

Abstract
Pin1 is a peptidyl prolyl cis-trans isomerase that only binds to and isomerizes phosphorylated serine/threonine-proline motifs, inducing conformational changes that alter target protein function and phosphorylation. We have shown previously that deficiency of another peptidyl prolyl isomerase, FK506 binding protein 12/12.6, alters endothelial NO synthase phosphorylation and causes endothelial dysfunction and hypertension. Endothelial NO synthase contains the Pin1 binding sequence at (p)serine 116-proline 117 and phosphorylation of endothelial NO synthase serine 116 inhibits NO production; however, whether Pin1 deficiency alters vascular function and blood pressure is unknown. We hypothesized that Pin1 isomerizes p-endothelial NO synthase serine 116, which enables dephosphorylation and stimulates NO production. Immunoprecipitation of endothelial NO synthase and probing for Pin1 in rat aortic endothelial cells confirmed the interaction between the two. Pin1 knockdown via small interfering RNA or inhibition by juglone increased endothelial NO synthase serine 116 phosphorylation and prevented vascular endothelial growth factor–induced serine 116 dephosphorylation in endothelial cells. Acute treatment of isolated mouse aortas with juglone increased endothelial NO synthase serine 116 phosphorylation and decreased NO production and relaxation responses. Mice treated with juglone for 2 weeks, as well as Pin1 knockout mice, exhibited increased aortic endothelial NO synthase serine 116 phosphorylation, endothelial dysfunction, and hypertension. These data demonstrate that Pin1 binds endothelial NO synthase and enables dephosphorylation of serine 116, which increases NO production and endothelium-dependent dilation, leading to blood pressure maintenance.
Keywords: endothelium, hypertension, experimental, NO synthase, Pin1, phosphorylation
Protein interacting with NIMA (never in mitosis A)-1 (Pin1) is a peptidyl prolyl isomerase expressed ubiquitously in cells and regulates a variety of cellular functions, including cell cycle progression, growth-signal responses, gene expression, and phosphorylation-dependent cell signaling.1,2 Pin1 is the only isomerase known to date that binds to phosphorylated serine/threonine-proline motifs and mediates a cis-trans conformational change of its substrate. This structural alteration after phosphorylation of the Ser/Thr residue can alter target protein function, interaction, stability, and phosphorylation, thus adding another level of posttranslational regulation. Pin1 has been reported to play a role in the pathogenesis of cancer, Alzheimer disease, and asthma, thus making it a likely target for future therapeutic development.3-5 However, the role of Pin1 in vascular physiology and pathology has only recently been examined, and whether inhibition of Pin1 exerts detrimental vascular effects is unknown.
It was shown recently that inhibition of Pin1 had an immunosuppressive effect in rodents and prevented both acute and chronic rejection of allogeneic lung transplants in rats.6 This was mediated by decreased inflammatory cytokine production by T cells. Current therapeutics used clinically for maintenance immunosuppression in organ transplant recipients include the prolyl isomerase-binding drugs cyclosporin A, tacrolimus (FK506), and sirolimus (rapamycin), which exert their immunosuppressive effects by inhibiting T-cell proliferation. However a major limitation with these drugs is the development of endothelial dysfunction and hypertension, which are significant risk factors for decreased allograft and patient survival.7-11 Cyclosporin A binds cyclophilin A and both tacrolimus and sirolimus bind FK506 binding proteins 12 and 12.6 (FKBP12/12.6), which all differ from Pin1 in that they bind and isomerize nonphosphorylated Xxx-Pro motifs. We and others have demonstrated that pharmacological inhibition or genetic deletion of FKBP12/12.6 leads to endothelial dysfunction and hypertension, and we have reported a molecular mechanism by which FKBP12/12.6 inhibition in the endothelium causes these adverse effects.12-14 Whether deficiency of Pin1, expressed in cells of the vasculature, also affects endothelial function and blood pressure regulation is unknown.
Endothelium-derived NO plays a major role in vascular tone and blood pressure regulation and is produced primarily by the enzyme endothelial NO synthase (eNOS). In addition to several cofactors, protein-protein interactions, and localization, eNOS activity is regulated by its phosphorylation status.15 NO generation is stimulated by phosphorylation of Ser1179, Ser635, Ser619, and Tyr83 but inhibited by Thr497 and Ser116 phosphorylation (bovine sequence). We have shown previously that vascular FKBP12/12.6 deficiency indirectly leads to increased eNOS Thr495 phosphorylation, as well as decreased NO production, endothelial dysfunction, and hypertension; therefore, it is possible that Pin1 deficiency might also alter eNOS phosphorylation and NO generation directly or indirectly.12,13 The genetic sequence of eNOS contains a potential Pin1 binding site at (p)Ser116-Pro117, suggesting that Pin1 may bind to and regulate the phosphorylation status of this site and modulate NO generation. Ruan et al16 confirmed recently that Pin1 binds to eNOS, and, through a series of elegant studies in bovine aortic endothelial cells, they demonstrated that this interaction occurs only when Ser116 is phosphorylated. Studies of Pin1 overexpression in endothelial cells and mouse aorta were reported to decrease NO production and decrease vasodilation16; however, the effects of Pin1 overexpression on eNOS Ser116 phosphorylation were not reported. In addition, Pin1 deficiency in blood vessels and in the whole animal was not studied.
