vascular dysfunction.12 Brachial artery flow-mediated dilatation (FMD) and arterial stiffness measured by arterial tonometry are used to assess vascular function and when they are impaired, they have been independently associated with increased cardiovascular risk.13–16 The effects of a short exposure to sedentarism on vascular function are poorly understood. Furthermore, an inflammatory response is implicated in the setting of vascular dysfunction and injury. Recent studies have shown that levels of certain pro-resolution lipid mediators, derived from fatty acid components of the red blood cell membrane, may suggest active resolution of inflammation.17–19 We hypothesized that short-term exposure to sedentarism in healthy subjects leads to endothelial dysfunction, an increase in arterial stiffness and an increase in inflammation. To test this hypothesis, we used a bed rest (BR) model in young healthy subjects.
METHODS
We selected a 5-day period for BR because our intent was to understand the effects of an acute, short-term period of sedentarism on vascular function and inflammatory parameters.
Subjects
Four healthy male subjects (age 22 ± 2) and one healthy female subject (age 23) were recruited. Screening procedures included a history and physical examination, 12-lead electrocardiogram, complete blood count with differential, chemistry profile, lipid profile, toxicology screen, β-human chorionic gonadotropin (in the woman), and psychological evaluation. Subjects were non-smokers and received no medication before the study. The exclusion criteria included documented peripheral arterial disease, vasculitis, evidence of active infection, other concurrent significant illness within 30 days of study initiation, a history or evidence of hypertension, coronary artery disease, diabetes, renal insufficiency, thyroid disease, hepatitis, anemia, current pregnancy, psychiatric disorder, obesity, hyperlipidemia, and alcohol or drug abuse. Additional exclusion criteria included known sleep disorders, shift work, and trans-meridian travel within the 6 months prior to starting the study. The female subject stopped taking oral or injected contraceptive agents 6 months prior to the beginning of the experiment and was not pregnant during conduction of the studies.
Protocol
The protocol consisted of three phases: pre-bed rest (BR), BR, and the 2-day post-BR recovery period (Figure 1). Measurements of vascular function and inflammation were made at five time points: pre-BR, Days 1, 3, and 5 (end-BR, last day), and 2 days after completion of BR (recovery). The remaining variables—including metabolic biomarkers, bone density, and body composition—were measured at the pre-BR and end-BR time points. Pre-BR included an equilibration period of 55 hours, during which subjects were admitted in a fasting state to the Clinical Research Center (CRC) and started on an isocaloric diet containing 200 mEq of sodium, 100 mEq of potassium, 1,000 mg of calcium, and 2,500 mL of fluid. BR was begun after the equilibration period and lasted 5 days; during this period, subjects wore prophylactic anti-embolism stockings. Sleep-wake cycles remained constant throughout BR, with each subject attaining 8 hours of sleep each day between 11:30 pm and 7:30 am. Subjects were confined to bed for the entire BR period. They were allowed to lie on their sides, in a supine position on their backs or in a prone position on their abdomens. They voided and defecated in the supine position. They ate meals while lying on their sides, propped up on one elbow. Smoking, alcohol, and caffeine were not permitted during the experimental period. The end-BR phase began immediately after BR and lasted 2 days, during which time subjects resumed ambulatory activities at the CRC, at the end of which testing took place. Various physiologic measurements were assessed pre-BR, during, at the completion of BR, and 2 days after the end of BR (recovery), including blood analysis, heart rate, blood pressure, brachial artery flow-mediated vasodilation ultrasound (FMD) and applanation tonometry. Dual-energy x-ray absorptiometry (DEXA) was completed during the pre-BR and again as soon as BR was completed. The study protocol was approved in advance by the Committee on Human Research (CHR) of the University of California, San Francisco (UCSF). Each subject provided written informed consent before study commencement.
Fig 1. 5-day Bed Rest Protocol.

FMD = measure of endothelium-dependent flow-mediated dilatation.
HOMA-IR= homeostatic model assessment of insulin resistance.
DEXA= Dual-energy x-ray absorptiometry.
Demographic Data
Demographic data, including age, race, gender, waist-hip ratio, body mass index, prior tobacco use and alcohol exposure, were recorded and are presented in Table 1.
Table 1.
Baseline characteristics of the study population.
| N=5 | Mean ± SD or n (%) |
|---|---|
| Age (years) | 22 ± 2 |
| Gender (male) | 4 (80) |
| Race (% subjects) | |
| Caucasian | 2 (40) |
| Hispanic | 1 (20) |
| Mixed ethnicity (Hispanic/Asian) | 2 (40) |
| Current tobacco use (% subjects) | 0 (0) |
| Occasional alcohol intake (% subjects) | 5 (100) |
|
General Laboratory Values
| |
| Cholesterol, total (mg/dL) | 161 ± 12 |
| HDL-C (mg/dL) | 59 ± 14 |
| LDL-C (mg/dL) | 81 ± 11 |
| Triglycerides (mg/dL) | 110 ± 50 |
| Prebeta-1 HDL (mg/dL) | 6 ± 2 |
| Glucose (mg/dL) | 87 ±5 |
| Insulin (mg/dL) | 6 ± 2 |
| Serum creatinine (mg/dL) | 1 ± 0.1 |
|
Anthropometric Measures
| |
| Height (cm) | 178 ± 9 |
| Weight (kg) | 76 ± 14 |
| BMI (kg/m2) | 24 ± 3 |
| Waist to Hip ratio | 1 ± 0.1 |
Hemodynamics and Vascular Reactivity of Brachial and Femoral Arteries
Systolic blood pressure (SBP), diastolic blood pressure (DBP), and heart rate (HR) were recorded by an indirect sphygmomanometer daily. To evaluate vascular function, brachial and superficial femoral artery (SFA) FMD was performed according to current guidelines and standards.20, 21 The specific protocol used in our laboratory included the following: subjects were asked to be fasting (≥ 8 hours) and free of nicotine (≥ 4 hours). A history of recent medications was recorded. Prior to the measurements, subjects rested for ten minutes in a supine position in a darkened room at 23°C. For brachial artery measurements, each subject’s right arm was extended onto a movement-constraining pillow with the palmar aspect oriented anteriorly. A 5-cm tourniquet blood pressure cuff was placed on the upper arm proximal to the insertion of the deltoid. For SFA measurements, the blood pressure cuff was placed on the upper right thigh distal to the region of the femoral triangle and the ultrasound probe was placed approximately near the junction of sartorius and vastus medialis. The length of the brachial and femoral arteries was surveyed by B-mode ultrasound (Philips IU 22) using a broadband linear array transducer with a 3–12 MHz range (Philips L12-3) until a straight segment with a visible registration structure could be located. The probe was oriented so that visibility of each artery was at least 3-cm deep to the surface of the skin, the focus aligned with the deep boundary of the vessel and clearly-demarcated intima/lumen boundaries were visible.
