

Abstract
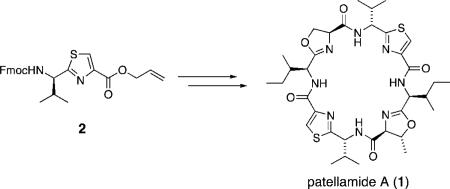
Patellamide A was efficiently synthesized from thiazole 2 via two complementary heterocyclization approaches to form the thiazole and oxazoline rings.
Patellamides A–C comprise a class of lipophilic cyclic peptides that were originally isolated from the marine tunicate Lissoclinum patella that contain oxazoline- and thiazole-containing subunits.1 Initial screening efforts revealed that the patellamides were cytotoxic, displaying IC50 values of 2–4 μg/mL against L1210 murine leukemia cells. In addition, patellamide A was found to be active versus the human ALL cell line (T cell acute leukemia) CEM with an ID50 of 0.028 μg/mL.
Subsequent to their initial disclosure by Ireland, synthetic efforts directed toward the synthesis of patellamides B and C resulted in a revision of their originally reported structures. Additional synthetic work by Hamada and Shiori resulted in a revison of the structure assigned to patellamide A.2 Their synthetic work determined that the actual structure of the patellamide A macrocycle contained oxazoline and thiazole subunits that were separated by intervening isoleucine residues whereas in the structure originally reported by Ireland, the oxazoline and thiazole heterocycles were directly fused (as in (3), Figure 1).
Figure 1.

Original and revised structures for patellamide A.
The patellamides have recently received renewed interest as a result of their intriguing biosynthetic pathway. Schmidt et al., have recently disclosed that the patellamides are not biosynthetic products of the marine tunicate L. patella but are instead the biosynthetic products of a marine symbiont, Prochloron didemni.3
Patellamide A is an octapeptide containing two oxazoline and two thiazole rings that arise from a ribosomally synthesized 71-residue precursor protein, PatE. This preprotein is then modified by other products of the pat gene cluster (patA–G) which results in proteolytic cleavage, macrocyclization, epimerization, heterocyclization, and dehydroge-nation that ultimately results in the production of patellamide A.
Sequencing of the P. didemni genome also uncovered similarities in the function of the gene products utilized for oxazoline and thiazole formation with those used for maturation of the microcins. Subsequent work by the Schmidt group demonstrated that pat pathway could be exploited for production of a diverse array of cyclic peptides through appropriate manipulation of the precursor proteins.4
Azole-containing cyclic peptides have garnered significant interest in recent years as a result of their multifarious nature. They have been shown to effect the reversal of multiple drug resistance (MDR) in cancer cells,5 to have a binding affinity for metals as pseudoporphyrin structures,6 and to effectively stabilize G-quadruplexes.7
With respect to the patellamides, although intriguing aspects of their biosynthetic pathway have been revealed,3 much of this pathway remains to be established.8 A reliable and efficient synthetic route to patellamide A, and its precursors, would enable more detailed study of this very interesting, and potentially pharmaceutically useful, biosynthetic pathway.
As part of our effort to support additional investigation into the biosynthetic pathway utilized for the production of patellamide A, and to gain additional insights into G-quadruplex binding ability9 of such natural products, we report herein our efforts that have culminated in the synthesis of patellamide A.
As mentioned above, it was the synthetic efforts of Hamada and Shiori that ultimately resulted in the strucutral revision for patellamide A. Although effective, the Hamada/ Shiori synthesis employs the use of harsh conditions and long reaction times for the development of the oxazoline and thiazolines moeities that characterize this family of natural products.
In view of the improved methodology available for the synthesis of amino acid-derived oxazolines10 and thiazo-lines,11 we sought to pursue an alternative synthesis of patellamide A that would utilize relatively mild conditions for formation of the oxazoline/thiazole heterocycles.
Our retrosynthesis is outlined in Figure 2. An important difference between our envisioned synthetic route and that utilized by Hamada and Shiori is that we hypothesized that installation of all four of the oxazoline and thiazole units into a linear precursor would enhance the rate and efficiency of the macrocyclization reaction. Thus, our first disconnection reveals the linear octapeptide fragment 4 shown below. The linear octapeptide fragment containing the mature oxazoline and thiazole subunits would be prepared via convergent condensation of two tetrapeptide fragments that differ from one another only by the presence of an allo-threonine- versus (as in 5) serine-derived (as in 4) precursor for their respective oxazoline subunits.
Figure 2.

