

Abstract
Scribble (SCRIB) localizes to cell-cell junctions and regulates establishment of epithelial cell polarity. Loss of expression of SCRIB functions as a tumor suppressor in Drosophila and mammals, conversely, overexpression of SCRIB promotes epithelial differentiation in mammals. Here, we report that SCRIB is frequently amplified, mRNA over-expressed and protein is mislocalized from cell-cell junctions in human breast cancers. High levels of SCRIB mRNA are associated with poor clinical prognosis identifying an unexpected role for SCRIB in breast cancer. We find that, transgenic mice expressing a SCRIB mutant (Pro 305 to Leu (P305L)) that fails to localize to cell-cell junctions, under the control of the mouse mammary tumor virus long terminal repeat promoter, develop multifocal hyperplasia that progresses to highly pleomorphic and poorly differentiated tumors with basal characteristics. SCRIB interacts with PTEN and the expression of P305L, but not wild-type SCRIB, promotes an increase in PTEN levels in the cytosol. Overexpression of P305L, but not wild type SCRIB, activates the Akt/mTOR/S6K signaling pathway. Human breast tumors overexpressing SCRIB have high levels of S6K but do not harbor mutations in PTEN or PIK3CA, identifying SCRIB amplification as a mechanism of activating PI3K signaling in tumors without mutations in PIK3CA or PTEN. Thus, we demonstrate that high levels of mislocalized SCRIB functions as a neomorph to promote mammary tumorigenesis by affecting subcellular localization of PTEN and activating an Akt/mTOR/S6kinase signaling pathway.
Introduction
Scribble (SCRIB) was identified in a Drosophila screen for maternal effects mutants that displayed defects in epithelial polarization and morphogenesis. Loss of SCRIB that results in uncontrolled proliferation and tissue growth identifying it as tumor suppressor (1). SCRIB is a scaffold protein containing 16 N-terminal leucine rich repeat (LRR) domains and four PDZ (PSD-95/Discs-large/ZO-1) domains, conserved from Drosophila to humans (1). In mammals, loss of SCRIB induces dysplastic growth in vivo (2, 3) identifying loss of expression as a tumor suppressive mechanism. While the precise mechanism of tumor suppression is unclear, SCRIB is known to inhibit apoptosis in a β-PIX (PAK-interacting exchange factor β)/Rac/JNK pathway-dependent manner, and promote proliferation in a Ras/MAPK-dependent manner in mammary and prostate epithelia, respectively (2, 3). Conversely, overexpression of SCRIB in mammary epithelial cells promotes epithelial differentiation by suppressing expression of epithelial mesenchymal transition regulators in a Ras/MAPK dependent manner (4). Conditional deletion of SCRIB in the corneal epithelium decreases E-cadherin expression and promotes mesenchymal transition suggesting that SCRIB is required for maintaining epithelial cell identity (5). In addition, SCRIB also regulates the Hippo signaling pathway (6), and signal transducer and activator of transcription (STAT) (7). SCRIB interacts with the Akt phosphatase PHLPP1 (8), the planar cell polarity protein VANGL1 (9), and the neuronal nitric oxide synthase adaptor protein NOS1AP (10) to regulate cancer cell migration and axon morphogenesis.
In epithelial cells, SCRIB localizes to cell-cell junctions and mislocalization of SCRIB phenocopies a complete null phenotype in Drosophila (11) demonstrating that subcellular localization is critical for SCRIB function. Flies carrying the scrib 1 allele, in which a conserved leucine within the tenth LRR domain is mutated to glutamine (L223Q) display epithelial defects, including disrupted cell shape and multilayered organization. Interestingly, overexpression of SCRIBL223Q fails to rescue the scrib null phenotype, demonstrating the importance of basolateral localization for normal function. In mammals, a genetic screen for cortical defects in mice uncovered a novel SCRIB allele, encoding an isoleucine to lysine mutation within an LRR domain, displaying an open neural tube and disorganized and hyperplastic neuroepithelium (12). A specific point mutation within LRR13 (Prol 305 to Leu, P305L) of the human SCRIB protein (hSCRIB) has also been shown to abrogate membrane localization (13), disrupting recruitment of βPIX, and exocytosis. Consistently, overexpression of wild-type hSCRIB in MCF-10A mammary epithelial cells grown in 3D had no impact on morphogenesis, whereas expression of hSCRIBP305L displayed defective morphogenesis (2). These observations demonstrate that basolateral membrane localization is required for normal SCRIB function in both Drosophila and mammals.
In addition to changes in SCRIB gene expression, hSCRIB protein is mislocalized from cell-cell junctions in multiple human cancers, including breast, prostate and colon (2, 3). In prostate cancer, hSCRIB mislocalization is correlated with poor patient survival (3). Mislocalization of other polarity proteins, including LGL and DLG, has also been associated with cancer progression (14), suggesting that mislocalization of polarity proteins is likely to have important implications for cancer in addition to changes in gene expression levels. However, it is not known if mislocalization of SCRIB is a consequence or if mislocalized SCRIB has a causal role in the cancer process.
To investigate the effect of expressing Scribble that does not localize to cell-cell junctions, we generated a transgenic mouse expressing hSCRIBP305L within the mammary epithelium under the control of the mouse mammary tumor virus (MMTV) long terminal repeat promoter. We show that expression of hSCRIBP305L induces mammary tumors, providing direct evidence that SCRIB mislocalization can have a causal role during tumorigenesis.
Materials and Methods
Plasmids and reagents
The hSCRIBP305L vector was previously described (2). The T7-SCRIB construct was generated by subcloning the SCRIB cDNA into the pET vector (Novagen). The T7-tagged SCRIB PDZ mutants were generated by site-directed mutagenesis using the QuikChange kit (Stratagene). Four conserved residues in each PDZ domain (PDZ1-R733/L738/I740/I742); PDZ2-A871/L877/F879/I881; PDZ3 – R1009/L1014/L1016/I1018; PDZ4 – K1105/L1111/I1113/I1115) were mutated to alanine. The WT-PTEN and PTEN399 plasmids were kind gifts from Dr. Nicholas Tonks (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY).
The following antibodies were purchased from commercial sources: Scribble (Santa Cruz), β-Actin (Sigma), CK14 (Covance), E-cadherin (BD Biosciences), PTEN (Cell Signaling), GFP (Invitrogen), c-Met (Cell signaling), pAKT308 (Cell Signaling), pAKT473 (Cell Signaling), AKT (Cell Signaling), p-ERK1/2 (pT202, pY204) (Biosource), ERK2 (BD Transduction Laboratories), pTSC2 (T1462) (Cell Signaling), TSC2 (Cell Signaling), pPRAS40 (T246) (Cell Signaling), PRAS40 (Cell Signaling), pp70S6K (T389) (Cell Signaling), p70S6K (Cell Signaling), pS6 (S235/236) (Cell Signaling), S6 (Cell Signaling), Ki67 (Invitrogen), cleaved caspase 3 (Cell Signaling), Vimentin (Abcam), CD49f-PE (eBioscience), CD61-647 (BioLegend), CD31-Biotin (BD Biosciences), CD140a-Biotin (eBioscience), FITC-Streptavidin (AnaSpec), Biotin mouse lineage panel (BD Biosciences) and p-(S/T)-AKT Substrate (Cell Signaling). The T7 antibody was a kind gift from Dr. Adrian Krainer (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY) and the caspase-3 antibody was from Dr. Yuri Lazebnik (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY).
Transgenic mouse generation
Full length SCRIB cDNA (from pBluescript SCRIBibble P305L) containing a P305L mutation (2) was subcloned into the MMTV p206 vector. Mice were generated by pronuclear injection. Screening of founder mice for insertion was confirmed by PCR from genomic DNA using the MMTV forward primer (5′-GGCCCCGGCCCCAAGCTT-3′) and SCRIB reverse primers (5′-GCAGAACTTGATGCTCTC-3′) and (5′-ATCATCCTCCGTCTGGAAC-3′) with predicted 300 and 1200 bp PCR products.
Cell culture
MCF-10A cells were grown as previously described (2). Stable cell lines were generated by retroviral infection as previously described (15). HEK293 cells were cultured in DMEM supplemented with 10% FBS. HEK293 cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s suggested protocol. All cell lines were obtained from ATCC.
Immunoprecipitation and fractionation
All immunoprecipitations were carried out on cells lysed with 0.7% CHAPS buffer. 1mg protein was incubated with 1μg antibody and 10μl Protein G Sepharose beads (GE Healthcare) overnight at 4°C. The beads were washed three times in lysis buffer and the bound proteins were eluted with sample buffer. The samples were subjected to SDS-PAGE on 8% acrylamide gels, the separated proteins transferred to PVDF membranes, and the membranes probed with the appropriate antibodies. Cell fractionation was performed with the subcellular protein fractionation kit for cultured cells (Thermo Scientific) according to the manufacturer’s suggested protocol.
Immunobloting
Cell lysates were prepared in radioimmunoprecipitation assay buffer and normalized as previously described (2).
Histology and immunohistochemistry
Histological and immunohistochemical analysis of paraffin embedded mouse tissue was performed as previously described (2). Mammary glands were harvested and stained as whole mounts with carmine alum.
Microscopy and image analysis
All fluorescence images were collected using an Axiovert 200M equipped with an Apotome imaging system. Images were analyzed using Axiovision software.
