

Abstract
Clostridium difficile associated diarrhea (CDAD) is a critical public health problem worldwide with over 300,000 cases every year in the United States alone. Clearly, a potent vaccine preventing the morbidity and mortality caused by this detrimental pathogen is urgently required. However, vaccine efforts to combat C. difficile infections have been limited both in scope as well as to efficacy, as such there is not a vaccine approved for use against C. difficile to date. In this study, we have used a highly potent Adenovirus (Ad) based platform to create a vaccine against C. difficile. The Ad-based vaccine was able to generate rapid and robust humoral as well as cellular (T-cell) immune responses in mice that correlated with provision of 100% protection from lethal challenge with C. difficile toxin A. Most relevant to the clinical utility of this vaccine formulation was our result that toxin A specific IgGs were readily detected in plasma of Ad immunized mice as early as 3 days post vaccination. In addition, we found that several major immunodominant T cell epitopes were identified in toxin A, suggesting that the role of the cellular arm in protection from C. difficile infections may be more significant than previously appreciated. Therefore, our studies confirm that an Adenovirus based-C. difficile vaccine could be a promising candidate for prophylactic vaccination both for use in high risk patients and in high-risk environments.
Keywords: Clostridium difficile, Recombinant Adenovirus, Vaccine, T cell responses, Antibody responses, Challenge model
1. Introduction
Clostridium difficile (C. difficile), a gram-positive, spore-forming, noninvasive enteric pathogen is a leading cause of nosocomial infections in the developed world, yet has no vaccine available for its prevention [1]. Life threatening manifestations of C. difficile-associated diarrhea (CDAD) include pseudomembranous colitis, toxic megacolon and systemic inflammatory response syndrome [2,3]. Mortality due to CDAD ranges from 6% to 30% in affected patients [4]. More than 300,000 cases of CDAD are reported every year in the United States, and this case rate is predicted to rise by at least 40% within the next several years [2,5,6]. The annual cost for CDAD in the U.S. alone is estimated to be over $1.1 billion [2,5,6].
Mildly symptomatic or asymptomatic patients harboring C. difficile account for the majority of infectious spreading, resulting in new outbreaks. C. difficile pores can be found on environmental surfaces, equipment and clothing years after being deposited. Several host factors including advanced age, pre-existing severe illness, and broad-spectrum antibiotic usage predispose individuals to acute symptomatic C. difficile infection [7]. Recently, a new, highly virulent strain of C. difficile (BI/NAP1/r027) has been associated with outbreaks of severe nosocomial CDAD [2].
The main virulence factors of the C. difficile bacterium are the toxins A (TA) and B (TB) [8]. These toxins belong to the large clostridial cytotoxin family and contain several distinct domains: (1) N-terminal enzymatic domain, (2) Central hydrophobic region, and (3) the C-terminal domain, which recognizes host cell surface carbohydrate receptors [7]. Both TA and TB are enteropathic and potent cytotoxic enzymes [3]. TA and TB are also glucosyltransferases, which catalyze the inactivation of Rho proteins that are involved in cellular signaling. Together, this leads to cytotoxicity, including actin cytoskeleton depolymerization and cell death by apoptosis. In addition, C. difficile infections induce massive cellular immune responses, including neutrophil and monocyte infiltrations, as well as cytokine and chemokine elevations, including IL-6, IL-8, IL-1β, IFNγ [4,5,9]. Moreover, following damage of the intestinal mucosa, systemic release of TA and TB from the lumen of the gut are typically observed in severe life threatening cases of CDAD, and is correlated with acute respiratory distress syndrome, liver damage, multiple organ failure and cardiopulmonary arrest [10–12].
Clearly, the problem of C. difficile is a significant one, as C. difficile is now recognized by the CDC as a Group II pathogen on the NIAID list of emerging and re-emerging infectious diseases (http://www.niaid.nih.gov/topics/emerging/pages/list.aspx). What is desperately required is a potent vaccine that can generate rapid immune responses against C. difficile infections. Such a vaccine could be utilized both as a therapeutic vaccine in patients recently diagnosed with C. difficile, as well as a prophylactic vaccine for use in at-risk patients. Vaccine efforts to combat C. difficile infection have been limited. A number of putative vaccines against C. difficile infection have been developed and tested on animal models. For example, mucosal immunization with C. difficile surface proteins showed moderate efficacy in reducing intestinal colonization by C. difficile in mouse challenge models [3]. Vaccination with a formalin inactivated C. difficile TA (or TA/TB mixture) induced both systemic and mucosal immunity by 14–28 days post immunization in mice, including induction of anti-TA IgG and IgA as well as TA neutralizing antibodies [2]. Hamsters, vaccinated with formalin inactivated TA/TB mixture, were protected from diarrhea and death in a C. difficile challenge model [13]. Mice, injected with high dose of DNA-based vaccine, encoding partial toxin A sequence was shown to induce high plasma IgG titers and protect from lethal TA challenge [14]. It was not studied, however, whether T cell immunity is playing any major role in protection from CDAD; but portions of TA and TB proteins are known to induce Th1/Th2 mixed responses and act as strong mucosal adjuvants [15,16]. Purified inactivated TA/TB toxoid vaccine was relatively well tolerated when tested in phase I clinical trial on healthy individuals; however, it demonstrated moderate efficiency in CDAD patients with recurrent C. difficile infection [13,17]. An inability to evoke rapid immune responses to important C. difficile antigens may in part be the cause of this lack of efficacy [13,17].
We have used a highly potent vaccine platform to create a novel vaccine against this pathogen, namely an Adenovirus (Ad) based C. difficile specific vaccine. Specifically, we constructed an Ad based vaccine expressing the C-terminal, highly immunogenic region of the C. difficile toxin A (amino acids 1870–2680). Our results suggest that moderate doses of this vaccine are able to generate rapid and robust C. difficile specific humoral as well as T cellular immune responses in mice, and provide 100% protection from lethal challenges with toxin A.
2. Materials and methods
2.1. Adenovirus vector construction, production and characterization
All Ads utilized in this study are human Ad type 5 derived replication deficient vectors (deleted for the E1 and E3 genes). To construct Ad5-TA we specifically selected, optimized for human expression, ordered synthetic gene (http://www.geneart.com, Regensburg, Germany) and subcloned the C-terminal region of TA (spanning amino acids 1870–2680) into pShuttle-CMV as previously described [18]. Region selection was based on previous studies [19–21], however, TA region we selected was unique and not utilized in previous publications. Importantly, this C terminal domain of TA is non-toxic and lack enzymatic activity [7,15,19,22]. Recombination and viral propagation were completed as described [18,23,24]. Ad5-Null vector was constructed by recombining pShuttle (with no transgene) with pAdEasyI and purified as described [23]. Propagation and characterization of all Ads was performed as previously described [18]. All viruses were found to be RCA free both by RCA PCR (E1 region amplification) and direct sequencing methods as previously described [25]. All Ads have also been tested for the presence of bacterial endotoxin as previously described [25] and were found to contain <0.15 EU per ml.
