

Fatty acids and their metabolites play critical roles in mammalian and nonmammalian cell biology as signaling molecules, components of membranes, and storage lipids.[1] Enzymatic oxidation of polyunsaturated fatty acids leads to structurally diverse metabolites including unstable products such as allene oxides, divinyl ethers, and endoperoxides.[2,3] For example, in plants 12-oxophytodienoic acid IV is produced by an initial oxidation of linolenic acid by 13-lipoxygenase, followed by a transformation to an unstable allene oxide III (Scheme 1).[4] In 1988 Brash and co-workers showed that brief exposure of 13S-hydroperoxide II to a preparation of an allene oxide synthase followed by rapid organic extraction and treatment with diazomethane resulted in the isolation and characterization of the delicate allene oxide III.[5] The advent of whole genome sequencing of microbes has further expanded the discovery of novel enzymatic transformations and characterization of unstable products.[6,7] More recently, Brash and co-workers[8] identified, by genomic analysis, a dual-function protein encoded in the cyanobacterium Anabaena PCC 7120; this protein consists of a lipoxygenase domain fused to a catalase-related domain. After in vitro reconstitution of this protein, linolenic acid (I) was examined as an enzyme substrate and found to yield bicyclobutane fatty acid 1, the structure of which was confirmed by the characterization of methyl ester 2. The novel two-step transformation was proposed to occur by lipoxygenase-mediated C9 oxidation (I→V, Scheme 1) followed by cyclization within the neighboring catalase-related domain to bicyclobutane 1 via intermediate bicyclobutonium ion VII. While ample precedent existed for the rearrangement of 9R-hydroperoxide V to an epoxy allylic carbocation,[9] the formation of a bicyclobutonium ion VII leading to the bicyclo[1.1.0]butane ring system was unprecedented. Interestingly, cyclopropyl carbinyl cations have been implicated as intermediates in the biosynthesis of oxylipins.[10] The unique structure of bicyclobutane fatty acid 1 and its unknown bioactivity drew our attention, and lead to the total synthesis of bicyclobutane 2 described herein.
Scheme 1.

Lipoxygenase/catalase-mediated modifications of linolenic acid in plants and cyanobacteria.
The high strain energy (66 Kcal) and acid lability of bicyclo[1.1.0]butanes makes them challenging structures for synthesis and isolation.[11] Indeed, the parent bicyclobutane fatty acid (1) was not directly isolable, thus esterification with diazomethane in anhydrous solvent was required followed by careful purification of the methyl ester 2 at pH 8. When considering the synthesis of 2 we recognized that the installation of the delicate array of a vinyl epoxide conjugated to a strained bicyclobutane would be best achieved simultaneously, as illustrated in Scheme 2. The proposed reaction cascade[12, 13] starts with the generation of an all-cis metallated cyclopropane, followed by an SN2′ opening of the neighboring vinyl epoxide with release of an alkoxide appropriately oriented for displacement, to deliver the desired trans ep-oxide.
Scheme 2.

Proposed cascade reaction leading to bicyclobutane 2.
Precedent[14, 15] for the SN2′ opening of a vinyl epoxide by a cyclopropyl metal to form a bicyclobutane ring system lead us to initially examine the cyclization of vinyl epoxide 7 (Scheme 3). Preparation of 7 started with dibromocarbene addition to the TBS ether, which was derived from cis-2-pentenol (3), followed by desilylation and oxidation to afford aldehyde 4. The Horner–Wadsworth–Emmons olefination of 4 and a subsequent stereoselective reduction of the exo-bromo group using triphenylstannane and triethylborane/oxygen[16] as an initiator at low temperature afforded all-cis bromocyclopropane 5 in 83% yield.[17] Conversion of ester 5 into aldehyde 6 was achieved by a standard two-step reduction/oxidation sequence. Aldehyde methylenation was accomplished using the protocol reported by Matteson and Sadhu,[18] starting with the addition of (chloromethyl)lithium to 6. The chlorohydrin products were treated with NaH in THF to give a 2:1 mixture of diastereomers (7); these diastereomers reacted convergently to give bicyclobutane 8 upon cyclization initiated by lithium–halogen exchange (nBuLi, diethyl ether, −78°C). Attempts to purify bicyclobutane 8 by chromatography led to decomposition, but 1H and 13C NMR analysis of crude 8 was in full agreement with the assigned structure, as relevant NMR signals of fatty acid bicyclobutane 2 and bicyclobutane 8 coincided.[8, 19]
Scheme 3.