Although the inhibition of Pin1 is a promising therapeutic target for immunosuppression and cancer, it is unknown whether inhibition of Pin1 might cause off-target effects, such as endothelial dysfunction and hypertension. We hypothesized that Pin1 binds to eNOS at pSer116-Pro117 and enables Ser116 dephosphorylation, NO generation, vasodilation, and blood pressure maintenance. To test this we examined the interaction of Pin1 and eNOS, as well as the effects of Pin1 knockdown or inhibition on eNOS Ser116 phosphorylation in endothelial cells. We also determined the direct vascular effects of Pin1 inhibition on eNOS Ser116 phosphorylation, NO production, and vascular reactivity in isolated mouse aortas. In addition, we tested the in vivo effects of Pin1 deficiency on aortic eNOS Ser116 phosphorylation, NO production, endothelial function, and blood pressure in mice treated with a specific Pin1 inhibitor, as well as Pin1 knockout (KO) mice.
Methods
An expanded Methods section is available in the online Data Supplement at http://hyper.ahajournals.org.
Endothelial Cell Studies
Immunoblotting using anti-Pin1 or anti-eNOS antibodies was performed on rat aortic endothelial cell lysates immunoprecipitated with anti-eNOS or IgG (negative control), as described previously.12 Immunoblotting was performed after Pin1 knockdown via small interfering RNA (siRNA). Immunoblotting was also performed in cells after treatment with the Pin1-specific inhibitor juglone (5-hydroxy-1,4-naphthoquinone) or vehicle (ethanol, <1% final concentration), as well as after treatment with the eNOS agonist vascular endothelial growth factor. The blots were identified simultaneously using near-infrared visualization, and densitometry was performed.
Acute Ex Vivo and In Vitro Vascular Studies
Male C57Bl/6 mice aged 10 to 12 weeks were anesthetized with isoflurane and euthanized by cervical dislocation. Endotheliumintact aortas were treated with juglone or vehicle, and immunoblotting was performed on vascular homogenates as described above. NO production was measured using the fluorescent dye 4-amino-5-methylamino-2′,7′-difluorofluorescein with and without NO synthase inhibition with Nω-nitro-l-arginine (LNNA). NO production was determined by measuring peak fluorescent counts in the absence of LNNA minus peak fluorescent counts in the presence of LNNA and expressed as a percentage of control.
Vascular reactivity was measured in endothelium-intact aortas, as described previously.12,13 Vessels were set at a passive tension of 0.75 g based on previous length tension studies, and indomethacin was present in all of the experiments. Endothelium-intact aortic rings were incubated with juglone or vehicle (ethanol, <1% final concentration), and concentration-force curves were obtained in a half-log, cumulative fashion to the endothelium-dependent dilator acetylcholine (ACh) and the endothelium-independent dilator sodium nitroprusside after contraction to phenylephrine (PE).
In Vivo Studies
Male C57BL/6 mice, Pin1 WT mice, and Pin1 KO mice aged 10 to 18 weeks were used in all of the experiments. Heterozygous Pin1 mice were obtained from Dr Anthony Means (Duke University) and are on a pure C57BL/6, as described previously.17 Tail-cuff systolic blood pressures in C57BL/6 control mice were measured at baseline and on days 7 and 14 of daily treatment with juglone or diluent (saline and ethanol, 20% final concentration), as described previously.6 Juglone-treated mice were euthanized on day 14 of treatment, whereas Pin1 WT and KO mice were euthanized after final blood pressure measurements. Aortic vascular reactivity, NO production, and immunoblotting were performed as above. All of the procedures were approved by the Institutional Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Statistical Analyses
Results are presented as mean ± SEM. The 2-tailed Student’s t test was used to compare variables between 2 groups. An ANOVA was used for multiple comparisons, followed by the Student-Newman-Keuls post hoc test when necessary. The significance level was 0.05.
Results
Pin1 Interaction With eNOS and Effects on eNOS Ser116 Phosphorylation
eNOS contains a Pin1-binding consensus sequence at pSer116-Pro117, and Ruan et al16 showed recently that this interaction occurs in bovine aortic endothelial cells; therefore, we first confirmed the Pin1-eNOS interaction in rat aortic endothelial cells. Endothelial cells treated with vehicle or the Pin1-specific inhibitor juglone for 30 minutes were immunoprecipitated with an anti-eNOS or anti-IgG antibody, followed by immunoblotting for Pin1. Figure 1 confirms that Pin1 interacts with eNOS, and juglone had no effect on this interaction, as was expected, because juglone inhibits the isomerase activity of Pin1 but not target binding. The negative control, consisting of only mouse IgG for immunoprecipitation, did not detect any Pin1.