Prior to cuff inflation, the baseline diameter of the blood vessel was recorded for 60 seconds using EKG-gated image capture software (Brachial Imager, Medical Imaging Applications LLC, Coralville, IA). Baseline blood-flow velocity was measured by Doppler and recorded for 60 seconds using an insonation angle of 60°. The doppler sample gate was positioned to cover the center, but not the edges, of the lumen. The probe was not moved between measurements.
The blood pressure cuff was inflated to either 250 mm Hg or 50 mm Hg above the subject’s SBP, whichever was greater, for a period of 5 minutes. Recording of the B-mode images was begun 10 seconds prior to cuff release. Blood-flow velocity was assessed for a period of 30 seconds post-cuff release using the methods described above. B-mode images were recorded for 3 minutes post-cuff release.
Analysis of the acquired images was performed using continuous edge-detection software (Brachial Analyzer, Medical Imaging Applications LLC, Coralville, IA). Baseline diameter was recorded as the mean of 60 seconds of data. From hyperemia recordings, the exact moment of cuff release was noted. Hyperemia diameter was calculated using a pre-determined time window (55–65 seconds post-cuff release). FMD% was calculated as ([60s Hyperemia diameter-Average Baseline diameter]/Average Baseline diameter)*100. Additional vascular reactivity measurements obtained from the software output included blood flow rate at baseline and with reactive hyperemia and shear stress.
Time-averaged velocity measurements and velocity-time integrals were obtained using the peak-velocity method. At baseline, average velocity was obtained from the mean of 60 seconds of data. Velocity of the hyperemia stimulus phase was calculated as the mean velocity of the first four heart beats after cuff-release. Both the mean velocity and velocity time integral were recorded.
Quality control was assessed at each point during recording of measurements. Image quality was evaluated by a second observer and graded on a 6-point scale, which included registration of structure (landmark), horizontally-directed artery, correct longitudinal alignment, a clearly visualized near-wall intimal medial thickness (IMT) and far-wall IMT and at least 5-mm of a clearly-visualized artery.
Tonometry
Measurements of arterial stiffness were performed with an applanation tonometer (SphygmoCor CP Tonometry System, Itasca, IL) and the provided analysis software. After measuring the subject’s blood pressure and applying EKG leads, the applanation tonometer probe was placed over the subject’s radial, carotid and femoral arteries for approximately 3 minutes at each site to capture pulse wave tracings of the artery of interest. The tracings were used in the Fourier Transform algorithm to calculate several parameters, including the augmentation index, ejection duration and pulse wave velocity between any two sites in the vasculature.
Bone Mineral Density and Body Fat Composition
DEXA scans assessed pre- and end-BR total BMD, as well as regional BMD: spine, ribs, pelvis, and legs. Bodily fat mass distribution was also obtained from DEXA scans [GE Healthcare Lunar, Madison, WI].
Blood Analysis
Blood samples were collected in a fasting state at 9:00 am after the subject had remained supine overnight on the last day of the pre-BR ambulatory baseline period, on days 1, 3 and 5 (end) of BR, and after 2 days of recovery.
Inflammatory Markers
Inflammation was measured through inflammatory biomarkers, including high sensitivity C-reactive protein (hsCRP), interleukin-6 (IL-6), soluble intracellular adhesion molecule-1 (sICAM-1), and tumor necrosis factor-α (TNF-α).22 Serum was isolated and frozen −80C° until assayed for IL-6, sICAM-1, TNF-α, per standard kit protocol (EMD Millipore Process Solutions, Billerica, MA). The typical coefficients of variation for IL-6, sICAM-1, and TNF-α are 7.4%, 4.6%, and 5.4%, respectively. The lower limits of detection are 0.04pg/ml, 0.1ng/ml, and 0.11pg/ml, respectively. Serum at room temperature was assayed for hsCRP on the same day as collection by UCSF Clinical Labs per standard methodology. The coefficient of variation for hsCRP using this procedure is 5.1%
Metabolo-Lipidomics Analysis
Lipid mediators in plasma samples were measured by LC-MS/MS as previously described.23 Briefly, plasma was diluted with 2 volumes of cold-methanol containing deuterium-labeled internal standards (i.e., d4PGE2 and d8-5-HETE). After solid phase extraction (C18), lipid mediators were profiled by LC-MS/MS using multiple reaction monitoring and established transitions to identify and quantitate lipid mediators generated from arachidonic acid (i.e., 15-hydroxyeicosatetraenoic acid (15-HETE), 12-HETE, 5-HETE, 20-HETE, LTB4, PGE2 and PGD2), eicosapentaenoic acid (12-HEPE and 15-HEPE) and dihomogamma-linolenic acid (15-HETrE).24 Lipid mediators were quantified based on external calibration curves for each mediator using authentic standards and after determination of extraction recovery based on internal standards.
Metabolic Hormones and Insulin Resistance
Metabolic hormones and changes in homeostatic model assessment of insulin resistance (HOMA-IR) were assessed to determine whether their levels correlated with endothelial dysfunction, vascular damage or inflammation. The established guidelines for metabolic syndrome25 were used to determine if any of the subjects met criteria. Adiponectin, leptin, resistin, and serum glucose were measured in mg/dL and plasma insulin in μU/ml. The typical coefficients of variation for adiponectin, leptin, and resistin are 3.0%, 2.6%, and 4.5%, respectively. The lower limits of detection are 0.195ng/ml, 0.2ng/ml, and 0.2ng/ml, respectively. Glucose and insulin assays were completed the same day as collection by UCSF Clinical Labs per standard methodology. HOMA-IR was calculated in the standard fashion by multiplying both values and dividing by 405.26 Serum was isolated and frozen until assayed for metabolic hormones.
Lipid Measurements
Total levels of cholesterol, triglycerides, low-density lipoprotein (LDL), and high-density lipoprotein (HDL) in serum were measured. Serum at room temperature was assayed for these analytes on the same day as collection by UCSF Clinical Labs per standard methodology. Prebeta-1 HDL27, 28 was assessed by an immune-fixation assay method: after rapidly thawing the plasma samples at 37°C, they were run on an automated electrophoresis machine (SPIFE 3000, Helena Laboratories, model # G1088001). The proteins were separated in agarose medium, and goat anti-human apolipoprotein A-1 (apo A-1) antiserum was used to react specifically with apo A-1 containing particles.29 After washing away all non-specific proteins in a NaCl (0.15M) - EDTA (1mM) - Triton X-100 (0.05% v/v) wash buffer, the apo A-1 immuno-precipitates were stained with Coomassie R250 blue dye [(1.2mM) in ethanol, acetic acid, and water] and visualized by scanning densitometry (Hardware: Epson Perfection V700 Photo Scanner; Software: QuickScan 2000 WIN Version 2 provided by Helena Labs, Beaumont, TX). A log-linear standard curve was created using calibrators of delipidated-HDL containing apo A-1.
