

Abstract
In order to compare the global gene expression profiles of different embryonic cell types, it is first necessary to isolate the specific cells of interest. The purpose of this chapter is to provide a step-by-step protocol to perform laser capture microdissection (LCM) on embryo samples and obtain sufficient amounts of high-quality RNA for microarray hybridizations. Using the LCM/microarray strategy on mouse embryo samples has some challenges, because the cells of interest are available in limited quantities. The first step in the protocol is to obtain embryonic tissue, and immediately cryoprotect and freeze it in a cryomold containing Optimal Cutting Temperature freezing media (Sakura Finetek), using a dry ice–isopentane bath. The tissue is then cryosectioned, and the microscope slides are processed to fix, stain, and dehydrate the cells. LCM is employed to isolate specific cell types from the slides, identified under the microscope by virtue of their morphology. Detailed protocols are provided for using the currently available ArcturusXT LCM instrument and CapSure® LCM Caps, to which the selected cells adhere upon laser capture. To maintain RNA integrity, upon removing a slide from the final processing step, or attaching the first cells on the LCM cap, LCM is completed within 20 min. The cells are then immediately recovered from the LCM cap using a denaturing solution that stabilizes RNA integrity. RNA is prepared using standard methods, modified for working with small samples. To ensure the validity of the microarray data, the quality of the RNA is assessed using the Agilent bioanalyzer. Only RNA that is of sufficient integrity and quantity is used to perform microarray assays. This chapter provides guidance regarding troubleshooting and optimization to obtain high-quality RNA from cells of limited availability, obtained from embryo samples by LCM.
Keywords: Laser capture microdissection, RNA, Microarray, Infrared laser
1 Introduction
As a prerequisite to comparing the global gene expression profiles of different embryonic cell types, it is necessary to purify the cells of interest. Laser capture microdissection (LCM) is a technique that allows the precise identification and isolation of homogeneous populations of cells from complex heterogeneous tissues. The principles and applications of the technique have recently been reviewed [1, 2]. In short, a microscope slide with a tissue cryosection is placed on the platform of the LCM instrument, and the specific cells of interest are identified by morphology using the inverted microscope. An LCM cap covered by a thermoplastic film is placed over the section. An infrared laser is directed through the cap to the cells of interest, and these cells are attached to the thermoplastic film. The cells can then be removed from the cap using a standard RNA preparation denaturing solution, and RNA can be isolated. This chapter provides a step-by-step protocol to perform LCM on embryo samples and obtain sufficient amounts of high-quality RNA for microarray hybridizations. With some modifications, the protocol described here could be applied in any case where limited tissue samples are available for LCM and subsequent microarray analyses, for example, for patient samples in research settings.
Recently, LCM followed by quantitative reverse transcriptase-PCR (qRT-PCR) has been employed to compare gene expression patterns in different embryonic cell types [3–5] or to compare mutant and normal embryonic cells [6]. In these cases, specific candidate genes were selected for qRT-PCR, to determine if they are differentially expressed. In order to obtain a more global profile of differential gene expression in embryos, however, an LCM/microarray strategy can be employed. This strategy requires more RNA than does qRT-PCR, but it is becoming increasingly more popular. In our experience, high-quality qRT-PCR data can be obtained with RNA prepared from hundreds of LCM-isolated cells (or less depending on the abundance of the mRNA), whereas high-quality Affymetrix GeneChip microarray assays require at least a few thousand cells [7].
In the past 3 years, the LCM/microarray approach has been used for several applications in developmental biology [7–13]. For example, Redmond et al. identified erythroid-enriched genes expressed in the mouse embryonic day 9.5 (E9.5) yolk sac, using Affymetrix GeneChips [7]. Xiao et al. identified genes that are differentially expressed in E10 and E12 mouse lens cells [10]. Williams et al. identified genes that are differentially expressed along the dorsal–ventral, medial–lateral, and anterior–posterior axes of the E17.5 mouse olfactory bulb. Bhattacherjee et al. isolated neural crest- and mesoderm-derived mesenchymal cells from the first brain branchial arch of E9.5 mouse embryos, and used Affymetrix GeneChip microarrays to determine which genes are differentially expressed in the two distinct lineages [9]. Brunskill et al. generated an atlas of gene expression in the developing mouse kidney at microanatomic resolution. They used LCM to isolate some kidney cell populations. To isolate other populations, the study employed transgenic mice expressing GFP in specific cells, followed by fluorescence-activated cell sorting (FACS) [13]. Our laboratory has used the LCM/microarray strategy to compare the genetic profiles of normal E9.5 yolk sac erythroid cells to cells from embryos with a null mutation in the gene encoding Krüppel-like factor 2 (KLF2−/−), embryos that are KLF1 null, and double null mutants [14, 15]. Clearly, the results from these LCM/microarray experiments can be used to develop hypotheses about the genes controlling development in specific tissues.
The LCM part of the protocol described here is designed for the ArcturusXT microdissection instrument, but could easily be adapted for use with other Arcturus instruments. Microdissection instruments from other manufacturers differ in that they employ specialized platforms for tissue attachment, rather than regular microscope slides. In any case, for microdissecting cells from mouse embryonic tissue, infrared (IR) laser capture as opposed to UV laser cutting is the best option, because a limited number of cells within a small area on the slide are being isolated. UV laser cutting is designed to capture larger numbers of cells than are available in mouse embryos. The tissue and RNA preparation methods and troubleshooting guides described here should be applicable with any LCM instrument. The methods described here do not duplicate a manufacturer’s protocol; rather they combine various manufacturer recommended kits, other kits and reagents found to be both reliable and economical. The protocol is written specifically for mouse embryonic yolk sac, but notes indicate how it can be adapted for use with other embryonic tissues, or any other tissue that has a limited number of cells of interest.