Figure 1.
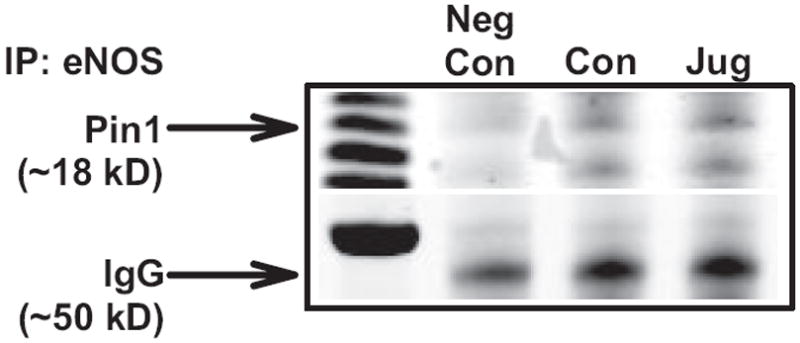
Pin1 and endothelial NO synthase (eNOS) interaction in endothelial cells. Rat aortic endothelial cells were treated with vehicle or juglone (10 μmol/L, 30 minutes), and lysates were immunoprecipitated with anti-eNOS or mouse IgG (negative control) followed by immunoblotting for Pin1. IgG was used as a loading control. Representative image of 3 independent experiments.
Because Ser116 phosphorylation decreases NO production and dephosphorylation of this inhibitory site occurs during NO production,16,18 we determined whether Pin1 knockdown with siRNA alters eNOS phosphorylation in endothelial cells. Figure 2A demonstrates that transfection of endothelial cells with Pin1 siRNA decreased Pin1 protein expression ≈75%, which resulted in a significant increase in eNOS Ser116 phosphorylation (Figure 2B). In addition, Pin1 knockdown significantly decreased eNOS Ser1177 phosphorylation (Figure 2C) and increased eNOS Thr495 phosphorylation (Figure 2D), consistent with a decrease in eNOS activity. Similarly, juglone elicited an increase in eNOS Ser116 phosphorylation (Figure 3A) and prevented eNOS Ser116 dephosphorylation in response to the eNOS agonist vascular endothelial growth factor (Figure 3B). These findings support our hypothesis that Pin1 binds to eNOS and negatively affects eNOS phosphorylation in endothelial cells.
Figure 2.
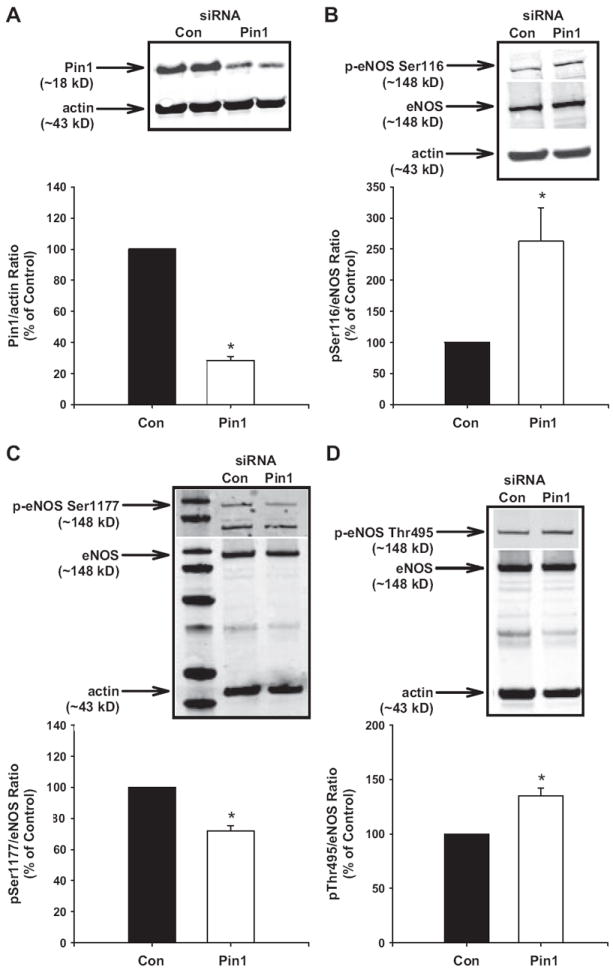
Effect of Pin1 small interfering RNA (siRNA) on endothelial NO synthase (eNOS) Ser116 phosphorylation. Rat aortic endothelial cells were transfected with Pin1 or scrambled siRNA. Representative Western blots showing (A) Pin1 expression, (B) eNOS Ser116 phosphorylation, (C) eNOS Ser1177 phosphorylation, and (D) eNOS Thr495 phosphorylation and densitometry for ratio of Pin1:actin or eNOS phosphorylation:eNOS as a percentage of control. Actin was used as a loading control. Results are expressed as mean±SEM (n>3 independent experiments). *P<0.05 vs control.