2.2. Animal procedures
Adult BALB/c WT mice were purchased from Jackson Laboratory (Bar Harbor, ME). Ad5 vectors were injected intramuscularly (IM, into the tibialis anterior of the right hindlimb, total volume 25 μl) into 8 weeks old male mice after performing proper anesthesia with isofluorane. A total of 1 × 1010 vp per mouse was administered IM. Number of animals used for each experiment is specified on corresponding figure legend. Toxin A challenge experiments were performed at 14 dpi as previously described [14] by injecting 300 ng of freshly reconstituted toxin A (List Biological Laboratories Inc., Campbell, CA or Calbiochem, San Diego, CA) intraperitoneally in 100 μl (in PBS). After the challenge mice were carefully and routinely monitored every 6 h by lab personnel and by technicians from MSU animal core facility for mortality and other parameters, all in accordance with MSU ORCBS and IACUC. Plasma and tissue samples were collected and processed at the indicated time points in accordance with Michigan State University Institutional Animal Care and Use Committee. All procedures with recombinant Ads and TA were performed under BSL-2, and all vector treated animals were maintained in ABSL-2 conditions. All animal procedures were reviewed and approved by the Michigan State University ORCBS and IACUC. Care for mice was provided in accordance with PHS and AAALAC standards.
2.3. Antibody titering assay
ELISA based titering experiments were essentially completed as previously described [14,24,26]. Briefly, 50–100 ng of purified toxin A (diluted in PBS) was used to coat wells of a 96 well plate overnight at 4 °C. Plates were washed with PBS–Tween (0.05%) solution, and blocking buffer (3% BSA in PBS) was added to each well and incubated for 1–3 h at room temperature. Total IgG antibodies, were measured in plasma samples collected from naïve, Ad5-Null or Ad5-TA injected mice. Collection of plasma samples was performed at 3, 7 or 14 dpi. Dilutions were made in blocking buffer (1:10 to 1:40,000). Following dilution, plasma was added to the wells, and incubated at RT for 1 h. Wells were washed using PBS–Tween (0.05%) and HRP-conjugated rabbit anti-mouse antibody (BioRad, Hercules, CA) was added at a 1:5000 dilution in PBS–Tween. TMB (Sigma–Aldrich, St. Louis, MO) substrate was added to each well, and the reaction was stopped with 2 N sulfuric acid. Plates were read at 450 nm in a microplate spectrophotometer.
2.4. ELISpot analysis
96-Well Multiscreen high protein binding Immobilon-P membrane plates (Millipore, Billerica, MA) were pre-treated with ethanol, coated with mouse anti-IFNγ(or IL-4, or IL-2) capture antibody, incubated overnight, and blocked with RPMI medium (with 10% FBS, 1% PSF) prior to the addition of 1.0 × 106 splenocytes/well [18,27]. Ex vivo stimulation included the incubation of splenocytes in 100 μL of media alone (unstimulated), or media containing (1) 2 μg/well of single peptides from a 15-mer-peptide library, spanning the C. difficile TA region, encoded by Ad-TA vaccine (15 aa peptides overlapping by 5 aa on both N- and C-termini); or (2) pool of 12 peptides from the library (each peptide 0.2 μg/well); or (3) 0.2 μg/well of single most immunogenic peptides from the library; or (5) Ad5 vector, inactivated at 56 °C for 45 min (100 vp/cell); for 18–24 h in a 37 °C, 5% CO2 incubator. The library was specifically produced utilizing http://www.jpt.com (JPT Peptide Technologies, Berlin, Germany). Ready-set Go IFNγ, IL-2 and IL-4 mouse ELISpot kits were purchased from eBioscience (San Diego, CA). Staining of plates was completed per the manufacturer’s protocol. Spots were counted and photographed by an automated ELISpot reader system (Cellular Technology, Cleveland, OH).
2.5. Cell staining and flow cytometry
Splenocytes from immunized mice were ex vivo stimulated with peptide #63 (2 μg/well) or with mixture of peptides (#9, #13, #51, #55, #63, all 0.4 μg/well) sequences of peptides as indicated in the text. Following stimulation splenocytes were stained with the following antibodies: PerCpCy5.5-CD3, Alexa Floure700-CD8a, PE-Cy7-CD4, FITC-IFNγ, PE-IL2, Alexa Fluor647-IL4 (4 μg/ml), as indicated in the text, all obtained from BD Biosciences (San Diego, CA). Cells were incubated on ice with the respective antibodies for surface staining for 30 min. After washing, intracellular staining was performed: cells were fixed with 2% formaldehyde (Polysciences, Warrington, PA), permeabilized with 0.2% Saponin (Sigma–Aldrich, St. Louis, MO), and stained. We included the violet fluorescent reactive dye (ViViD, Invitrogen) as a viability marker to exclude dead cells from the analysis. Cells were sorted using an LSR II instrument and analyzed using FlowJo software [28].
2.6. Hematoxylin and eosin staining (hepatic inflammation)
Upon sacrifice, liver tissues were fixed in 10% neutral buffer formalin for 12 h, washed in 70% ethanol, embedded in paraffin and 6-μm sectioned were stained with H&E, exactly as previously described [25,29]. We have adapted a previously developed semi-quantitative scoring system, which allows the level of hepatic pathology between different liver sections to be quantified and statistically compared [25,29]. For every mouse, liver sections obtained at different portions of the liver (0–1000 μm from liver surface) were analyzed and given a numerical score (0–3) for three different categories of liver pathology (portal, periportal, lobular) for at least 15 fields per mouse. We have consulted MSU qualified pathologist prior to performing scoring in blind manner (performed by 2 researchers with similar results). The sum of scores (all fields) for each mouse was taken and individual category scores were averaged for each group. Total inflammation index was computed by averaging the sum of all three individual category scores for each mouse as described previously [25,29].
2.7. Statistical analysis
For every experiment, pilot trials were performed with N = 3 per group. This allowed us to determine effect size and sample variance so that power analysis could be performed to correctly determine the number of subjects per group required to achieve a statistical power >0.8 at the 95% confidence level. Statistically significant differences in C. difficile specific adaptive immune responses were determined using statistical analyses specified in figure legends. Kaplan–Meier survival analysis was performed for challenge experiments. Graphs in this paper are presented as mean of the average ± SD, unless otherwise specified. GraphPad Prism software was utilized for statistical analysis.