2 Materials
2.1 Preparation and Freezing of Tissue
Aluminum foil.
RNase AWAY (ISC BioExpress, Kaysville, UT, USA).
2-Methylbutane (isopentane).
Freshly made 20% (w/v) sucrose cryoprotection buffer (2 g of sucrose per 10 ml of 1× PBS).
Optimal Cutting Temperature freezing media (OCT, Sakura Finetek USA, Inc., Torrance, CA, USA).
Watchmaker’s forceps.
Metal crucible.
Intermediate 15 × 15 × 5 mm cryomolds.
Plastic disposable 1-ml sterile Pasteur pipettes.
60 × 15 mm plastic Petri dishes.
Dissecting microscope.
Zip-lock sandwich bags.
2.2 Cryosectioning of Tissue
RNase AWAY (ISC BioExpress).
Low-Profile Accu-Edge Disposable Microtome Blades (Sakura Finetek).
Vibratome UltraPro 5000 (Global Medical Instrumentation, Inc., Ramsey, Minnesota, USA).
Paintbrushes with 0.5–1 cm bristles.
Silane-Prep microscope slides (Sigma-Aldrich).
Rapid Stain (American Master Tech Scientific, Inc., Lodi, CA).
Uni-Cassette® (Sakura Finetek).
Desiccant.
Plastic 25-slide boxes.
2.3 Dehydration and Staining of Microscope Slides for LCM
HistoGene™ LCM Frozen Section Staining Kit (MDS Analytical Technologies, Sunnyvale, CA, USA). This kit is purchased mainly for the stain, items 2–4 are purchased separately to extend the kit.
Anhydrous ethanol (VWR, West Chester, PA, USA, catalog number IB15720).
Xylene.
Nuclease-free and proteinase-free H2O.
SUPERaseIn™ (20 U/μl) (Ambion, Foster City, CA, USA).
75% EtOH and 95% EtOH dilutions (made with anhydrous ethanol and nuclease-free H2O).
50 ml sterile conical polypropylene screw cap tubes.
RNase AWAY (ISC BioExpress).
Glass Staining Dishes for 20 slides, including two slide-staining trays (Fisher).
Molecular sieves, 4 Å (Sigma-Aldrich).
2.4 Laser Capture Microdissection Using the ArcturusXT
ArcturusXT Microdissection Instrument (MDS Analytical Devices).
CapSure® HS LCM Caps (MDS Analytical Technologies).
GeneAmp™ 500 μl Thin-walled PCR Reaction Tubes (Applied Biosystems, Foster City, CA, USA).
Post-it® Notes (3 M). Keep them in their original packaging or a clean zip-lock bag to avoid RNase contamination.
Denaturing solution from ToTALLY RNA™ kit (Ambion).
2.5 RNA Preparation
1.5 ml round bottom screw cap microcentrifuge tubes with O-rings (Fisher Scientific).
3 M sodium acetate and Acid Phenol from the Totally RNA™ kit (Ambion).
Glycogen (5 mg/ml, nucleic acid- and nuclease-free).
Isopropanol.
75 % EtOH made with anhydrous EtOH and nuclease-free H2O.
SUPERaseIn™ (20 U/μl) (Ambion).
Nuclease-free and proteinase-free H2O (USB).
2.6 Assessing RNA Quality
Agilent 2100 bioanalyzer with chip priming station (Agilent Biotechnologies, Santa Clara, California, USA).
Pico RNA Chip, reagents and electrode cleaner (Agilent).
RNA 6000 Pico ladder (Ambion).
RNase AWAY (ISC BioExpress).
Nuclease-free H2O.
Pipette accurate for measuring 1 μl volume, such as Rainin P10.
RNase-free 1.5 ml snap cap microcentrifuge tubes.
Vortex mixer with Agilent adapter.
3 Methods
3.1 Preparation and Freezing of Tissue
Test the OCT media to make sure that is in good condition for freezing specimens (see Note 1).
Ensure that all reagents, dissection equipment, instruments, pipettes, slides, and storage vessels that will contact samples are RNase free (e.g., handle only with gloved hands, treat with RNase AWAY) to maintain RNA quality. Use sterile pipette tips designated for RNA use only.
Chill the isopentane in a metal crucible surrounded by dry ice in a Styrofoam box.
The uterine horns from staged pregnant mice are removed and placed in a Petri dish containing 1× PBS at 4°C.
Individual E9.5 yolk sacs are dissected from the embryo and immediately placed in 1.5 ml microcentrifuge tubes containing 1 ml of cryoprotection buffer at 4°C. Ensure yolk sac is not floating at the top of the cryoprotection buffer by gently tapping the tube on a tabletop.
After 25–30 min, when the yolk sac sinks to the bottom of the tube, use a sterile 1 ml plastic disposable Pasteur pipette to transfer yolk sac into a Petri dish containing 1:1 20% sucrose PBS:Optimal Cutting Temperature freezing media. Rinse well in the solution. It is helpful to cut off the tip of the pipette with a clean razor blade to avoid shearing the tissue.
Carefully transfer the yolk sac into another Petri dish containing OCT. A watchmaker’s forceps can be used to scoop and transfer the yolk sac to prevent bubbles from forming in the OCT. Rinse yolk sac in OCT and examine Petri dish under the dissecting scope to ensure that there is no sucrose residue, which will negatively affect the integrity of the yolk sac upon freezing.
