

SUMMARY
Sonic hedgehog (Shh), a soluble ligand overexpres sed by neoplastic cells in pancreatic ductal adenocarcinoma (PDAC), drives formation of a fibroblast-rich desmoplastic stroma. To better understand its role in malignant progression, we deleted Shh in a well-defined mouse model of PDAC. As predicted, Shh-deficient tumors had reduced stromal content. Surprisingly, such tumors were more aggressive and exhibited undifferentiated histology, increased vascularity, and heightened proliferation – features that were fully recapitulated in control mice treated with a Smoothened inhibitor. Furthermore, administration of VEGFR blocking antibody selectively improved survival of Shh-deficient tumors, indicating that Hedgehog-driven stroma suppresses tumor growth in part by restraining tumor angiogenesis. Together, these data demonstrate that some components of the tumor stroma can act to restrain tumor growth.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is notable for its profuse desmoplastic stroma comprised of activated fibroblasts, leukocytes, and extracellular matrix (Olive et al., 2009; Theunissen and de Sauvage, 2009). Studies utilizing in vitro assays and transplantation models have concluded that various stromal elements can enhance cancer cell proliferation and invasion (Hwang et al., 2008; Ikenaga et al., 2010; Lonardo et al., 2012; Vonlaufen et al., 2008; Xu et al., 2010). Various stromal cells can also contribute to immune suppression, further supporting tumor survival and growth. Together these observations have led to the paradigm that tumor stroma functions to support and promote the growth of cancer (Hanahan and Weinberg, 2011). Based on this paradigm, the concept of “anti-stromal” therapy has emerged as a promising, albeit unproven, therapeutic approach (Engels et al., 2012).
The Hedgehog (Hh) signaling pathway contributes to stromal desmoplasia in multiple solid tumor systems. Though normally absent in the adult pancreas, this developmental morphogen pathway is reactivated during inflammation and neoplasia. Both sonic hedgehog (Shh) ligand and downstream signaling are induced de novo in pre-neoplastic lesions, and increase significantly during PDAC progression as the stromal compartment enlarges (Thayer et al., 2003). Although ectopic activation of Hh signaling within pancreatic epithelial cells can accelerate tumorigenesis (Mao et al., 2006; Morton et al., 2007; Pasca di Magliano et al., 2006), deletion of the Hh signaling mediator Smoothened (Smo) from the epithelium has no impact on PDAC progression (Nolan-Stevaux et al., 2009). Hence, canonical Hh signaling in PDAC is likely to occur in a paracrine fashion, whereby Shh ligand secreted from epithelial cells activates Smoothened (Smo)-dependent downstream signaling in adjacent stromal cells, promoting desmoplasia (Bailey et al., 2008; Tian et al., 2009). The notion that Hh-dependent tumor stroma facilitates tumorigenesis is supported by the finding that inhibiting Hh signaling retards pancreatic tumor growth and metastasis in transplantation models (Bailey et al., 2008; Feldmann et al., 2008a; Feldmann et al., 2008b), and through our own study of the effects of acute inhibition of Smo in genetically engineered mouse models (Olive et al., 2009). In this study, we sought to interrogate the role of the tumor stroma by using both genetic deletion and long-term pharmacologic inhibition to eliminate stroma-promoting Hh signaling.
RESULTS
Shh loss accelerates PDAC progression
To explore the role of paracrine Hh signaling in an autochthonous mouse model of PDAC, we conditionally deleted Shh, the predominant Hh ligand expressed in the diseased pancreas, by breeding Shhfl alleles into the Pdx1-Cre;KrasLSL-G12D/+;p53fl/+;Rosa26LSL-YFP/+ (PKCY) model (Rhim et al., 2012). As Pdx1-Cre mediates recombination exclusively in the epithelial cells of the pancreas (Rhim et al., 2012), this combination of alleles results in the simultaneous activation of mutant Kras and deletion of Shh and p53 within this tissue compartment (Fig. 1A). Shh deletion had no effect on pancreatic development (Fig. S1A), and the resulting Shhfl/fl;Pdx1-Cre;KrasLSL-G12D/+;p53fl/+;Rosa26LSL-YFP (ShhPKCY) mice were born at expected Mendelian ratios and were phenotypically normal at birth.
Figure 1. Sonic hedgehog behaves as a tumor suppressor in a genetically engineered mouse model of PDAC.
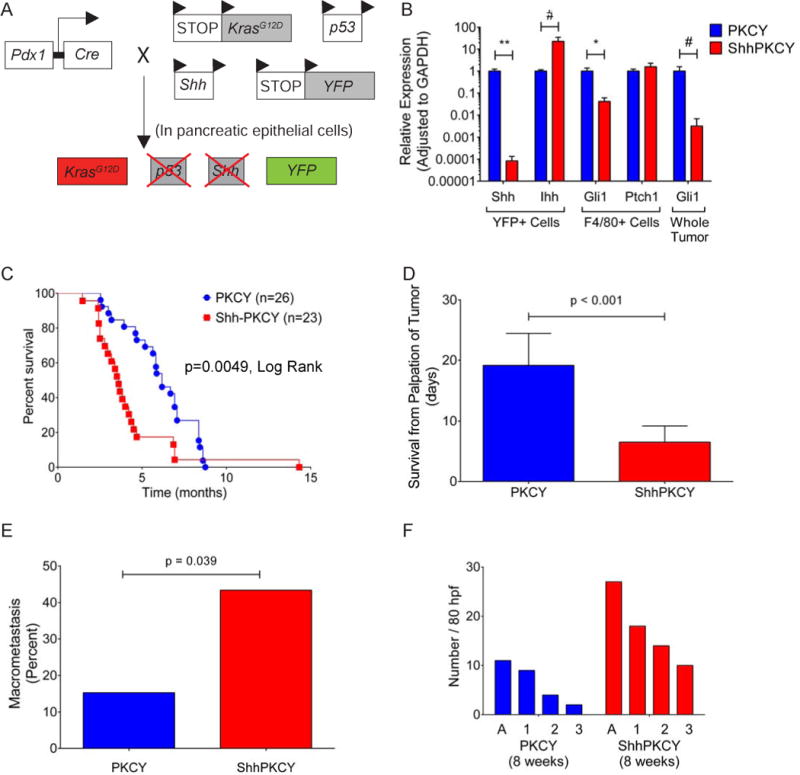
(A) Schematic of the ShhPKCY mouse model used in this study, which employs the KrasG12D, (“K”), Pdx1-Cre (“C”), p53 (P”), RosaYFP (“Y”) and Shh alleles. Cre-mediated deletion results in simultaneous activation of Kras, deletion of one allele of p53 and both alleles of Shh, and recombination of the YFP lineage label.
(B) Confirmation of Shh knockdown in ShhPKCY animals. qPCR analysis of Hedgehog signaling components in YFP+ sorted pancreatic epithelial derived cells and F4/80+ cells from tumors as well as whole tumor derived from PKCY (blue) and ShhPKCY (red) mice (n=5 for each group; bars represent mean +/− SD).
(C) Kaplan-Meier survival analysis for PKCY (n=26) and ShhPKCY mice (n=23). p<0.005 by Mantel-Cox (log-rank) test.
(D) Survival of mice from first clinical palpation of tumor. Presence of tumor was confirmed by ultrasound. Bars represent mean +/− SD; p<0.001.
(E) Fraction of mice with any macrometastatic lesion by visual inspection at the time of tissue harvest by genotype (n=26 and 23 for PKCY and ShhPKCY mice, respectively). p=0.039.
(F) Quantitation of acinar to ductal metaplasia (A) and PanIN lesions by grade (1, 2, or 3) in 8 week-old PKCY and ShhPKCY mice. Eighty non-overlapping high powered fields in which pancreas tissue covered at least 90% of the entire field were analyzed (n=3 for each group). Data are presented as the aggregate number of ADMs and PanINs (by grade) for each genotype.
#, p<0.05; *, p<0.01; **, p<0.001 by two-tailed Student’s t test.
See also Figure S1.
To confirm the deletion of Shh in the pancreatic epithelial compartment, we performed transcriptional analysis on FACS-sorted YFP+ cells from 10- to 16-week old PKCY and ShhPKCY mice (Rhim et al., 2012). As predicted, Shh transcripts were markedly reduced in YFP+ pancreatic epithelial cells from ShhPKCY mice (Fig. 1B). Interestingly, this decrease in Shh transcription was accompanied by a ten-fold increase in the expression of Indian hedgehog (Ihh), another Hh ligand, although absolute levels of Ihh remained significantly lower than Shh. Desert hedgehog (Dhh) was undetectable under all conditions (data not shown). We then determined the impact of Shh deletion on signaling within the stromal compartment by measuring the expression of the Hh target genes Ptch1 and Gli1 in sorted PDAC-associated F4/80+ monocytes and whole pancreas, as previously described (El-Zaatari et al., 2013). Although Ptch1 expression was similar, transcript levels for Gli1 were significantly decreased in ShhPKCY samples as compared to PKCY samples, indicating that overall Hh signaling was reduced following Shh deletion (Fig. 1B).