3. Results
Adenovirus based vaccine against C. difficile toxin A is able to induce rapid and robust TA-specific humoral responses in mice. C. difficile toxin A is widely considered the best candidate antigen for use in vaccine development against this detrimental pathogen [2,19,20,22,30]. Numerous studies have been completed to identify the most immunogenic portions of toxin A. The receptor binding C-terminal domain of TA contains a series of repeating units; there are 39 tandem repeats. The C-terminal repeat domain of TA was found to be the most effective in generating immunity to this protein when used in conventional vaccine formulations [19,20].
We constructed a recombinant, E1 and E3 gene deleted Ad based vaccine expressing the C-terminal, highly immunogenic region of C. difficile toxin A (amino acids 1870–2680) which we refer to herein as Ad5-TA (see Section 2 for full details). To investigate if Ad5-TA vaccine is able to induce C. difficile specific humoral responses (known to be critical for protection from CDAD), we intramuscularly (IM) injected adult BALB/c mice with moderate dose (1010 vp/mouse) of Ad5-TA or a control vaccine, referred to as Ad5-Null. Elevated plasma levels of TA-specific antibodies were detected at 3, 7 and 14 days post injection (dpi). More specifically, we confirmed that significant (p < 0.05) IgG titers were present as early as 3 dpi (Fig. 1A); IgG titers were further increased by 7 dpi (Fig. 1B) and further increased by 14 dpi (Fig. 1C and data not shown).
Fig. 1.

Adenovirus based vaccine against Clostridium difficile toxin A is able to induce rapid and robust TA-specific humoral responses in mice. WT BALB/c mice were IM injected (1010 vp/mouse) with Ad5-Null (n = 5) or Ad5-C. difficile-TA (n = 5). At 3 dpi (A), 7 dpi (B) and 14 dpi (C) plasma samples were collected and total TA-specific IgG measured by ELISA as described in Section 2. Naïve mice (n = 5) were utilized as baseline control and these values were subtracted Ad-injected values. The error bars represent ±SD. Statistical analysis was completed using two-tailed Student’s t-test to compare 2 groups of virus-injected animals (*p < 0.05). One of two representative experiments shown.
3.1. Adenovirus based vaccine against Clostridium difficile toxin A is able to induce robust TA-specific T cell responses
To examine if T cell responses to C. difficile TA can be elicited by the potent Ad5-TA vaccine platform, we analyzed cellular immune responses by ELISPOT assays, utilizing a 15-mer-peptide library (overlapping each other by 5 amino acids from both N- and C-termini) that spanned the portions of the C. difficile TA protein, encoded by the Ad5-TA vaccine. By utilizing this TA peptide library, we identified several clusters of immunogenic epitopes in the TA non-enzymatic region, with two being major immunodominant epitopes: VNGSRYYFDTDTAIA and YYFNTNTSIASTGYT (>400 IFNγ SFCs per 106 splenocytes) (Fig. 2A and B, Table 1). Interestingly, two 9-mer peptides were suggested as being the top 2 for binding to the BALB/c H2Kd allele when utilizing a popular MHC I epitope prediction program (http://www.syfpeithi.de/scripts/MHCServer.dll/): these predicted 9-mers are indeed present in the 15-mers identified in our ELISPOT analyses (Table 1). However, use of the Ad5-TA vaccine iden-tified several other epitopes as also being highly immunogenic subsequent to Ad5-TA vaccinations, including 3 epitopes yielding 150–400 SFCs, 4 yielding 100–150 SFCs and 2 epitopes yielding 50–100 SFCs per 106 splenocytes in the IFNγ ELISpot (Fig. 2A and B, Table 1).
Fig. 2.

Adenovirus based vaccine against Clostridium difficile toxin A is able to induce robust TA-specific T cell responses, several clusters of immunogenic T cell epitopes revealed in non-enzymatic TA domain. (A) WT BALB/c mice were IM injected (1010 vp/mouse) with Ad5-C. difficile-TA (n = 3). Mice were sacrificed at 14 dpi, splenocytes prepared, pooled and stimulated with 2 μg/well of single peptides from a 15-mer-peptide library, spanning the C. difficile TA region, encoded by Ad-TA vaccine, followed by IFNγ ELISPOT, performed as described in Section 2. Naïve (n = 3) pooled mice were stimulated with same individual peptides as baseline control. Six clusters of immunogenic T cell epitopes was identified (grey) in non-enzymatic TA domain, two major immunodominant epitopes determined were #13 and #63. One of two representative experiments shown. (B) Representative pictures of wells from ELISpot are shown.
Table 1.
C. difficile toxin A specific major T cell epitopes.a
| 400+ SFCs | 150–400 SFCs | 100–150 SFCs | 50–100 SFCs | |
|---|---|---|---|---|
| Number of responders | 2 | 3 | 4 | 2 |
| Peptides | #13 VNGSRYYFDTDTAIA | #9 NEKYYFNPN-NAIAAV | #27 YFNTNTA-IASTGYTI #30 IGVFKGP-NGFEYFAP |
#66 QIGVFKG-PDGFEYFA |
| #63 YYFNTNTSIASTGYT | #51 QTIDGKKYYFNT-NTF | #36 KYYFNPN-NAIAAIHL | #74 NKN-FYFRNGLPQIGV | |
| #55 VFKGP-NGFEYFAPAN | #37 AAIHLCTINND-KYYF |
C. difficile toxin A specific major T cell epitopes. Splenocytes derived from Ad5-TA vaccinated BALB/c mice were stimulated with 15-mer library spanning the C. difficile TA region, encoded by Ad5-TA vaccine, followed by ELISPOT assay. Peptides with most dramatic SFC (spot forming cells) count are presented (most immunogenic epitopes). Note that H2Kd (9-mers) epitope prediction (http://www.syfpeithi.de/scripts/MHCServer.dll/) yields derivatives of peptides #13 and #63 as 2 top hits (highlighted in bold and underlined).
To more fully characterize T cell responses induced by the Ad5-TA C. difficile vaccine, we have stimulated splenocytes derived from Ad5-TA vaccinated (at 14 dpi) or naïve mice with pools each containing twelve TA-specific peptides. Utilizing these peptide pools, we performed IFNγ (Fig. 3A) or IL-2 (Fig. 3B) based ELISPOT analyses as well, and again confirmed that significant TA specific T cell responses could be detected in TA-vaccinated mice, but not in control mice. This study confirmed that use of the Ad5-TA vaccine induced a robust and broad T cell immune response to this target antigen, as immunogenic T cell epitope clusters were located in several regions within the TA C-terminal non-enzymatic domain, rather than being concentrated in only one particular region subsequent to Ad5-TA vaccination.