Gently place the yolk sac in the middle of a cryomold containing OCT. Examine under a dissecting microscope, and remove or move away any bubbles near the yolk sac, using a forceps or Pasteur pipette. With the edge of dry forceps, manually sink the yolk sac close to the bottom of the cryomold (see Note 2).
Lower cryomold into the metal crucible containing isopentane chilled with dry ice until cryomold floats on the surface of the isopentane. Aluminum foil or gauze strips can be used as a sling to position the cryomold while slowly lowering it. Allow OCT media to completely cover the cryomold over the course of 2 min, and then drop cryomold into the crucible to completely submerge in cold isopentane, and freeze for one additional minute (see Note 3).
Remove cryomold from isopentane and wrap with chilled aluminum foil, storing in a chilled zip-lock sandwich bag. Store temporarily on dry ice until transferring to −80°C for storage.
3.2 Cryosectioning of Tissue
Clean the knife, brushes and slide box with RNase AWAY. Equilibrate the cryostat, metal chuck, knife, slide box, and brushes to −23°C (see Note 4). The cryostat should be completely defrosted and thoroughly cleaned with RNase AWAY at least once per month.
Mount OCT block onto metal chuck using OCT and let freeze completely. The block should be mounted so the yolk sac is facing away from the metal chuck.
Ensure that knife and OCT block are firmly in place. Adjust relative knife-to-block angle. Lower angles can cause sections to curl more severely while steeper angles can cause chattering of sections.
Orient the OCT block so that the knife cuts evenly on both the left and right sides of the block.
Place 8-μm sections on room temperature Silane-Prep microscope slides, number slides with a pencil, and store slides in the chilled 25-slide box in the cryostat. Place tissue in the middle (at least 0.5 cm away from the edges) of slide for effective LCM (see Notes 5 and 6).
Stain periodic landmark slides (every tenth section) with Rapid Stain to observe cellular morphology.
Place a cassette containing dust-free desiccant behind the 23rd slide of a 25-slide box.
Wrap slide box with chilled aluminum foil and store at −80°C.
3.3 Dehydration and Staining of Microscope Slides for LCM
Place molecular sieves into stock solution of 100% EtOH to ensure that the 100% EtOH bath will completely dehydrate the slides. Incomplete dehydration will result in poor transfer from slide to cap during LCM.
Aliquot and prepare Histogene LCM stain by adding 1 μl of SuperaseIn™ for every of 20 μl of stain to be used in the session, keep on ice.
Frozen sections are processed for LCM using reagents from the HistoGene LCM frozen staining kit, supplemented with anhydrous ethanol, xylene, and nuclease-free water purchased separately. All instructions assume that slides are blotted on absorbent paper between solutions to avoid carryover of solution. To maintain good RNA quality, there should be as little delay as possible in placing slides in subsequent solutions.
Remove two slides at a time from the slide box and place back-to-back into staining dish filled with 75% EtOH. Transfer all slides into dish within 2 min of removing slide box from −80°C. Alternatively, keep the foil-wrapped slide box on dry ice while transferring slides. If processing only 2–4 slides at a time, this can be done back-to-back in one or two 50 ml tubes, rather than in slide dishes.
One slide at a time is vigorously dipped 8–10 times in a 50 ml conical tube containing nuclease-free H2O for about 10–15 s to remove OCT residue. Use fresh tubes with fresh H2O after every ten slides that are processed.
Pipette 20 μl of Histogene LCM stain directly onto the yolk sac tissue on the slide, leave for 10 s, and wash off stain by pipetting H2O on the slide and discarding.
The slide is dipped in H2O several times to remove stain residue and placed into another staining dish with 75% EtOH. Repeat H2O and staining steps with the rest of the slides. All slides should remain in the second 75% EtOH wash for at least 30 s.
The slide-staining rack is transferred to a staining dish containing 95% EtOH for 30 s and then to a dish with 100% EtOH for 3 min.
The rack is dipped twice in a dish containing xylene, and then placed in another staining dish with xylene for 10 min.
To maintain RNA integrity, stained slides should remain in xylene no longer than 2 h total time prior to LCM.
3.4 Laser Capture Microdissection Using the ArcturusXT
Wipe the cap loading dock, slide holders, and the QC station with Kimwipes sprayed with RNase AWAY before using the LCM instrument. One to two slides may be removed from the xylene and allowed to air-dry for 2 min by standing an edge on absorbent paper. Since xylene dissolves the LCM cap polymer, the slide should be completely dry. To preserve RNA integrity, duration of LCM session should be no longer than 20 min per LCM cap, and no longer than 20 min per slide.
Selecting “macro” as the cap type will allow cell capture outside of the cap black ring. At 2× setting, position the yolk sac under the cap. Avoid overlapping the black ring, another captured region, or the 12-μm railing circumscribing the black ring. Examples of suitable cap placements are illustrated in Fig. 1. When the “place cap” icon is clicked, the automated robot arm will place the cap in the center of the current view.
Set the power and duration of laser pulses as low as possible, ensuring however that there is good contact between the thermoplastic cover on the cap and the slide after the pulse (Fig. 2). The following parameters are used for LCM of yolk sac erythroid cells on the ArcturusXT Microdissection Instrument: 10 μm spot size, 70 power setting (coarse adjustment), 30 duration (fine adjustment), 80 IR (infrared) spot spacing. In 20× magnification, fire several test pulses by double clicking on a blank slide region to check that the laser melts the cap polymer to the slide making a well-defined black ring as illustrated in Fig. 2. For the Arcturus PixCell II Laser Capture Microdissection system, the parameters are as follows: 7.5 μm laser spot size, power setting at 80 mW, and laser pulse duration of 1.2 ms.