Given the important role of Shh in promoting the desmoplastic stroma of PDAC, we expected that Shh loss would impair tumorigenesis. Surprisingly, however, pancreatic tumors arose in both PKCY and ShhPKCY mice, demonstrating that Shh is dispensable for tumorigenesis. Remarkably, ShhPKCY mice developed tumors earlier and had a significantly decreased survival compared to PKCY mice (p<0.001 by Log-rank (Mantel-Cox) test; Fig. 1C). Specifically, ShhPKCY mice had a median survival of 3.61 ± 1.97 months as compared to a median survival of 6.17 ± 2.65 months for PKCY mice (Fig. S1B). Heterozygous Shhfl/+;Pdx-Cre; KrasLSL-G12D/+;p53fl/+;Rosa26LSL-YFP (Shhfl/+PKCY) mice that retained one copy of Shh also had reduced median survival compared to PKCY mice (4.14 ± 1.57 months, p=0.004; Fig. S1B). ShhPKCY tumors were more aggressive than PKCY tumors, as mean survival from first detection of tumor was significantly shorter in ShhPKCY mice (19.2 ± 5.27 v. 6.5 ± 2.7 days, p<0.001; Fig. 1D), and the frequency of gross metastasis was higher in ShhPKCY mice than in PKCY mice (43.4 v. 15.3%, p=0.039 by Chi-square test; Fig. 1E) although the tissue distribution of macrometastases was similar (Fig. S1C). Moreover, histological analysis revealed a higher frequency of acinar-to-ductal metaplasia (ADM) and pancreatic intraepithelial neoplasia (PanIN) of all grades in 8 week-old ShhPKCY compared to PKCY mice (Fig. 1F). These data indicate that Shh is not merely dispensable for pancreatic tumorigenesis, but that it somehow restrains tumor progression and aggressiveness.
Shh loss is associated with changes in stromal composition
Next, we compared the histology of ShhPKCY and PKCY tumors. In contrast to the well- to moderately-differentiated histology of most PKCY tumors, ShhPKCY tumors exhibited predominantly undifferentiated and poorly differentiated histology, with few of the ductal elements observed in most PKCY and human pancreatic tumors (Fig. 2A–B, Fig. S2A). ShhPKCY tumors also exhibited a significant increase in Zeb1 and Slug expression, two markers of epithelial-to-mesenchymal transition (EMT), consistent with the predominance of poorly differentiated and undifferentiated histology (Singh et al., 2009; Watanabe et al., 2009) (Fig. S2B; p<0.05).
Figure 2. Loss of Shh leads to a shift in pancreatic tumor histopathology.
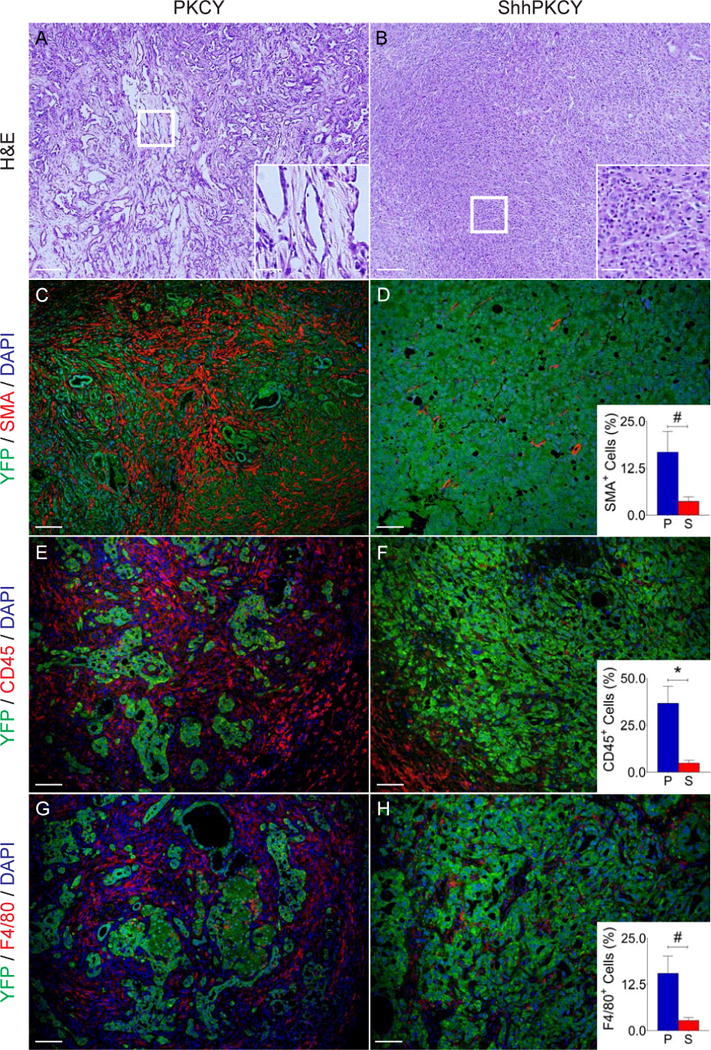
(A and B) H&E staining showing representative histology from PKCY (A) and ShhPKCY (B) tumors. Insets show higher magnified view of sections marked by the box.
(C–H) Multicolor immunofluorescence (IF) images of PKCY (left column) and ShhPKCY tumors (right column) assessed for myofibroblasts (C–D), total leukocytes (E–F), or macrophages (G–H).
(C and D) Fluorescent images showing staining with the pancreas epithelial lineage label YFP (green) and the myofibroblast marker alpha smooth muscle actin (SMA, red). Inset, quantitation of SMA+ cells as a percentage of all nucleated (DAPI+) cells within PKCY (blue) and ShhPKCY (red) tumors (n=3–5; #, p=0.016; bars represent mean +/− SD).
(E and F) Fluorescent images showing staining with YFP (green) and the pan-leukocyte marker CD45 (red). Inset, quantitation of CD45+ cells as a percentage of all nucleated (DAPI+) cells (n=3–5; *, p=0.0039; bars represent mean +/− SD).
(G and H) Fluorescent images showing staining with YFP (green) and the macrophage marker F4/80 (red). Inset, quantitation of F4/80+ cells as a percentage of all nucleated (DAPI+) cells (n=3–5; #, p=0.010; bars represent mean +/− SD).
Scale bars = 40 μm for larger images and 20 μm for insets.
See also Figure S2.
Using the YFP lineage label to distinguish epithelial-derived cancer cells from mesenchyme-derived stromal cells, we found that Shh-deficient tumors had significantly reduced stroma, as indicated by decreased numbers of YFP-negative alpha smooth muscle actin (SMA)-positive myofibroblasts (3.7 ± 0.7 v. 16.7 ± 3.2% of all DAPI+ cells within pancreas tumors; p=0.016; Fig. 2C–D). Despite their increased aggressiveness (but consistent with stromal loss) ShhPKCY tumors exhibited a trend towards decreased weight (Fig. S1B). In addition, ShhPKCY tumors had fewer CD45+ myeloid cells (4.9 ± 0.9 v. 36.7 ± 5.2%; p=0.0039; Fig. 2E–F) and F4/80+ monocytes (2.8 ± 0.5 v. 15.5 ± 2.7%; p=0.010; Fig. 2G–H). Indeed, the stromal cell composition of ShhPKCY tumors was similar to that of normal pancreas tissue from Pdx1-Cre;Rosa26LSL-YFP/+ mice (data not shown). These results demonstrate that robust tumor formation can occur in the absence of a fibroblast- and leukocyte-rich desmoplastic stroma.
Because ShhPKCY tumors progressed more rapidly than their PKCY counterparts, we hypothesized that parallel but opposite changes in tumor vasculature might influence tumor growth. Hence, we examined the endothelial compartment in tumors with and without Shh by CD31 staining. Consistent with this hypothesis, ShhPKCY tumors exhibited a substantial increase in the number of blood vessels within the tumor (32.4 ± 7.2 v. 11.2 ± 3.8 CD31+ vessels per high powered field; p=0.0004; Fig. 3A–B). In addition, the autofluorescent drug doxorubicin was delivered more effectively to ShhPKCY tumors, suggesting that increased vascular density was accompanied by greater perfusion (Fig. 3C–D). To assess whether this increase in vasculature was associated with changes in autophagy or proliferation, we stained for the autophagosome marker LC3 and the proliferation marker PCNA. This analysis revealed a decrease in YFP+LC3+ cells in ShhPKCY tumors (Fig. 3E–F) and an increase in the frequency of YFP+PCNA+ proliferating tumor cells (Fig. 3G–H). These data therefore suggest that undifferentiated ShhPKCY tumors are better perfused than PKCY tumors, a change that was associated with enhanced nutrient delivery, decreased autophagy, and increased proliferation.