Statistical Analysis
Values are reported as mean ± SD unless otherwise specified. Paired t-tests were performed to compare differences in patients’ vascular reactivity and functional measurements, biomarkers of inflammation and metabolism, and bone density between baseline (pre-BR) and completion (end-BR), as well as between baseline and after 2 recovery days. As the paired t-test requires the data to be normally distributed, especially with small sample sizes, transformations consistent with the distributions of certain variables in larger studies were utilized. For example, the distributions of adiponectin and hsCRP are known to be right-skewed and therefore, log transformations were taken to satisfy the normality assumption required in utilization of the paired t-test. A generalized estimated effect test was used to compare repeated measures of daily FMD and 15-HETE among the 5 participants (Figures 2–3). Statistical significance was accepted at a p-value of ≤0.05. Statistical analyses were performed using Stata/SE 12 (StataCorp, College Station, TX).
Fig 2.

Fig 2a,b. Temporal Pattern for Brachial and Femoral Artery Flow-Mediated Dilatation (FMD) during the 5-day Bed Rest Intervention, N=5
FMD = measure of endothelium-dependent flow-mediated dilatation. Each bar represents daily average FMD% for 5 subjects, with SD.
P-value calculated from a generalized estimated effect test for repeated measures comparing daily FMD from Pre-BR though the Recovery period is 0.03 for the brachial artery and 0.31 for the femoral artery.
Fig 3. Temporal Pattern for 15-HETE during the 5-day Bed Rest Intervention, N=4.
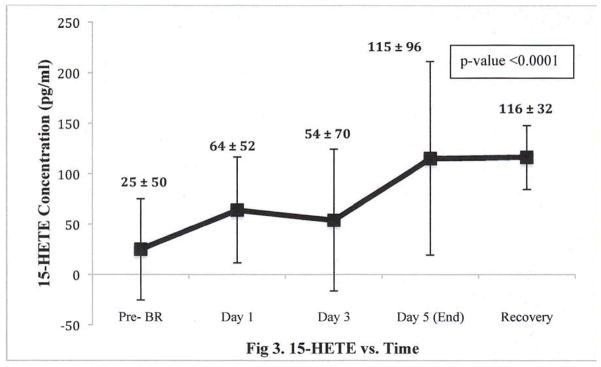
15-HETE = an arachidonic acid metabolite, a product of enzymatic degradation by 15-lipoxygenase (15-LOX).
P-value calculated from a generalized estimated effect test for repeated measures comparing change in 15-HETE from Pre-BR though the Recovery period is <0.0001.
RESULTS
A total of five healthy patients (4 males, 1 female) participated in the BR protocol. Mean age was 22 ± 2, weight was 76 ± 14kg, and BMI was 24 ± 3 kg/m2 (Table 1). Blood lipid and electrolyte profiles were within normal limits (Table 1). At baseline, none of the subjects met criteria for metabolic syndrome, according to established guidelines.25
The hemodynamic and non-invasive vascular function measurements from pre- to end-BR are presented in Table 2 and Figure 2. BR led to a decrease in brachial artery FMD (pre-BR: 11 ± 3% vs. end-BR: 9 ± 2%, P=0.04). Decreasing trends were observed for brachial artery diameter at rest (pre-BR: 3.6 ± 0.7 vs. end-BR: 3.8 ± 0.7 mm, P=0.09) and RH diameter (pre-BR: 3.9 ± 0.7 vs. end-BR: 4.2 ± 0.7 mm, P=0.06). After 2 days of recovery, brachial artery FMD appeared to improve from end-BR values but did not fully return to baseline (recovery: 9 ± 2 % vs. pre-BR: 11 ± 3%, P=0.18).
Table 2.
Non-invasive vascular reactivity and hemodynamic measurements, pre- to completion of (end) bed rest analysis, N=5
| Pre- BR | End- BR | p-value* | |
|---|---|---|---|
|
Brachial artery FMD
| |||
| Baseline diameter, mm | 4 ± 1 | 4 ± 1 | 0.09 |
| Baseline velocity, m/s | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.56 |
| Baseline flow, mL/s | 112 ± 47 | 130 ± 95 | 0.74 |
| Baseline shear stress | 13 ± 6 | 16 ± 6 | 0.41 |
| Reactive hyperemia diameter, mm | 4 ± 1 | 4 ± 1 | 0.06 |
| Reactive hyperemia velocity, m/s | 1 ± 0.4 | 1 ± 0.2 | 0.67 |
| Reactive hyperemia flow, mL/s | 1,058 ± 401 | 874 ± 288 | 0.22 |
| Reactive hyperemia shear stress | 86 ± 30 | 89 ± 29 | 0.72 |
| Flow-mediated vasodilation, FMD (%) | 11 ± 3 | 9 ± 2 | 0.04 |
|
Femoral artery FMD
| |||
| Baseline diameter, mm | 6 ± 1 | 5 ± 1 | 0.15 |
| Baseline velocity, m/s | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.77 |
| Baseline flow, mL/s | 244 ± 76 | 241 ± 131 | 0.96 |
| Baseline shear stress | 8 ± 4 | 8 ± 2 | 0.58 |
| Reactive hyperemia diameter, mm | 6 ± 0.1 | 6 ± 1 | 0.06 |
| Reactive hyperemia velocity, m/s | 0.4 ± 0.2 | 1 ± 0.1 | 0.40 |
| Reactive hyperemia flow, mL/s | 709 ± 352 | 648 ± 96 | 0.79 |
| Reactive hyperemia shear stress | 17 ± 8 | 26 ± 12 | 0.19 |
| Flow-mediated vasodilation, FMD (%) | 4 ± 1 | 2 ± 1 | 0.04 |
|
Applanation Tonometry
| |||
| Central Augmentation Index (Aix), % | −4 ± 9 | 5 ± 11 | 0.03 |
| Central Aix, 75BPM**, % | −15 ± 11 | −6 ± 11 | 0.05 |
| Carotid-Femoral | 5 ± 1 | 5 ± 1 | 0.09 |
| Pulse-Wave Velocity, m/s | |||
|
Hemodynamics
| |||
| Heart rate, bpm | 53 ± 11 | 52 ± 7 | 0.97 |
| SBP, mm Hg | 113 ± 14 | 110 ± 19 | 0.52 |
| DBP, mm Hg | 58 ± 7 | 62 ± 7 | 0.02 |
p-value calculated from paired t-test.
Central Augmentation Index is adjusted for heart rate, at 75 beats per minute.
BR also led to a decrease in femoral artery FMD (pre-BR: 4 ± 1% vs. end-BR: 2 ± 1%, P=0.04). No changes were observed in femoral artery resting diameter, though there was a trend for decrease in RH diameter (pre-BR: 6.3 ± 0.9 vs. end-BR: 5.5 ± 1.2 mm, P=0.06). Femoral artery FMD slightly improved from end-BR values after 2 recovery days though did not fully return to baseline (recovery: 2 ± 1% vs. 4 ± 1% pre-BR, P=0.04). No changes were observed in arterial flow rate, shear stress, or velocity in either the brachial or femoral arteries.