Synthesis of bicyclobutane 8. a) TBSCl, Et3N, CH2Cl2, RT, 94%; b) CHBr3, BnEt3NCl, 50% NaOH (aq), RT, 80%; c) TBAF, THF, RT, 95%; d) SO3·Pyr, DMSO, iPr2EtN, CH2Cl2, −20°C, 79%; e) (EtO)2P(O)CH2CO2CH3, NaH, THF −78 °C, 87%; f) Et3B, Ph3SnH, PhMe, −78 °C, 95%; g) DIBAl-H, CH2Cl2, −78°C, 95%; h) MnO2, CH2Cl2, RT, 99%; i) ICH2Cl, nBuLi, THF, −78°C, 51%; j) NaH, THF, −78 °C, 84%; k) i. nBuLi, THF, −78 °C, ii. [CuI·2 (LiCl)], THF, −78 to −20 °C. DIBAl-H=diisobutylaluminium hydride, DMSO=dimethylsulfoxide, Pyr=pyridine, TBAF=tetra-n-butylammonium fluoride, TBS=tert-butyldimethylsilyl, THF=tetrahydrofuran.
Having demonstrated the key carbanion-mediated cyclization we turned our attention to the synthesis of bicyclobutane fatty acid 2, starting with the condensation of β-keto phosphonate 9[20] and aldehyde 6 (Scheme 4). A Luche[21] reduction of the resulting keto ω-ester gave allylic alcohol 10. Epoxidation of 10 using either metal-catalysis or oxidation using a peracid[22] failed to afford the desired epoxy alcohol and led instead to presumed acid-promoted decomposition. Oxidation using dimethyldioxirane[23] provided the desired epoxide as a 1:1 mixture of the syn (11) and the desired anti (12) epoxy alcohols.[24] The syn isomer (11) was converted into the required anti diastereomer (12) using the Mitsunobu two-step alcohol inversion protocol.[25] Mesylation of alcohol 12 provided sulfonate 13, poised for the key cyclization cascade. Employing reaction conditions that were successful for the conversion of epoxide 7 into bicyclobutane 8 (nBuLi then CuI·2 LiCl) resulted in only decomposition of 13. Addition of mesylate 13 to a solution of nBuLi and subsequent warming from −78 to −20°C lead to bicyclobutane formation, as determined by 1H NMR analysis, but the epoxide formation did not occur, and there was an accompanying loss of the methyl ester group. However, treatment of 13 with 4 equivalents of tert-butyllithium in THF at −78°C did lead to the desired reaction cascade, unfortunately the cyclization was accompanied by conversion of the terminal methyl ester into the corresponding tert-butyl ketone. Altering the number of equivalents of tert-butyllithium did not provide a satisfactory solution, but by using the corresponding carboxylic acid 14 the formation of the tert-butyl ketone was avoided and the desired methyl ester was obtained after treatment of the crude product with diazomethane.[26] Attempts to isolate the parent fatty acid 1 failed, as observed in the original isolation work.[8] A solution of methyl ester 2 in [D6]DMSO was estimated to have a half life of 3 days.
Scheme 4.