Fig. 3.
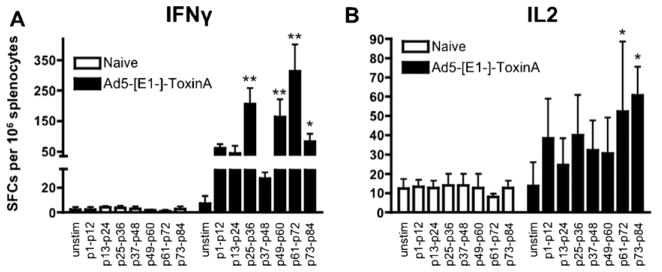
Adenovirus based vaccine against Clostridium difficile toxin A is able to induce robust TA-specific T cell responses. WT BALB/c mice were IM injected (1010 vp/mouse) with Ad5-C. difficile-TA (n = 5). Mice were sacrificed at 14 dpi splenocytes prepared, and individually stimulated with pool of 12 peptides from the TA peptide library (each peptide 0.2 μg/well) and (A) IFNγ or (B) IL-2 ELISPOT was performed as described in Section 2. Naïve (n = 5) mice were stimulated with same individual peptides as baseline control. One of two representative experiments shown. Bars represent mean ± SD. Statistical analysis was completed using Two-Way ANOVA with a Bonferroni post hoc test (stimulations × treatments), p < 0.05 was deemed a statistically significant difference. *, **Statistically different from those in naïve mice (for the same stimulation), p < 0.05, p < 0.001, respectively.
To assess for potential, non-specific Ad-mediated effects of vaccination, we similarly analyzed these responses after administration of an Ad5-Null control vaccine. IFNγ ELISpot revealed a lack of any significant responses in both Ad-naïve and Ad5-Null injected mice, relative to the robust TA-specific T cell responses again noted in Ad5-TA vaccinated mice (Fig. 4). Moreover, we detected significant Ad5-specific T cell responses in Ad5-Null and Ad5-TA injected mice, which were identical between the 2 groups of Ad5-injected animals, indirectly confirming that both the Ad5 control and Ad5-TA vaccinations were performed identically and therefore C. difficile TA-specific T cell responses are truly derived only from Ad vaccine mediated expression of the TA protein (Fig. 4).
Fig. 4.
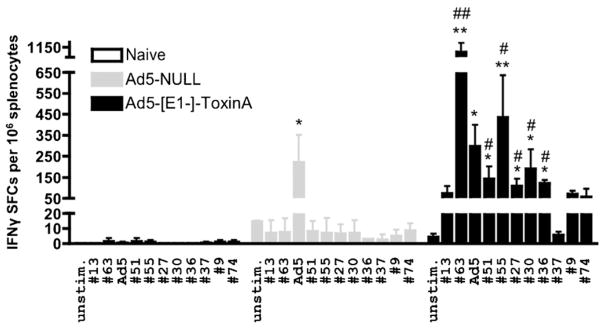
Ad5-Clostridium difficile-TA vaccine induces robust TA-specific T cell responses in contrast to Ad5-Null. WT BALB/c mice were IM injected (1010 vp/mouse) with Ad5-C. difficile-TA (n = 5) or Ad5-Null (n = 5). Mice were sacrificed at 14 dpi splenocytes prepared, and individually stimulated with 0.2 μg/well of single most immunogenic peptides from the TA library (or with inactivated Ad5 vector). IFNγ ELISPOT was performed as described in Section 2. Naïve (n = 3) mice were stimulated with same peptides as baseline control. Bars represent mean ± SD. Statistical analysis was completed using Two-Way ANOVA with a Bonferroni post hoc test (stimulations × treatments), p < 0.05 was deemed a statistically significant difference. *, **Statistically different from those in naïve mice (for the same stimulation), p < 0.05, p < 0.001, respectively; #, ##Significant inductions over Ad5-Null group within the same stimulation, p < 0.05, p < 0.001, respectively. One of two representative experiments shown.
Ad5 vaccines are known to preferentially induce CD8 specific effector T cell responses, however, CD4 responses are also induced by Ad based vaccines [31,32]. IL-4 and IL-2 TA specific ELISpot results strongly suggest that Ad5-TA vaccination is able to induce a pleiotropic CD8 (and likely CD4) as well as potentially Th1/Th2 immunity to the C. difficile TA protein (Fig. 5A and B). We have shown that Ad5-TA vaccine is able to induce IFNγ (but not other cytokines) production from CD8+ T cells in a recall response to TA-specific antigens (Fig. 6A and B) as determined by ICS. We did not however detect any significant IL2, IL4 or IFNγ production by CD4+ T cells in Ad5-TA vaccinated mice (data not shown).
Fig. 5.

Ad5-Clostridium difficile-TA vaccine induces pleiotropic TA-specific T cell responses in contrast to Ad5-Null. WT BALB/c mice were IM injected (1010 vp/mouse) with Ad5-C. difficile-TA (n = 5) or Ad5-Null (n = 5). Mice were sacrificed at 14 dpi splenocytes prepared, and individually stimulated with 0.2 μg/well of single most immunogenic peptides from the TA library (or with inactivated Ad5 vector). (A) IL-4 and (B) IL-2 ELISPOT was performed as described in Section 2. Naïve (n = 3) mice were stimulated with same peptides as baseline control. Bars represent mean ± SD. Statistical analysis was completed using Two-Way ANOVA with a Bonferroni post hoc test (stimulations × treatments), p < 0.05 was deemed a statistically significant difference. *, **Statistically different from those in naïve mice (for the same stimulation), p < 0.05, p < 0.001, respectively; #, ##Significant inductions over Ad5-Null group within the same stimulation, p < 0.05, p < 0.001, respectively. One of two representative experiments shown.
Fig. 6.

Ad5-Clostridium difficile-TA vaccine induces robust TA-specific CD8 T cell specific responses in contrast to Ad5-Null. WT BALB/c mice were IM injected (1010 vp/mouse) with Ad5-C. difficile-TA (n = 3) or Ad5-Null (n = 3). Mice were sacrificed at 14 dpi splenocytes prepared, and individually stimulated with 2 μg/well of peptide #63 or with mixture of peptides (#9, #13, #51, #55, #63, all 0.4 μg/well), stained and FACS sorted as described in Section 2. The bars represent mean ± SEM. Statistical analysis was completed using a two tailed homoscedatic Student’s t-test.