The blue cross marks where the IR beam is focused. After firing a test pulse, check that the blue cross is centered within the pulse region. If not, right-click in the center and select “locate IR Beam” from the drop-down menu to align the laser and the blue cross.
Right-click on the viewing screen and select “Draw line of IR pulses.” Laser pulses are fired in the same order that they are drawn (see Note 7). An example of the steps in cell collection is illustrated in Fig. 3.
After marking the laser pulses on the viewing screen, click the “IR laser pulse” icon to proceed with firing the laser.
The ArcturusXT Microdissection Instrument tracks the number of pulses fired as well as surface area melted in the “I” menu of the “Select” toolbar. At least 300–400 erythroid cells were collected from three slides on a single LCM cap within 20 min.
After the 20 min of LCM for the cap, click the “Move to QC” icon to inspect for purity of collected cell population. Click “present stage” icon and remove cap.
Place cap directly on three different clean sticky regions of a 3 M Post-it® to remove nonspecific binding to the cap (Fig. 4). Place cap back on the LCM QC and right-click the respective cap and select “view cap.” Check for purity of cell population. Place cap on the 3 M as necessary until nonspecific adherents are removed.
Immediately place the cap on top of a GeneAmp™ 500 μl Thin-walled PCR Reaction Tube containing 200 μl of 4°C denaturing solution. Invert tube and vortex for 3 min, place tube on ice.
Carefully zip spin the tube with attached LCM cap at a low speed to recover solution from the cap (make sure that the LCM cap on top of the tube does not obstruct the centrifuge rotor from spinning normally; it will be taller than a tube alone). Remove LCM cap from the GeneAmp™ tube and store the tube at −80°C.
Fig. 1.
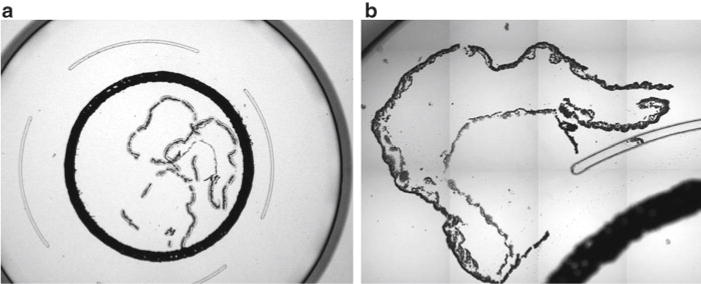
Positioning of the LCM cap on the yolk sac A: The yolk sac can be placed within the black ring on the LCM cap (a, 20× magnification) or outside of it (b, 100× magnification). Avoid overlapping the yolk sac with previously collected cells, with the black ring, and with the clear railing. Because our RNA extraction method exposes the entire cap to the denaturing solution, RNA from all captured cells inside and outside of the black ring will be obtained
Fig. 2.

Various laser pulse intensities and melting of the LCM cap polymer. The cap polymer contains a dye that allows visualization of the melted polymer. In (a, b), the appropriate laser pulse power, duration, and size is indicated, and the polymer is making good contact with the slide. In (c), the laser pulse was fired with too much power, resulting in blackening inside the ring. (d) Depicts inadequate power of the laser, and the cap film is not firmly contacting the slide. If this occurs, increase the power and/or duration, or adjust the positioning of the cap. If good contact is still not achievable, the LCM cap may be defective
Fig. 3.
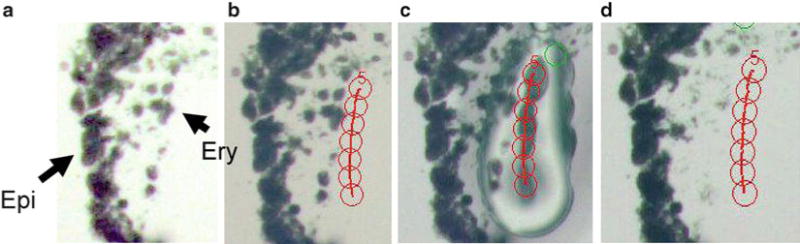
Embryonic yolk sac tissue sample before and after laser capture microdissection. (a) Stained slide photographed at 200× magnification with erythroid (Ery) and epithelial cells (Epi) indicated. (b) The seven concentric circles represent the position of the intended line of IR pulses in relation to the erythroid cells (cells of interest). (c) The actual line of IR pulses, showing the width and intensity of the melted polymer. The single circle at the top right marks the center of the microscope field. (d) Erythroid cells have been removed with the LCM cap, leaving the epithelial cells remaining on the slide. In this specialized case, the position of the laser pulses was offset from the erythroid cells of interest, to allow an increase of power to remove these very adherent cells from the slide, without collecting contaminating epithelial cells
Fig. 4.