An increase in arterial stiffness was seen during the BR intervention, as indicated by an increase in the central augmentation index (pre-BR: −4 ± 9% vs. end-BR: 5 ± 11%, P=0.03). When adjusted for heart rate, the central augmentation index still showed an increase (pre-BR: −15 ± 11% vs. end-BR: −6 ± 11%, P=0.05). The carotid-femoral pulse-wave velocity did not change. Hemodynamics were affected in that DBP increased (pre-BR: 58 ± 7 vs. end-BR: 62 ± 7 mmHg, P=0.02). However, neither SBP nor HR changed (Table 2).
BR did not yield detectable changes in inflammatory or metabolic biomarkers (Table 3). Compared to the pre-BR average 0.5 ± 0.3 mg/dL, the mean end-BR hsCRP was 0.3 ± 0.1 mg/dL (P=0.06; Table 3). Levels of adiponectin, a marker inversely associated with inflammation, appeared to be lower after BR (pre-BR: 55 ± 34 vs. end-BR: 46 ± 31 mg/dL, P=0.16) and remained low after recovery (pre-BR: 55 ± 34 mg/dL vs. recovery: 46 ± 33, P=0.24). The reduction in adiponectin levels was statistically significant when a log transformation was taken to meet normality assumptions of the paired t-test. BR did not significantly alter blood lipid or lipoprotein levels, degree of insulin sensitivity, bone density, or regional fat distribution. Interestingly, when taking into account daily values of 15-HETE, a metabolite of arachidonic acid (AA), an increase was observed during the course of the 5-day BR intervention (Figure 3). When comparing only the pre-BR and end-BR 15-HETE values, however, there was no significant difference. Levels of the other lipid mediators were not significantly altered, although a general trend toward an increase in 20-HETE, LTB4 and 15-HEPE were noted (Table 4).
Table 3.
Inflammatory and metabolic biomarkers, pre- to completion of (end) bed rest analysis, N=5
| Inflammatory Biomarker | Pre- BR | End- BR | p-value* |
|---|---|---|---|
| hsCRP | 0.5 ± 0.3 | 0.3 ± 0.1 | 0.18 |
| IL-6 | 0.4 ± 0.2 | 0.4 ± 0.3 | 0.62 |
| sICAM | 7 ± 2 | 6 ± 2 | 0.58 |
| TNF-α | 2 ± 1 | 2 ± 1 | 0.28 |
| Metabolic Biomarker | |||
|
| |||
| Adiponectin | 55 ± 3 | 46 ± 31 | 0.16 |
| logAdiponectin | 1.6 ± 0.5 | 1.5 ± 0.5 | 0.05 |
| Resistin | 7 ± 2 | 8 ± 3 | 0.18 |
| logResistin | 1 ± 0.2 | 1 ± 0.2 | 0.23 |
| Leptin | 2 ± 2 | 1 ± 1 | 0.25 |
| logLeptin | −0.02 ± 1 | −0.1 ± 1 | 0.59 |
| HOMA-IR | 1 ± 1 | 1 ± 0 | 0.96 |
p-value calculated from paired t-test.
Table 4.
Plasma LC-MS/MS metabo-lipidomics, pre- to completion of (end) bed rest analysis, N=4
| Metabolite (pg/ml) | Pre- BR | End- BR | p-value* |
|---|---|---|---|
| 12-HETE | 80 ± 20 | 68 ± 61 | 0.78 |
| 15-HETE | 25 ± 50 | 115 ± 96 | 0.29 |
| 19-HETE | 114 ± 139 | 89 ± 177 | 0.66 |
| 20-HETE | 49 ± 59 | 127 ± 120 | 0.11 |
| LTB4 | 188 ± 188 | 240 ± 70 | 0.71 |
| PGE2 | 93 ± 30 | 98 ± 31 | 0.84 |
| PGD2 | 90 ± 42 | 91 ± 52 | 0.98 |
| 12-HEPE | 5 ± 9 | 17 ± 28 | 0.49 |
| 15-HEPE | 9 ± 18 | 166 ± 312 | 0.40 |
| 15-HETrE | 97 ± 81 | 46 ± 36 | 0.40 |
p-value calculated from paired t-test.
DISCUSSION
In this study of five young and healthy individuals, we demonstrate that a 5-day BR intervention leads to a significant decrease in endothelial function, increased arterial stiffness, and increased DBP. These findings were accompanied by an increase in 15-HETE but no changes in other inflammatory and metabolic biomarkers. In light of these results, we propose that short-term inactivity promotes a vascular “deconditioning” state characterized by a weakened endothelial cell response and diminished vasodilation, leading to arterial stiffness and increased basal arterial tone.
Endothelial cells serve as an interface between blood and tissues and play an integral role in the maintenance of homeostasis via the release of biologic agents affecting vascular tone and function as well as responses to inflammatory, mechanical and hormonal stimuli.30,31 FMD is a non-invasive, ultrasound-guided tool readily used as a reliable measure of nitric-oxide (NO)–mediated vascular endothelial function; decreasing FMD is a surrogate for worsening endothelial function.32, 33 Dysfunctional endothelium has been recognized as an initial step in the development of atherosclerosis.16,30 In a recent meta-analysis, Inaba et al. found that a 1% decline in FMD was associated with an 8% increased risk of future cardiovascular events, highlighting the utility of this tool for cardiovascular risk prediction.34 Our results demonstrate that when young and healthy subjects were inactive for only 5 days, FMD declined by an average 2% in both the brachial and femoral arteries. Furthermore, the lower values persisted after 2 recovery days. These findings raise the important concern that if endothelial function is markedly affected in young and healthy individuals, that of older people who are prone to more comorbidities and sedentary living or hospitalized patients who are immobilized may also be reduced, and perhaps to a greater extent. Mechanisms that may explain how physical inactivity weakens endothelial function include promotion of vascular NADPH oxidase expression and activity, and increased production of vascular reactive oxygen species (ROS), which attenuate the effects of the potent vasodilator NO.1, 35 Increased production of asymmetric dimethylarginine species36 may also contribute to reduced vasodilation. In addition, a decline in resting blood flow resulting from inactivity and subsequent decrease in local shear stress may inhibit expression of endothelial NO-synthase, thereby inhibiting vasodilation.12
In addition to reductions in FMD in the upper and lower extremities, the 5-day BR intervention in our study led to declines in brachial artery resting diameter and RH diameter, as well as femoral artery RH diameter, changes that were nearly statistically significant (Table 2). Prior studies that assessed prolonged inactivity demonstrated that BR leads to reductions in conduit artery diameter,7 decreased RH8 and possibly vascular dysfunction.12 Hamburg and colleagues, in their 5-day BR study of 19 healthy non-smoking subjects, found that this duration of inactivity led to decreased brachial artery diameter and RH diameter as well as increased SBP.12 Methods used to assess vascular function included endothelium-dependent FMD of the brachial artery and endothelium-independent dilation with sublingual nitroglycerin (also brachial artery), as well as RH (a measure of flow velocity) in the brachial artery and venous occlusion plethysmography in the lower extremities to evaluate microvascular function. They found a significant reduction in RH in the upper and lower extremities though no changes in FMD, suggesting a preferential impairment in microvascular function. Their results also showed that insulin resistance developed, and total cholesterol and triglycerides increased after 5 days. They did not report a change in circulating inflammatory markers. Although a similar BR model and testing period were employed, the study by Hamburg et al. differs from ours in that participants were allotted 30 minutes out of bed for each 24 hour period, they were not maintained on a similar diet, femoral artery FMD and arterial stiffness measurements were not completed, and metabolo-lipidomics measurements were not performed.