Figure 3. Shh deletion results in greater vascular density and proliferation within pancreatic tumors.
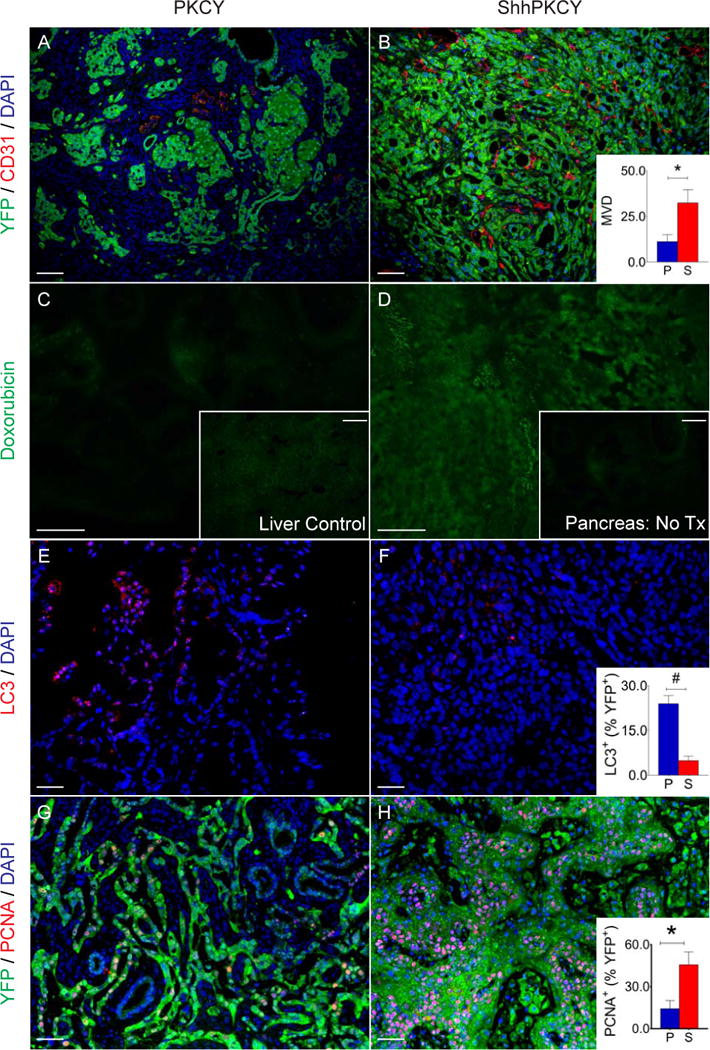
(A and B) Blood vessel density in PKCY and ShhPKCY tumors was determined by staining for the endothelial marker CD31 (red) and the tumor cell lineage marker YFP (green). Inset, measurement of mean vascular density within PKCY (blue) and ShhPKCY (red) tumors (quantified as number of CD31+ vessels per high powered field; n=3–5; *, p=0.004; bars represent mean +/− SD).
(C and D) Cellular perfusion in PKCY and ShhPKCY tumors was determined by intravascular delivery of the autofluorescent drug doxorubicin. Inset in C shows fluorescence of a liver section from the same PKCY mouse (positive control). Inset in D shows fluorescence of a ShhPKCY tumor injected with PBS (negative control).
(E–F) Autophagy in PKCY and ShhPKCY tumors was determined by staining for the autophagosomal protein LC3 (red). Inset, percentage of LC3+ cells within the YFP+ tumor cell population in PKCY (blue) and ShhPKCY (red) tumors (n=3–5; #, p=0.002; bars represent mean +/− SD).
(G–H) Proliferation in PKCY and ShhPKCY tumors was determined by staining for the cell cycle marker PCNA (red). Inset, percentage of PCNA+ cells within the YFP+ tumor cell population in PKCY (blue) and ShhPKCY (red) tumors (n=3–5; *, p=0.004; bars represent mean +/− SD).
Scale bars = 40 μm.
Chronic smoothened inhibition phenocopies Shh deletion
We next sought to learn whether the effect of Shh deletion in pancreatic cancer is mediated by canonical Hh signaling. We utilized IPI-926 (Infinity Pharmaceuticals), a targeted inhibitor of Smo, to inhibit canonical Hh signaling in KPC mice (a PDAC model closely related to the PKCY model). We previously performed a preclinical evaluation of IPI-926 in KPC mice harboring large (6–9mm) pancreatic tumors and found that the combination of IPI-926 and the nucleoside analog gemcitabine (gem) resulted in extension of overall survival (Olive et al., 2009), a finding at odds with the observed effect of genetic Shh deletion. We reasoned that long-term, chronic exposure to Smo inhibition might unveil indirect responses related to the depletion of stroma from tumors rather than the acute response to improved drug delivery. Therefore, we treated KPC mice with IPI-926 alone or vehicle beginning at 8 weeks of age, a time-point at which ADM and PanIN lesions are present but mice have not yet developed tumors (Fig. S3A)(see Supplementary Experimental Procedures for additional details).
Strikingly, IPI-926-treated KPC mice exhibited a reduction in overall survival compared to vehicle treated mice (121 vs. 156 days, p< 0.0001 by Log Rank test; Fig. 4A). This result diverged dramatically from those obtained in our previous intervention study (Olive et al., 2009) despite the fact that the model, drug, dose, route, and schedule were all identical. In an effort to replicate this result and also determine whether co-administration of gemcitabine might change the dynamics of tumor response, we treated a separate cohort of mice with the combination of gemcitabine + IPI-926 or gemcitabine + vehicle. As shown in Fig. 4B, the addition of gemcitabine provided a minor extension of survival over IPI-926 monotherapy (p=0.01, Log Rank), but the IPI-926-gem combination therapy still resulted in shortened survival compared to vehicle-treated from the previous cohort. These results suggest that any benefit afforded by improved drug availability following Smo inhibition is outweighed by other effects on tumor biology in the chronic setting.
Figure 4. Smoothened inhibition accelerates pancreatic tumor development.
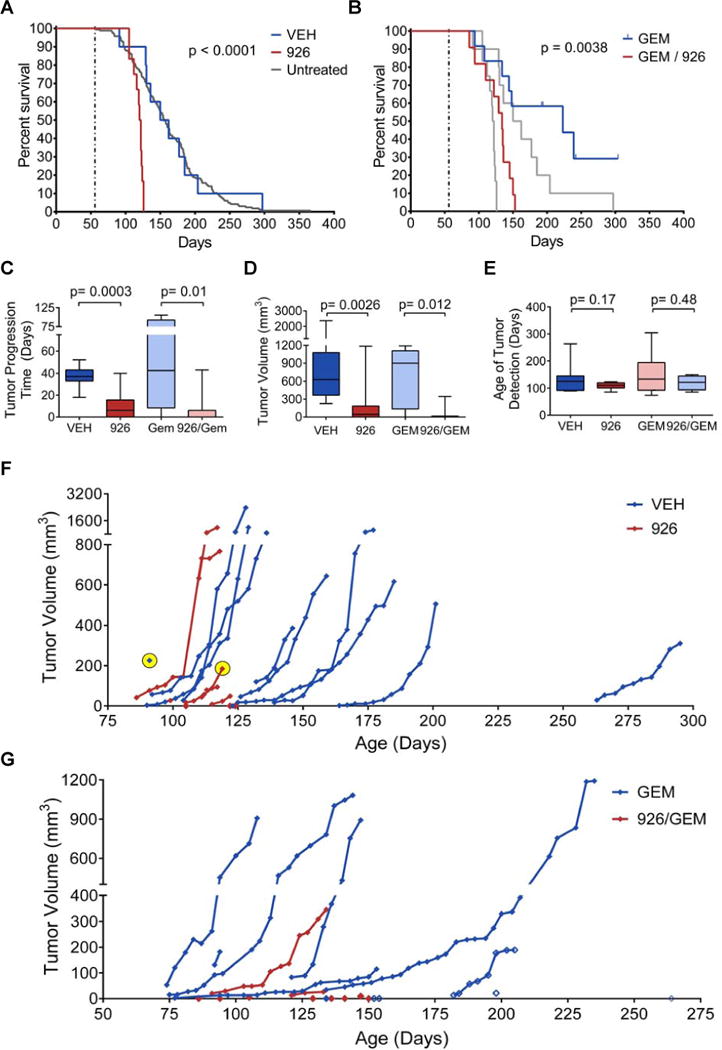
(A–G) Two separate cohorts of KPC mice were treated with vehicle (VEH) vs. IPI-926 (926), or gemcitabine (GEM) vs. IPI-926 + gemcitabine (926/GEM), beginning at 8 weeks of age, as described in Supplementary Online Methods. High resolution 3D ultrasound was used to monitor tumor development and to quantify tumor volumes. Treatment continued until mice met endpoint criteria.