Quantitative polymerase chain reaction
RNA was Trizol extracted from NDL and P305L tumors and reverse transcribed as previously described (2). Primer sets were as follows: CK5 5′-ACAGGAAGCTGCTGGAGGGC, 5′-GGTGGAGACAAATTTGACACTGG; CK6 5′-AGAGAGGGGTCGCATGAACT, 5′-TCATCTGTTAGACTGTCTGCCTT; CK14 5′- GATCTGCAGGAGGACATTGG, 5′- GGCCCACTGAGATCAAAGAC; CK18 5′- CTTGCTGGAGGATGGAGAAG, 5′- CTGGCTGGAGCAGGAGATTCG; GATA3 5′-AGCCACATCTCTCCCTTCAG, 5′-AGGGCTCTGCCTCTCTAACC; Actin 5′-TCTTGGGTATGGAATCCTGTGGCA, 5′-ACAGCACTGTGTTGGCATAGAGGT.
Flow cytometry analysis
Mammary glands were isolated from 30-week old mice, placed in wash buffer (DMEM/F12, 5% FBS), chopped into a paste, then digested with collagenase A. After a one-hour incubation at 37°C, cells were separated from fat by centrifugation at 600 × g for 10 minutes, and epithelial organoids were isolated by short spins at 450 × g. After a wash in PBS the organoids were resuspended in trypsin-EDTA and incubated at 37°C for 20 minutes. Wash buffer was then added and the cells passed through a 70μm filter. Single cells were resuspended in HBSS+ (HBSS, 2%FBS, 100mM Hepes) and prepared for FACS analysis. One million cells (in 200μL HBSS+) were incubated with primary antibodies (1:100) on ice for one hour. Cells were washed in HBSS+ and then incubated with secondary antibodies (1:100) on ice for 20 minutes. Cells were washed in HBSS+ and then filtered into 5mL flow tubes.
Gene expression analysis
SCRIB gene expression was analyzed in an available microarray dataset of 266 breast tumors with defined molecular subtypes (16). Comparison of levels of SCRIB expression in molecular subtype groups was performed using the Kruskal-Wallis nonparametric method with post-hoc Dunn’s test and plotted using Graphpad Prism version 6.0.
The relationship of SCRIB gene expression and overall survival was evaluated in an integrated multi-study breast cancer transcriptomic dataset using Kmplotter (17). Kaplan-Meier estimates of 10-year overall survival were calculated with upper quartile of SCRIB expression used to dichotomise data into high and low expression groups. Differences in survival curves were evaluated by log-rank test and cox-regression analysis.
For the TNBC subtype analysis, gene expression (GE) profiles were obtained from 21 publicly available data sets that contained 3,247 primary human breast cancers (18). Using a 2-component Gaussian mixture distribution model we estimated the posterior probability of negative expression (< 0.5) for ER, PR, and HER2 within each dataset and identified 587 triple negative breast cancer cases by mRNA. Raw microarray data were collectively RMA normalized, summarized and log-transformed using the combined raw GE from each data set. For genes containing multiple probes, the probe with the largest interquartile range across the samples was chosen to represent the gene. TNBC subtypes were assigned to each of the cases according to previously published methods (19). Gene expression for SCRIB (212556_at) was extracted and used for all comparisons.
Results
Genomic and gene expression changes in SCRIB associated with breast cancer
To investigate if the SCRIB gene is altered in human breast cancer, we analyzed the data from the Cancer Genome Atlas (TCGA), curated in cBioPortal (20). Among the 748 breast cancer samples analyzed, SCRIB was amplified in 116 tumors (15.6%), homozygous deleted in two (0.3%) and contained a somatic frame shift mutation in one (Fig. 1A). SCRIB mRNA was upregulated, in the absence of genomic amplification, in 68 samples (9.1%). These observations demonstrated that the SCRIB gene is frequently amplified or overexpressed and not lost or downregulated in breast tumors.
Figure 1. Genomic changes in SCRIB associated with human breast cancer.
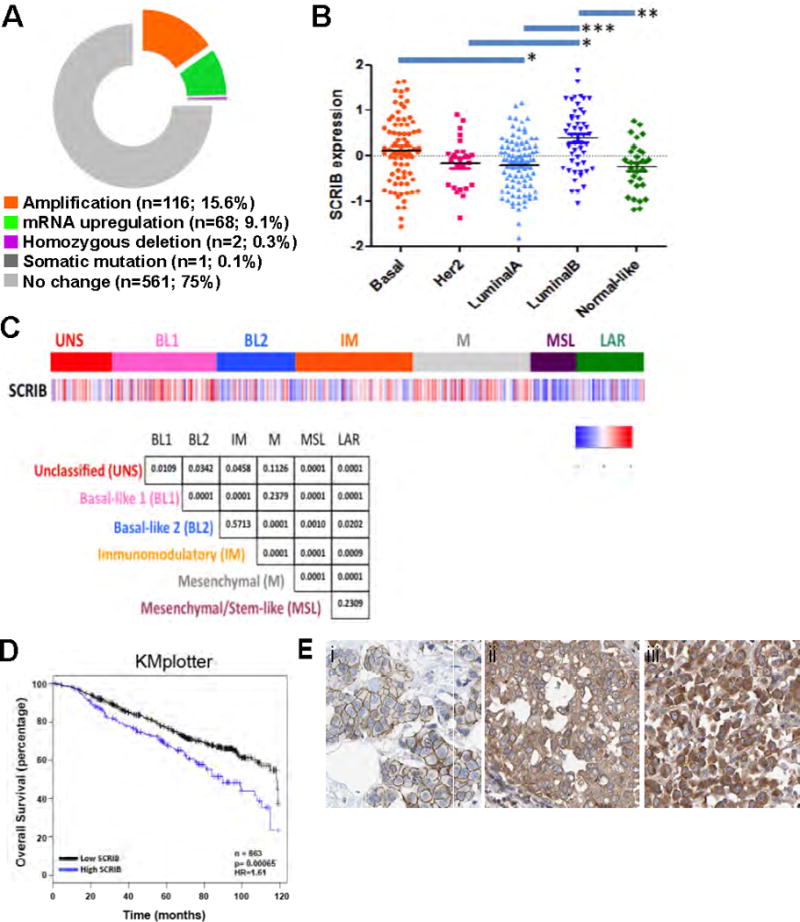
(A) The cBioPortal was used to detect genomic alterations and gene expression changes (mutations, copy number alterations, mRNA expression) in the SCRIB gene among 748 human breast tumors. (B) SCRIB gene expression in breast cancer molecular subtypes. Normalized and median-centered SCRIB gene expression levels were compared in breast cancer molecular subtypes as defined in a publically available dataset of breast tumors. SCRIB expression is expressed as log2 values. Mean SCRIB expression between tumors within each subtype is represented by a black line. Lines above the graph represent statistical significance of variance between subtypes as determined by Kruskall-Wallace non-parametric method. Asterisks represent p values; *** = p<0.001; ** = p<0.01; * = p<0.05; n.s. = p>0.05. (C) Analysis of SCRIB mRNA across TNBC subtypes. Gene expression (GE) profiles were obtained from 21 publicly available data sets that contained 3,247 primary human breast cancers. Gene expression for SCRIB (212556_at) was extracted and used for all comparisons. The lower panel displays p-values representing the difference between SCRIB expression in each subgroup (D) SCRIB expression stratifies overall survival in breast cancer. Kaplan-Meier estimates of overall survival were calculated in a publically available integrated multi-study breast cancer transcriptomic dataset through the KMplotter tool. The upper quartile of SCRIB expression used to dichotomise data into high versus low expression groups. Number of total patients are shown, significance of differences in survival curves are determined by log-rank p-values and hazard ratios are calculated by cox regression analysis. (E) (i) Human breast tumor with predominantly membrane SCRIB localization. (ii) Human breast tumor with membrane and cytosolic SCRIB localization. (iii) Human breast tumor with predominantly cytosolic SCRIB localization.
We next sought to determine if SCRIB overexpression was variable among breast cancer subtypes. In four publically available datasets (16, 21–23) using normalized and median-centered SCRIB gene expression (Fig. 1B, Supp. Figs. 1A,B,C), we observed that SCRIB expression was significantly higher in the basal and luminal B tumors, as compared with luminal A, Her2 and normal-like tumors. We have recently reported that triple negative breast cancer (TNBC) can be subdivided into six types that differ in cancer signaling pathway activation and drug sensitivity (18). Here we find that SCRIB was significantly upregulated in Basal-like 1 (BL1) and Mesenchymal (M), as compared with Basal-like 2 (BL2), Immunomodulatory (IM), Mesenchymal stem-like (MSL) and Luminal androgen receptor (LAR) (Fig. 1C, Supp. Figs. 1D,E) suggesting that high SCRIB levels are selected-for in certain breast cancer subtypes while not in others.
To determine the relationship between SCRIB upregulation and patient outcome, we evaluated Kaplan-Meier estimates of 10-year overall survival in a series of 663 breast tumors and find that high SCRIB expression predicts for worse overall survival (HR = 1.61, p < 0.0007; Fig 1D). These observations demonstrate that SCRIB overexpression is associated with the aggressive forms of breast cancers and with poor clinical prognosis.