In our study, changes in serum levels of inflammatory biomarkers were negligible relative to those of vascular measurements. In contrast to our study, inflammation—and hsCRP in particular—has been correlated with impaired endothelial function in vivo and in vitro,37, 38 and hsCRP levels have been inversely correlated with acetylcholine-induced, endothelium-dependent forearm blood flow.39 Furthermore, the role of increased inflammatory markers as a result of physical inactivity and its relation to cardiovascular risk have been described previously.3 Despite these associations, in their 5-day BR study similar to ours, Hamburg and colleagues did not observe changes in inflammation.12 The fact that inflammatory biomarkers were not altered in their study or ours may reflect the relatively small sample sizes or that changes in inflammatory markers in response to inactivity develop over a longer time period.
One notable exception is the significant increase in 15-HETE, a 15-lipoxygenase (15-LOX) metabolite, over the duration of the study period (Figure 3). Production of 15-HETE has been shown to inhibit neutrophil migration across cytokine-activated endothelium in vitro,40 as well as prevent superoxide production neutrophil degranulation,41 and is therefore thought to be anti-inflammatory. 15-HETE has also been observed to affect vascular reactivity, though it remains unclear whether it promotes vasoconstriction or vasodilation. One view holds that 15-HETE may have a direct vasoconstrictive effect, as evidenced by its role in pulmonary vasculature,42, 43 promote leukocyte activation and inhibit prostacyclin production.44 The steady and sustained increase of 15-HETE in our study may help explain the concurrent increase in DBP and decreases in FMD. An alternate explanation is that after enzymatic degradation by 15-LOX, 15-HETE can be further metabolized to anti-inflammatory lipoxins,19 and these metabolites promote vasodilation and regulate endothelial-derived nitric oxide production.45, 46 The rise in 15-HETE that we observed may then be an early compensatory response to vasoconstriction.
In regards to metabolic changes, we have previously demonstrated that BR leads to important alterations in the renal, cardio-endocrine and cardiovascular systems,47–50 and others have shown that prolonged BR leads to development of insulin resistance,9, 12 a precursor state to metabolic syndrome12 and type 2 diabetes.10 A number of circulating hormones, cytokines, and metabolic fuels that originate from the adipocyte modulate insulin action,51 thereby augmenting basal metabolic expenditure. Specifically, the non-esterifed fatty acids, leptin, adiponectin, and resistin, as well as TNF-alpha and IL-6, released by visceral adipose tissues affect the insulin cascade.51–53 These adipokines play crucial roles in the homeostasis of body weight by modulating food intake and energy expenditure.54 In the current study, we observed statistically significant decreases in levels of adiponectin, which were not accompanied by changes in other metabolic markers. Adiponectin in particular is an anti-atherogenic adipocyte-derived protein.55 Decreased serum levels have been associated with development of inflammatory states over time and increased cardiovascular risk.56 This association requires further exploration given that in our study, the time course was relatively short and the other metabolic biomarkers had fluctuating trends.
We did not find that 5 days on BR led to changes in bone density or fat distribution, though previous studies have found that prolonged BR leads to loss of bone density.57, 58 Dyslipidemia and associated fatty plaque deposition in the arterial wall are additional factors implicated in the progression of cardiovascular disease and may develop as a result of sedentary behavior. Interestingly, Laufs et al showed that after 6 weeks, sedentary mice experienced accelerated atherosclerotic lesion formation as compared to physically active counterparts.1 These findings suggest that a state of dyslipidemia may develop secondary to inactivity. In their 5-day BR intervention, Hamburg et al found that total cholesterol and triglycerides increased.12 In our cohort, there were no changes in lipid levels after BR. We also measured a relatively novel biomarker, prebeta-1 HDL, that reflects cholesterol efflux from the artery wall and has recently been shown to be a sensitive predictor of risk for atherosclerosis.27, 28 To our knowledge, its association with physical inactivity has not been examined previously. Levels of this metabolite did not change notably during our 5-day BR period.
Sedentarism, also termed physical inactivity, is identified as the leading cause of excessive and preventable cardiovascular risk.55 As per the 2010 US Physical Activity Statistics of the Centers for Disease Control and Prevention, less than half of US adults met recommended physical activity standards in 2007, and nearly one-quarter of the US population had no leisure time physical activity in 2008, with rates of sedentarism exceeding 30% for those over the age of 65.59 Overall, there is evidence that the health care costs of sedentary individuals are close to one-third higher than those of individuals who are physically active, representing at least 100 billion US dollars in excess healthcare costs.60 Although some effects of sedentarism relate to the development of obesity, it appears that some of the detrimental changes occur independently of changes in body fat.61
Exercise, in contrast to physical inactivity, has been shown to exert cardio-protective effects.62, 63 Thijssen and colleagues observed that approximately 60% of risk reduction is related physical activity attenuating the effects of traditional cardiovascular risk factors, including dyslipidemia, hypertension, obesity, and diabetes.64 They reasoned that the remaining 40% of risk reduction is due to direct “conditioning” effects on the vascular endothelium.
Physical inactivity may too have direct physiologic effects on vascular remodeling and function—the opposite of exercise. It may compromise endothelial function and lead to impaired hemodynamics,65 increase propensity to inflammation,3 accelerate atherosclerotic plaque formation,1 and cause changes in skeletal muscle characteristics.66 Our findings suggest that inactivity leads to quantifiable impairment in vascular function and arterial wall stiffening (or increased vascular resistance), and we speculate that these are a result of endothelial dysfunction. Arterial stiffness, like FMD, has been shown to be an independent predictor for increased cardiovascular risk.14, 67, 68 A dynamic relationship between these two physiologic parameters has been suggested,14, 69–72 and an association could explain how both endothelial cell dysfunction and arterial stiffness are related to worse cardiovascular outcomes. It should be emphasized that our evaluation of arterial resistance is based on non-invasive measurements made with an applanation tonometry device; consequently, the overall arterial stiffening that we observed during the 5 days of BR is likely functional and reversible. This is in contrast to true arterial stiffness that results from years of deleterious insults to the vasculature, caused by chronic inflammatory conditions such as atherosclerosis and vasculitis. In addition, we speculate that the arterial stiffness elicited in this study may have been caused by an increase in basal arterial tone – evidenced by the increase in DBP — that typically occurs with muscular inactivity.