Synthesis of bicyclobutane 2. a) KHMDS, THF, −78°C→RT, then 6, 92%; b) NaBH4, CeCl3, MeOH, RT, 98%; c) DMDO, CH2Cl2, 0°C, 95%; d) PPh3, DIAD, p-nitrobenzoic acid, RT then K2CO3, MeOH, 74%; e) MsCl, Et3N, DMAP, CH2Cl2, 0°C to RT, 56%; f) LiOH (aq), THF, 60°C; g) tBuLi, THF, −78 to −20°C, then CH2N2, 20%. DIAD=diisopropyl azodicarboxylate, DMAP=4-dimethylaminopyridine, DMDO=dimethyl dioxirane, HMDS=hexamethyldisilazide, Ms=methanesulfonyl.
In conclusion we have developed a 13-step synthesis of bicyclobutane fatty acid methyl ester (±)-2. Other related structurally novel and/or unstable lipids such as thromboxane A2 and pentacycloanammoxic acid (ladderane) have important biological functions.[1, 27] Future work will be directed toward the preparation of the unstable parent fatty acid 1 and studies aimed at defining the biological properties of this unusual oxylipin natural product.
Supplementary Material
Footnotes
We thank the National Institutes of Health (5RO1CA059515) for support of this research. S.M.D. acknowledges the support of the Vanderbilt Chemical Biology Interface (CBI) training program (T32 GM065086). We gratefully acknowledge Prof. Alan Brash for valuable discussion.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201104366.
Contributor Information
Sean M. DeGuire, Departments of Chemistry and Biochemistry, Vanderbilt University Vanderbilt Institute of Chemical Biology, Nashville, TN 37235 (USA)
Shutao Ma, Department of Medicinal chemistry School of Pharmaceutical Sciences, Shandong University 44 West Culture Road, Jinan, 250012 (P.R. China).
Gary A. Sulikowski, Departments of Chemistry and Biochemistry, Vanderbilt University Vanderbilt Institute of Chemical Biology, Nashville, TN 37235 (USA) gary.a.sulikowski@vanderbilt.edu
References
- 1.Jahn U, Galano J-M, Durand T. Angew. Chem. 2008;120:5978–6041. doi: 10.1002/anie.200705122. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:5894–5955. doi: 10.1002/anie.200705122. [DOI] [PubMed] [Google Scholar]
- 2.Buist PH. Nat. Prod. Rep. 2007;24:1110–1127. doi: 10.1039/b508584p. [DOI] [PubMed] [Google Scholar]
- 3.Andreou A, Brodhun F, Feussner I. Prog. Lipid Res. 2009;48:148–170. doi: 10.1016/j.plipres.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Hamberg M, Gardner H. Biochim. Biophys. Acta Lipids Lipid Metab. 1993;1165:1–18. doi: 10.1016/0005-2760(92)90069-8. [DOI] [PubMed] [Google Scholar]
- 5.Brash A, Baertschi S, Ingram C, Harris T. Proc. Natl. Acad. Sci. USA. 1988;85:3382–3386. doi: 10.1073/pnas.85.10.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corre C, Challis GL. Nat. Prod. Rep. 2009;26:977–986. doi: 10.1039/b713024b. [DOI] [PubMed] [Google Scholar]
- 7.Feussner I, Wasternack C. Annu. Rev. Plant Biol. 2002;53:275–297. doi: 10.1146/annurev.arplant.53.100301.135248. [DOI] [PubMed] [Google Scholar]
- 8.Schneider C, Niisuke K, Boeglin WE, Voehler M, Stec DF, Porter NA, Brash AR. Proc. Natl. Acad. Sci. USA. 2007;104:18941–18945. doi: 10.1073/pnas.0707148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerwick W. Lipids. 1996;31:1215–1231. doi: 10.1007/BF02587906. [DOI] [PubMed] [Google Scholar]
- 10.Gerwick W. Chem. Rev. 1993;93:1807–1823. [Google Scholar]
- 11.de Meijere A, Kozhushkov SI, Schill H. Chem. Rev. 2006;106:4926–4996. doi: 10.1021/cr0505369. [DOI] [PubMed] [Google Scholar]
- 12.Tietze L. Chem. Rev. 1996;96:115–136. doi: 10.1021/cr950027e. [DOI] [PubMed] [Google Scholar]
- 13.Nicolaou KC, Edmonds DJ, Bulger PG. Angew. Chem. 2006;118:7292–7344. doi: 10.1002/anie.200601872. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2006;45:7134–7186. doi: 10.1002/anie.200601872. [DOI] [PubMed] [Google Scholar]
- 14.Bentley T, Engels B, Hupp T, Bogdan E, Christl M. J. Org. Chem. 2006;71:1018–1026. doi: 10.1021/jo0519918. [DOI] [PubMed] [Google Scholar]
- 15.Bailey W, Tao Y. Tetrahedron Lett. 1997;38:6157–6158. [Google Scholar]
- 16.Miura K, Ichinose Y, Nozaki K, Fugami K, Oshima K, Utimoto K. Bull. Chem. Soc. Jpn. 1989;62:143–145. [Google Scholar]
- 17.Product stereochemistry was assigned based on coupling constants and NOE analysis.
- 18.Sadhu K, Matteson D. Tetrahedron Lett. 1986;27:795–798. [Google Scholar]
- 19.The yield of the crude product 8 was >100% and >70% purity as judged by 1H and 13C NMR analysis.
- 20.Delamarche I, Mossett P. J. Org. Chem. 1994;59:5453–5457. [Google Scholar]
- 21.Luche J-L. J. Am. Chem. Soc. 1978;100:2226–2227. [Google Scholar]
- 22.Hoveyda AH, Evans DA, Fu GC. Chem. Rev. 1993;93:1307–1370. [Google Scholar]
- 23.Murray RW. Chem. Rev. 1989;89:1187–1201. [Google Scholar]
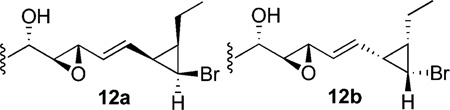
- 24.For simplicity only one syn (11) and one anti (12) diastereomer is represented in Scheme 4. Upon cyclization (via mesylate 13), both anti diastereomers (12a and 12b) react convergently to give 2 with formation of the achiral [1.1.0]bicyclobutane ring. When starting from the assigned syn diasteromers (11) only decomposition was observed under the same reaction conditions. This observation supports the assigned relative stereochemistry.
-
25.
Mitsunobu O. Synthesis. 1981:1–28.
- 26.The yield of the crude product 2 was near quantitative (see the Supporting Information for a 1H NMR spectrum of the crude product 2). Extensive decomposition occurs upon purification leading to a yield of 20%.
- 27.Sinninghe Damsté JS, Strous M, Rijpstra WIC, Hopmans EC, Geenevasen JAJ, van Duin ACT, van Niftrik LA, Jetten MSM. Nature. 2002;419:708–712. doi: 10.1038/nature01128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.