Characterization of SCRIB localization in human breast tumors
Although it is counterintuitive that a tumor suppressor is amplified and overexpressed in breast cancer, previous studies in Drosophila (11) and in mammals (13), demonstrate that expression of a mislocalized form of SCRIB fails to rescue a loss of function phenotype, suggesting that, mislocalizing SCRIB phenocopies a complete loss of function. We analyzed the SCRIB immunohistochemistry reported in 20 breast cancer samples with moderate to strong SCRIB expression from the Human Protein Atlas (Fig. 1E) to determine if SCRIB is mislocalized in breast cancers. 11 of the 20 samples showed predominantly cytosolic SCRIB localization (Fig. 1Eiii), while the remaining nine showed a combination of membrane and cytosolic localization (Fig. 1Ei,ii). This is consistent with our previous observation that primary breast tumors show intra-tumor heterogeneity in Scribble localization (2). As SCRIB localization is mediated by the LRR domains, we sequenced the SCRIB LRR region in several human breast cell lines, including the non-transformed mammary epithelial cell line MCF-10A, as well as the cancer cell lines MDA-MB-231, T47D and MCF7. While the cancer cell lines all showed mislocalized SCRIB, no mutations within the Pro 305-containing LRR domain were found (data not shown). It is likely that SCRIB localization is dysregulated by multiple mechanisms in cancer cell lines and human tumors. While detailed investigation is needed to understand the mechanisms by which SCRIB is mislocalized in cancers, we sought to investigate if expression of mislocalized SCRIB plays a causal role for promoting cancer in the mammary gland. To accomplish this, we developed a transgenic mouse model expressing a SCRIB Pro 305 to Leu mutant, which fails to localize to cell membranes (13).
Generation and characterization of MMTV-hSCRIBP305L mice
To investigate the consequence of expressing a mislocalized mutant version of SCRIB in vivo, we generated transgenic mice expressing hSCRIBP305L in the mammary epithelium under the control of the mouse mammary tumor virus (MMTV) long terminal repeat promoter (Fig. 2A). Two distinct lines (P305L1, P305L2) were shown to transmit the transgene and used for further characterization. Both lines harbor 8 – 10 copies of the transgene as determined by quantitative polymerase chain reaction (QPCR) analysis (data not shown). Cell lysates from mammary glands isolated from FVB and hSCRIBP305L mice were analyzed for expression of SCRIB (Fig. 2B). As expected, hSCRIBP305L mice displayed higher levels of SCRIB protein in the mammary gland than control mice.
Figure 2. Generation and characterization of MMTV-hSCRIBP305L mice.
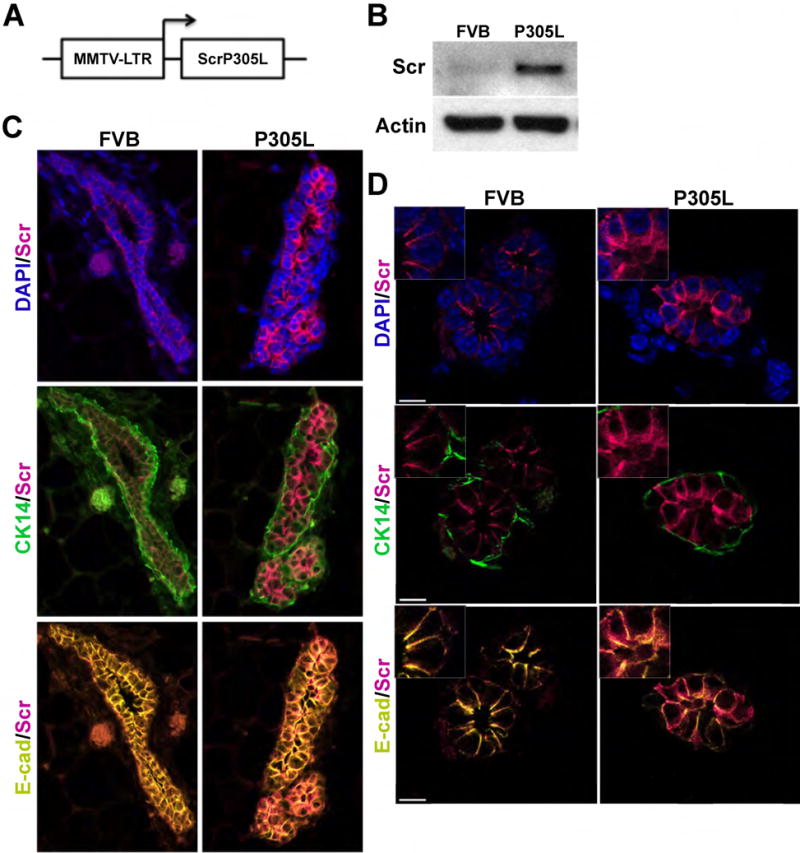
(A) Schematic of MMTV-hSCRIBP305L targeting construct. (B) Cell lysates from mammary glands isolated from FVB and hSCIBP305L mice were analyzed for expression of SCRIB and actin (loading control) by Western blot. (C) Expression of SCRIB, CK14 and E-cad was analyzed by IHC in mammary glands isolated from FVB and hSCRIBP305L mice. (D) Expression of SCRIB, CK14 and E-cad was analyzed by IHC in mammary glands isolated from FVB and hSCRIBP305L mice. Scale bars = 10μm.
To determine the spatial distribution of hSCRIBP305L expression in vivo, we analyzed localization of SCRIB, E-cadherin (E-cad), a luminal epithelial marker and cytokeratin 14 (CK14), a basal epithelial marker, by immunohistochemistry (IHC) (Fig. 2C,D). In adult virgin mammary glands of FVB mice, SCRIB co-localizes with E-cad to the basolateral surface of luminal epithelial cells (Fig. 2C,D, left panels). In hSCRIBP305L mice, SCRIB expression in the mammary gland was primarily expressed in E-cad positive luminal epithelial cells (Fig. 2C,D, right panels). However, within the luminal epithelial cells of hSCRIBP305L mice, SCRIB is localized throughout the cytosol (Fig. 2D).
hSCRIBP305L mice display mammary morphogenesis defects
To investigate the early changes in mammary morphogenesis in hSCRIBP305L mice, mammary glands were excised from virgin FVB and hSCRIBP305L mice at different ages and their branching patterns analyzed by whole mount carmine alum staining (Fig. 3A). In FVB mice, tertiary side branches were visible by 20 weeks and prominent at all subsequent time points analyzed (up to 50 weeks) (Fig. 3B). In contrast, hSCRIBP305L mice displayed fewer side branches, a difference first apparent by 30 weeks of age and sustained over subsequent time points (Fig. 3A, right panels). Quantitative analysis of hSCRIBP305L mice through 50 weeks of age demonstrated a significant impairment in tertiary side branching as compared with control mice (Fig. 3B). These results demonstrate that overexpression of mislocalized SCRIB disrupts mammary gland morphogenesis.
Figure 3. Mislocalization of SCRIB in the mouse mammary gland disrupts morphogenesis.
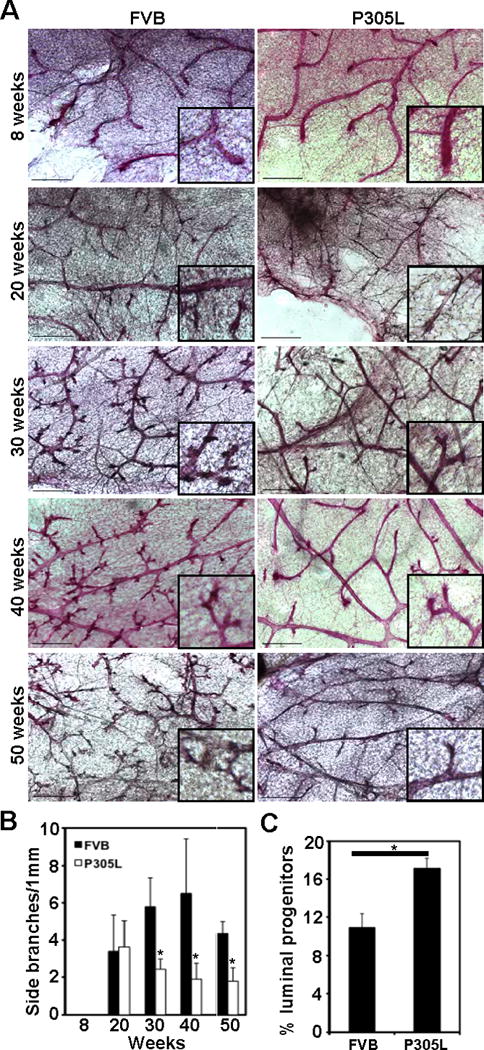
(A) Whole mount analysis of FVB and hSCRIBP305L mammary glands collected at the indicated time points. (B) Quantitative analysis of the results presented in (A), from three mice per condition. Error bars represent standard error of the mean (SEM). * denotes p<0.05. (C) FACS analysis of luminal progenitor cell (CD49flow/CD61+) content in mammary epithelial cells isolated from FVB and hSCRIBP305L mice. * denotes p<0.05.
The lack of tertiary structures within the adult virgin hSCRIBP305L mice led us to speculate that these glands may show defects in differentiation of luminal progenitor cells. To test this directly, we isolated epithelial organoids from FVB and hSCRIBP305L mammary glands, dissociated them into individual cells, and then performed flow cytometry analysis with antibodies for CD49f (integrin α6) and CD61 (integrin β3). In 30-week old virgin FVB mice, 10.9% of organoid-derived cells were found in the CD49flo/CD61+ population (Fig. 3C). This population accounted for 17.1% of cells in hSCRIBP305L mice, a 57% increase over FVB controls. Since mammary epithelial cells within the CD49flo/CD61+ population have been functionally characterized as luminal progenitors (24), our results suggest that expression of mislocalized SCRIB results in accumulation of luminal progenitor cells in the mammary gland.
hSCRIBP305L mice develop multifocal hyperplasia
Virgin hSCRIBP305L mice, but not FVB control mice, developed regions of hyperplastic growth, as determined by whole mount carmine alum-stained mammary glands (Fig. 4A) and H&E-stained sections (Fig. 4B). The hyperplastic nodules were widespread and filled the entire mammary fat pad (Fig. 4B). In addition, the hyperplasia phenotype was highly penetrant and observed in 8/10 transgenic mice older than one year of age (Fig. 4C). Histological analysis of the hyperplastic glands showed evidence of scattered inflammatory and squamous nodules and secretory lobuloalveolar hyperplasia (Supp. Fig 2A).