The decreases in endothelial function that we observed, coupled with increases in arterial resistance at baseline and adjusted for heart rate, as well as DBP, imply a relation. The precise mechanism by which short-term exposure to physical inactivity alters healthy vascular reactivity, tone and structure requires more detailed study, as does the clinical relevance of these physiologic findings.
LIMITATIONS
Our study has several limitations. First, our sample size was small, which could have altered our ability to detect changes in inflammatory markers or metabolic measures. However, clear changes were seen in vascular function. Second, definitive or causal conclusions cannot be made given the observational nature of the study. Third, our findings apply only to young and healthy individuals and cannot be extrapolated to older individuals or patients. Fourth, we did not evaluate nitroglycerine-mediated vasodilation (endothelium-independent), which could have determined if subjects’ arterial vessels were capable of dilating to their baseline diameter. Another limitation is that the cohort was predominantly male, and gender may have played a role in the physiological results observed. Lastly, physical inactivity likely exerts a large spectrum of physiologic effects; we limited our study to observing alterations in vascular physiology, inflammation, and metabolic changes.
CONCLUSIONS
In a cohort of five healthy patients, 5 days of BR was associated with endothelial dysfunction, arterial wall stiffening, and a narrowing of conduit artery diameter, implying a state of impaired vascular function. This study builds on previous ones by demonstrating that a short-term exposure to sedentarism disturbs healthy vascular reactivity patterns and affects basal arterial tone. These findings may be especially relevant to hospitalized and post-operative patients as their physical activity is limited and their vascular healing may be compromised as a result. Further investigation into how sedentary behavior affects endothelial function, metabolic regulation and inflammatory patterns will facilitate a better understanding of their effects on the development of vascular disease.
Table 5.
Body composition biomarkers, pre- to completion of (end) bed rest analysis, N=5
| DEXA measurements | Pre- BR | End- BR | p-value |
|---|---|---|---|
| BMD, total | 1 ± 0.1 | 1 ± 0.1 | 1.0 |
| BMD, spine | 1 ± 0.2 | 1 ± 0.2 | 0.48 |
| BMD, ribs | 1 ± 0.1 | 1 ± 0.1 | 0.64 |
| BMD, pelvis | 1 ± 0.3 | 1 ± 0.2 | 0.19 |
| BMD, legs | 2 ± 0.3 | 2 ± 0.2 | 0.65 |
| Android (%fat distribution) | 22 ± 6 | 22 ± 7 | 0.85 |
| Gynoid (%fat distribution) | 25 ± 8 | 26 ± 9 | 0.73 |
| Android/Gynoid ratio | 1 ± 0.2 | 1 ± 0.2 | 0.76 |
Footnotes
Author Contributions
S.M.G., M.S.C., and E.V.N. formulated the hypothesis and analysis plan. K.C.C., H.F.A., A.Q., J.H. and E.V.N. were involved in data acquisition. S.M.G., M.S., P.Y., J.H. and E.V.N. performed the statistical analysis. All authors interpreted the data. E.V.N. primarily drafted the manuscript. All authors have read and approved the final version of the manuscript. There are no conflicts of interest related to this submission.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Laufs U, et al. Physical inactivity increases oxidative stress, endothelial dysfunction, and atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:809–14. doi: 10.1161/01.ATV.0000158311.24443.af. [DOI] [PubMed] [Google Scholar]
- 2.Manson JE, et al. Walking compared with vigorous exercise for the prevention of cardiovascular events in women. N Engl J Med. 2002;347:716–25. doi: 10.1056/NEJMoa021067. [DOI] [PubMed] [Google Scholar]
- 3.Pedersen BK. The diseasome of physical inactivity--and the role of myokines in muscle--fat cross talk. J Physiol. 2009;587:5559–68. doi: 10.1113/jphysiol.2009.179515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pate RR, O’Neill JR, Lobelo F. The evolving definition of “sedentary”. Exerc Sport Sci Rev. 2008;36:173–8. doi: 10.1097/JES.0b013e3181877d1a. [DOI] [PubMed] [Google Scholar]
- 5.Activity, CfDCaPUSP. http://apps.nccd.cdc.gov/PASurveillance/DemoComparev.asp., S.
- 6.Brownson RC, Boehmer TK, Luke DA. Declining rates of physical activity in the United States: what are the contributors? Annu Rev Public Health. 2005;26:421–43. doi: 10.1146/annurev.publhealth.26.021304.144437. [DOI] [PubMed] [Google Scholar]
- 7.Bleeker MW, et al. Vascular adaptation to deconditioning and the effect of an exercise countermeasure: results of the Berlin Bed Rest study. J Appl Physiol. 2005;99:1293–300. doi: 10.1152/japplphysiol.00118.2005. [DOI] [PubMed] [Google Scholar]
- 8.Shoemaker JK, et al. Head-down-tilt bed rest alters forearm vasodilator and vasoconstrictor responses. J Appl Physiol. 1998;84:1756–62. doi: 10.1152/jappl.1998.84.5.1756. [DOI] [PubMed] [Google Scholar]
- 9.Stuart CA, Shangraw RE, Prince MJ, Peters EJ, Wolfe RR. Bed-rest-induced insulin resistance occurs primarily in muscle. Metabolism. 1988;37:802–6. doi: 10.1016/0026-0495(88)90018-2. [DOI] [PubMed] [Google Scholar]
- 10.Manson JE, et al. A prospective study of exercise and incidence of diabetes among US male physicians. JAMA. 1992;268:63–7. [PubMed] [Google Scholar]
- 11.Grenon SM, et al. Renal, endocrine, and cardiovascular responses to bed rest in male subjects on a constant diet. J Investig Med. 2004;52:117–28. doi: 10.1136/jim-52-02-21. [DOI] [PubMed] [Google Scholar]
- 12.Hamburg NM, et al. Physical inactivity rapidly induces insulin resistance and microvascular dysfunction in healthy volunteers. Arterioscler Thromb Vasc Biol. 2007;27:2650–6. doi: 10.1161/ATVBAHA.107.153288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gokce N, et al. Predictive value of noninvasively determined endothelial dysfunction for long-term cardiovascular events in patients with peripheral vascular disease. J Am Coll Cardiol. 2003;41:1769–75. doi: 10.1016/s0735-1097(03)00333-4. [DOI] [PubMed] [Google Scholar]
- 14.Schnabel RB, et al. Noninvasive vascular function measurement in the community: cross-sectional relations and comparison of methods. Circ Cardiovasc Imaging. 2011;4:371–80. doi: 10.1161/CIRCIMAGING.110.961557. [DOI] [PubMed] [Google Scholar]