(A) Kaplan-Meier curve showing IPI-926 treated KPC mice (red, n=12), vehicle treated KPC mice (blue, n=12) (p<0.0001, Log Rank test, vehicle versus IPI-926) and an historical collection of untreated KPC mice (gray, n=165) (p<0.0001, Log Rank test, historical cohort versus IPI-926).
(B) Kaplan-Meier curve showing IPI-926 + gemcitabine treated KPC mice (red, n=11) and vehicle + gemcitabine treated KPC mice (blue, n=12) (p< 0.004, Log Rank test, gemcitabine + vehicle versus gemcitabine + IPI-926). Data from panel A are overlaid in gray (p=0.01, Log Rank test, IPI-926 versus IPI-926 + gemcitabine).
(C) Graph of time from first detection of tumor (by 3D ultrasound) to death in animals among the four treatment groups, comparing animals that received IPI-926 to those that did not (p=0.0003 in monotherapy group, p=0.006 in combination group). Animals with microscopic tumors on necropsy but no measurable tumor on ultrasound were included as 0 days. Data are presented as standard box and whisker plots.
(D) Final tumor volumes (measured by 3D ultrasound) among the four treatment groups, comparing animals that received IPI-926 to those that did not (p=0.0026 in monotherapy group, p=0.012 in the combination group). Several IPI-926 treated mice met endpoint criteria prior to the detection of tumors by ultrasound and are included as 0 mm3. Data are presented as standard box and whisker plots.
(E) Age of tumor detection (by 3D ultrasound) among the four treatment groups (p=0.17 for monotherapy, p=0.48 for combination). Data are presented as standard box and whisker plots.
(F) Graph showing tumor volumes of mice treated with IPI-926 (red) or vehicle (blue) plotted versus the mouse’s age in days. Two exceptional tumors noted in the text are highlighted in yellow.
(G) Graph showing tumor volumes of mice treated with gemcitabine + IPI-926 (red) or gemcitabine + vehicle (blue) are plotted versus the mouse’s age in days. Lethal tumors that were undetectable by ultrasound were assigned a volume of 0 on the day of death. Animals still alive at the time of submission are denoted with open diamonds.
See also Figure S3.
Analysis of 3D high resolution ultrasound data (Sastra and Olive, 2013) demonstrated that KPC mice treated with IPI-926 or IPI-926-gemcitabine succumbed more rapidly following initial tumor detection (Fig. 4C), and that tumor size was significantly smaller at sacrifice (Fig. 4D), similar to the reduced tumor weight observed in ShhPKCY mice (Fig. S1B). Indeed, several mice met endpoint criteria before it was possible to detect tumors in their pancreas by ultrasound. However, all but one of these mice was found to have tumors upon careful histopathological analysis. There was no difference in the age at which tumors developed between the two groups, suggesting that IPI-926 treatment accelerates tumor progression after initiation without reducing latency (Fig. 4E). Complete tumor volume data for these mice are presented in Fig. 4F, G.
To better understand why IPI-926-treated mice with very small tumors were dying, we performed detailed necropsies on each mouse and assigned a “proximal cause of death” to each animal. Vehicle- or gem-treated KPC mice typically succumbed to the consequences of locally destructive disease (e.g. local invasion into the gut or abdominal hemorrhage) or high metastatic burden, with only a small subset exhibiting severe weight loss. By contrast, nearly all of the 926-treated mice were euthanized following a period of rapid and severe weight loss (Fig. S3B, C), a phenomenon also observed in ShhPKCY animals (data not shown).
The tumors arising in IPI-926-treated mice were more poorly differentiated than those arising in controls treated with vehicle or gem alone (Fig. 5A–F), consistent with the observation that ShhPKCY mice developed poorly differentiated tumors. It is worth highlighting a pair of noteworthy exceptions that emphasize the relationship between differentiation state and tumor progression: one IPI-926-treated tumor was extremely well differentiated and progressed slowly, while one vehicle-treated mouse succumbed at an early time-point from a small tumor that was 50% poorly differentiated (Fig. 4F, highlighted). Examination of pancreatic tissues adjacent to the tumors revealed an exceptionally high content of acinar-to-ductal metaplasias (ADMs) and pancreatic intraepithelial neoplasias (PanINs) in IPI-926 treated mice (Fig. S4A–D). This feature was not shared in IPI-926 / gemcitabine treated mice, possibly due to the impact of genotoxic chemotherapy on the proliferation of pre-neoplastic lesions. Immunohistochemical analyses revealed that IPI-926-treated tumors were more highly proliferative (Fig. 5G, Fig. S4E–H) and had increased vascular content (Fig. 5H, Fig. S4I–L). Together, these data demonstrate that pharmacologic inhibition of canonical Hedgehog signaling accelerates tumor growth, phenocopying the effect of genetic deletion of Shh in pancreatic tumors.
Figure 5. Long-term Smoothened inhibition yields poorly differentiated pancreatic tumors with increased proliferation and vascularity.
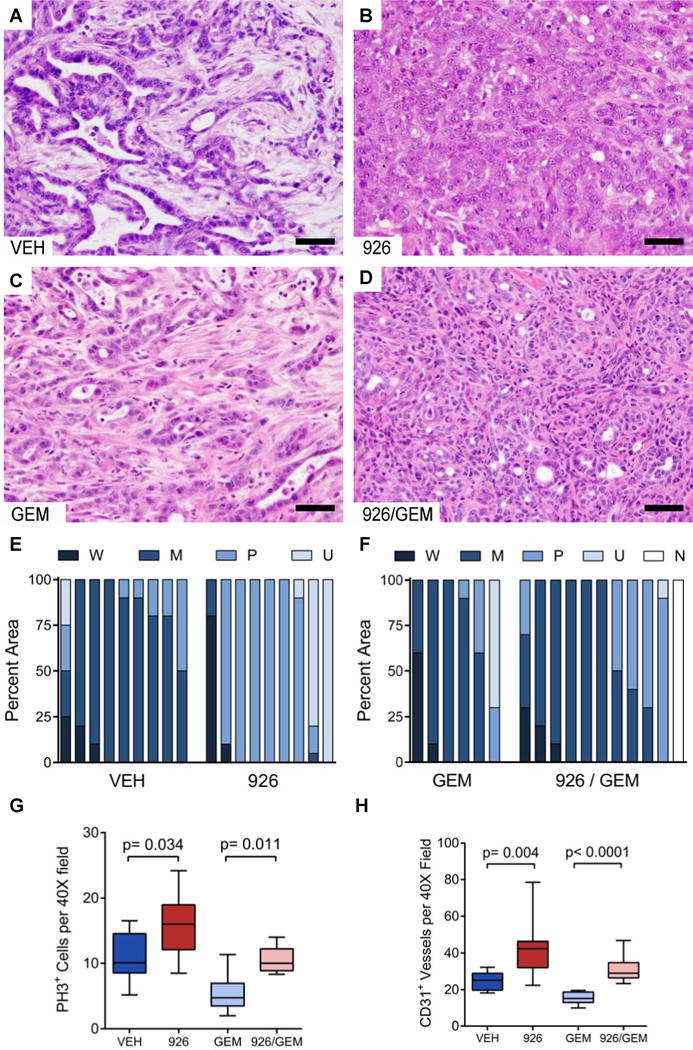
(A–D) Representative histology (hematoxylin and eosin stain) of KPC tumors arising after long-term treatment with vehicle (A), IPI-926 (B), gemcitabine + vehicle (C), or gemcitabine + IPI-926 (D).
(E and F) Quantification of the differentiation state for each cohort. The fraction of each tumor that was observed to be well-differentiated (W), moderately differentiated (M), poorly-differentiated (P), or undifferentiated (U), was scored in a blinded manner, and compared between the treatments. No tumor could be located in one IPI-926 + gemcitabine treated mouse (N).
(G) Quantification of phospho-histone H3+ cells per 40X field in each treatment group (p=0.034 for monotherapy, p=0.011 for combination). Data are presented as standard box and whisker plots.
(H) Quantification of CD31+ vessel structures by IHC in each treatment cohort (p=0.004 for monotherapy, p<0.0001 for combination). Data are presented as standard box and whisker plots.
Two-tailed Mann-Whitney U was used for all unpaired tests. Scale bars = 50 μm.
See also Figure S4.