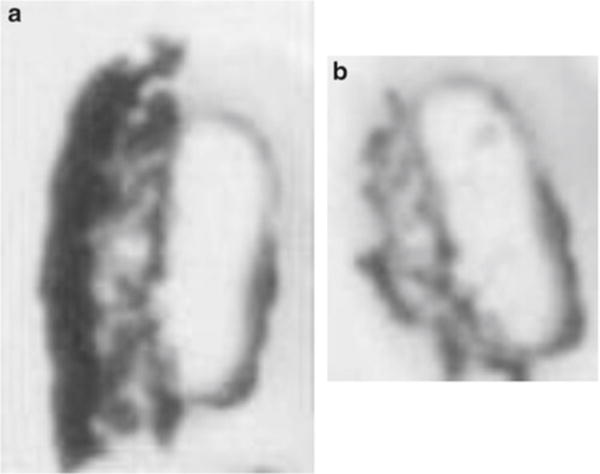
Removing nonspecifically bound cells contaminating the LCM cap. (a) Both erythroid and epithelial cells on the LCM cap at the QC station, prior to placing the cap on the 3 M Post-It adhesive region. (b) Captured erythroid cells remain on the cap and nonspecifically bound epithelial cells have been removed by pressing on a Post-It adhesive region. It is difficult to accurately focus images on the QC station
3.5 RNA Preparation
Make sure that all reagents, working environment and pipettes are RNase free. Allow the 0.5 ml tubes containing the LCM material (200 μl) to thaw on ice. Once thawed, mix the cellular extract by pipetting twice and transfer to a 1.5 ml microcentrifuge tube with O-ring screw cap. If there are any bubbles, zip spin tubes again. (Please note that material of the same cell type collected on three LCM caps can be combined for a total of 600 μl, and RNA can be extracted in a single tube.)
Add 1/10 volume of 3 M NaAC (sodium acetate) pH 4.50 (ToTALLY RNA isolation kit; Ambion) and mix by pipetting up and down (for every 200 μl of denaturing solution, use 20 μl of 3 M sodium acetate pH 4.50).
Add 200 μl of acid-phenol:chloroform (5:1, pH 4.5, ToTALLY RNA isolation kit; Ambion) per LCM cap to the tube (i.e., 600 μl if three caps are combined per tube).
Vortex the tubes for 2 min and then centrifuge at 12,000 × g (11,400 rpm) at 4°C for 5 min.
Transfer the aqueous phase to a fresh 1.5 ml screw cap tube leaving behind 20 μl at the interphase, to avoid DNA contamination (for three LCM caps combined, leave behind at least 60 μl).
To 180 μl of the aqueous phase, add 1.8 μl of 5 mg/ml glycogen as a carrier, and 180 μl of chilled isopropanol.
Vortex the tube and store overnight at −80°C.
Thaw the tube on ice, vortex, and centrifuge at 12,000 × g (11,400 rpm) at 4°C for 30 min. During this time, prepare the nuclease-free water containing 1/20 volume of SuperaseIn™.
Discard the isopropanol supernatant without disturbing the RNA pellet. The pellet should be readily visible because of the use of glycogen as a carrier. Wash the pellet with 1 ml of cold 75% ethanol (made with nuclease-free H2O). Add the ethanol down the sides of the tubes, washing to remove any residual salt. Briefly vortex, zip spin and remove all of the ethanol. Wash the pellet again with 1 ml of cold 75% ethanol, using the same technique as above. Vortex, zip spin the tube, and remove 900 μl of the ethanol. Zip spin again and remove the remaining ethanol. Allow the pellet to air-dry for 1 min. RNA can become difficult to resuspend if pellet is left to dry longer.
Redissolve the RNA by adding 12 μl of nuclease-free water with 1/20 volume of SuperaseIn™ to the same side of the tube as the pellet, zip spin, and mix by pipetting up and down ten times. Keep tubes on ice or store at −80°C. If combining RNA from several tubes for a single microarray assay or for qRT-PCR, see Note 8. RNA collected for qRT-PCR can be used for assays at this step; typically not enough cells are collected to assess RNA quality using the Agilent bioanalyzer.
3.6 Assessing RNA Quality
Allow the reagents from the Pico kit to equilibrate to room temperature for at least 30 min.
Prepare dilutions of RNA and of the RNA 6000 ladder (Ambion). For RNA samples, prepare a 1:4 and a 1:8 dilution using nuclease-free H2O (see Note 9). Undiluted RNA can contain too much salt to run true to size on the Agilent chip; the dilution amount can be adjusted depending on the number of cells collected. Transfer 5 μl (150 ng/μl) of the RNA ladder into a 1.5 ml microcentrifuge tube and heat at 70°C for 2 min. Place the tube on ice and dilute the ladder to 1 ng/μl (1,000 pg/μl) using nuclease-free H2O. Use the ladder immediately, or aliquot and store the prepared ladder at −80°C for later use.
Prepare the gel mix by filtering 550 μl in the supplied filter column. Centrifuge at 1,500 × g for 10 min. Aliquot 65 μl of the filtered gel into RNase-free microcentrifuge tubes. The filtered gel can be stored and used for up to 2 months.
Decontaminate the electrodes on the bioanalyzer before each run. First slowly pipette 350 μl of RNase AWAY into any well of the electrode cleaner chip, so that it covers the bottom of all of the wells. Place the electrode cleaner in the bioanalyzer for 5 min. Open the lid, remove the chip, and empty the RNase AWAY. Add 350 μl of RNase-free H2O to the electrode cleaner and place in the bioanalyzer for about 1 min but no more than 5 min. Allow the bioanalyzer to dry for about 1 min with the lid open.
Vortex the dye concentrate for 10 s and zip spin. Add 1 μl of dye to 65 μl of the filtered gel-matrix aliquot. Vortex thoroughly and centrifuge at 13,000 × g for 10 min. The dye is light sensitive, so care should be taken to return it quickly to the kit box and close the lid securely.
Take a Pico chip out of the bag and place in the chip priming station. Carefully pipette 9 μl of the gel-dye mix without introducing any bubbles into the well marked G with a black dot. Make sure that the syringe on the chip priming station is set to the 1 ml mark. Close the chip priming station so that the latch audibly clicks. Depress the plunger so that it is tucked under the clip for 30 s. Release the plunger using the clip release and wait about 15 s before pulling the plunger to the 1 ml mark. Slowly pull the plunger to the 1 ml mark and open the chip priming station. If the syringe gasket is properly sealed, the plunger should quickly be at the 0.7 ml mark before time expires.