3.2. Ad5-TA C. difficile vaccination completely protects mice from lethal TA challenge
To determine if the rapid and robust induction of humoral and T cell responses elicited by vaccination with Ad-TA is clinically meaningful, we challenged vaccinated mice with 300 ng (6 × LD50) of purified TA 14 days post A5-TA vaccination [14,33]. We have found that Ad5-Null injected mice have a 40% survival rate with this challenge, whereas Ad5-TA vaccinated mice have 100% survival for the duration of experiment (Fig. 7A), a significantly improved outcome (p < 0.05) relative to use of the Ad5-Null control vaccine. It was unclear, however, why some of Ad5-Null injected mice had survived the challenge, since similar challenges previously published resulted in 100% death of unvaccinated mice [14]. We repeated the experiment, and this time utilized TA from a source identical to the one used in the previously published studies [14]. Again, we found that 100% of the Ad5-TA vaccinated mice survived TA challenge, a significantly increased survival (p < 0.05) as compared to the Ad5-Null challenged mice (~50% survival) and naïve unvaccinated mice (~30% survival) (Fig. 7B). Other technical experimental variabilities may be accounting for the lack of 100% death in our TA challenged mice.
Fig. 7.

Ad5-Clostridium difficile-TA vaccination completely protects mice from lethal TA challenge. (A) WT BALB/c mice were IM injected (1010 vp/mouse) with Ad5-Null (n = 5) or Ad5-C. difficile-TA (n = 5). At 14 dpi mice were IP challenged with 300 ng (6 × LD50 ) of purified toxin A, purchased from Calbiochem. Kaplan–Meyer survival curves are shown. Curves were compared by log-rank analysis and were found to be significantly (p < 0.05) different. (B) WT BALB/c mice were left uninjected (naïve, n = 11) or were IM injected (1010 vp/mouse) with Ad5-Null (n = 8) or Ad5-C. difficile-TA (n = 7). At 14 dpi mice were IP challenged with 300 ng (6 × LD50 ) of purified toxin A, purchased from List Biological Laboratories Inc. Kaplan–Meyer survival curves are shown. Curves were pair-wise compared by log-rank analysis and the following results were obtained: naïve versus Ad5-TA curve were significantly (p = 0.0043) different, Ad5-Null versus Ad5-TA curves were significantly (p = 0.036) different, however naïve versus Ad5-Null curves were not significantly different (p = 0.486).
The severity of hepatic inflammation in Ad5-TA vaccinated mice after TA challenge was also assessed. We first confirmed that there is a dramatic level of portal, periportal and lobular inflammation in livers of unvaccinated control mice that underwent TA challenge, data that serve as a positive control (unvaccinated censored mice). In contrast, we found that Ad5-TA vaccinated mice that were also (TA-)challenged had reduced levels of portal, periportal and lobular hepatic inflammation (as compared to unvaccinated mice, challenged with TA), indicating another indirect benefit of Ad5-TA vaccination (Fig. 8A and B). Specifically, Ad5-TA vaccinated mice showed almost no signs of leukocyte invasion, lobular disarray, or necrosis. There was no apparent difference between the Ad5-TA group and the naïve mice. Unvaccinated censored mice had complete lobular disarray, with pooling of erythrocytes subsequent to hemorrhage throughout the tissue. The unvaccinated group of mice that survived had lobular disarray with focal areas of glassy eosinophilic hyalin deposition and necrosis. These mice also had areas of cellular swelling indicating some cell stress (Fig. 8B).
Fig. 8.

Ad5-TA vaccinated mice had overall reduced portal, periportal and lobular hepatic inflammation as compared to unvaccinated mice, when challenged with toxin A. WT BALB/c mice were IM injected (1010 vp/mouse) with Ad5-Null or Ad5-C. difficile-TA. At 14 dpi mice were challenged with 300 ng (6 × LD50 ) of purified toxin A (IP). At 14 dpi survivors from both groups were sacrificed, liver sections were stained with H&E and morphometric evaluation of these sections was performed as described in Section 2. (A) Representative sections from each treated animal were analyzed, scored and averaged for the levels of portal, periportal and lobular inflammation, as described in Section 2. The sum of averages for each category was computed to obtain a total inflammation index score. The error bars represent ±SD. Unvaccinated censored mice (n = 2) were used as positive control (high inflammation). Statistical analysis was completed using two-tailed Student’s t-test to compare 2 groups of virus-injected animals: Ad5-Null injected (n = 4) and Ad5-TA injected (n = 4). There was a trend of reduced inflammation in vaccinated mice, however, no significant differences was detected. (B) Representative liver sections for each group of mice are shown. Arrows indicate the following: (1) complete lobular disarray, with pooling of erythrocytes throughout the tissue subsequent to hemorrhage; (2) glassy eosinophilic hyalin deposition; (3) necrosis; (4) areas of cellular swelling indicating cell stress.
4. Discussion
A number of indirect CDAD treatments are currently in clinical trials, including use of new antibiotics, toxin sequestration compounds, monoclonal antibodies to C. difficile toxins and active probiotics [3,4,6,34]. It is well known that recurrence of CDAD is associated with a lack of protective immunity to C. difficile toxins TA and TB. The incidence of CDAD reoccurrence ranges from 8 to 50% of cases with a trend to increase [4]. Patients developing high serum IgG antibodies titers against TA during their first episode of CDAD are 48-fold less likely to develop recurrent CDAD [2,7,13,30]. These findings suggest a great potential for efficacy if a potent vaccine against C. difficile could be identified. Currently, clinically tested C. difficile vaccines lack efficacy [13,17]. Although several C. difficile immunoreactive surface proteins have been identified (including Cwp2, cwp84, cwp66), they contribute modestly to overall C. difficile associated immunity [1,8]. Moreover, the highly pathogenic strain BI/NAP1/r027 produces larger quantities (16–23 times) of C. difficile toxins A and B as compared to other strains [5]. These strong correlations have prompted research into the development of a vaccine targeting these (TA and TB) C. difficile antigens [2,22,30].
Recently, a DNA based vaccine targeting receptor-binding domain of TA was developed and tested in murine model. A two dose vaccination scheme utilizing high doses (25 μg each) of the plasmid DNA encoding the TA antigen have yielded significant TA-specific plasma IgG titers, which correlated with 100% protection from subsequent TA lethal challenge, however, it was unclear if T cell responses played any role in protection from TA challenge [14]. Despite this protection, it is difficult to envision rapid translation of this vaccine to human trial, largely due to common problems associated with plasmid DNA based vaccines: lack of immunogenicity, high cost of large-scale production and inability to solely induce beneficial robust immune responses in humans relative to virus based vaccine platforms such as Ad [35–37]. Our study, however, clearly demonstrated that Ad based vaccines encoding immunogenic portions of TA could be efficacious.