- 15.Soga J, et al. Relationship between augmentation index and flow-mediated vasodilation in the brachial artery. Hypertens Res. 2008;31:1293–8. doi: 10.1291/hypres.31.1293. [DOI] [PubMed] [Google Scholar]
- 16.Vita JA. Endothelial function. Circulation. 2011;124:e906–12. doi: 10.1161/CIRCULATIONAHA.111.078824. [DOI] [PubMed] [Google Scholar]
- 17.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–7. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 18.Serhan CN, et al. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med. 2000;192:1197–204. doi: 10.1084/jem.192.8.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spite M, Serhan CN. Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circ Res. 2010;107:1170–84. doi: 10.1161/CIRCRESAHA.110.223883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corretti MC, et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. Journal of the American College of Cardiology. 2002;39:257–65. doi: 10.1016/s0735-1097(01)01746-6. [DOI] [PubMed] [Google Scholar]
- 21.Thijssen DH, et al. Assessment of flow-mediated dilation in humans: a methodological and physiological guideline. American journal of physiology. Heart and circulatory physiology. 2011;300:H2–12. doi: 10.1152/ajpheart.00471.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho KJ, et al. Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. The American journal of pathology. 2010;177:2116–23. doi: 10.2353/ajpath.2010.091082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hellmann J, et al. Increased saturated fatty acids in obesity alter resolution of inflammation in part by stimulating prostaglandin production. J Immunol. 2013;191:1383–92. doi: 10.4049/jimmunol.1203369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu Y, Hong S, Gotlinger K, Serhan CN. Lipid mediator informatics and proteomics in inflammation resolution. Scientific World Journal. 2006;6:589–614. doi: 10.1100/tsw.2006.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grundy SM, et al. Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Arterioscler Thromb Vasc Biol. 2004;24:e13–8. doi: 10.1161/01.ATV.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- 26.Matthews DR, et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 27.Guey LT, et al. Relation of increased prebeta-1 high-density lipoprotein levels to risk of coronary heart disease. Am J Cardiol. 2011;108:360–6. doi: 10.1016/j.amjcard.2011.03.054. [DOI] [PubMed] [Google Scholar]
- 28.Kane JP, Malloy MJ. Prebeta-1 HDL and coronary heart disease. Curr Opin Lipidol. 2012;23:367–71. doi: 10.1097/MOL.0b013e328353eef1. [DOI] [PubMed] [Google Scholar]
- 29.Kunitake ST, O’Connor P, Naya-Vigne J. Heterogeneity of high-density lipoproteins and apolipoprotein A-I as related to quantification of apolipoprotein AI. Methods Enzymol. 1996;263:260–7. doi: 10.1016/s0076-6879(96)63018-3. [DOI] [PubMed] [Google Scholar]
- 30.Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol. 2004;15:1983–92. doi: 10.1097/01.ASN.0000132474.50966.DA. [DOI] [PubMed] [Google Scholar]
- 31.Vita JA, Hamburg NM. Does endothelial dysfunction contribute to the clinical status of patients with peripheral arterial disease? Can J Cardiol. 2010;26 (Suppl A):45A–50A. doi: 10.1016/s0828-282x(10)71062-x. [DOI] [PubMed] [Google Scholar]
- 32.Inoue T, et al. Flow-mediated vasodilation as a diagnostic modality for vascular failure. Hypertens Res. 2008;31:2105–13. doi: 10.1291/hypres.31.2105. [DOI] [PubMed] [Google Scholar]
- 33.Al-Qaisi M, Kharbanda RK, Mittal TK, Donald AE. Measurement of endothelial function and its clinical utility for cardiovascular risk. Vasc Health Risk Manag. 2008;4:647–52. doi: 10.2147/vhrm.s2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inaba Y, Chen JA, Bergmann SR. Prediction of future cardiovascular outcomes by flow-mediated vasodilatation of brachial artery: a meta-analysis. Int J Cardiovasc Imaging. 2010;26:631–40. doi: 10.1007/s10554-010-9616-1. [DOI] [PubMed] [Google Scholar]
- 35.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–4. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 36.Cooke JP. Asymmetrical dimethylarginine: the Uber marker? Circulation. 2004;109:1813–8. doi: 10.1161/01.CIR.0000126823.07732.D5. [DOI] [PubMed] [Google Scholar]
- 37.Verma S, et al. A self-fulfilling prophecy: C-reactive protein attenuates nitric oxide production and inhibits angiogenesis. Circulation. 2002;106:913–9. doi: 10.1161/01.cir.0000029802.88087.5e. [DOI] [PubMed] [Google Scholar]
- 38.Owens CD, et al. Elevated C-reactive protein levels are associated with postoperative events in patients undergoing lower extremity vein bypass surgery. J Vasc Surg. 2007;45:2–9. doi: 10.1016/j.jvs.2006.08.048. discussion 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fichtlscherer S, et al. Elevated C-reactive protein levels and impaired endothelial vasoreactivity in patients with coronary artery disease. Circulation. 2000;102:1000–6. doi: 10.1161/01.cir.102.9.1000. [DOI] [PubMed] [Google Scholar]
- 40.Takata S, et al. 15-Hydroxyeicosatetraenoic acid inhibits neutrophil migration across cytokine-activated endothelium. Am J Pathol. 1994;145:541–9. [PMC free article] [PubMed] [Google Scholar]
- 41.Smith RJ, Justen JM, Nidy EG, Sam LM, Bleasdale JE. Transmembrane signaling in human polymorphonuclear neutrophils: 15(S)-hydroxy-(5Z,8Z,11Z,13E)-eicosatetraenoic acid modulates receptor agonist-triggered cell activation. Proc Natl Acad Sci U S A. 1993;90:7270–4. doi: 10.1073/pnas.90.15.7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burhop KE, Selig WM, Malik AB. Monohydroxyeicosatetraenoic acids (5-HETE and 15-HETE) induce pulmonary vasoconstriction and edema. Circ Res. 1988;62:687–98. doi: 10.1161/01.res.62.4.687. [DOI] [PubMed] [Google Scholar]
- 43.Zhu D, et al. Chronic hypoxia activates lung 15-lipoxygenase, which catalyzes production of 15-HETE and enhances constriction in neonatal rabbit pulmonary arteries. Circ Res. 2003;92:992–1000. doi: 10.1161/01.RES.0000070881.65194.8F. [DOI] [PubMed] [Google Scholar]