Figure 3.

Effect of Pin1 inhibition on endothelial NO synthase (eNOS) Ser116 phosphorylation. Experiments were performed in rat aortic endothelial cells in the absence or presence of juglone without (A) or with (B) the eNOS agonist vascular endothelial growth factor (VEGF). Representative Western blots showing eNOS Ser116 phosphorylation and densitometry for ratio of eNOS Ser116 phosphorylation: eNOS as a percentage of control. Actin was used as a loading control. Results are expressed as mean±SEM (n=5 independent experiments). *P<0.05 vs “0,” +P<0.05 vs previous time point.
Acute Inhibition of Pin1 Causes Endothelial Dysfunction
To examine the direct vascular effects of Pin1 inhibition and eNOS Ser116 phosphorylation, we treated endothelium-intact aortas from control mice acutely with juglone and measured Ser116 phosphorylation, NO production, and relaxation responses. Juglone increased aortic eNOS Ser116 phosphorylation (Figure 4A), similar to that seen in endothelial cells and decreased NO production ≈60% (P<0.05 versus controls; Figure 4B). Next, we measured aortic endothelium-dependent and endothelium-independent relaxation responses after acute treatment with vehicle or juglone. There were no differences in PE-induced contractions between juglone-treated aortas and vehicle-treated aortas (contraction to PE 1 μmol/L in grams: juglone: 1.0±0.3 g versus controls: 1.1±0.3 g; P>0.05). Juglone significantly decreased maximal ACh-induced relaxation responses compared with vehicle-treated controls in a dose-dependent manner (Figure 4C). The NO synthase inhibitor LNNA (10 μmol/L) abolished relaxation responses to ACh in all of the groups (data not shown). Juglone had no effect on relaxation responses or sensitivity to the NO donor sodium nitroprusside, suggesting a detrimental effect on endothelium-derived NO generation (Figure S1A, available in the online Data Supplement at http://hyper.ahajournals.org).
Figure 4.
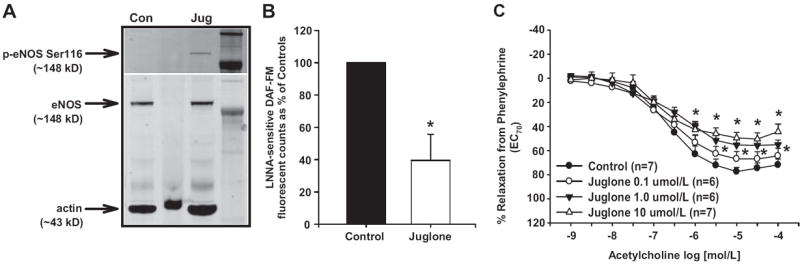
Effect of acute Pin1 inhibition on vascular endothelial NO synthase (eNOS) function. Isolated aortas from control mice were treated with either juglone (A and B: 10 μmol/L; C: 0.1 to 10.0 μmol/L) or vehicle for 30 minutes. A, Representative Western blot showing eNOS Ser116 phosphorylation. B, NO production as measured by Nω-nitro-l-arginine (LNNA)–sensitive 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM) fluorescence. C, Relaxation responses to acetylcholine after contraction with phenylephrine. Relaxation to 100% represents a full return to a passive tension of 0.75 g. Results are expressed as mean±SEM (n=3 independent experiments for A and n=6 to 7 aortas in each group for B and C). *P<0.05 vs control.
Pin1 Deficiency In Vivo Causes Endothelial Dysfunction and Hypertension
To test the in vivo effects of Pin1 inhibition, we first treated mice daily for 2 weeks with vehicle or juglone at a dose used previously to suppress the immune system.6 Aortas from juglone-treated mice exhibited increased eNOS Ser116 phosphorylation (Figure 5A), decreased NO production (≈51% decrease versus controls; Figure 5B), and decreased LNNA-sensitive, ACh-induced relaxation responses compared with aortas from vehicle-injected controls (Figure 5C). There were no differences in sodium nitroprusside–induced relaxation responses in aortas from juglone-treated mice compared with vehicle-treated mice (Figure S1B). Also, there were no differences in PE-induced contractions between aortas from juglone-treated mice and vehicle-treated mice (contraction to PE 1 μmol/L in grams: juglone-IP: 0.9±0.2 g versus controls: 1.0±0.2 g; P>0.05). Systolic blood pressures were significantly increased in juglone-treated mice at day 7 of treatment compared with controls (juglone: 124±2 mm Hg versus control: 103±4 mm Hg) and were further increased at day 14 of treatment (juglone: 140±1 mm Hg versus control: 107±3 mm Hg; Figure 5D).
Figure 5.