Figure 4. Mislocalization of SCRIB induces hyperplastic lesions in the mammary gland.
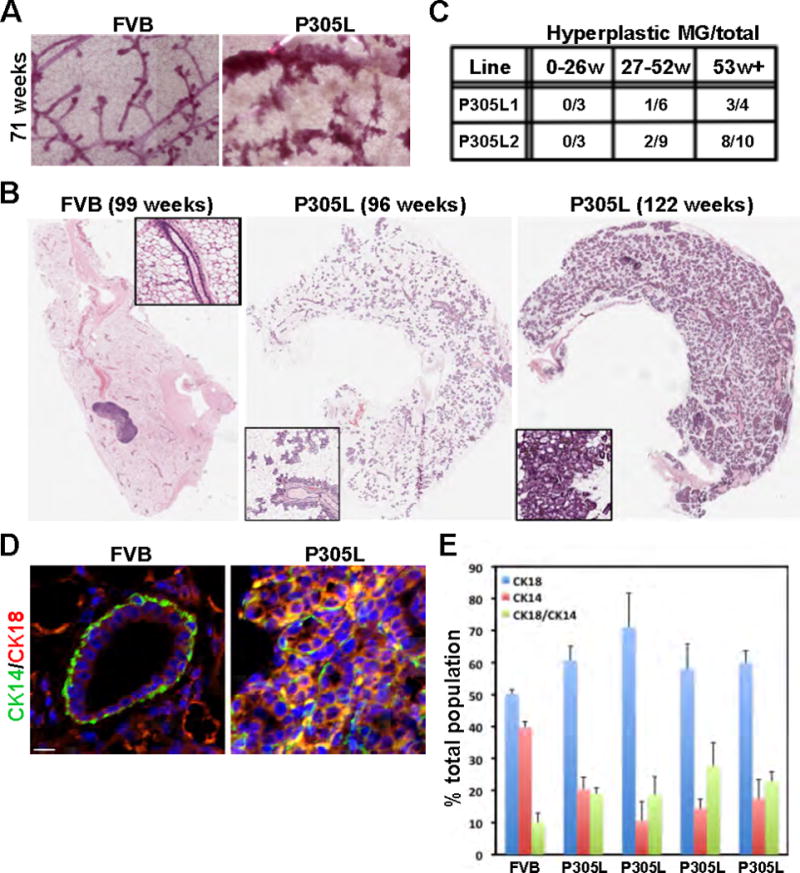
(A) Whole mount analysis of FVB and hSCRIBP305L mammary glands collected at the indicated time point. (B) H&E staining of mammary glands collected from FVB and P305L mice. (C) Analysis of hyperplastic phenotype in two distinct hSCRIBP305L lines. (D) Expression of CK18 and CK14 were analyzed by IHC in mammary glands isolated from FVB and hSCRIBP305L mice. Scale bars = 20μm. (E) Quantitative analysis of the results presented in (D).
To understand the differentiation status of the epithelial cells in hyperplastic glands, we performed IHC analysis for the luminal marker cytokeratin-18 (CK18) and the basal marker CK14 (Fig. 4D,E). In aged, virgin FVB mice, 50% of the ductal cells were CK18-positive, while 40% were CK14-positive and the remaining 10% were double positive (Fig. 4E). In contrast, hyperplastic hSCRIBP305L mammary glands showed increased levels of CK18-positive and double positive cells, with a decrease in the percentage of CK14-positive cells (Fig. 4D,E).
hSCRIBP305L mice develop spontaneous mammary tumors
To determine if the hyperplasia can progress to carcinoma, we allowed cohorts of virgin FVB and hSCRIBP305L mice to age, checking for tumor onset weekly by palpation. Within one and a half years of age, virgin hSCRIBP305L mice from two distinct lines developed focal mammary tumors (Fig. 5A). Penetrance of the tumor phenotype reached as high as 55% by two years of age in P305L2 demonstrating that overexpression of mislocalized SCRIB can initiate tumorigenesis in the mouse mammary gland.
Figure 5. Mislocalization of SCRIB generates tumors with basal characteristics.
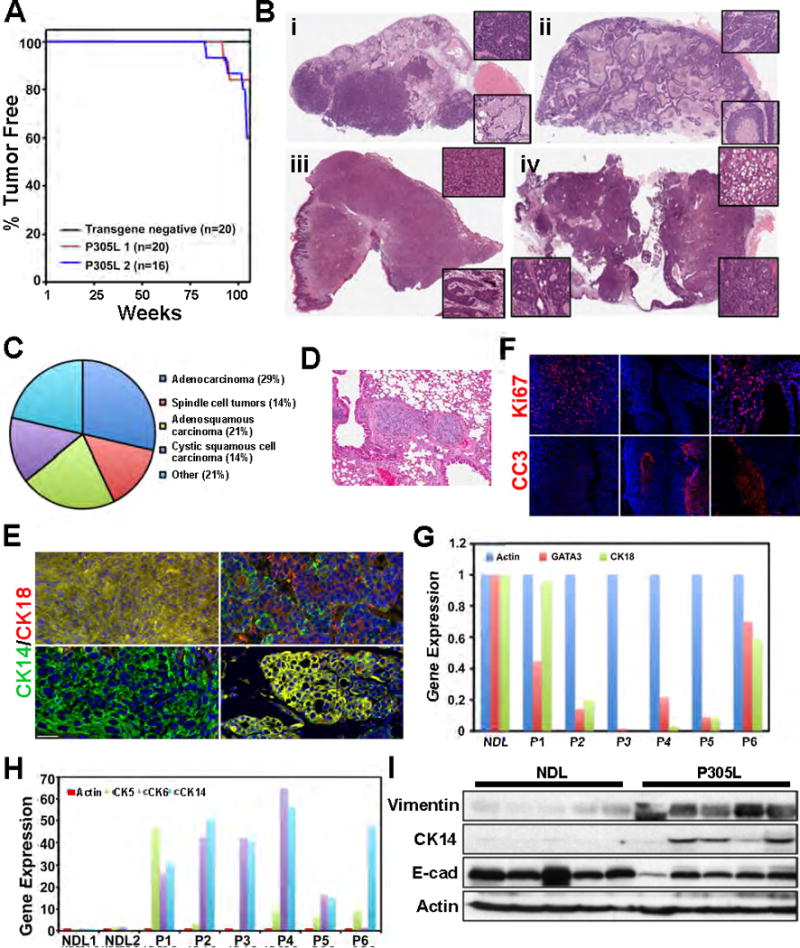
(A) Kaplan-Meier curve of tumor onset in FVB and two distinct hSCRIBP305L lines. (B) H&E staining of mammary tumors collected from hSCRIBP305L mice. (i) Glandular adenocarcinoma, (ii) microacinar tumor, (iii) spindle cell tumor, (iv) adenosquamous carcinoma. (C) Pathological classification of hSCRIBP305L tumors. (D) H&E staining of tumor emboli within lung from hSCRIBP305L tumor bearing mouse. (E) Expression of CK18 and CK14 were analyzed by IHC in mammary tumors isolated from hSCRIBP305L mice. Scale bars = 50μm. (F) Expression of Ki67 and cleaved caspase 3 was analyzed by IHC in mammary tumors isolated from hSCRIBP305L mice. Each image was taken from a different tumor. (G) Quantitative PCR analysis was utilized to examine expression of luminal markers (GATA3, CK18) on cDNA generated from NDL and hSCRIBP305L tumors. (H) Quantitative PCR analysis was utilized to examine expression of basal markers (CK5, CK6, CK14) on cDNA generated from NDL and hSCRIBP305L tumors. (I) Western blot analysis was utilized to examine expression of the luminal marker E-cad, and the basal markers Vimentin and CK14 in NDL and P305L tumors.
SCRIB mislocalization generates mammary tumors with high pathologic diversity
To characterize the hSCRIBP305L tumor phenotype we analyzed the pathological features of 14 hSCRIBP305L tumors [six from P305L1 and eight from P305L2] (Fig. 5B, Supp. Fig. 2B). The tumors were broadly classified as adenocarcinomas (29%), spindle cell tumors (14%), adenosquamous carcinoma (21%), and cystic squamous cell carcinoma (14%), while the remainder did not fall into simple classification (21%) (Fig. 5C). We observed both inter-tumor and intra-tumor heterogeneity for histological characteristics (Fig. 5B, insets). Interestingly, one of the 14 hSCRIBP305L tumor-bearing mice revealed intravascular tumor emboli in lung tissue (Fig. 5D), suggesting that the tumor cells are not benign and have the capacity to extravasate from the primary site.
To determine if the pathological heterogeneity was maintained at the level of lineage specific markers, we performed IHC for CK18 and CK14 in the hSCRIBP305L tumors (Fig. 5E, Supp. Fig. 3A). In concordance with the pathological analysis, no clear unifying pattern emerged. While several tumors were composed entirely of CK18/CK14 double positive cells, others were mostly CK18 or CK14 positive (Fig. 5E, Supp. Fig. 3A). Intra-tumoral heterogeneity was also observed, as different staining patterns were evident throughout individual tumors. In addition, the tumors displayed varying levels of proliferation and regions of extensive apoptosis, as measured by Ki67 and cleaved caspase-3 IHC, respectively (Fig. 5F, Supp. Fig. 3B).