- 44.Wittwer J, Hersberger M. The two faces of the 15-lipoxygenase in atherosclerosis. Prostaglandins Leukot Essent Fatty Acids. 2007;77:67–77. doi: 10.1016/j.plefa.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 45.von der Weid PY, Hollenberg MD, Fiorucci S, Wallace JL. Aspirin-triggered, cyclooxygenase-2-dependent lipoxin synthesis modulates vascular tone. Circulation. 2004;110:1320–5. doi: 10.1161/01.CIR.0000140985.89766.CB. [DOI] [PubMed] [Google Scholar]
- 46.Paul-Clark MJ, Van Cao T, Moradi-Bidhendi N, Cooper D, Gilroy DW. 15-epi-lipoxin A4-mediated induction of nitric oxide explains how aspirin inhibits acute inflammation. J Exp Med. 2004;200:69–78. doi: 10.1084/jem.20040566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grenon SM, et al. Why is orthostatic tolerance lower in women than in men? Renal and cardiovascular responses to simulated microgravity and the role of midodrine. J Investig Med. 2006;54:180–90. doi: 10.2310/6650.2006.05064. [DOI] [PubMed] [Google Scholar]
- 48.Xiao X, et al. Bed rest effects on human calf hemodynamics and orthostatic intolerance: a model-based analysis. Aviat Space Environ Med. 2005;76:1037–45. [PubMed] [Google Scholar]
- 49.Grenon SM, et al. Simulated microgravity induces microvolt T wave alternans. Ann Noninvasive Electrocardiol. 2005;10:363–70. doi: 10.1111/j.1542-474X.2005.00654.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grenon SM, et al. Readaptation from simulated microgravity as a stimulus for improved orthostatic tolerance: role of the renal, cardioendocrine, and cardiovascular systems. J Investig Med. 2005;53:82–91. doi: 10.2310/6650.2005.00203. [DOI] [PubMed] [Google Scholar]
- 51.Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: pathogenesis and treatment. Lancet. 2008;371:2153–6. doi: 10.1016/S0140-6736(08)60932-0. [DOI] [PubMed] [Google Scholar]
- 52.Rajala MW, Scherer PE. Minireview: The adipocyte--at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology. 2003;144:3765–73. doi: 10.1210/en.2003-0580. [DOI] [PubMed] [Google Scholar]
- 53.Ravussin E, Smith SR. Increased fat intake, impaired fat oxidation, and failure of fat cell proliferation result in ectopic fat storage, insulin resistance, and type 2 diabetes mellitus. Ann N Y Acad Sci. 2002;967:363–78. doi: 10.1111/j.1749-6632.2002.tb04292.x. [DOI] [PubMed] [Google Scholar]
- 54.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–70. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 55.Colditz GA. Economic costs of obesity and inactivity. Med Sci Sports Exerc. 1999;31:S663–7. doi: 10.1097/00005768-199911001-00026. [DOI] [PubMed] [Google Scholar]
- 56.Kawano J, Arora R. The role of adiponectin in obesity, diabetes, and cardiovascular disease. J Cardiometab Syndr. 2009;4:44–9. doi: 10.1111/j.1559-4572.2008.00030.x. [DOI] [PubMed] [Google Scholar]
- 57.Beller G, et al. WISE-2005: bed-rest induced changes in bone mineral density in women during 60 days simulated microgravity. Bone. 2011;49:858–66. doi: 10.1016/j.bone.2011.06.021. [DOI] [PubMed] [Google Scholar]
- 58.Zerwekh JE, Ruml LA, Gottschalk F, Pak CY. The effects of twelve weeks of bed rest on bone histology, biochemical markers of bone turnover, and calcium homeostasis in eleven normal subjects. J Bone Miner Res. 1998;13:1594–601. doi: 10.1359/jbmr.1998.13.10.1594. [DOI] [PubMed] [Google Scholar]
- 59.https://apps.nccd.cdc.gov/PASurveillance/DemoCompare-v.asp.
- 60.Pratt M, Macera CA, Wang G. Higher direct medical costs associated with physical inactivity. Phys Sportsmed. 2000;28:63–70. doi: 10.3810/psm.2000.10.1237. [DOI] [PubMed] [Google Scholar]
- 61.Bergouignan A, et al. Regulation of energy balance during long-term physical inactivity induced by bed rest with and without exercise training. J Clin Endocrinol Metab. 2010;95:1045–53. doi: 10.1210/jc.2009-1005. [DOI] [PubMed] [Google Scholar]
- 62.Sesso HD, Paffenbarger RS, Jr, Lee IM. Physical activity and coronary heart disease in men: The Harvard Alumni Health Study. Circulation. 2000;102:975–80. doi: 10.1161/01.cir.102.9.975. [DOI] [PubMed] [Google Scholar]
- 63.Myers J, et al. Exercise capacity and mortality among men referred for exercise testing. N Engl J Med. 2002;346:793–801. doi: 10.1056/NEJMoa011858. [DOI] [PubMed] [Google Scholar]
- 64.Thijssen DH, et al. Impact of inactivity and exercise on the vasculature in humans. Eur J Appl Physiol. 2010;108:845–75. doi: 10.1007/s00421-009-1260-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Joyner MJ, Green DJ. Exercise protects the cardiovascular system: effects beyond traditional risk factors. J Physiol. 2009;587:5551–8. doi: 10.1113/jphysiol.2009.179432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McDermott MM, et al. Greater sedentary hours and slower walking speed outside the home predict faster declines in functioning and adverse calf muscle changes in peripheral arterial disease. J Am Coll Cardiol. 2011;57:2356–64. doi: 10.1016/j.jacc.2010.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mitchell GF. Arterial Stiffness and Wave Reflection: Biomarkers of Cardiovascular Risk. Artery Res. 2009;3:56–64. doi: 10.1016/j.artres.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mitchell GF, et al. Arterial stiffness and cardiovascular events: the Framingham Heart Study. Circulation. 2010;121:505–11. doi: 10.1161/CIRCULATIONAHA.109.886655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Widlansky ME, Gokce N, Keaney JF, Jr, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol. 2003;42:1149–60. doi: 10.1016/s0735-1097(03)00994-x. [DOI] [PubMed] [Google Scholar]
- 70.McEniery CM, et al. Endothelial function is associated with pulse pressure, pulse wave velocity, and augmentation index in healthy humans. Hypertension. 2006;48:602–8. doi: 10.1161/01.HYP.0000239206.64270.5f. [DOI] [PubMed] [Google Scholar]
- 71.Kuvin JT, Mammen A, Mooney P, Alsheikh-Ali AA, Karas RH. Assessment of peripheral vascular endothelial function in the ambulatory setting. Vasc Med. 2007;12:13–6. doi: 10.1177/1358863X06076227. [DOI] [PubMed] [Google Scholar]
- 72.Heffernan KS, Karas RH, Mooney PJ, Patel AR, Kuvin JT. Pulse wave amplitude is associated with brachial artery diameter: implications for gender differences in microvascular function. Vasc Med. 2010;15:39–45. doi: 10.1177/1358863X09349523. [DOI] [PMC free article] [PubMed] [Google Scholar]