Pipette 9 μl of the gel-dye matrix to the other two wells marked G and discard the remaining mixture. Pipette 9 μl of the conditional solution to the well marked CS.
Pipette 5 μl of Pico marker into the well marked ladder and to each of the sample wells to be used for the experimental run. Place 6 μl of marker in the unused sample wells or the chip will not run properly.
Pipette 1 μl of the prepared RNA ladder into the designated well and 1 μl of the diluted RNA samples to each of the test wells. A pipette that is accurate for measuring 1 μl of volume should be used, to obtain results that are as accurate as possible with respect to the concentration of the RNA.
Vortex the chip for 1 min. It is recommended that the chip is held down with a finger or with tape, so it does not become dislodged from the adapter. Make certain that the chip is flat and stable, or samples in different wells may become mixed together. The chip run should begin within 5 min after mixing, to avoid evaporation.
Activate the software before loading the chip. In the instrument context, select Assay → Electrophoresis → RNA → Eukary ote Total RNA and press start to initiate the run. Sample names can be added while the chip is running. At the end of the run, discard the chip, clean the electrodes as above, and return the reagents to 4°C.
After the run is completed, select results to examine the electropherograms and virtual agarose gel for the RNA samples and the RNA ladder. For interpreting the electropherograms, refer to Note 10 and Fig. 5. Only RNA of high quality is processed for subsequent microarray hybridizations. For processing LCM-prepared RNA for Affymetrix microarray hybridizations, see Note 11. For troubleshooting RNA samples that are degraded, see Note 12.
Fig. 5.
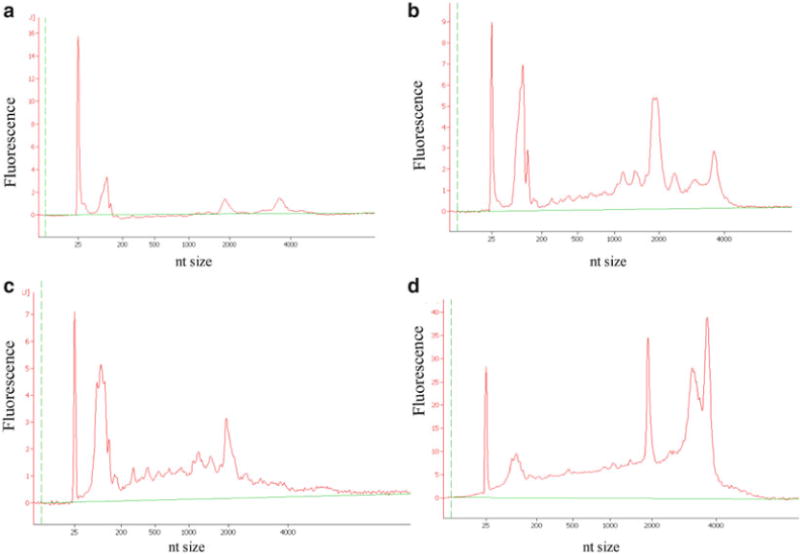
Electropherograms to assess quality of RNA prepared from microdissected cells. RNA integrity is tested using the Agilent bioanalyzer, and only intact RNA is used for performing microarray assays. (a) Intact RNA that is properly diluted, though of a low concentration (see Note 8). The 18S rRNA fluorescent peak appears at about 2,000 nucleotides, and the 28S rRNA peak is just under 4,000 nucleotides. (b) Intact RNA that is not diluted can appear as though it is partially degraded, with peaks between the 18S and 28S rRNA peaks. (c) Partially degraded RNA may contain an apparent 18S rRNA peak, but no 28S rRNA peak. (d) RNA that is contaminated with DNA will have a broad 28S rRNA peak, or two peaks near the 28S rRNA peak
Footnotes
Test the OCT media to verify that it has not undergone a freeze/thaw cycle prior to arrival. Gently pour the OCT into an intermediate cryomold, and freeze the cryomold in a dry ice–isopentane bath. Cut 8-μm sections of the OCT media and place on a Silane-Prep slide; use a permanent marker to identify the area. If the media has a stringy appearance during cryosectioning, or appears dirty or contains holes when slides are examined on the microscope, replace it with a new batch.
Correct placement of the tissue in the cassette prior to freezing is important. Make certain that the tissue is completely surrounded by OCT media on all sides. It may be necessary to place the tissue/embryo at the correct angle to obtain optimal sections. To obtain the maximum amount of tissue from each cryosection, unfold and flatten out yolk sac tissue in the OCT medium prior to freezing in isopentane.
Cryoprotection with sucrose ensures that no ice crystal frozen artifacts form while freezing the tissue. This is essential for yolk sac which is a very thin tissue, and easily subject to freezer burn. Cryoprotection is not required for freezing some other tissues for LCM. Some tissues can simply be dried by blotting on Whatman 3M filter paper, to remove moisture and allow the OCT to directly contact tissue, prior to freezing in OCT in a dry ice–isopentane bath. Some tissues can be dissected without rinsing in 1× PBS, and immediately frozen in OCT in a dry ice–isopentane bath.
Using the appropriate cryostat temperature can influence the quality of the sections, and the optimal temperature is specific to the tissue type and may also depend on the cryostat. For example, bone marrow frozen cryosections are typically prepared at −25°C, and adult brain cryosections are prepared at −18°C. Using a temperature as cold as possible helps maintain RNA integrity. If the temperature is too cold, however, there will be shattering throughout the section.
To keep cryosections flat, cut 8-μm sections until only one edge is still attached to the block. Use paintbrushes to unravel section and push the free edges inwards opposing the axis of curling.