Ads offer tremendous capabilities as potent vaccine platforms for use against C. difficile, primarily reflected by their superior ability to induce both humoral and cellular (predominantly CD8) immune responses to desired antigens expressed by the vectors [38–41]. It is thought that Ads induce a unique activation of the innate immune system resulting in rapid induction of antigen specific T or B cell responses [42–45]. Ad base vaccines have shown superior efficacy in HIV, Ebola and H5N1 influenza applications, relative to other DNA based as well other viral platforms [46–48]. Ad5-based vaccines outperform other vaccine platforms not only in animal model settings [40,49–52], but also in human trials [37]. Specifically, the Merck® sponsored STEP trial used an Ad5 gag, pol, and nef encoding cocktail of vaccines, and it was the first human clinical trial reporting induction of HIV specific T cell responses in vaccinees, surpassing previous attempts utilizing poxvirus or plasmid DNA based vaccines [53].
The dose regimen we utilized in our study (1010 vp/mouse) is considered moderate [54,55], since at least 20-fold higher doses have been utilized in mice for identical intramuscular immunizations [56]. Moreover, 1011 vp/person is a currently FDA approved dose of Ad5-based vectors, a dose utilized in clinical trials (http://clinicaltrials.gov/ct2/show/NCT01147965). At moderate dosages we have directly shown that Ad5-TA C. difficile vaccine can induce rapid (detectable as early as 3 dpi) and robust humoral and robust specific T cell responses to the C. difficile TA antigen. Due to the potency of this vaccine, we have also identified important C. difficile TA specific immunogenic T cell epitopes. Furthermore, we confirmed that the TA specific immune responses induced by the Ad5-TA vaccine positively correlated with the ability of the Ad5-TA vaccine to fully protect mice in a well-established C. difficile TA challenge model, even when the challenge is performed as early as 14 days after a single, Ad5-TA vaccination, a time point much earlier than reported previously [14].
5. Conclusions
In conclusion, we have developed an efficacious, Adenovirus based vaccine against C. difficile, a vaccine that encodes the C-terminal, highly immunogenic non-enzymatic region of the toxin A protein expressed by C. difficile. We have shown that moderate doses of this vaccine result in rapid and robust inductions of both humoral and cellular C. difficile specific immune responses in mice. T cell responses were also identified, suggesting that clusters of immunogenic T cell epitopes may contribute significantly to immunity against C. difficile. These findings might lend insight into future studies investigating what role T cells may have in CDAD recurrence risk.
Acknowledgments
We wish to thank the Michigan State University Laboratory Animal support facility for their assistance in the humane care and maintenance of the animals utilized in this work and Amy S. Porter, Kathy A. Joseph, Rick A. Roseburry from MSU Investigative Histopathology Laboratory for their assistance in performing H&E stains of liver tissues. A.A. was supported by the National Institutes of Health grants RO1DK-069884, P01 CA078673, the MSU Foundation as well the Osteopathic Heritage Foundation.
Footnotes
Conflict of interest statement: No competing financial interests exist.
References
- 1.Pechine S, Janoir C, Collignon A. Variability of Clostridium difficile surface proteins and specific serum antibody response in patients with Clostridium difficile-associated disease. J Clin Microbiol. 2005;43(October 10):5018–25. doi: 10.1128/JCM.43.10.5018-5025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghose C, Kalsy A, Sheikh A, Rollenhagen J, John M, Young J, et al. Transcutaneous immunization with Clostridium difficile toxoid A induces systemic and mucosal immune responses and toxin A-neutralizing antibodies in mice. Infect Immun. 2007;75(June 6):2826–32. doi: 10.1128/IAI.00127-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pechine S, Janoir C, Boureau H, Gleizes A, Tsapis N, Hoys S, et al. Diminished intestinal colonization by Clostridium difficile and immune response in mice after mucosal immunization with surface proteins of Clostridium difficile. Vaccine. 2007;25(May 20):3946–54. doi: 10.1016/j.vaccine.2007.02.055. [DOI] [PubMed] [Google Scholar]
- 4.Aslam S, Hamill RJ, Musher DM. Treatment of Clostridium difficile-associated disease: old therapies and new strategies. Lancet Infect Dis. 2005;5(September 9):549–57. doi: 10.1016/S1473-3099(05)70215-2. [DOI] [PubMed] [Google Scholar]
- 5.Hookman P, Barkin JS. Clostridium difficile associated infection, diarrhea and colitis. World J Gastroenterol. 2009;15(April 13):1554–80. doi: 10.3748/wjg.15.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerding DN, Muto CA, Owens RC., Jr Treatment of Clostridium difficile infection. Clin Infect Dis. 2008;46(January Suppl 1):S32–42. doi: 10.1086/521860. [DOI] [PubMed] [Google Scholar]
- 7.Giannasca PJ, Warny M. Active and passive immunization against Clostridium difficile diarrhea and colitis. Vaccine. 2004;22(February 7):848–56. doi: 10.1016/j.vaccine.2003.11.030. [DOI] [PubMed] [Google Scholar]
- 8.Wright A, Drudy D, Kyne L, Brown K, Fairweather NF. Immunoreactive cell wall proteins of Clostridium difficile identified by human sera. J Med Microbiol. 2008;57(June Pt 6):750–6. doi: 10.1099/jmm.0.47532-0. [DOI] [PubMed] [Google Scholar]
- 9.Savidge TC, Pan WH, Newman P, O’Brien M, Anton PM, Pothoulakis C. Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine. Gastroenterology. 2003;125(August 2):413–20. doi: 10.1016/s0016-5085(03)00902-8. [DOI] [PubMed] [Google Scholar]
- 10.Hamm EE, Voth DE, Ballard JD. Identification of Clostridium difficile toxin B car-diotoxicity using a zebrafish embryo model of intoxication. Proc Natl Acad Sci USA. 2006;103(September 38):14176–81. doi: 10.1073/pnas.0604725103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson S, Kent SA, O’Leary KJ, Merrigan MM, Sambol SP, Peterson LR, et al. Fatal pseudomembranous colitis associated with a variant clostridium difficile strain not detected by toxin A immunoassay. Ann Intern Med. 2001;135(September 6):434–8. doi: 10.7326/0003-4819-135-6-200109180-00012. [DOI] [PubMed] [Google Scholar]