Hedgehog signaling acts in a paracrine fashion in PDAC
During embryonic endoderm development, Hedgehog ligand is secreted by gut epithelial cells and acts on adjacent mesenchymal cells to pattern the submucosal layers of the gut (Roberts et al., 1998; Sukegawa et al., 2000). Given strong evidence from previous literature that Hedgehog signaling acts in a paracrine fashion during pancreatic carcinogenesis (Bailey et al., 2008; Tian et al., 2009), we sought to determine whether a similar signaling relay was operating in the autochthonous models. To this end, we introduced a Gli1GFP reporter (Brownell et al., 2011) into the KPC background and assessed the distribution of GFP+ cells in the resulting KPC-Gli1GFP mice by immunofluorescence. GFP staining was readily observed in the E-cadherin-negative (stromal) portions of KPC-Gli1GFP mice, but was absent from E-cadherin-positive (epithelial) cells (Fig. 6A, B). Consistent with this observation, spheroid formation of a KPC pancreatic tumor cell line was unaffected by treatment with recombinant Shh protein or IPI-926 (Fig. S5A). In contrast, nearly all alpha-SMA+ myofibroblasts were found to be Gli1GFP positive, indicating active Hh pathway signaling in this mesenchymally-derived cell type (Fig. 6A, C). This is consistent with previous reports demonstrating a proliferative effect from Hh pathway signaling in fibroblasts (Walter et al., 2010). However, further examination of these tissue sections revealed that many Gli1GFP positive cells (43%) were alpha-SMA negative (Fig. 6A, C), indicating that multiple stromal cell types can respond to Hh signaling. Importantly, treatment of KPC-Gli1GFP mice with IPI-926 for 10 days completely abrogated GFP staining, indicating that GFP staining was accurately reporting canonical Hedgehog signaling (Fig. 6D, E). Taken together, these experiments confirm that canonical Hh signaling operates in a paracrine fashion in PDAC, as it does during embryonic development.
Figure 6. Hh pathway activity is restricted to mesenchymally-derived stromal cells.
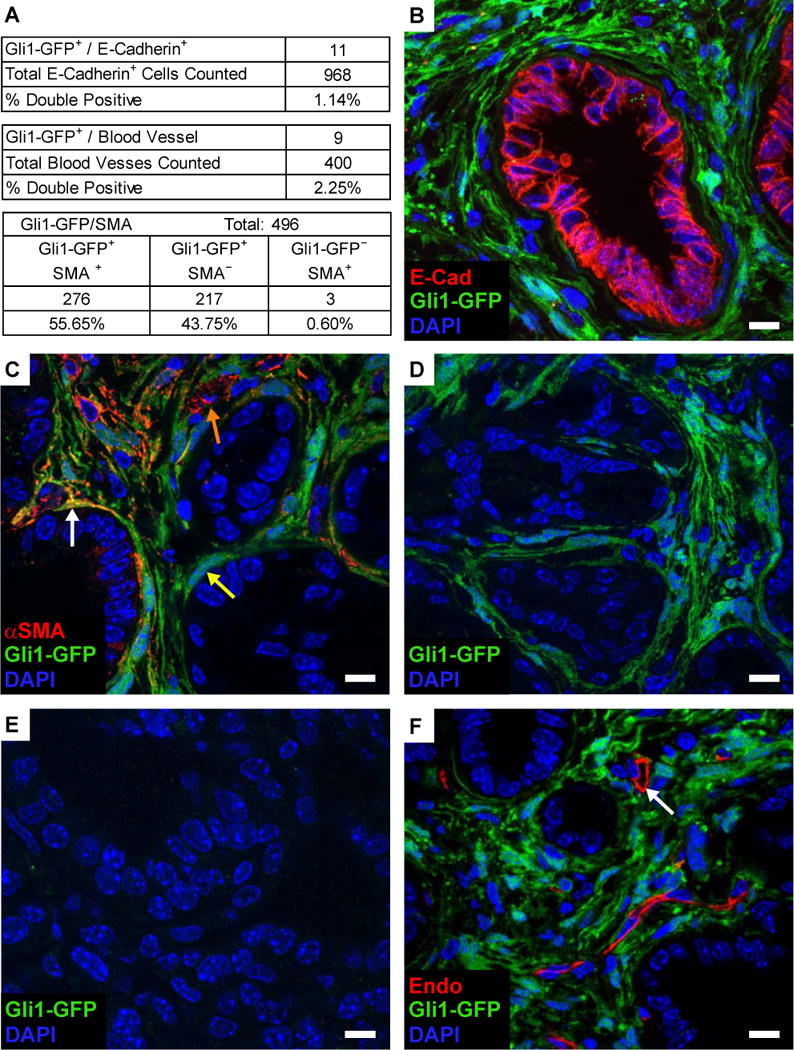
(A) Quantification of co-immunofluorescence (Co-IF) for the Gli1GFP Hh reporter and markers of various tumor and stromal populations.
(B) Co-IF for Gli1GFP (green) and E-Cadherin (red) in KPC-Gli1GFP tumors.
(C) Co-IF for Gli1GFP (green) and alpha-smooth muscle actin (αSMA, red) in in KPC-Gli1GFP tumors.
(D–E) Co-IF for Gli1GFP reporter on an untreated KPC-Gli1GFP tumor (D) or after 10 days of IPI-926 treatment (E).
(F) Co-IF for Gli1GFP (green) and the endothelial marker endomucin (red) in KPC-Gli1GFP tumors.
Scale bars = 10 μm.
See also Figure S5.
Shh-deficient tumors are sensitive to VEGF inhibition
We next investigated the surprising increase in vasculature in ShhPKCY mice and in IPI-926-treated KPC mice. We first sought to determine whether increased angiogenesis was a direct result of reduced Hh pathway signaling in endothelial cells. We performed co-immunofluorescence on untreated KPC-Gli1GFP mice for GFP and the endothelial marker endomucin but found that Hh pathway signaling is absent in nearly all tumor endothelial cells (Fig. 6A, F). Consistent with this observation, capillary sprouting of cultured human umbilical venous endothelial cells (HUVECs) was unaffected by exposure to recombinant Shh or IPI-926 (Fig. S5B). These results suggest that the pro-angiogenic effect of Hh pathway inhibition is mediated indirectly by signals from mesenchyme-derived stromal cells.
VEGF is a well-known soluble angiogenic factor. As a group, mouse and human PDAC are unaffected by treatment with VEGF receptor inhibitors, consistent with their poor vascularization (Singh et al., 2010). However, undifferentiated tumors comprise approximately 5–10% of all advanced PDAC (Iacobuzio-Donahue et al., 2009). We hypothesized that the higher vessel density and paucity of stroma of undifferentiated tumors might render them sensitive to angiogenesis inhibitors. To test this hypothesis, we treated tumor-bearing ShhPKCY and control PKCY mice with DC101, a blocking antibody against VEGFR2. Similar to previous studies (Singh et al., 2010), VEGFR inhibition had little effect on tumor size or survival in PKCY mice (Fig. 7A; Fig. S6A). However, treatment of ShhPKCY mice with DC101 led to significantly improved overall survival compared to treatment with Ig control (median survival 22.4 v. 12.5 days from enrollment; p=0.028; Fig. 7B). DC101 treatment was associated with the appearance of large areas of necrosis on H&E (Fig. 7C, D), and immunofluorescence staining demonstrated a reduced number of CD31+ blood vessels (Fig. 7E; Fig. S6B, C), an increase in the percentage of tumor cells with LC3+ autophagosomes (Fig. 7F; Fig. S6D, E), and reduced proliferation (Fig. 7G; Fig. S6F, G) in DC101-treated ShhPKCY mice. Interestingly, there was no difference in tumor VEGF expression between ShhPCKY vs. PKCY mice, or between IPI-926 vs. vehicle treated KPC tumors (data not shown). Thus, the depletion of Hh-dependent stroma from pancreatic tumors leads to a greater utilization and dependence on existing VEGF-mediated angiogenic pathways rather than a de novo induction of VEGF ligand.
Figure 7. VEGFR2 antagonism leads to selective inhibition of tumor growth in ShhPKCY mice.