SCRIB mislocalization promotes mammary tumors with basal characteristics
To define if the hSCRIBP305L tumors belong to tumor subtype, we probed the tumors for expression of luminal and basal markers by QPCR (Fig. 5G,H). As the hSCRIBP305L tumors possessed significant intra-tumor heterogeneity, we chose to utilize a candidate gene approach, measuring the expression of known luminal and basal markers, rather than whole genome expression arrays. Previous studies have demonstrated that mammary tumors induced by expression of a constitutively active ErbB2 (NDL) under the control of the MMTV promoter, expressed high levels of the luminal markers GATA3 and CK18 and are classified as luminal tumors (25). Therefore, we used RNA from NDL tumors as a reference for luminal-type gene expression. Although relative expression levels of GATA3 and CK18 varied among different hSCRIBP305L tumors, all samples analyzed (6/6) expressed decreased levels of these markers compared with NDL tumors (Fig. 5G). As expected, two NDL tumors analyzed expressed low levels of the basal markers cytokeratin-5 (CK5), cytokeratin-6 (CK6), and CK14 (Fig. 5H). In contrast, all six hSCRIBP305L tumors analyzed expressed dramatically enhanced levels (10- to 60-fold increase) of at least two of these basal markers. These observations were extended by Western blot analysis of protein lysates from five NDL and hSCRIBP305L tumors (Fig. 5I). NDL tumors expressed high levels of the luminal marker E-cadherin and low levels of the basal markers Vimentin and CK14. In contrast, hSCRIBP305L tumors expressed lower levels of E-cadherin and high levels of Vimentin and CK14. Taken together, our data provide evidence that SCRIB mislocalization induces histologically diverse mammary tumors with basal characteristics.
SCRIB mislocalization promotes activation of Akt
To gain mechanistic insight into pathways activated by mislocalized SCRIB, we generated MCF-10A cells stably expressing SCRIBWT or hSCRIBP305L (Fig. 6A). Overexpressed SCRIBWT localizes to the cell membrane and is concentrated at cell-cell junctions, whereas hSCRIBP305L is predominantly localized to the cytosol (Fig. 6B). Others and we have shown that knockdown of SCRIB regulates Rac-JNK and RAS-MAPK signaling pathways in mammary epithelial cells (2). We analyzed if expression of hSCRIBP305L affects epidermal growth factor (EGF)-induced activation of extracellular signal-related kinase (ERK), c-Jun N-terminal kinase (JNK), p38, and Akt. EGF-stimulation of MCF-10A cells promoted ERK phosphorylation within 2.5 minutes, which was maintained through 15 minutes (Fig. 6C). The kinetics and amplitude of EGF-stimulated ERK phosphorylation was similar in cells expressing either SCRIBWT or hSCRIBP305L. As with ERK, neither SCRIBWT nor hSCRIBP305L modified EGF-stimulated JNK or p38 kinase phosphorylation (data not shown).
Figure 6. Mislocalization of SCRIB enhances EGF-induced AKT activation.
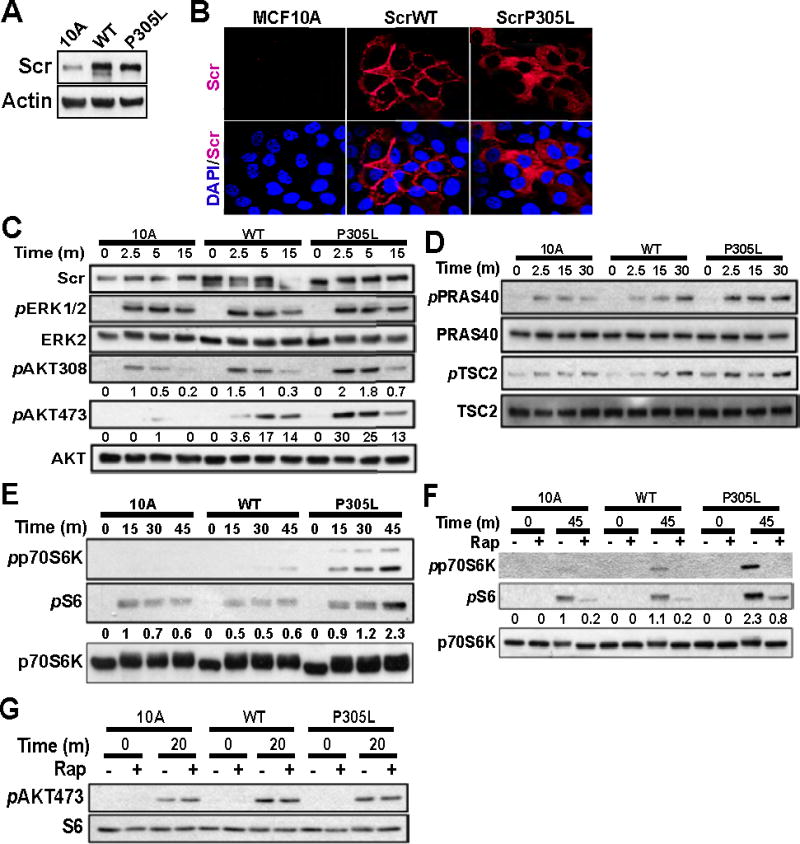
(A) Expression of SCRIB was analyzed in MCF-10A stably expressing SCRIBWT or hSCRIBP305L. (B) MCF-10A cells, either control or overexpressing SCRIBWT or hSCRIBP305L, were analyzed by IF for SCRIB localization. (C) MCF-10A cells, either control or overexpressing SCRIBWT or hSCRIBP305L, were grown in the absence of EGF overnight, then allowed to recover in EGF-containing medium for the indicated lengths of time. Protein lysates from each condition were analyzed for phosphorylation of AKT at Thr308 and Ser473, and ERK1/2 at Thr202/Tyr204. (D) MCF-10A cells, either control or overexpressing SCRIBWT or hSCRIBP305L, were grown in the absence of EGF overnight, then allowed to recover in EGF-containing medium for the indicated lengths of time. Protein lysates from each condition were analyzed for phosphorylation of TSC2 at Thr1462 and PRAS40 at Thr246. (E) MCF-10A cells, either control or overexpressing SCRIBWT or hSCRIBP305L, were grown in the absence of EGF overnight, then allowed to recover in EGF-containing medium for the indicated lengths of time. Protein lysates from each condition were analyzed for phosphorylation of p70S6K at Thr389 and S6 at Ser235/236. (F) MCF-10A cells, either control or overexpressing SCRIBWT or hSCRIBP305L, were grown in the absence of EGF overnight, pretreated with rapamycin for 30 minutes, then allowed to recover in EGF-containing medium for 45 minutes. Protein lysates from each condition were analyzed for phosphorylation of p70S6K at Thr389 and S6 at Ser235/236. (G) MCF-10A cells, either control or overexpressing SCRIBWT or hSCRIBP305L, were grown in the absence of EGF overnight, pretreated with rapamycin for 30 minutes, then allowed to recover in EGF-containing medium for 30 minutes. Protein lysates from each condition were analyzed for phosphorylation of Akt at Ser473.
We next focused on Akt, a potent oncogene in human breast cancer. Akt is phosphorylated within the activation loop at Thr308 by PDK1 (3-phosphoinositide dependent protein kinase-1) in response to insulin and other growth and survival factors (26). In MCF-10A cells grown in the absence of EGF, phosphorylation of Akt at Thr308 was undetectable (Fig. 6C). Addition of EGF to the cells led to an increase in Akt308 phosphorylation within 2.5 minutes, reduced after 5 minutes and returned to basal levels by 15 minutes. In MCF-10A cells expressing SCRIBWT, addition of EGF led to higher levels of Akt308 phosphorylation (Fig. 6C). MCF-10A cells expressing hSCRIBP305L displayed a marked increase in Akt308 phosphorylation as compared with either control or SCRIBWT cells (Fig. 6C).
Akt is also phosphorylated within the carboxyl-terminal hydrophobic motif at Ser473 by the serine/threonine kinase mammalian target of rapamycin complex 2 (mTORC2), within a rapamycin-insensitive protein complex (27). In parental MCF-10A cells, Akt is phosphorylated at Ser473 within 5 minutes of EGF stimulation (Fig. 6C). In cells expressing SCRIBWT, addition of EGF led to three-fold increase Ser473 phosphorylation within 2.5 minutes, with a peak at 5 minutes after stimulation. MCF-10A cells expressing hSCRIBP305L displayed a nine fold increase of Ser473 phosphorylation at 2.5 minutes that was maintained through 5 minutes and decreased by 15 minutes, suggesting that Akt kinase is activated early in both SCRIBWT and hSCRIBP305L expressing cells but sustained longer in cells expressing hSCRIBP305L.
Phosphorylation of Akt at 308 and 473 is associated with an increase in its kinase activity. To directly monitor changes in Akt activity, we probed the cell lysates using an Akt-substrate antibody that recognizes p-Ser/Thr within the Akt phosphorylation motif RxRxxS/T (Supp. Fig. 4A). In parental MCF-10A cells, an EGF stimulation-induced increase in substrate phosphorylation was detectable by 30 minutes. However, cells expressing hSCRIBP305L showed a greater increase in signal compared to those observed in parental or SCRIBWT expressing cells (Supp. Fig 4A), suggesting that expression of hSCRIBP305L activates Akt in MCF-10A cells.