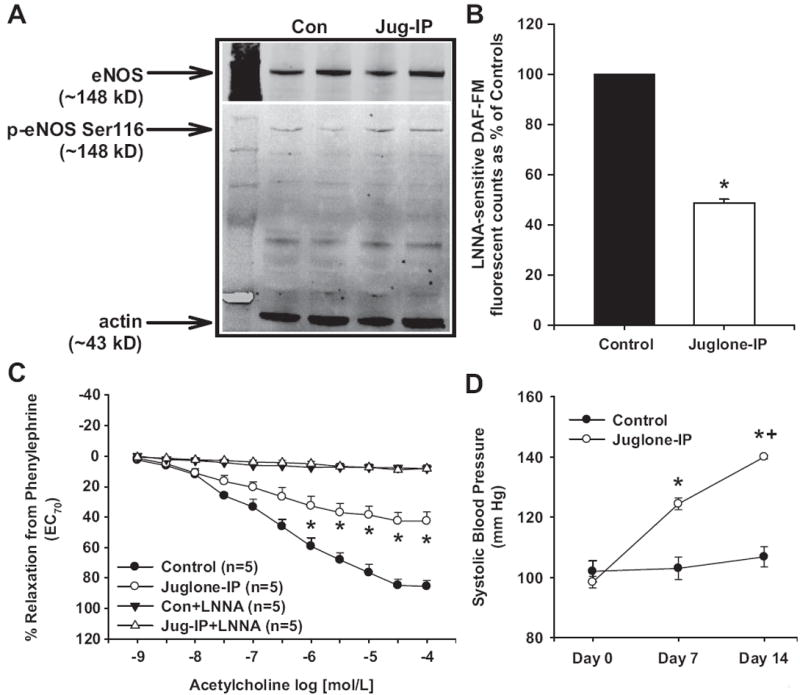
Effect of chronic Pin1 inhibition on vascular endothelial NO synthase (eNOS) function and systolic blood pressure in mice. Control mice were treated daily for 2 weeks with either juglone (1 mg/kg, IP) or vehicle. A, Representative Western blot showing aortic eNOS Ser116 phosphorylation. B, Aortic NO production as measured by Nω-nitro-l-arginine (LNNA)–sensitive 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM) fluorescence. C, Aortic relaxation responses to acetylcholine after contraction with phenylephrine in the absence and presence of NO synthase (NOS) inhibition (LNNA). Relaxation to 100% represents a full return to a passive tension of 0.75 g. D, Systolic blood pressure at baseline, day 7, and day 14 of juglone or vehicle treatment. Results are expressed as mean±SEM (n>5 mice for each group). *P<0.05 vs control, +P<0.05 vs day 7 blood pressure.
To confirm these in vivo effects, we measured aortic eNOS Ser116 phosphorylation, NO production, and relaxation responses, as well as systolic blood pressure, in Pin1 WT and KO mice. Aortas from Pin1 KO mice had increased eNOS Ser116 phosphorylation (Figure 6A), decreased NO production (≈42% decrease versus controls; Figure 6B), and decreased LNNA-sensitive, ACh-induced relaxation responses compared with aortas from Pin1 WT littermate controls (Figure 6C). There were no differences in sodium nitroprusside–induced relaxation responses in aortas from Pin1 KO mice compared with Pin1 WT mice (Figure S1C). There were no differences in PE-induced contractions between aortas from Pin1 KO mice and Pin1 WT mice (contraction to PE 1 μmol/L in grams: Pin1 KO: 1.1±0.2 g versus Pin1 WT: 1.0±0.2 g; P>0.05). Systolic blood pressures were significantly increased in Pin1 KO mice compared with Pin1 WT mice (Pin1 KO: 143±3 mm Hg versus Pin1 WT: 99±4 mm Hg; Figure 6D).
Figure 6.
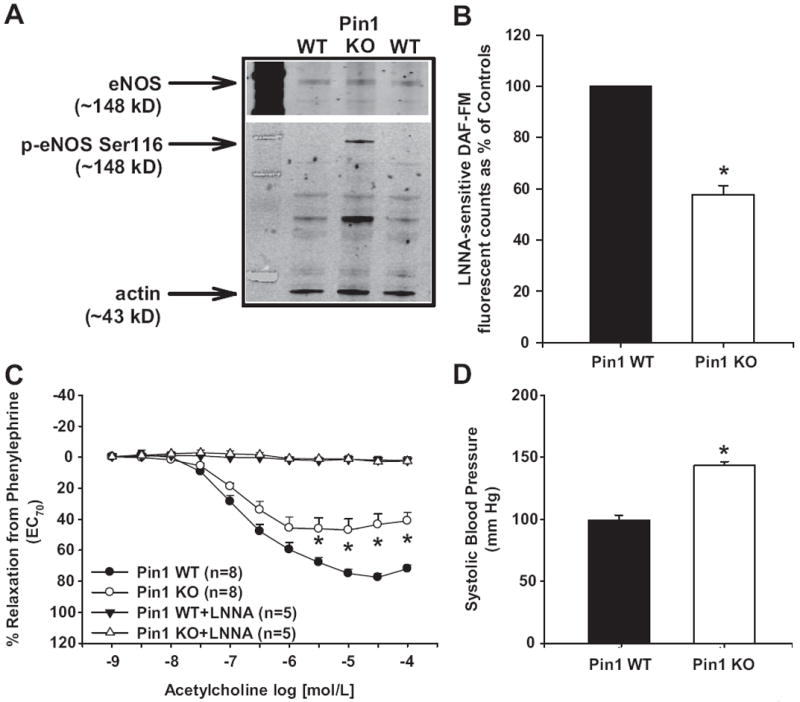
Effect of Pin1 gene deletion on vascular endothelial NO synthase (eNOS) function and systolic blood pressure in mice. A, Representative Western blot showing aortic eNOS Ser116 phosphorylation. B, Aortic NO production as measured by Nω-nitro-l-arginine (LNNA)–sensitive 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM) fluorescence. C, Aortic relaxation responses to acetylcholine after contraction with phenylephrine in the absence and presence of NO synthase (NOS) inhibition (LNNA). Relaxation to 100% represents a full return to a passive tension of 0.75 g. D, Systolic blood pressure in Pin1 wild-type (WT) and knockout (KO) mice. Results are expressed as mean±SEM (n>5 mice for each group). *P<0.05 vs Pin1 WT.