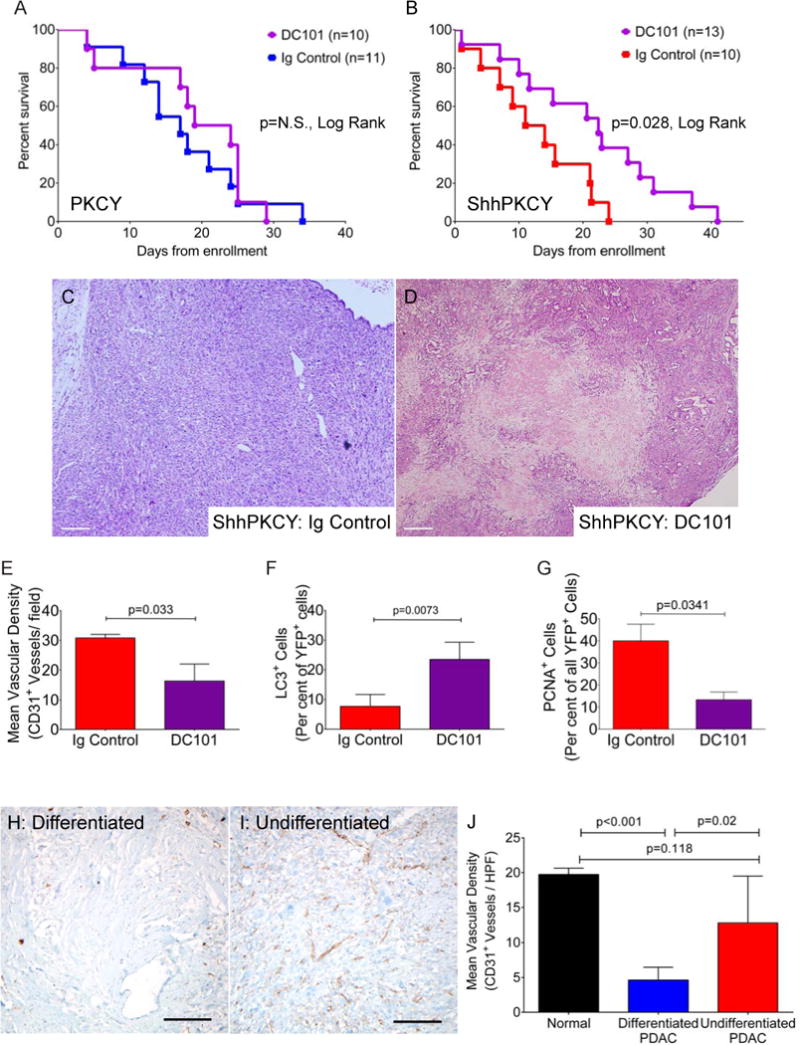
(A) Kaplan-Meier survival curve for tumor-bearing PKCY mice treated with bi-weekly DC101 (purple, n=10) or Ig control (blue, n=11).
(B) Kaplan-Meier survival curve for tumor-bearing ShhPKCY mice treated with bi-weekly DC101 (purple, n=13) or Ig control (red, n=10).
(C and D) H&E analysis of ShhPKCY tumors showing a large area of necrosis upon DC101 treatment (D). Scale bars = 250 μm.
(E) Quantification of CD31+ vessel density in ShhPKCY mice treated with DC101 compared to Ig control, depicted as mean (± SD) number of CD31+ vessels per high powered field (n=3–5).
(F) Quantification of YFP+ cells containing LC3+ autophagosomes in ShhPKCY mice treated with DC101 compared to Ig control, depicted as percentage (± SD) of YFP+ tumor cells exhibiting LC3 staining (n=3–5).
(G) Quantification of YFP+ cells staining with the proliferation in ShhPKCY mice treated with DC101 compared to Ig control, depicted as percentage (± SD) of YFP+ tumor cells that were PCNA positive (n=3–5).
(H and I) IHC for CD31 in differentiated (H) and undifferentiated (I) portions of human pancreas tumor A21. Scale bars = 200 μm.
(J) Mean vascular density of normal pancreas (n=3), differentiated human PDAC (n=5) and undifferentiated human PDAC (n=8). Bars represent p-values by two-sided Student’s t-test.
See also Figure S6.
Finally, we examined human pancreatic tumors to determine if similar relationships between histology, Hedgehog signaling and vascularity were present. In a set of 225 prospectively collected and analyzed human PDAC, undifferentiated tumors had significantly less Gli1 expression compared to all other tumors with well-, moderate- and poorly-differentiated histology (1.42 v. 2.99; p=0.0102 by Student’s t-test, two-tailed; Fig. S6H). However, no statistically significant difference was detected in Shh expression. Thus, undifferentiated human PDAC may be associated with attenuated canonical Hedgehog signaling, as predicted by our studies of genetically engineered mice. In a separate, independently collected and analyzed set of human PDAC, we examined 13 total human pancreatic tumors stratified into differentiated (n=5) and undifferentiated (n=8) histology to determine if differentiation status correlated with vessel density in patients. Similar to mice, undifferentiated human pancreatic tumors exhibited significantly greater vascular density and less stroma compared to differentiated tumors even when both components were present within the same primary carcinoma (Fig. 7H–J). Surprisingly, undifferentiated PDAC had a similar vascular density as normal human pancreas tissue (Fig. 7J). These data indicate that like murine ShhPKCY tumors, undifferentiated pancreatic tumors in patients also have a more prominent vasculature.
DISCUSSION
Nearly fifty years have passed since Stoker’s pioneering studies of epithelial/stromal interactions in cancer demonstrated that normal fibroblasts restrain the growth of transformed baby hamster kidney cells (Stoker et al., 1966). This “neighbor suppression” effect may be part of an evolved microenvironment surveillance against the development of preneoplasia (Klein, 2014). Nevertheless, in the context of established tumors, the prevailing paradigm of the tumor microenvironment field has been that tumor stroma supports, rather than inhibits, neoplastic growth and progression (Hanahan and Weinberg, 2011). This concept has been bolstered by work on pancreatic tumors, which are associated with a particularly dense “desmoplastic” stroma. Such studies, which have mostly relied on cell transplantation or in vitro assays, have affirmed that the stroma plays a supportive role in of pancreatic cancer progression (Bailey et al., 2008; Feldmann et al., 2008a; Feldmann et al., 2008b; Hwang et al., 2008; Ikenaga et al., 2010; Lonardo et al., 2012; Xu et al., 2010). In this study, we have examined the effects of perturbing the tumor microenvironment by genetically deleting Sonic hedgehog or pharmacologically inhibiting its essential signaling mediator Smoothened. These interventions greatly reduced stromal desmoplasia, but such tumors unexpectedly exhibited accelerated tumor growth, increased systemic morbidity and increased metastasis, ultimately leading to earlier mortality. Thus, our findings demonstrate that at least some stromal constituents can act to restrain, rather than promote, tumor progression.
We previously reported that acute administration of IPI-926 to KPC mice bearing large tumors leads to stromal collapse and increased vascularity, consistent with the results shown here (Olive et al., 2009). In that study, treatment with Smo inhibitor alone had minimal effects on tumor size or survival, while combined treatment with the Smo inhibitor and gemcitabine led to transient stabilizations and regressions, producing a modest survival benefit (Olive et al., 2009). These results were interpreted as an indication that stromal inhibition could lead to improved drug delivery without a direct effect on tumor growth. However, as most animals in our previous study experienced less than three weeks of IPI-926 treatment, there was little opportunity to detect accelerated tumor progression in this acute setting. Indeed, despite the success of IPI-926 in treating basal cell carcinoma (Jimeno et al., 2013), the poor clinical performance of Smo inhibitors in pancreatic cancer trials has led to uncertainty regarding the approach of stromal targeting. Our current data suggest that the short-term, beneficial effects of increased drug delivery are eventually overcome by the negative effects of long-term Smo inhibition.
What is responsible for the increase in tumor cell proliferation and overall mortality in ShhPKCY (and IPI-926-treated) tumors? At present, the precise mechanism for increased tumor cell proliferation remains unknown and is likely to be complex. Nevertheless, the nearly 3-fold increase in blood vessel density in ShhPKCY tumors compared to PKCY tumors – an association that has been previously observed in IPI-926-treated KPC mice (Olive et al., 2009) – is likely to be a major contributor to this effect. This inference is supported by the observation that the more vascular ShhPKCY tumors had a nearly five-fold decrease in autophagy, a process of cellular autodigestion used by nutrient-deprived cells (Kondo et al., 2005). Moreover, ShhPKCY tumors exhibited a responsiveness to anti-angiogenic therapy that was absent in PKCY tumors, suggesting that the more highly vascular Shh-deficient tumors were dependent on this enhanced blood supply. Thus, our study suggests that the interaction between the tumor and its microenvironment is complex, with certain components of the microenvironment (i.e., vasculature) having a tumor-promoting role and other components (i.e., myofibroblasts) having an inert or tumor-suppressive role.
A paradoxical result of our study was the observation that the tumors lacking Hedgehog signaling – following either genetic ablation or treatment with IPI-926 – were smaller despite their more aggressive and lethal phenotype, a finding that may reflect the fact that the stroma normally comprises a large percentage of PDAC tumor volume (Chu et al., 2007). Although we do not fully understand the accelerated mortality of Shh-deficient tumors, one possibility is suggested by the finding that tumor-bearing animals treated with IPI-926 exhibited significantly more weight loss and wasting prior to death than vehicle- or gem-treated tumors. It is conceivable that stromal inhibition (and associated changes in tumor metabolism) may lead to increased cachexia, a wasting syndrome common in human PDAC patients. Importantly, we do not believe that IPI-926 contributes to increased mortality or wasting independent of PDAC, as extensive clinical follow-up (Jimeno et al., 2013) and our own observations treating tumor-free animals (data not shown) failed to demonstrate any such toxicity.