To probe the effects of SCRIB mislocalization on specific downstream targets of Akt activation, we analyzed phosphorylation of PRAS40 (40kDa proline-rich protein) and TSC2 (tuberin) in EGF-stimulated MCF-10A cells (28, 29). Akt directly phosphorylates PRAS40 at Thr246, relieving the PRAS40 inhibitory effect on the mTOR1 complex (mTORC1) (30). In MCF-10A cells, EGF stimulation induced PRAS40 phosphorylation within 2.5 minutes, returning to near basal levels by 30 minutes (Fig. 6D). In MCF-10A cells expressing SCRIBWT, EGF induced PRAS40 phosphorylation with similar kinetics and intensity but greater at 30 minutes after EGF-stimulation. In MCF-10A cells expressing hSCRIBP305L, PRAS40 phosphorylation was both enhanced and sustained at all time points tested, as compared with control cells or those expressing SCRIBWT. Akt directly phosphorylates TSC2 at Thr1462, resulting in activation of mTORC1 (29). Expression of hSCRIBP305L enhanced EGF-induced phosphorylation of TSC2, as compared with MCF-10A cells and those expressing SCRIBWT (Fig. 6D).
As PRAS40 and TSC2 regulate the activity of mTOR, we investigated the effect of SCRIB mislocalization on mTOR-induced signaling events. mTOR phosphorylates p70S6 kinase (p70S6K) at several residues, resulting in increased kinase activity (31). Active p70S6K phosphorylates the ribosomal protein S6, enhancing translation of mRNA transcripts involved in cell cycle progression (32). MCF-10A cells expressing hSCRIBP305L displayed phosphorylation of p70S6K as early as 15 minutes following EGF stimulation, which was maintained through 45 minutes (Fig 6E). In contrast, in MCF10A cells, and those expressing SCRIBWT, EGF stimulation did not significantly increase p70S6K phosphorylation up to 45 minutes after stimulation. Consistently, MCF-10A cells expressing hSCRIBP305L displayed enhanced phosphorylation of S6, as compared with parental MCF-10A cells or MCF-10A cells expressing SCRIBWT (Fig. 6E). Therefore, SCRIB mislocalization specifically induces activation of Akt/mTOR pathway that leads to activation of downstream targets in mammary epithelial cells.
To investigate if the increase in phosphorylation of p70S6K and S6 requires mTORC1 activity, we inhibited mTOR with rapamycin. MCF-10A cells expressing hSCRIBP305L displayed enhanced phosphorylation of p70S6K and S6 45 minutes after EGF stimulation, as compared with MCF-10A cells or MCF-10A cells expressing SCRIBWT (Fig. 6F). In the presence of rapamycin, mTORC1-mediated phosphorylation of p70S6K and S6 proteins was inhibited (Fig 6F). As expected, mTORC2 mediated phosphorylation of Akt at 473 was not affected in rapamycin treated cells (Fig 6G). The above results, taken together, identify mTOR and Akt signaling as downstream effectors of expression of hSCRIBP305L.
SCRIB interacts with PTEN in a PDZ domain-dependent manner
To define the mechanism by which SCRIB mislocalization activates Akt, we investigated the hypothesis that SCRIB interacts with phosphatase and tensin homolog (PTEN), a tumor suppressor frequently mutated or deleted in human cancers, including those of the breast (33, 34). PTEN is a phosphatase that dephosphorylates phosphatidylinositol (3,4,5)-trisphosphate (PIP3), a second messenger generated by phosphoinositide 3-kinase (PI3K) (35). Loss of PTEN enhances PIP3-mediated signaling, including phosphorylation and activation of Akt (36). PTEN is localized to cell-cell junctions and binds through its C-terminus to the PDZ domain of the basolateral polarity protein DLG1 as well as the PDZ2 and PDZ3 domains of the apical polarity determinant PARD3. Furthermore, knockdown of PTEN in mammary epithelial cells grown in 3D culture results in hyperproliferation and disrupted acinar organization (37). Finally, disruption of PTEN membrane localization, through removal of the C-terminal PDZ binding site, has been shown to disrupt its ability to suppress Akt activation.
To investigate a possible interaction between SCRIB and PTEN, we utilized a SCRIB antibody to immunoprecipitate (IP) endogenous SCRIB from luminal epithelial Eph4 cells and analyzed PTEN interaction by immunoblot (Fig. 7A). Whereas no PTEN was pulled down in the control IP (IgG), endogenous SCRIB was able to pull down endogenous PTEN.
Figure 7. SCRIB is found in a protein complex with PTEN.
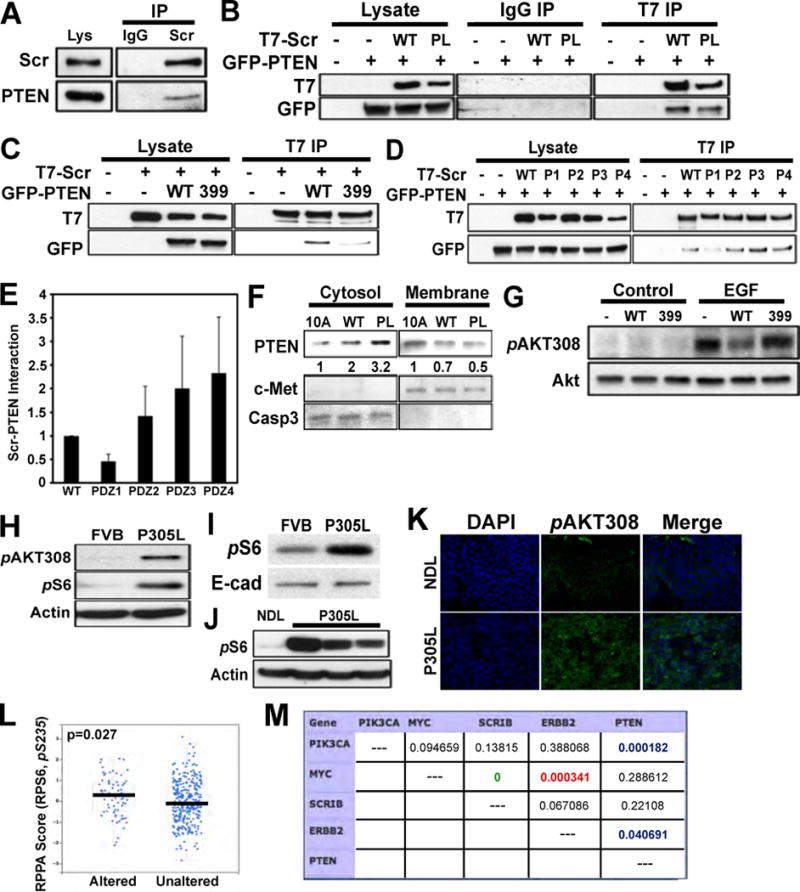
(A) Control (IgG) and SCRIB immunoprecipitations were performed on Eph4 protein lysates. (B) HEK293 cells were transfected with an empty vector, T7-tagged SCRIBWT (WT), T7-tagged hSCRIBP305L (PL), and GFP-WT-PTEN. Immunoprecipitations were carried out from total cell lysates utilizing the T7 antibody or control IgG. (C) HEK293 cells were transfected with an empty vector, T7-tagged SCRIB, GFP-WT-PTEN or GFP-PTEN399. Immunoprecipitations were carried out from total cell lysates utilizing the T7 antibody. (D) HEK293 cells were transfected with an empty vector, T7-tagged SCRIB or T7-tagged SCRIB PDZ mutants, and GFP-WT-PTEN. Immunoprecipitations were carried out from total cell lysates utilizing the T7 antibody. (E) Quantification of results presented in (D), from three independent experiments. (F) MCF-10A cells, either control or overexpressing SCRIBWT or hSCRIBP305L, were fractionated to separate the membrane and cytosol fractions. Protein lysates from each condition were analyzed for PTEN, caspase 3 (cytosolic marker) and c-Met (membrane marker). (G) MCF-10A cells were transfected with an empty vector, WT-PTEN (WT) or PTEN-399 (399), grown in the absence of EGF overnight, then allowed to recover in EGF-containing medium for 10 minutes. Protein lysates from each condition were analyzed for phosphorylation of AKT at Thr308. (H) Phosphorylation of Akt and S6 was analyzed by WB in mammary glands isolated from FVB and hSCRIBP305L mice. (I) Phosphorylation of S6 was analyzed by WB in mammary glands isolated from FVB and hSCRIBP305L mice. (J) Western blot analysis was utilized to examine phosphorylation of S6 in NDL and P305L tumors. (K) Phosphorylation of Akt was analyzed by IHC in NDL and P305L tumors. (L) Analysis of phosphorylation of S6 at Ser235 in human tumor samples with amplification of SCRIB. (M) Mutual exclusivity calculations of alterations in selected genes in human breast cancer. p>0.05 for pairs outlined in red. Orange=strong tendency towards co-occurrence (odds ratio>10); Yellow=tendency towards co-occurrence (2<odds ratio<10); Blue=strong tendency towards mutual exclusivity (0<odds ratio<0.1); White=no association (0.5<odds ratio<2).
To determine if the hSCRIBP305L mutation altered PTEN binding, we immunoprecipitated T7-epitope tagged SCRIB from HEK293 cells transfected with T7-SCRIBWT, T7-hSCRIBP305L, GFP-PTEN, or an empty vector (Fig. 7B). Importantly, both SCRIBWT and hSCRIBP305L were able to pull down PTEN. Next, we utilized GFP-tagged wild type PTEN (WT) and a PTEN mutant lacking the PDZ binding motif (PTEN-399) to determine if the SCRIB-PTEN interaction is PDZ-dependent. T7-SCRIB was immunoprecipitated from HEK293 cells transfected with T7-SCRIB, WT-PTEN, PTEN-399, or an empty vector (Fig. 7C). T7-SCRIB was able to pull down WT-PTEN, but significantly less PTEN-399, demonstrating that the C-terminal PDZ binding site of PTEN is critical for the SCRIB/PTEN interaction. To determine which of the SCRIB PDZ domains was required for PTEN binding, we generated four T7-SCRIB mutants (see methods section for details), each with a mutation in only one of the four PDZ domains (P1, P2, P3, P4). T7-SCRIB, either WT or PDZ mutant, was immunoprecipitated from HEK293 cells co-transfected with GFP-PTEN, or an empty vector (Fig. 7D). Whereas GFP-PTEN was able to bind T7-WTSCRIB, or PDZ mutants P2, P3 and P4, mutation of the first PDZ domain of SCRIB significantly reduced the SCRIB/PTEN interaction (Fig. 7D,E). Taken together, our results suggest that SCRIB and PTEN are present in a protein complex requiring the PDZ-binding motif of PTEN and the first PDZ domain of SCRIB.