- 12.Jacob SS, Sebastian JC, Hiorns D, Jacob S, Mukerjee PK. Clostridium difficile and acute respiratory distress syndrome. Heart Lung. 2004;33(July–August 4):265–8. doi: 10.1016/j.hrtlng.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 13.Sougioultzis S, Kyne L, Drudy D, Keates S, Maroo S, Pothoulakis C, et al. Clostridium difficile toxoid vaccine in recurrent C. difficile-associated diarrhea. Gastroenterology. 2005;128(March 3):764–70. doi: 10.1053/j.gastro.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Gardiner DF, Rosenberg T, Zaharatos J, Franco D, Ho DD. A DNA vaccine targeting the receptor-binding domain of Clostridium difficile toxin A. Vaccine. 2009;27(June 27):3598–604. doi: 10.1016/j.vaccine.2009.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castagliuolo I, Sardina M, Brun P, DeRos C, Mastrotto C, Lovato L, et al. Clostridium difficile toxin A carboxyl-terminus peptide lacking ADP-ribosyltransferase activity acts as a mucosal adjuvant. Infect Immun. 2004;72(May 5):2827–36. doi: 10.1128/IAI.72.5.2827-2836.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brun P, Scarpa M, Grillo A, Palu G, Mengoli C, Zecconi A, et al. Clostridium difficile TxAC314 and SLP-36 kDa enhance the immune response toward a co-administered antigen. J Med Microbiol. 2008;57(June Pt 6):725–31. doi: 10.1099/jmm.0.47736-0. [DOI] [PubMed] [Google Scholar]
- 17.Kotloff KL, Wasserman SS, Losonsky GA, Thomas W, Jr, Nichols R, Edelman R, et al. Safety and immunogenicity of increasing doses of a Clostridium difficile toxoid vaccine administered to healthy adults. Infect Immun. 2001;69(February 2):988–95. doi: 10.1128/IAI.69.2.988-995.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seregin SS, Aldhamen YA, Appledorn DM, Hartman ZC, Schuldt NJ, Scott J, et al. Adenovirus capsid-display of the retro-oriented human complement inhibitor DAF reduces Ad vector-triggered immune responses in vitro and in vivo. Blood. 2010;116(September 10):1669–77. doi: 10.1182/blood-2010-03-276949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kink JA, Williams JA. Antibodies to recombinant Clostridium difficile toxins A and B are an effective treatment and prevent relapse of C. difficile-associated disease in a hamster model of infection. Infect Immun. 1998;66(May 5):2018–25. doi: 10.1128/iai.66.5.2018-2025.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ward SJ, Douce G, Figueiredo D, Dougan G, Wren BW. Immunogenicity of a Salmonella typhimurium aroA aroD vaccine expressing a nontoxic domain of Clostridium difficile toxin A. Infect Immun. 1999;67(May 5):2145–52. doi: 10.1128/iai.67.5.2145-2152.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahearn JM, Fischer MB, Croix D, Goerg S, Ma M, Xia J, et al. Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B cell response to T-dependent antigen. Immunity. 1996;4(March 3):251–62. doi: 10.1016/s1074-7613(00)80433-1. [DOI] [PubMed] [Google Scholar]
- 22.Aboudola S, Kotloff KL, Kyne L, Warny M, Kelly EC, Sougioultzis S, et al. Clostridium difficile vaccine and serum immunoglobulin G antibody response to toxin A. Infect Immun. 2003;71(March 3):1608–10. doi: 10.1128/IAI.71.3.1608-1610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng P, Graham FL. Construction of first-generation adenoviral vectors. Methods Mol Med. 2002;69:389–414. doi: 10.1385/1-59259-141-8:389. [DOI] [PubMed] [Google Scholar]
- 24.Seregin SS, Aldhamen YA, Appledorn DM, Schuldt NJ, McBride AJ, Bujold M, et al. CR1/2 is an important suppressor of Adenovirus-induced innate immune responses and is required for induction of neutralizing antibodies. Gene Ther. 2009;16(October 10):1245–59. doi: 10.1038/gt.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seregin SS, Appledorn DM, McBride AJ, Schuldt NJ, Aldhamen YA, Voss T, et al. Transient pretreatment with glucocorticoid ablates innate toxicity of systemically delivered adenoviral vectors without reducing efficacy. Mol Ther. 2009;17(April 4):685–96. doi: 10.1038/mt.2008.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hensley SE, Cun AS, Giles-Davis W, Li Y, Xiang Z, Lasaro MO, et al. Type I inter-feron inhibits antibody responses induced by a chimpanzee adenovirus vector. Mol Ther. 2007;15(February 2):393–403. doi: 10.1038/sj.mt.6300024. [DOI] [PubMed] [Google Scholar]
- 27.Weaver EA, Barry MA. Effects of shielding adenoviral vectors with polyethylene glycol (PEG) on vector-specific and vaccine-mediated immune responses. Hum Gene Ther. 2008;19(12):1369–82. doi: 10.1089/hum.2008.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aldhamen YA, Appledorn DM, Seregin SS, Liu CJ, Schuldt NJ, Godbehere S, et al. Expression of the SLAM family of receptors adapter EAT-2 as a novel strategy for enhancing beneficial immune responses to vaccine antigens. J Immunol. 2011;186(January 2):722–32. doi: 10.4049/jimmunol.1002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu H, Serra D, Amalfitano A. Persistence of an [E1-, polymerase-] adenovirus vector despite transduction of a neoantigen into immune-competent mice. Hum Gene Ther. 1999;10(February 3):355–64. doi: 10.1089/10430349950018805. [DOI] [PubMed] [Google Scholar]
- 30.Kyne L, Warny M, Qamar A, Kelly CP. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N Engl J Med. 2000;342(February 6):390–7. doi: 10.1056/NEJM200002103420604. [DOI] [PubMed] [Google Scholar]
- 31.Appledorn DM, Aldhamen YA, Depas W, Seregin SS, Liu CJ, Schuldt N, et al. A new adenovirus based vaccine vector expressing an Eimeria tenella derived TLR agonist improves cellular immune responses to an antigenic target. PLoS One. 2010;5(3):e9579. doi: 10.1371/journal.pone.0009579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seregin SS, Aldhamen YA, Appledorn DM, Zehnder J, Voss T, Godbehere S, et al. Use of DAF-displaying adenovirus vectors reduces induction of transgene-and vector-specific adaptive immune responses in mice. Hum Gene Ther. 2011;22(9):1083–94. doi: 10.1089/hum.2010.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen X, Katchar K, Goldsmith JD, Nanthakumar N, Cheknis A, Gerding DN, et al. A mouse model of Clostridium difficile-associated disease. Gastroenterology. 2008;135(December 6):1984–92. doi: 10.1053/j.gastro.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 34.Taylor CP, Tummala S, Molrine D, Davidson L, Farrell RJ, Lembo A, et al. Open-label, dose escalation phase I study in healthy volunteers to evaluate the safety and pharmacokinetics of a human monoclonal antibody to Clostridium difficile toxin A. Vaccine. 2008;26(June 27–28):3404–9. doi: 10.1016/j.vaccine.2008.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seregin SS, Amalfitano A. Gene therapy for lysosomal storage diseases: progress, challenges and future prospects. Curr Pharm Des. 2011;17(24):2558–74. doi: 10.2174/138161211797247578. [DOI] [PubMed] [Google Scholar]