Discussion
The ubiquitously expressed peptidyl-prolyl isomerases play numerous roles in protein configuration and function.1,2 In addition to acting as molecular chaperones, isomerases also regulate ion channels, growth factor receptors, and protein-protein interactions. The relatively recent discovery of Pin1 has also shown that proline-directed protein phosphorylation can be regulated by this isomerase. Pin1-mediated cis-trans isomerization of p(Ser/Thr)-Pro residues has been shown to both increase or decrease protein stability, increase or inhibit enzyme activity, and promote dephosphorylation or phosphorylation of its targets.1,2 Although its role in transplant immunology, cancer, and Alzheimer disease has been better elucidated, there is little information on the role of Pin1 in vascular function and no information on blood pressure regulation.
Examination of the eNOS genetic sequence revealed a potential Pin1 binding motif at the inhibitory site (p)Ser116-Pro117, and Ruan et al16 were the first to show in endothelial cells that Pin1 interacts with eNOS when Ser116 is phosphorylated. We confirmed the Pin1-eNOS interaction in endothelial cells and demonstrate that pharmacological inhibition or siRNA-mediated knockdown of Pin1 increased phosphorylation of eNOS at Ser116 in endothelial cells and prevented agonist-induced dephosphorylation of eNOS Ser116 without altering eNOS protein expression. Uncoupling of eNOS after Pin1 inhibition/deletion is possible; however, given the known effects of eNOS Ser116 phosphorylation inhibiting eNOS activity, we doubt that this is sufficient to explain the decreased aortic NO production. In addition, our data demonstrate that Pin1 siRNA significantly decreases eNOS Ser1177 phosphorylation, a measure of eNOS activity, and significantly increases eNOS Thr495 phosphorylation, which inhibits eNOS activity. Together, the increased phosphorylation of Ser116 and Th495, known inhibitory sites, and decreased phosphorylation of Ser1177, a known stimulatory site, induced by Pin1 deficiency suggest that the primary mechanism by which NO production decreases is by reduced eNOS activity. Acute treatment of isolated mouse aortas with the Pin1-specific inhibitor juglone increased eNOS Ser116 phosphorylation, decreased NO production, and decreased endothelium-dependent relaxation responses, demonstrating a direct vascular effect. Juglone has been shown to be specific for Pin1 and does not affect other isomerases.19,20 Aortas from Pin1 KO mice also exhibited increased eNOS Ser116 phosphorylation, decreased NO production, and decreased endothelium-dependent relaxation responses. It remains to be determined whether these effects also occur in resistance vessels and whether microvascular endothelial dysfunction contributes to the hypertension caused by inhibition of Pin1.
Our results showing that Pin1 deficiency negatively affects NO production are in contrast to those of Ruan et al,16 who reported that inhibition of Pin1 with juglone in bovine aortic endothelial cells increased NO production. In addition, they found that Pin1 overexpression decreased NO production and expression of a dominant-negative form of Pin1 increased NO production in bovine aortic endothelial cells. However, eNOS Ser116 phosphorylation was not reported after the various modulations of Pin1, thus it is possible that the cellular manipulations may have altered NO production by other mechanisms. The only other examination of the interaction between Pin1 and NOS to our knowledge is a report by Liu et al,21 showing that Pin1 interacts directly with inducible NO synthase in murine aortic endothelial cells and that the WW domain of Pin1 is necessary for this interaction. Similar to the study by Ruan et al,16 they did not report the effects on Ser116 phosphorylation and reported that either short hairpin RNA or siRNA knockdown of Pin1 increased inducible NO synthase protein expression and NO production after treatment with lipopolysaccharide and interferon-γ. However, in the absence of lipopolysaccharide and interferon-γ, Pin1 knockdown did not increase NO production in endothelial cells. If Pin1 negatively affects eNOS and NO production, then it is curious why Pin1 is upregulated in prostate cancer cells.22 eNOS-derived NO has been shown to protect prostate cancer cells from tumor necrosis factor–related apoptosis-inducing ligand–induced apoptosis, suggesting that Pin1 up-regulation aids in the generation of NO from eNOS and tumor cell survival.23 Our data support this view that Pin1 positively affects eNOS by enabling Ser116 dephosphorylation and increasing NO generation. The differences between our findings and those of previous groups may be because of the cellular and tissue environments in which Pin1 is acting. External and internal signals may differentially regulate Pin1, which has been shown to promote phosphorylation of some proteins yet lead to dephosphorylation of other proteins.1 More studies are needed to determine how Pin1 might affect other eNOS interacting proteins.