Patellamide A retrosynthetic analysis.
Thus, the synthesis of both peptide fragments (Scheme 1) began with the known thiazole 2, which was readily prepared in five steps and in high yield from Fmoc-Cys(Trt)-OH according to the procedure outlined by Kelly and co-workers using the combination of Ph3PO and Tf2O.11 Thiazole amino acids, such as 2, have proven to be valuable starting materials for the synthesis of many marine-derived natural products, including dendroamide A12 tenuecyclamides A-D,13 bistratamides E-J,14 and didmolamides A and B.15
Synthesis of the left-hand fragment 5 began with removal of the Fmoc protecting group (20% piperidine/DMF) of thiazoline 2. Coupling of the free amine with Fmoc-aThr(Trt)-OH (HOBt, HBTU, DIEA, CH2Cl2) provided thiazolyl tripeptide 7 in 86% yield. Subsequent Fmoc deprotection (20% piperidine/DMF) and coupling with Fmoc-Ile-OH (HOBt, HBTU, DIEA, CH2Cl2) provided thiazolyl tetrapeptide 8. Cleavage of the C-terminal allyl ester (Pd-(PPh3)4, PhSiH3, CH2Cl2) provided the target thiazolyl tetrapeptide 5.
Coupling of the free amine, derived from Fmoc deprotection of 2, with Fmoc-Ser(Trt)-OH (HOBt, HBTU, DIEA, CH2Cl2) cleanly provided thiazolyl tripeptide 9 in 92% overall yield. Fmoc cleavage (20% piperidine/DMF) followed by coupling with Fmoc-Ile-OH (HOBt, HBTU, DIEA, CH2Cl2) provided thiazolyl tetrapeptide 10. A final Fmoc cleavage (20% piperidine/DMF) provided the final thiazolyl tetrapeptide 6 required for elaboration of the linear octapep-tide.
Activation of carboxylic acid 6 and coupling with 2 (HOBt, HBTU, DIEA, CH2Cl2) provided the linear octapep-tide 11 in 84% yield. Exposure of the octapeptide to 2% TFA in dichloromethane with a thiophenol capture reagent cleanly removed the trityl protecting groups and set the stage for introduction of the allo-threonine- and serine-derived oxazoline subunits.
Cyclodehydration of 12 was readily achieved through treatment of the intermediate diol with the Burgess reagent (inner salt) and provided the fully elaborated linear precursor 4 to patellamide A.16 It was found that reagent purity and carefully controlled reaction times were critical in order to achieve optimal reaction yields as formation of an unidentified reaction byproduct would compromise the yield of the desired reaction product, possibly due to the presence of triethylamine as a byproduct of the decomposition of the Burgess reagent over time.
Palladium(0)-mediated cleavage of the C-terminal allyl ester was achieved through use of Pd(PPh3)4 using phenylsilane as an allyl cation capture reagent. Cleavage of the N-terminal Fmoc protecting group (20% piperidine/DMF) and macrocyclization (PyBOP, DMAP, DIEA, CH2Cl2/DMF) provided the target compound, patellamide A in 55% overall yield.
In summary, the synthesis of patellamide A has been accomplished employing contemporary methods for the formation of oxazolines and thiazolines. This methodology can be easily extended to the synthesis of other members of the patellamide class of cyclic peptides. Further efforts directed toward this end, as well as construction of putative biosynthetic intermediates utilized in the patellamide bio-synthetic pathway will be presented in due course.
Supplementary Material
Scheme 1.

Synthesis of Tetrapeptide Fragments 5 and 6
Scheme 2.

Patellamide A End Game
Acknowledgment
Financial support for this work was provided by the Regents of the University of California and Indiana University. MSV acknowledges Eli Lilly and Company for support in the form of an Eli Lilly New Faculty Award. P.G.-R. acknowledges the National Science Foundation for an NSF Bridge to the Doctorate Fellowship.
Footnotes
Supporting Information Available: Experimental details and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ireland CM, Durso AR, Jr., Newman RA, Hacker MP. J. Org. Chem. 1982;47:1807. [Google Scholar]
- 2.Hamada Y, Shibata M, Shiori T. Tetrahedron Lett. 1985;26:6501. [Google Scholar]
- 3.Schmidt EW, Nelson JT, Rasko DA, Sudek S, Eisen JA, Haygood MG, Ravel J. Proc. Natl. Acad. Sci. U.S.A. 2005;102:7315. doi: 10.1073/pnas.0501424102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donia MS, Hathaway BJ, Sudek S, Haygood MG, Rosovitz MJ, Ravel J, Schmidt EW. Nature Chem. Biol. 2006;2:729. doi: 10.1038/nchembio829. [DOI] [PubMed] [Google Scholar]
- 5.a Williams AB, Jacobs RS. Cancer Lett. 1993;71:97. doi: 10.1016/0304-3835(93)90103-g. [DOI] [PubMed] [Google Scholar]; b Fu X, Do T, Schmitz FJ, rusevich V, Engel MH. J. Nat. Prod. 1998;61:1547. doi: 10.1021/np9802872. [DOI] [PubMed] [Google Scholar]
- 6.For a general review, see: Bertram A, Pattenden G. Nat. Prod. Rep. 2007;24:18. doi: 10.1039/b612600f.
- 7.Jantos K, Rodriguez R, Ladame S, Shirude P, Balasubramanian S. J. Am. Chem. Soc. 2006;128:13662. doi: 10.1021/ja064713e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milne BF, Long PF, Starcevic A, Daslav H, Jaspars M. Org. Biomol. Chem. 2006;4:631. doi: 10.1039/b515938e. [DOI] [PubMed] [Google Scholar]
- 9.The results of these studies will be disseminated in due course.
- 10.Wipf P, Miller CP. Tetrahedron Lett. 1992;33:907. [Google Scholar]
- 11.You S-L, Razavi H, Kelly JW. Angew. Chem., Int. Ed. 2003;42:83. doi: 10.1002/anie.200390059. [DOI] [PubMed] [Google Scholar]
- 12.You S-L, Kelly JW. J. Org. Chem. 2003;68:9506. doi: 10.1021/jo0302657. [DOI] [PubMed] [Google Scholar]
- 13.You S-L, Deechongkit S, Kelly JW. Org. Lett. 2004;6:2627. doi: 10.1021/ol049020m. [DOI] [PubMed] [Google Scholar]
- 14.a You S-L, Kelly JW. Chem. Eur. J. 2004;10:71. [Google Scholar]; b You S-L, Kelly JW. Tetrahedron. 2005;61:241. [Google Scholar]
- 15.You S-L, Kelly JW. Tetrahedron Lett. 2005;46:2567. [Google Scholar]
- 16.Wipf P, Fritch PC. J. Am. Chem. Soc. 1996;118:12358. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.