- 36.Wang R, Doolan DL, Le TP, Hedstrom RC, Coonan KM, Charoenvit Y, et al. Induction of antigen-specific cytotoxic T lymphocytes in humans by a malaria DNA vaccine. Science. 1998;282(October 5388):476–80. doi: 10.1126/science.282.5388.476. [DOI] [PubMed] [Google Scholar]
- 37.Asmuth DM, Brown EL, Dinubile MJ, Sun X, Del Rio C, Harro C, et al. Comparative cell-mediated immunogenicity of DNA/DNA, DNA/Adenovirus type 5 (Ad5), or Ad5/Ad5 HIV-1 Clade B gag vaccine prime-boost regimens. J Infect Dis. 2010;201(January 1):132–41. doi: 10.1086/648591. [DOI] [PubMed] [Google Scholar]
- 38.Liu J, O’Brien KL, Lynch DM, Simmons NL, La Porte A, Riggs AM, et al. Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature. 2009;457(January 7225):87–91. doi: 10.1038/nature07469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shott JP, McGrath SM, Pau MG, Custers JH, Ophorst O, Demoitie MA, et al. Adenovirus 5 and 35 vectors expressing Plasmodium falciparum circumsporozoite surface protein elicit potent antigen-specific cellular IFN-gamma and antibody responses in mice. Vaccine. 2008;26(June 23):2818–23. doi: 10.1016/j.vaccine.2008.03.080. [DOI] [PubMed] [Google Scholar]
- 40.Abbink P, Lemckert AA, Ewald BA, Lynch DM, Denholtz M, Smits S, et al. Comparative seroprevalence and immunogenicity of six rare serotype recombinant adenovirus vaccine vectors from subgroups B and D. J Virol. 2007;81(May 9):4654–63. doi: 10.1128/JVI.02696-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barouch DH, Pau MG, Custers JH, Koudstaal W, Kostense S, Havenga MJ, et al. Immunogenicity of recombinant adenovirus serotype 35 vaccine in the presence of pre-existing anti-Ad5 immunity. J Immunol. 2004;172(May 10):6290–7. doi: 10.4049/jimmunol.172.10.6290. [DOI] [PubMed] [Google Scholar]
- 42.Allaker RP. Host defence peptides-a bridge between the innate and adaptive immune responses. Trans R Soc Trop Med Hygiene. 2008;102(January 1):3–4. doi: 10.1016/j.trstmh.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 43.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5(October 10):987–95. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 44.Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev Immunol. 2007;7(January 1):9–18. doi: 10.1038/nri1994. [DOI] [PubMed] [Google Scholar]
- 45.Morgan BP, Marchbank KJ, Longhi MP, Harris CL, Gallimore AM. Complement: central to innate immunity and bridging to adaptive responses. Immunol Lett. 2005;97(March 2):171–9. doi: 10.1016/j.imlet.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 46.Catanzaro AT, Koup RA, Roederer M, Bailer RT, Enama ME, Moodie Z, et al. Phase 1 safety and immunogenicity evaluation of a multiclade HIV-1 candidate vaccine delivered by a replication-defective recombinant adenovirus vector. J Infect Dis. 2006;194(December 12):1638–49. doi: 10.1086/509258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao W, Soloff AC, Lu X, Montecalvo A, Nguyen DC, Matsuoka Y, et al. Protection of mice and poultry from lethal H5N1 avian influenza virus through adenovirus-based immunization. J Virol. 2006;80(February 4):1959–64. doi: 10.1128/JVI.80.4.1959-1964.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sullivan NJ, Sanchez A, Rollin PE, Yang ZY, Nabel GJ. Development of a preventive vaccine for Ebola virus infection in primates. Nature. 2000;408(November 6812):605–9. doi: 10.1038/35046108. [DOI] [PubMed] [Google Scholar]
- 49.Pinto AR, Fitzgerald JC, Giles-Davis W, Gao GP, Wilson JM, Ertl HC. Induction of CD8+ T cells to an HIV-1 antigen through a prime boost regimen with heterologous E1-deleted adenoviral vaccine carriers. J Immunol. 2003;171(December 12):6774–9. doi: 10.4049/jimmunol.171.12.6774. [DOI] [PubMed] [Google Scholar]
- 50.Reyes-Sandoval A, Sridhar S, Berthoud T, Moore AC, Harty JT, Gilbert SC, et al. Single-dose immunogenicity and protective efficacy of simian adenoviral vectors against Plasmodium berghei. Eur J Immunol. 2008;38(March 3):732–41. doi: 10.1002/eji.200737672. [DOI] [PubMed] [Google Scholar]
- 51.Shiver JW, Fu TM, Chen L, Casimiro DR, Davies ME, Evans RK, et al. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature. 2002;415(January 6869):331–5. doi: 10.1038/415331a. [DOI] [PubMed] [Google Scholar]
- 52.Schirmbeck R, Reimann J, Kochanek S, Kreppel F. The immunogenicity of adenovirus vectors limits the multispecificity of CD8 T-cell responses to vector-encoded transgenic antigens. Mol Ther. 2008;16(September 9):1609–16. doi: 10.1038/mt.2008.141. [DOI] [PubMed] [Google Scholar]
- 53.Buchbinder SP, Mehrotra DV, Duerr A, Fitzgerald DW, Mogg R, Li D, et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372(November 9653):1881–93. doi: 10.1016/S0140-6736(08)61591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weaver EA, Nehete PN, Buchl SS, Senac JS, Palmer D, Ng P, et al. Comparison of replication-competent, first generation, and helper-dependent adenoviral vaccines. PLoS One. 2009;4(3):e5059. doi: 10.1371/journal.pone.0005059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gabitzsch ES, Xu Y, Yoshida LH, Balint J, Amalfitano A, Jones FR. Novel Adenovirus type 5 vaccine platform induces cellular immunity against HIV-1 Gag, Pol, Nef despite the presence of Ad5 immunity. Vaccine. 2009;27(46):6394–8. doi: 10.1016/j.vaccine.2009.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pichla-Gollon SL, Lin SW, Hensley SE, Lasaro MO, Herkenhoff-Haut L, Drinker M, et al. Effect of preexisting immunity on an adenovirus vaccine vector: in vitro neutralization assays fail to predict inhibition by antiviral antibody in vivo. J Virol. 2009;83(June 11):5567–73. doi: 10.1128/JVI.00405-09. [DOI] [PMC free article] [PubMed] [Google Scholar]