The peptidyl-prolyl isomerases also play an important role in immunosuppression mediated by the drugs cyclosporin A, tacrolimus (FK506), and sirolimus (rapamycin). The binding of cyclophilin A by cyclosporin A, FKBP12/12.6 by tacrolimus, and FKBP12/12.6 by sirolimus forms a complex that then binds and inhibits calcineurin or mammalian target of rapamycin to prevent T-cell proliferation, which is independent of isomerase activity. However, these drugs promote hypertension and endothelial dysfunction, of which the binding of peptidyl-prolyl isomerases in off-target cells, such as endothelial cells, contributes greatly. Because of the detrimental cardiovascular effects elicited by these drugs, there is a need for the development of new immunosuppressants. Esnault et al6 reported recently that inhibition of Pin1 prevented allograft rejection in rats and decreased proinflammatory cytokine production and stability. In addition, they showed that lower concentrations of cyclosporin A combined with low concentrations of juglone were synergistic in preventing allograft rejection. Although these findings are very promising, blood pressure and endothelial function were not measured in transplanted animals treated with juglone. To determine whether in vivo Pin1 deficiency affects blood pressure and endothelial function, we treated control mice with the same concentration of juglone for 2 weeks and performed measures in Pin1 KO mice. Based on our in vitro work in both endothelial cells and isolated aortas showing increased eNOS Ser116 phosphorylation, decreased NO production, and decreased endothelium-dependent relaxation responses, we hypothesized that juglone treatment or Pin1 gene deletion would cause hypertension and endothelial dysfunction in mice. We saw a significant time-dependent increase in blood pressure in juglone-treated mice, as well as increased aortic eNOS Ser116 phosphorylation and decreased NO production and relaxation responses. Pin1 KO mice also exhibited increased aortic eNOS Ser116 phosphorylation, decreased relaxation responses and NO production, and hypertension. Taken together, these results demonstrate that Pin1 deficiency causes endothelial dysfunction and hypertension. Altered eNOS phosphorylation, especially increased eNOS Ser116 phosphorylation, after Pin1 deficiency contributes to these effects and is one pathway by which Pin1 inhibition negatively affects endothelial function and blood pressure regulation. Studies to examine other Pin1 targets that contribute to cardiovascular homeostasis are underway.
Perspectives
This is the first study demonstrating that Pin1 deficiency negatively affects eNOS Ser116 phosphorylation, NO production, endothelial function, and blood pressure regulation. The potential development of Pin1 inhibitors as antitumor and immunosuppressive agents should be target cell specific so as to avoid the adverse effects of endothelial dysfunction and hypertension, similar to that seen with current peptidyl-prolyl isomerase-binding immunosuppressive drugs.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by National Institutes of Health grant HL084299 and a Scott & White Development Grant (to B.M.M.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
Footnotes
Disclosures
None.