To determine the significance of the SCRIB/PTEN interaction, we analyzed the localization of endogenous PTEN in MCF-10A cells expressing SCRIBWT and hSCRIBP305L. Cell fractionation was performed, effectively separating the cytosolic fraction from the membrane (Fig. 7F). MCF-10A cells expressing hSCRIBP305L had a more than three-fold increase in cytosolic PTEN than the parental or WTSCRIB expressing cells (Fig. 7F, left panels). We then analyzed the effect of disrupting the SCRIB/PTEN interaction on EGF-induced signaling in MCF-10A cells. MCF-10A cells, either expressing an empty vector, WT-PTEN or PTEN-399, were probed for Akt308 phosphorylation in response to EGF (Fig. 7G). Expression of WT-PTEN, but not PTEN-399, attenuated EGF-induced phosphorylation of Akt (Fig. 7G). These results suggest that PTEN localization modulates EGF-induced activation of Akt.
hSCRIBP305L mammary tumors and human breast cancers with amplified SCRIB show activation of S6 kinase but no correlation with mutations in PTEN or PIK3CA
To determine if expression of mislocalized SCRIB activates Akt/mTOR signaling in vivo, we analyzed FVB and hSCRIBP305L mammary glands for phosphorylation of S6 by IHC (Supp. Fig. 4B). At the eight-week time point, no change was seen in the intensity of pS6 staining between FVB and hSCRIBP305L mice. However, ductal structures from 20-week old hSCRIBP305L mice displayed markedly higher pS6 staining than controls, and maintained this enhanced signal through 50 weeks. Western blot analysis of mammary glands isolated from FVB and hSCRIBP305L mice at 30 weeks (Fig. 7H), and hyperplastic glands at 75 weeks (Fig. 7I) showed higher levels of phospho-S6 in hSCRIBP305L glands. Finally, we evaluated Akt pathway activation in primary tumors through analysis of phospho-S6 levels (Fig. 7J). hSCRIBP305L tumors displayed enhanced phosphorylation of S6, as compared with NDL controls. In addition, IHC analysis showed enhanced phospho-Akt308 in hSCRIBP305L tumors (Fig. 7K). To determine if human breast tumors with SCRIB amplification display enhanced Akt pathway activation, we analyzed RPPA data from the TCGA dataset (20). Similar to our findings with mouse tumors induced by expressing mislocalized SCRIB, tumors with SCRIB amplification had higher levels of phosphorylated S6 as compared to non-amplified tumors (Fig. 7L). Interestingly, there was no correlation between amplification of SCRIB and mutations in PTEN (P=0.2, n=760) and PIK3CA (p=0.3, n=482), regulators of the PI3K signaling pathway, suggesting that SCRIB amplification is mutually exclusive with alternate mechanisms of activating the PI3K signaling pathway (Fig. 7M).
We have previously shown that expression of hSCRIBP305L cooperates with Myc during transformation of mammary epithelial cells (2). Consistent with this observation, there was a strong correlation between SCRIB and MYC co-amplification in human breast cancer (p=0, n=760) (Fig. 7M). By contrast, SCRIB amplification did not correlate with HER2 amplification in human breast cancer (p=0.06, n=760). Consistently, we find that transgenic mice co-expressing hSCRIBP305L and NEU/HER2 under the control of MMTV promoter did not show any change in latency of tumor onset (Supp. Fig. 4C), demonstrating lack of cooperation between hSCRIBP305L and HER2 during mouse mammary tumorigenesis. To gain a better understanding of possible genetic changes associated with SCRIB amplification, we compared the genetic interaction between SCRIB and 40 breast cancer driver genes (38) using the TCGA data archived in the cBioPortal analysis platform. SCRIB amplification correlated with alterations in TP53, BRCA1, BRCA2, FGFR1, EGFR, MAP3K1 and ZNF217, demonstrating a correlation between SCRIB amplification and genes frequently altered in aggressive forms of breast cancer. Taken together our observations demonstrate that amplification or overexpression of SCRIB is associated with aggressive subtypes of human breast cancer and that mice overexpressing SCRIB activate a PI3K/S6K pathway and induce formation of mammary tumors with the basal subtype.
Discussion
Herein, we identify an unexpected role for the polarity protein Scribble in the pathogenesis of cancer. We demonstrate that overexpression of a mislocalized form of SCRIB functions as a neomorph by interacting with another tumor suppressor, PTEN, and activating an mTORC1/Akt/S6K signaling pathway in culture and in vivo.
Others and we have previously reported that downregulation (or loss) of SCRIB expression functions as a tumor suppressor by inhibiting cell death or inducing proliferation (2, 3). In addition, Scribble is required for directional migration, promotes epithelial identity and suppresses epithelial to mesenchymal transition in mammary and corneal epithelial cells (4, 5, 39). Together, these observations suggest a working model that SCRIB normally promotes epithelial differentiation and loss of expression results in mesenchymal transition and tumorigenesis. As we report here, large-scale cancer genome analysis identifies SCRIB gene amplification and overexpression as a frequent and clinically relevant event in breast cancer and not deletion or decrease in gene expression, raising new questions on how SCRIB may regulate the biology of cancer cells. We report that mislocalization of SCRIB results in acquisition of a new function (neomorph), it mislocalizes PTEN and activates a mTORC1/Akt/S6K pathway and thus results in a gain of function. It is likely that the gain of function associated with hSCRIBP305L drives tumorigenesis and contributes to the invasive behavior observed in mammary tumors.
It is not clear how SCRIB localization would be altered in breast cancer. Our sequencing efforts and those reported by TCGA do not show evidence of frequent mutations in SCRIB suggesting that changes in Scribble localization must involve alternate mechanisms. Trafficking of SCRIB may be impaired by defects in the intracellular trafficking machinery. In support of this hypothesis, proper vesicle trafficking is critical for maintenance of epithelial polarity (40). Phosphorylation of serine residues in the carboxy terminal domain of SCRIB induces mislocalization to the cytosol (41). Furthermore, alterations in SCRIB binding partners, including E-cadherin, may disrupt SCRIB membrane localization (42). Therefore, multiple distinct mechanisms may account for the high prevalence of SCRIB mislocalization observed human tumors. Together, these observations begin to explain why high levels of SCRIB are frequently selected-for in breast cancers.
Our results showing that SCRIB mislocalization enhances Akt signaling and results in mammary morphogenesis defects are consistent with a mouse model of Akt pathway activation (43). Knockdown of PTEN in normal mammary epithelial cells implanted into humanized mouse fat pads resulted in disorganized, hyperplastic lesions (44). These outgrowths displayed increased basal markers (smooth muscle actin, CK5, CK6) and decreased luminal markers (estrogen receptor, CK18), similar to our findings with mislocalization of SCRIB. Furthermore, a mouse model in which direct activation of PI3K was accomplished by recruitment of p110α to the membrane resulted in alveolar hyperplasia and low penetrance tumorigenesis (45). Mice in which PTEN was deleted within the mammary epithelium developed pathologically heterogeneous tumors with high expression of basal markers (46). Therefore, our results are consistent with a model in which aberrant Akt activation within the mouse mammary gland results in basal-like tumors.
The observation that SCRIB mislocalization leads to basal tumors and that SCRIB mRNA is higher in basal-like and mesenchymal human breast tumors raises interesting clinical perspectives. A recent analysis, combining mutation, mRNA expression and protein levels, determined that the PI3K/Akt pathway was elevated in basal-like cancers as compared with luminal or HER2+ (47). Despite high pathway activity, basal-like cancers rarely display PIK3CA (PI3K catalytic domain) mutations (48). Our observations raise that possibility that mislocalization of SCRIB, and other PTEN interacting polarity proteins, play a critical role in modulation of Akt/mTOR signaling breast cancer. Thus, understanding the mechanisms that regulate subcellular localization of SCRIB, and possibly other cell polarity proteins can provide novel insights into cancer biology.
Supplementary Material
Acknowledgments
We would like to thank the members of the Muthuswamy and Tonks laboratories for helpful discussions and Pamela Moody from the CSHL Flow Cytometry Shared resource for FACS assistance. This work was supported by a postdoctoral fellowship (PF-11-026-01-CSM) from the American Cancer Society to MF; CA098830 and CA105388 grants from NCI; BC075024, Era of Hope Scholar award from DOD Breast Cancer Research Program; Rita Allen Foundation; FACT Foundation, Lee K Margaret Lau Chair for breast cancer research and Campbell Family Institute for breast cancer research to SKM. This work was also funded in part by the Ontario Ministry of Health and Long Term Care. The views expressed do not necessarily reflect those of the OMOHLTC.
Footnotes
The authors declare no conflict of interest.