Positioning multiple sections on the slide during cryosectioning can lead to further efficiency in the LCM staining steps as well as the actual LCM steps. Since the first cryosection needs to remain inside the cryostat, place other sections on the same slide within 15–20 s to ensure that they will adhere firmly to the slide.
Drawing a line of pulses is the preferred method for marking IR spots for collecting erythroid cells from embryonic yolk sac blood islands, about two cells are collected per pulse. Use the interactive pen to mark the touch-screen for further precision when drawing lines of pulses. For LCM collection of other cell types, other options for marking IR spots are available, including single pulses, defined circles, and rectangles. Optimal collection method, spot size, and power and duration settings need to be empirically determined for different cell types. Some cell types (erythroid precursors, for example) adhere more steadfastly to the slide and require higher power to remove than others (epithelial cells, for example). One method to avoid contamination from other nearby cells, when collecting very adherent cells by LCM, is to offset the laser pulses from the cells of interest, as shown in Fig. 3. There are several measures that can be taken to maximize cell collection efficiency in the 20 min LCM session. First, mark large blood islands and skip blood islands containing fewer erythroid cells. Second, mark up multiple yolk sac sections on an individual slide, then position the cap prior to laser capture to collect from consecutive sections simultaneously. Third, transfer the cap with collected cells more quickly onto the GeneAmp™ tube by shifting the slide platform so that the cap is exposed from underneath the LCM machine cover, and manually transfer the cap to the QC station using RNAse-free tweezers. The LCM software can then be restarted to “reset” the robot arm.
RNA prepared from the same cell type but extracted in multiple tubes can be pooled for microarray or qRT-PCR assays. Dissolve the first RNA pellet, then use the 12 μl in the first tube to resuspend the RNA pellets from subsequent tubes. Dissolve the first pellet, zip spin, and transfer to the next tube containing an RNA pellet. Resuspend the second pellet and continue this process until all pellets have been combined. This process has been used to successfully combine and obtain high-quality RNA from eight tubes. Hundreds of cells can be collected per LCM cap, and RNA from three caps can be extracted in a single tube, so RNA from 24 LCM caps can be combined in a single tube using this method to resuspend pellets. In this way, RNA from the thousands of cells necessary for a single microarray can be combined. This RNA is re-precipitated with isopropanol and used to prepare labeled cRNA for Affymetrix GeneChip hybridizations, as described in Note 11. At least 50 ng of RNA can typically be obtained from 10,000 yolk sac erythroid cells collected by LCM.
To dilute RNA from LCM samples for the Agilent bioanalyzer assay, add 2 μl of RNA to a 1.5 ml tube containing 6 μl of nuclease-free H2O and mix by pipetting (1:4 dilution). Prepare a 1:8 dilution from the 1:4 dilution. Diluting the RNA properly is essential for the bioanalyzer assay. If the RNA is not diluted enough, samples with a higher salt content will migrate incorrectly and appear to be degraded. If the RNA is too dilute, the bioanalyzer is not able to detect the ribosomal species, and the profile may suggest that the sample has no RNA or is degraded. A 1:4 and 1:8 dilution are recommended for the bioanalyzer assays. A 1:2 dilution may be used for lower yields.
Because LCM RNA microdissected samples contain very little RNA, the 28S:18S ribosomal ratios may not be as high as 2.0. The 28S:18S ribosomal ratios for yolk sac microdissected cells are typically between 0.3 and 1.2 and have produced successful microarray hybridizations. To determine whether the RNA is of high quality, two important quality assessment parameters should be considered. The parameters are the detection of strong 18S and 28S rRNA peaks in the electropherogram, and the area under the 18S and 28S peaks should be greater than 15% of the total area under the curve, as determined by the Agilent software. RNA preparations not meeting these quality control criteria should not be used for subsequent microarray hybridizations.
To process LCM-prepared RNA for microarray assays, re-precipitate the RNA collected from multiple tubes. After centrifugation at 12,000 × g (11,400 rpm) at 4°C for 30 min, carefully wash the pellet with 500 μl of chilled 70% EtOH, allowing it to air-dry for 1 min and then resuspend it in 3 μl of nuclease-free H2O. Proceed with the Affymetrix protocol using the Two-Cycle cDNA Synthesis Kit (following the manufacturer’s protocol). This protocol claims to create enough labeled cRNA from 10 ng of total RNA, but a good yield of labeled cRNA can reliably be obtained from at least 50 ng of RNA. Labeled cRNA yields exceeding 40 μg are considered to be good. As a quality control parameter, the 3′/5′ ratios for housekeeping genes such as GAPDH and β-actin are expected to be higher for the Two-Cycle than for the standard One-Cycle cDNA Synthesis protocol, which was developed to be used with at least 1 μg of RNA. This is due to a more accentuated 3′ bias introduced during the cDNA and cRNA synthesis steps of the Two-Cycle protocol.