References
- 1.Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signaling and disease. Nat Rev Mol Cell Biol. 2007;8:904–916. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- 2.Lu KP, Finn G, Lee TH, Nicholson LK. Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol. 2007;3:619–629. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- 3.Lu KP. Prolyl isomerase Pin1 as a molecular target for cancer diagnostics and therapeutics. Cancer Cell. 2003;4:175–180. doi: 10.1016/s1535-6108(03)00218-6. [DOI] [PubMed] [Google Scholar]
- 4.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 5.Shen ZJ, Esnault S, Malter JS. The peptidyl-prolyl isomerase Pin1 regulates the stability of granulocyte-macrophage colony-stimulating factor mRNA in activated eosinophils. Nature Immunol. 2005;6:1280–1287. doi: 10.1038/ni1266. [DOI] [PubMed] [Google Scholar]
- 6.Esnault S, Braun RK, Shen ZJ, Xiang Z, Heninger E, Love RB, Sandor M, Malter JS. Pin1 modulates the Type 1 immune response. PLoS One. 2007;2:e226. doi: 10.1371/journal.pone.0000226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindenfeld J, Miller GG, Shakar SF, Zolty R, Lowes BD, Wolfel EE, Mestroni L, Page RL, Kobashigawa J. Drug therapy in the heart transplant recipient–Part II: Immunosuppressive drugs. Circulation. 2004;110:3858–3865. doi: 10.1161/01.CIR.0000150332.42276.69. [DOI] [PubMed] [Google Scholar]
- 8.Lindenfeld J, Page RL, Zolty R, Shakar SF, Levi M, Lowes BD, Wolfel EE, Miller GG. Drug therapy in the heart transplant recipient–part III: common medical problems. Circulation. 2005;111:113–117. doi: 10.1161/01.CIR.0000151609.60618.3C. [DOI] [PubMed] [Google Scholar]
- 9.Opelz G, Wujciak T, Ritz E. Association of chronic kidney graft failure with recipient blood pressure: Collaborative Transplant Study. Kidney Int. 1998;53:217–222. doi: 10.1046/j.1523-1755.1998.00744.x. [DOI] [PubMed] [Google Scholar]
- 10.Mange KC, Cizman B, Joffe M, Feldman HI. Arterial hypertension and renal allograft survival. JAMA. 2000;283:633–638. doi: 10.1001/jama.283.5.633. [DOI] [PubMed] [Google Scholar]
- 11.The US Multicenter FK506 Liver Study Group. A comparison of tacrolimus for immunosuppression in liver transplantation. N Engl J Med. 1994;331:1110–1115. doi: 10.1056/NEJM199410273311702. [DOI] [PubMed] [Google Scholar]
- 12.Long C, Cook LG, Hamilton SL, Wu GY, Mitchell BM. FK506 binding protein 12/12.6 depletion increases endothelial nitric oxide synthase threonine 495 phosphorylation and blood pressure. Hypertension. 2007;49:569–576. doi: 10.1161/01.HYP.0000257914.80918.72. [DOI] [PubMed] [Google Scholar]
- 13.Long C, Cook LG, Wu GY, Mitchell BM. Removal of FKBP12/12.6 from endothelial ryanodine receptors leads to an intracellular calcium leak and endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2007;27:1580–1586. doi: 10.1161/ATVBAHA.107.144808. [DOI] [PubMed] [Google Scholar]
- 14.Xin HB, Senbonmatsu T, Cheng DS, Wang YX, Copello JA, Ji GJ, Collier ML, Deng KY, Jeyakumar LH, Magnuson MA, Inagami T, Kotlikoff MI, Fleischer S. Oestrogen protects FKBP12.6 null mice from cardiac hypertrophy. Nature. 2002;416:334–337. doi: 10.1038/416334a. [DOI] [PubMed] [Google Scholar]
- 15.Fulton D, Gratton JP, Sessa WC. Post-translational control of endothelial nitric oxide synthase: why isn’t calcium/calmodulin enough? J Pharmacol Exp Ther. 2001;299:818–824. [PubMed] [Google Scholar]
- 16.Ruan L, Torres CM, Qian J, Chen F, Mintz JD, Stepp DW, Fulton D, Venema RC. Pin1 prolyl isomerase regulates endothelial nitric oxide synthase. Arterioscler Thromb Vasc Biol. 2011;31:392–398. doi: 10.1161/ATVBAHA.110.213181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atchison FW, Capel B, Means AR. Pin1 regulates the timing of mammalian primordial germ cell proliferation. Development. 2003;130:3579–3586. doi: 10.1242/dev.00584. [DOI] [PubMed] [Google Scholar]
- 18.Li C, Ruan L, Sood SG, Papapetropoulos A, Fulton D, Venema RC. Role of eNOS phosphorylation at Ser-116 in regulation of eNOS activity in endothelial cells. Vascul Pharmacol. 2007;47:257–264. doi: 10.1016/j.vph.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hennig L, Christner C, Kipping M, Schelbert B, Rucknagel KP, Grabley S, Kullertz G, Fischer G. Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry. 1998;37:5953–5960. doi: 10.1021/bi973162p. [DOI] [PubMed] [Google Scholar]
- 20.Siepe D, Jentsch S. Prolyl isomerase Pin1 acts as a switch to control the degree of substrate ubiquitylation. Nat Cell Biol. 2009;11:967–972. doi: 10.1038/ncb1908. [DOI] [PubMed] [Google Scholar]
- 21.Liu T, Huang Y, Likhotvorik RI, Keshvara L, Hoyt DG. Protein never in mitosis gene A interacting-1 (PIN1) regulates degradation of inducible nitric oxide synthase in endothelial cells. Am J Physiol Cell Physiol. 2008;295:C819–C827. doi: 10.1152/ajpcell.00366.2007. [DOI] [PubMed] [Google Scholar]
- 22.Ayala G, Wang D, Wulf G, Frolov A, Li R, Sowadski J, Wheeler TM, Lu KP, Bao L. The prolyl isomerase Pin1 is a novel prognostic marker in human prostate cancer. Cancer Res. 2003;63:6244–6251. [PubMed] [Google Scholar]
- 23.Tong X, Li H. eNOS protects prostate cancer cells from TRAIL-induced apoptosis. Cancer Lett. 2004;210:63–71. doi: 10.1016/j.canlet.2003.12.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.