There are several models that could account for the finding that ShhPKCY tumors have a reduction in myofibroblasts and leukocytes and an increase in blood vessels. Our data demonstrate that Hh signaling is nearly absent in the endothelial population, arguing against a direct role in angiogenesis. Rather, our findings are consistent with a model in which mesenchymal stromal cells exert an anti-angiogenic effect on endothelial cells, possibly acting either directly or indirectly to elevate interstitial fluid pressure which then represses blood vessel growth (Jacobetz et al., 2013; Provenzano et al., 2012). Importantly, these possibilities are not mutually exclusive, and future studies will be needed to understand the nature of cellular crosstalk between different components of the microenvironment and the role that Shh plays in each.
It is worth noting that global deletion of Gli1, a known mediator of Hedgehog signaling, completely blocks the development of KrasG12D-driven pancreatic tumors (Mills et al., 2013). However, as Gli1 can be activated by signals other than Smoothened, it unclear whether this reported requirement for Gli1 is due to so-called “canonical” (Smo-mediated) Hedgehog signaling or is mediated by other Gli1-dependent signaling pathways. Moreover, as the Gli1GFP reporter showed little if any activity within the tumor epithelium, it is unlikely that the effects we observed are due to a Hh-Smo-Gli1 signaling relay within the tumor cells themselves. Importantly, we employed autochthonous models that harbor mutant Kras and p53 – mutations that are present in 95% and 75–90% of human PDAC patients, respectively – and it remains to be determined whether the observed effects of Hedgehog deficiency on tumor biology occur only in the context of these genetic lesions or are more generalizable.
It is remarkable that the genetic or pharmacologic manipulation of Hedgehog signaling was associated with a dramatic change in tumor histology, illustrating the high-degree of plasticity of differentiation status that exists within tumors in vivo. These data are corroborated by our analysis of human pancreatic tumors. Since poorly differentiated histopathology is strongly associated with poor outcome in PDAC (Han et al., 2006; Yonemasu et al., 2001), this finding has potential clinical implications. Specifically, the observation that inhibition of the Hedgehog pathway leads to the development of less differentiated and more aggressive tumors may explain the lack of benefit observed in clinical trials.
Finally, our discovery that undifferentiated tumors were sensitive to VEGFR inhibition points to a possible biomarker of sensitivity to angiogenesis inhibitors in pancreatic cancer. The marked response of ShhPKCY tumors to an anti-angiogenesis agent suggests that such undifferentiated tumors are dependent on a plentiful vascular network. While clinical trials of anti-angiogenesis agents failed to show benefit in PDAC, a portion of patients treated with bevacizumab did exhibit a durable response (Kindler et al., 2010). Approximately 5–10% of advanced human pancreatic tumors exhibit an undifferentiated histology (Iacobuzio-Donahue et al., 2009), and the data we present here suggest that such tumors have a higher blood vessel density. Given our finding that poorly differentiated but well-vascularized pancreatic tumors respond to VEGF receptor blockade, it may be worth reconsidering anti-angiogenesis treatment strategies for the subset of patients who harbor predominantly undifferentiated PDAC.
EXPERIMENTAL PROCEDURES
Mouse models
All studies were conducted in compliance with the institutional guidelines of their respective locations. Two genetically engineered mouse models were used in these studies: p53fl/+;KrasLSL-G12D/+;Pdx1-Cre;Rosa26YFP (PKCY; (Rhim et al., 2012)) and KrasLSL-G12D/+;p53LSL-R172H;Pdx1-Cre (KPC; (Hingorani et al., 2005)). ShhPKCY and PKCY mice were used to determine the effect of Shh deletion in tumorigenesis and cancer progression. KPC mice were used in a chronic treatment trial of IPI-926. The Gli1eGFP/+ allele was crossed into the KPC mouse for immunofluorescence studies (Brownell et al., 2011).
Analysis of tumor progression and survival in PKCY and ShhPKCY mice
All experiments involving mice were performed in accordance with relevant institutional and national guidelines and were approved by the institutional animal care and use committees at the University of Pennsylvania, Columbia University and University of Michigan prior to experimentation. Starting at two months of age, mice were palpated twice weekly for evidence of tumor. If a potential mass was appreciated, transabdominal ultrasound was performed using a SonoSite M-Turbo ultrasound. If the presence of a tumor was confirmed, mice were examined weekly using ultrasound and general physical exam. If the mouse appeared moribund (decreased spontaneous physical activity, decreased toe pinch reflex, tachycardia, tachypnea, failure to groom, and ruffled coat) indicating low probability of surviving for greater than 24 hours, it was sacrificed for analysis. Tumors were immediately removed, weighed, and dimensions measured and animals were analyzed for evidence of macrometastatic disease.
In vivo DC101 trial
Upon detection of tumor by ultrasound, ShhPKCY and PKCY mice were randomized to two treatment arms: DC101 or IgG1 control (800μg per mouse; Bio X Cell, West Hanover, NH) administered intraperitoneally every Monday and Thursday. Technicians were blinded to treatment group.
Analysis of human pancreatic tumors
Studies involving all human pancreas tumors were approved by the institutional review boards of Johns Hopkins University and Mayo School of Medicine, and informed consent were obtained from all patients prior to tissue procurement and subsequent analysis. Paraffin embedded sections (4μm thickness) of eight human ductal adenocarcinomas with undifferentiated features from the Johns Hopkins Gastrointestinal Rapid Medical Donation Program (GICRMDP; (Iacobuzio-Donahue et al., 2009)) were used and compared to five conventional PDACs and three samples of normal pancreas. Immunolabeling for CD31 was performed using standard histologic methods with prediluted anti-human CD31 monoclonal antibody (Ventana, clone JC70) and detected using the Dako universal Liquid DAB+ Substrate Chromagen System per manufacturer’s instructions (catalog K3468). Slides were counterstained with Hematoxylin for 30 seconds. Microvascular density was calculated as the number of CD31+ vessels per field using a 40× objective and a minimum of 4 fields per sample.
Statistics
All statistics, including Kaplan-Meier statistics (log-rank), Chi-squared tests, Mann-Whitney U-tests and Student’s t-tests were calculated using GraphPad Prism v.5.04 or v.6. p-values from Student’s t-tests are listed unless otherwise specified. In all graphs, means (bars) and standard deviations (lines) are denoted.
Supplementary Material
Significance.
Numerous therapies are being developed based on the premise that tumor stroma functions to promote cancer growth and invasion while simultaneously limiting the delivery of chemotherapy. Here, we demonstrate that depletion of stromal cells from pancreatic tumors – through genetic or pharmacological targeting of the Hh pathway – results in a poorly differentiated histology, increased vascularity and proliferation, and reduced survival. The study thus provides insight into the failure of Smoothened inhibitors, an anti-stromal therapy, in pancreatic cancer clinical trials. Moreover, we report that Hh-deficient tumors exhibit an increased sensitivity to VEGFR inhibition. As poorly differentiated human pancreatic tumors are well-vascularized, in contrast to most pancreatic cancers, our results suggest that this patient subset may be susceptible to angiogenesis inhibitors.
Highlights.
Shh-deficient tumors lacked stroma but were more aggressive and highly vascular.
Differentiation status is plastic and may be dependent in part upon Hh signaling.
Unlike differentiated tumors, undifferentiated PDAC is highly vascular.
Undifferentiated pancreas tumors may be susceptible to VEGFR inhibition.