References
- 1.Bilder D, Perrimon N. Localization of apical epithelial determinants by the basolateral PDZ protein Scribble. Nature. 2000;403(6770):676–80. doi: 10.1038/35001108. [DOI] [PubMed] [Google Scholar]
- 2.Zhan L, Rosenberg A, Bergami KC, Yu M, Xuan Z, Jaffe AB, Allred C, Muthuswamy SK. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell. 2008;135(5):865–78. doi: 10.1016/j.cell.2008.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pearson HB, Perez-Mancera PA, Dow LE, Ryan A, Tennstedt P, Bogani D, Elsum I, Greenfield A, Tuveson DA, Simon R, et al. SCRIB expression is deregulated in human prostate cancer, and its deficiency in mice promotes prostate neoplasia. J Clin Invest. 2011;121(11):4257–67. doi: 10.1172/JCI58509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elsum IA, Martin C, Humbert PO. Scribble regulates an EMT polarity pathway through modulation of MAPK-ERK signaling to mediate junction formation. J Cell Sci. 2013;126(Pt 17):3990–9. doi: 10.1242/jcs.129387. [DOI] [PubMed] [Google Scholar]
- 5.Yamben IF, Rachel RA, Shatadal S, Copeland NG, Jenkins NA, Warming S, Griep AE. Scrib is required for epithelial cell identity and prevents epithelial to mesenchymal transition in the mouse. Dev Biol. 2013 doi: 10.1016/j.ydbio.2013.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Skouloudaki K, Puetz M, Simons M, Courbard JR, Boehlke C, Hartleben B, Engel C, Moeller MJ, Englert C, Bollig F, et al. Scribble participates in Hippo signaling and is required for normal zebrafish pronephros development. Proc Natl Acad Sci U S A. 2009;106(21):8579–84. doi: 10.1073/pnas.0811691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Werme K, Wigerius M, Johansson M. Tick-borne encephalitis virus NS5 associates with membrane protein scribble and impairs interferon-stimulated JAK-STAT signalling. Cellular microbiology. 2008;10(3):696–712. doi: 10.1111/j.1462-5822.2007.01076.x. [DOI] [PubMed] [Google Scholar]
- 8.Li X, Yang H, Liu J, Schmidt MD, Gao T. Scribble-mediated membrane targeting of PHLPP1 is required for its negative regulation of Akt. EMBO Rep. 2011;12(8):818–24. doi: 10.1038/embor.2011.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anastas JN, Biechele TL, Robitaille M, Muster J, Allison KH, Angers S, Moon RT. A protein complex of SCRIB, NOS1AP and VANGL1 regulates cell polarity and migration, and is associated with breast cancer progression. Oncogene. 2012;31(32):3696–708. doi: 10.1038/onc.2011.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richier L, Williton K, Clattenburg L, Colwill K, O’Brien M, Tsang C, Kolar A, Zinck N, Metalnikov P, Trimble WS, et al. NOS1AP associates with Scribble and regulates dendritic spine development. J Neurosci. 2010;30(13):4796–805. doi: 10.1523/JNEUROSCI.3726-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeitler J, Hsu CP, Dionne H, Bilder D. Domains controlling cell polarity and proliferation in the Drosophila tumor suppressor Scribble. J Cell Biol. 2004;167(6):1137–46. doi: 10.1083/jcb.200407158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zarbalis K, May SR, Shen Y, Ekker M, Rubenstein JL, Peterson AS. A focused and efficient genetic screening strategy in the mouse: identification of mutations that disrupt cortical development. Plos Biol. 2004;2(8):E219. doi: 10.1371/journal.pbio.0020219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Audebert S, Navarro C, Nourry C, Chasserot-Golaz S, Lecine P, Bellaiche Y, Dupont JL, Premont RT, Sempere C, Strub JM, et al. Mammalian Scribble forms a tight complex with the betaPIX exchange factor. Curr Biol. 2004;14(11):987–95. doi: 10.1016/j.cub.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 14.Huang L, Muthuswamy SK. Polarity protein alterations in carcinoma: a focus on emerging roles for polarity regulators. Current opinion in genetics & development. 2010;20(1):41–50. doi: 10.1016/j.gde.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30(3):256–68. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 16.Sabatier R, Finetti P, Cervera N, Lambaudie E, Esterni B, Mamessier E, Tallet A, Chabannon C, Extra JM, Jacquemier J, et al. A gene expression signature identifies two prognostic subgroups of basal breast cancer. Breast cancer research and treatment. 2011;126(2):407–20. doi: 10.1007/s10549-010-0897-9. [DOI] [PubMed] [Google Scholar]
- 17.Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast cancer research and treatment. 2010;123(3):725–31. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- 18.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–67. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen X, Li J, Gray WH, Lehmann BD, Bauer JA, Shyr Y, Pietenpol JA. TNBCtype: A Subtyping Tool for Triple-Negative Breast Cancer. Cancer informatics. 2012;11:147–56. doi: 10.4137/CIN.S9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jonsson G, Staaf J, Vallon-Christersson J, Ringner M, Gruvberger-Saal SK, Saal LH, Holm K, Hegardt C, Arason A, Fagerholm R, et al. The retinoblastoma gene undergoes rearrangements in BRCA1-deficient basal-like breast cancer. Cancer Res. 2012;72(16):4028–36. doi: 10.1158/0008-5472.CAN-12-0097. [DOI] [PubMed] [Google Scholar]
- 22.Pawitan Y, Bjohle J, Amler L, Borg AL, Egyhazi S, Hall P, Han X, Holmberg L, Huang F, Klaar S, et al. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 2005;7(6):R953–64. doi: 10.1186/bcr1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, Lapuk A, Neve RM, Qian Z, Ryder T, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer cell. 2006;10(6):529–41. doi: 10.1016/j.ccr.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 24.Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zurrer-Hardi U, Bell G, et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012;148(5):1015–28. doi: 10.1016/j.cell.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson KS, Roberts H, Leek R, Harris AL, Geradts J. Differential gene expression patterns in HER2/neu-positive and -negative breast cancer cell lines and tissues. The American journal of pathology. 2002;161(4):1171–85. doi: 10.1016/S0002-9440(10)64394-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7(4):261–9. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 27.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 28.Kovacina KS, Park GY, Bae SS, Guzzetta AW, Schaefer E, Birnbaum MJ, Roth RA. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem. 2003;278(12):10189–94. doi: 10.1074/jbc.M210837200. [DOI] [PubMed] [Google Scholar]
- 29.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Molecular cell. 2002;10(1):151–62. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 30.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Molecular cell. 2007;25(6):903–15. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 31.Pullen N, Dennis PB, Andjelkovic M, Dufner A, Kozma SC, Hemmings BA, Thomas G. Phosphorylation and activation of p70s6k by PDK1. Science. 1998;279(5351):707–10. doi: 10.1126/science.279.5351.707. [DOI] [PubMed] [Google Scholar]
- 32.Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, Thomas G. Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70s6k. The EMBO journal. 1997;16(12):3693–704. doi: 10.1093/emboj/16.12.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275(5308):1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 34.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15(4):356–62. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 35.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273(22):13375–8. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 36.Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1998;95(26):15587–91. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stiles B, Groszer M, Wang S, Jiao J, Wu H. PTENless means more. Dev Biol. 2004;273(2):175–84. doi: 10.1016/j.ydbio.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 38.Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486(7403):400–4. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dow LE, Kauffman JS, Caddy J, Zarbalis K, Peterson AS, Jane SM, Russell SM, Humbert PO. The tumour-suppressor Scribble dictates cell polarity during directed epithelial migration: regulation of Rho GTPase recruitment to the leading edge. Oncogene. 2007;26(16):2272–82. doi: 10.1038/sj.onc.1210016. [DOI] [PubMed] [Google Scholar]
- 40.Mellman I, Nelson WJ. Coordinated protein sorting, targeting and distribution in polarized cells. Nature reviews Molecular cell biology. 2008;9(11):833–45. doi: 10.1038/nrm2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Metodieva G, Nogueira-de-Souza NC, Greenwood C, Al-Janabi K, Leng L, Bucala R, Metodiev MV. CD74-dependent deregulation of the tumor suppressor scribble in human epithelial and breast cancer cells. Neoplasia. 2013;15(6):660–8. doi: 10.1593/neo.13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Navarro C, Nola S, Audebert S, Santoni MJ, Arsanto JP, Ginestier C, Marchetto S, Jacquemier J, Isnardon D, Le Bivic A, et al. Junctional recruitment of mammalian Scribble relies on E-cadherin engagement. Oncogene. 2005;24(27):4330–9. doi: 10.1038/sj.onc.1208632. [DOI] [PubMed] [Google Scholar]
- 43.Schwertfeger KL, Richert MM, Anderson SM. Mammary gland involution is delayed by activated Akt in transgenic mice. Mol Endocrinol. 2001;15(6):867–81. doi: 10.1210/mend.15.6.0663. [DOI] [PubMed] [Google Scholar]
- 44.Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, Clouthier SG, Wicha MS. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009;7(6):e1000121. doi: 10.1371/journal.pbio.1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Renner O, Blanco-Aparicio C, Grassow M, Canamero M, Leal JF, Carnero A. Activation of phosphatidylinositol 3-kinase by membrane localization of p110alpha predisposes mammary glands to neoplastic transformation. Cancer Res. 2008;68(23):9643–53. doi: 10.1158/0008-5472.CAN-08-1539. [DOI] [PubMed] [Google Scholar]
- 46.Li G, Robinson GW, Lesche R, Martinez-Diaz H, Jiang Z, Rozengurt N, Wagner KU, Wu DC, Lane TF, Liu X, et al. Conditional loss of PTEN leads to precocious development and neoplasia in the mammary gland. Development. 2002;129(17):4159–70. doi: 10.1242/dev.129.17.4159. [DOI] [PubMed] [Google Scholar]
- 47.Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68(15):6084–91. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.