Samples can be tested prior to experimental LCM, to determine whether tissues and reagents will give intact RNA preparations. These steps can be used at any time to troubleshoot if RNA preparations are degraded. To troubleshoot, RNA samples should be prepared in three ways, and then tested for RNA integrity on the Agilent bioanalyzer. First, remove a slide from the −80°C freezer and place 200 μl of denaturing solution directly onto the tissue on the slide, allow it to thaw just enough to become liquid, pipette up and down to dissolve all of the tissue, and proceed with an RNA preparation (Subheading 3.5). Second, stain, fix, and dehydrate a slide as described in Subheading 3.3, then dissolve the tissue on the slide using denaturing solution, as described for the first slide and continue RNA preparation. Finally, stain, fix, and dehydrate a slide, and blanket the slide with laser pulses to adhere all of the tissue on the slide onto an LCM cap, then continue with the RNA preparation starting at step 10 of Subheading 3.4. Typically, entire cryosections from slides need to be diluted more than do LCM samples for the Agilent bioanalyzer assay, therefore a 1:100 and a 1:500 dilution is recommended. If the RNA from the first slide is intact, then the frozen tissue samples are of high quality. If not, then troubleshooting of the procedures for dissection, freezing and cryosectioning needs to be performed. If the RNA from the second slide preparation is intact, then the tissue sample is good, and the stain, solutions, and methods used for slide processing are producing intact RNA. If the first slide gives intact RNA and the second does not, then the slide processing steps need troubleshooting. If the RNA preparation from the final slide is the only one that is degraded, then the LCM steps need to be improved.
References
- 1.Chimge NO, Ruddle F, Bayarsaihan D. Laser-assisted microdissection (LAM) in developmental biology. J Exp Zool B Mol Dev Evol. 2007;308:113–118. doi: 10.1002/jez.b.21133. [DOI] [PubMed] [Google Scholar]
- 2.Murray GI. An overview of laser micro-dissection technologies. Acta Histochem. 2007;109:171–176. doi: 10.1016/j.acthis.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Nawshad A, LaGamba D, Olsen BR, Hay ED. Laser capture microdissection (LCM) for analysis of gene expression in specific tissues during embryonic epithelial-mesenchymal transformation. Dev Dyn. 2004;230:529–534. doi: 10.1002/dvdy.20064. [DOI] [PubMed] [Google Scholar]
- 4.Sainson RC, Johnston DA, Chu HC, Holderfield MT, Nakatsu MN, Crampton SP, Davis J, Conn E, Hughes CC. TNF primes endothelial cells for angiogenic sprouting by inducing a tip cell phenotype. Blood. 2008;111:4997–5007. doi: 10.1182/blood-2007-08-108597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canto-Soler MV, Huang H, Romero MS, Adler R. Transcription factors CTCF and Pax6 are segregated to different cell types during retinal cell differentiation. Dev Dyn. 2008;237:758–767. doi: 10.1002/dvdy.21420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toba Y, Tiong JD, Ma Q, Wray S. CXCR4/SDF-1 system modulates development of GnRH-1 neurons and the olfactory system. Dev Neurobiol. 2008;68:487–503. doi: 10.1002/dneu.20594. [DOI] [PubMed] [Google Scholar]
- 7.Redmond LC, Dumur CI, Archer KJ, Haar JL, Lloyd JA. Identification of erythroid-enriched gene expression in the mouse embryonic yolk sac using microdissected cells. Dev Dyn. 2008;237:436–446. doi: 10.1002/dvdy.21426. [DOI] [PubMed] [Google Scholar]
- 8.Spencer MW, Casson SA, Lindsey K. Transcriptional profiling of the Arabidopsis embryo. Plant Physiol. 2007;143:924–940. doi: 10.1104/pp.106.087668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhattacherjee V, Mukhopadhyay P, Singh S, Johnson C, Philipose JT, Warner CP, Greene RM, Pisano MM. Neural crest and mesoderm lineage-dependent gene expression in orofacial development. Differentiation. 2007;75:463–477. doi: 10.1111/j.1432-0436.2006.00145.x. [DOI] [PubMed] [Google Scholar]
- 10.Xiao W, Liu W, Li Z, Liang D, Li L, White LD, Fox DA, Overbeek PA, Chen Q. Gene expression profiling in embryonic mouse lenses. Mol Vis. 2006;12:1692–1698. [PubMed] [Google Scholar]
- 11.Plummer S, Sharpe RM, Hallmark N, Mahood IK, Elcombe C. Time-dependent and compartment-specific effects of in utero exposure to Di(n-butyl) phthalate on gene/protein expression in the fetal rat testis as revealed by transcription profiling and laser capture micro-dissection. Toxicol Sci. 2007;97:520–532. doi: 10.1093/toxsci/kfm062. [DOI] [PubMed] [Google Scholar]
- 12.Williams EO, Xiao Y, Sickles HM, Shafer P, Yona G, Yang JY, Lin DM. Novel subdomains of the mouse olfactory bulb defined by molecular heterogeneity in the nascent external plexiform and glomerular layers. BMC Dev Biol. 2007;7:48. doi: 10.1186/1471-213X-7-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brunskill EW, Aronow BJ, Georgas K, Rumballe B, Valerius MT, Aronow J, Kaimal V, Jegga AG, Yu J, Grimmond S, McMahon AP, Patterson LT, Little MH, Potter SS. Atlas of gene expression in the developing kidney at microanatomic resolution. Dev Cell. 2008;15:781–791. doi: 10.1016/j.devcel.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Redmond LC, Dumur CI, Archer KJ, Grayson DR, Haar JL, Lloyd JA. Krüppel-like factor 2 regulated gene expression in mouse embryonic yolk sac erythroid cells. Blood Cell Mol Dis. 2011;47(1):1–11. doi: 10.1016/j.bcmd.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pang CJ, Lemsaddek W, Alhashem YN, Bondzi C, Redmond LC, Ah-Son N, Dumur CI, Archer KJ, Haar JL, Lloyd JA, Trudel M. Krüppel-like factor 1 (KLF1), KLF2, and myc control a regulatory network essential for embryonic erythropoiesis. Mol Cellular Biol. 2012;32(13):2628–2644. doi: 10.1128/MCB.00104-12. [DOI] [PMC free article] [PubMed] [Google Scholar]