Acknowledgments
We thank E. Collisson, R. Hruban, A. Rustgi, R. Vonderheide, and T. Wang for helpful discussions, as well as B. Orelli, M. Badgley, and J. Eberle for assistance in preparing the manuscript. We also thank Infinity Pharmaceuticals for providing IPI-926 and A. Joyner for providing Gli1-GFP mice. This study was funded by the NIH (DK088945, CA177857, DK034933 and CA046952 (ADR), T32CA009503 (DHT), CA136526 (MEFZ), CA157980 (KPO) and CA169123, DK083355, and DK083111 (BZS)) and AGA/FDHN (Fellow to Faculty Transition Award (ADR) and Bernard L. Schwartz Designated Research Award in Pancreatic Cancer (KPO)). This work was supported in part by the NIH/NIDDK Center for Molecular Studies in Digestive and Liver Diseases (P30DK050306) and its core facilities (Molecular Pathology and Imaging Core, Molecular Biology/Gene Expression Core, Transgenic and Chimeric Mouse Core, and Cell Culture Core) at UPenn and by the Molecular Pathology Shared Resource, the Confocal and Specialized Imaging Shared Resource, and the Small Animal Imaging Shared Resource within the Columbia University Herbert Irving Comprehensive Cancer Center (P30CA013696). The confocal microscope was purchased with grant S10RR025686.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bailey JM, Swanson BJ, Hamada T, Eggers JP, Singh PK, Caffery T, Ouellette MM, Hollingsworth MA. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res. 2008;14:5995–6004. doi: 10.1158/1078-0432.CCR-08-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownell I, Guevara E, Bai CB, Loomis CA, Joyner AL. Nerve-derived sonic hedgehog defines a niche for hair follicle stem cells capable of becoming epidermal stem cells. Cell stem cell. 2011;8:552–565. doi: 10.1016/j.stem.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu GC, Kimmelman AC, Hezel AF, DePinho RA. Stromal biology of pancreatic cancer. Journal of cellular biochemistry. 2007;101:887–907. doi: 10.1002/jcb.21209. [DOI] [PubMed] [Google Scholar]
- El-Zaatari M, Kao JY, Tessier A, Bai L, Hayes MM, Fontaine C, Eaton KA, Merchant JL. Gli1 deletion prevents Helicobacter-induced gastric metaplasia and expansion of myeloid cell subsets. PloS one. 2013;8:e58935. doi: 10.1371/journal.pone.0058935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engels B, Rowley DA, Schreiber H. Targeting stroma to treat cancers. Semin Cancer Biol. 2012;22:41–49. doi: 10.1016/j.semcancer.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann G, Fendrich V, McGovern K, Bedja D, Bisht S, Alvarez H, Koorstra JB, Habbe N, Karikari C, Mullendore M, et al. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol Cancer Ther. 2008a;7:2725–2735. doi: 10.1158/1535-7163.MCT-08-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann G, Habbe N, Dhara S, Bisht S, Alvarez H, Fendrich V, Beaty R, Mullendore M, Karikari C, Bardeesy N, et al. Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut. 2008b;57:1420–1430. doi: 10.1136/gut.2007.148189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SS, Jang JY, Kim SW, Kim WH, Lee KU, Park YH. Analysis of long-term survivors after surgical resection for pancreatic cancer. Pancreas. 2006;32:271–275. doi: 10.1097/01.mpa.0000202953.87740.93. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB, Logsdon CD. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–926. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, Vilardell F, Wang Z, Keller JW, Banerjee P, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol. 2009;27:1806–1813. doi: 10.1200/JCO.2008.17.7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenaga N, Ohuchida K, Mizumoto K, Cui L, Kayashima T, Morimatsu K, Moriyama T, Nakata K, Fujita H, Tanaka M. CD10+ pancreatic stellate cells enhance the progression of pancreatic cancer. Gastroenterology. 2010;139:1041–1051. 1051 e1041–1048. doi: 10.1053/j.gastro.2010.05.084. [DOI] [PubMed] [Google Scholar]
- Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112–120. doi: 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimeno A, Weiss GJ, Miller WH, Jr, Gettinger S, Eigl BJ, Chang AL, Dunbar J, Devens S, Faia K, Skliris G, et al. Phase I Study of the Hedgehog Pathway Inhibitor IPI-926 in Adult Patients with Solid Tumors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:2766–2774. doi: 10.1158/1078-0432.CCR-12-3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindler HL, Niedzwiecki D, Hollis D, Sutherland S, Schrag D, Hurwitz H, Innocenti F, Mulcahy MF, O’Reilly E, Wozniak TF, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303) J Clin Oncol. 2010;28:3617–3622. doi: 10.1200/JCO.2010.28.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein G. Evolutionary aspects of cancer resistance. Seminars in cancer biology. 2014;25C:10–14. doi: 10.1016/j.semcancer.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nature reviews Cancer. 2005;5:726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- Lonardo E, Frias-Aldeguer J, Hermann PC, Heeschen C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle. 2012;11:1282–1290. doi: 10.4161/cc.19679. [DOI] [PubMed] [Google Scholar]
- Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, McMahon AP. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer research. 2006;66:10171–10178. doi: 10.1158/0008-5472.CAN-06-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills LD, Zhang Y, Marler RJ, Herreros-Villanueva M, Zhang L, Almada LL, Couch F, Wetmore C, Pasca di Magliano M, Fernandez-Zapico ME. Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating KRAS oncogene-induced transformation. J Biol Chem. 2013;288:11786–11794. doi: 10.1074/jbc.M112.438846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton JP, Mongeau ME, Klimstra DS, Morris JP, Lee YC, Kawaguchi Y, Wright CV, Hebrok M, Lewis BC. Sonic hedgehog acts at multiple stages during pancreatic tumorigenesis. Proc Natl Acad Sci U S A. 2007;104:5103–5108. doi: 10.1073/pnas.0701158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan-Stevaux O, Lau J, Truitt ML, Chu GC, Hebrok M, Fernandez-Zapico ME, Hanahan D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009;23:24–36. doi: 10.1101/gad.1753809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca di Magliano M, Sekine S, Ermilov A, Ferris J, Dlugosz AA, Hebrok M. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes & development. 2006;20:3161–3173. doi: 10.1101/gad.1470806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DJ, Smith DM, Goff DJ, Tabin CJ. Epithelial-mesenchymal signaling during the regionalization of the chick gut. Development. 1998;125:2791–2801. doi: 10.1242/dev.125.15.2791. [DOI] [PubMed] [Google Scholar]
- Sastra SA, Olive KP. Quantification of murine pancreatic tumors by high-resolution ultrasound. Methods Mol Biol. 2013;980:249–266. doi: 10.1007/978-1-62703-287-2_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, Settleman J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer cell. 2009;15:489–500. doi: 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Lima A, Molina R, Hamilton P, Clermont AC, Devasthali V, Thompson JD, Cheng JH, Bou Reslan H, Ho CC, et al. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat Biotechnol. 2010;28:585–593. doi: 10.1038/nbt.1640. [DOI] [PubMed] [Google Scholar]
- Stoker MG, Shearer M, O’Neill C. Growth inhibition of polyoma-transformed cells by contact with static normal fibroblasts. Journal of cell science. 1966;1:297–310. doi: 10.1242/jcs.1.3.297. [DOI] [PubMed] [Google Scholar]
- Sukegawa A, Narita T, Kameda T, Saitoh K, Nohno T, Iba H, Yasugi S, Fukuda K. The concentric structure of the developing gut is regulated by Sonic hedgehog derived from endodermal epithelium. Development. 2000;127:1971–1980. doi: 10.1242/dev.127.9.1971. [DOI] [PubMed] [Google Scholar]
- Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernandez-del Castillo C, Yajnik V, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–856. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen JW, de Sauvage FJ. Paracrine Hedgehog signaling in cancer. Cancer research. 2009;69:6007–6010. doi: 10.1158/0008-5472.CAN-09-0756. [DOI] [PubMed] [Google Scholar]
- Tian H, Callahan CA, DuPree KJ, Darbonne WC, Ahn CP, Scales SJ, de Sauvage FJ. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci U S A. 2009;106:4254–4259. doi: 10.1073/pnas.0813203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonlaufen A, Phillips PA, Xu Z, Goldstein D, Pirola RC, Wilson JS, Apte MV. Pancreatic stellate cells and pancreatic cancer cells: an unholy alliance. Cancer research. 2008;68:7707–7710. doi: 10.1158/0008-5472.CAN-08-1132. [DOI] [PubMed] [Google Scholar]
- Walter K, Omura N, Hong SM, Griffith M, Vincent A, Borges M, Goggins M. Overexpression of smoothened activates the sonic hedgehog signaling pathway in pancreatic cancer-associated fibroblasts. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:1781–1789. doi: 10.1158/1078-0432.CCR-09-1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Ueda Y, Akaboshi S, Hino Y, Sekita Y, Nakao M. HMGA2 maintains oncogenic RAS-induced epithelial-mesenchymal transition in human pancreatic cancer cells. Am J Pathol. 2009;174:854–868. doi: 10.2353/ajpath.2009.080523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Vonlaufen A, Phillips PA, Fiala-Beer E, Zhang X, Yang L, Biankin AV, Goldstein D, Pirola RC, Wilson JS, et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am J Pathol. 2010;177:2585–2596. doi: 10.2353/ajpath.2010.090899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemasu H, Takashima M, Nishiyama KI, Ueki T, Yao T, Tanaka M, Tsuneyoshi M. Phenotypical characteristics of undifferentiated carcinoma of the pancreas: a comparison with pancreatic ductal adenocarcinoma and relevance of E-cadherin, alpha catenin and beta catenin expression. Oncol Rep. 2001;8:745–752. doi: 10.3892/or.8.4.745. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.