

Abstract
Purpose
To identify the MTD of AT9283, an inhibitor of Aurora kinases A and B, in patients with relapsed or refractory leukemias. Other endpoints included pharmacokinetics, safety and tolerability, pharmacodynamics and preliminary evidence of efficacy.
Patients and Methods
AT9283 was administered as a continuous 72h infusion every 21 days. Doses were escalated by a standard 3+3 design. After the MTD for the 72h infusion was identified, infusion duration was increased incrementally to 96h and 120h. In total, 48 patients received ≥1 cycle of AT9283. Median age was 61 years (range 22–86 years), with 56% men, 75% diagnosed with AML, and 89% having received ≥3 (up to 16) prior lines of therapy.
Results
324 mg/m2/72h AT9283 was determined to be the MTD. DLTs were myocardial infarction, hypertension, cardiomyopathy, tumor lysis syndrome, pneumonia and multiorgan failure. Other AT9283-related toxicities (non-DLT) included myelosuppression, predominantly leucopenia and mucositis. Bone marrow blasts decreased ≥38% after AT9283 treatment in approximately one-third of patients with relapsed/refractory AML; however, this effect was transient and no objective responses were achieved, despite evidence of aurora kinase B inhibition. Two patients with accelerated phase CML showed evidence of benefit, manifest as a cytogenetic response in one case; one patient completed 6 cycles of treatment. Exposure to AT9283 was generally dose proportional.
Conclusion
AT9283 tolerability was strongly dose dependent with reversible myelosuppression predominating at lower doses and events such as cardiovascular toxicities manifesting at higher doses. Clinical trials with AT9283 are ongoing in alternative patient populations.
Keywords: blood cancer, clinical, dose escalation, response, tolerability
Introduction
This article describes an open-label, dose-escalation study of AT9283, a small molecule inhibitor of Aurora kinases A and B, c-ABL, JAK2, and other kinases considered to be therapeutic targets in a variety of leukemias. The study assessed the safety and tolerability of AT9283, and identified the maximum tolerated dose (MTD), in patients with relapsed or refractory leukemias. On the basis of preclinical studies, AT9283 was initially administered as a 72-hour infusion, but following identification of the MTD, the infusion time was increased up to 96 and 120 hours to provide more sustained kinase inhibition.
Aurora kinases are key regulators of mitosis and have roles in centrosome function, mitotic spindle formation, chromosome segregation and cytokinesis.1 Overexpression of Aurora kinases A and B has been linked to genetic instability and cancer, due to dysregulation of the process of cell division. Both have been found overexpressed in solid tumors and leukemias.2–7 Under experimental conditions in vitro, inhibition of aurora kinase B with sRNA-induced proliferation arrest and apoptosis, whereas inhibition of aurora kinase A gave rise only to transient growth delay.8 The potent cell cycle inhibitory and proapoptotic effects of aurora kinase B may provide therapeutic benefit in diseases typified by rapid cell proliferation such as acute leukemias or chronic myeloid leukemia (CML) in accelerated or blast phase.
A number of small molecule aurora kinase inhibitors are undergoing clinical evaluation in adult leukaemia patients (e.g. AZD1152, PF-03814735, MLN8054, MK-0457, and AMG 900). In a Phase I/II clinical trial in adult AML patients, the aurora kinase B inhibitor AZD1152 yielded a complete response in one subject with relapsed disease that was refractory to conventional treatment, and eight of 32 patients (25%) had a haematologic response according to IWG AML criteria.9
In addition to its aurora kinase inhibitory properties, AT9283 also exhibits other protein kinase inhibitory properties of potential therapeutic value, including inhibition of ABL. AT9283 also inhibits the effect of a number of commonly observed kinase domain mutations that induce resistance to clinically validated existing tyrosine kinase inhibitors such as T315I in CML.10 AT9283 is also a potent inhibitor of JAK2 including the V617F mutation, which plays a pathogenic role in myeloproliferative disorders, especially myelofibrosis for which few effective therapies exist (Table 111).12–14
Table 1.
AT9283 in vitro kinase inhibition11
| Protein kinase | IC50 (nM) |
|---|---|
| Aurora A | 52% @ 3 nM |
| Aurora B | 58% @ 3 nM |
| JAK2 | 1.2 |
| JAK3 | 1.1 |
| T315I ABL | 4 |
| Fit3 | 57% @ 15 nM |
| c-kit | 46% @ 250 nM |
| RSK-1 | 37 |
| Lck | 63 |
| Src | 97 |
| c-abl | 110 |
These properties suggest that AT9283 has potential as a therapeutic agent in a variety of hematological malignancies, including resistant and refractory acute leukemias, high-risk myelodysplasias, imatinib-resistant CML and myelofibrosis.
Consistent with its kinase inhibition profile, AT9283 exhibited an improved therapeutic index following sustained exposure in preclinical in vivo studies, so the initial Phase I administration schedule was planned as a continuous 72h infusion. After the 72h infusion was demonstrated to be tolerable, the duration of the infusion was increased to 96h, and then 120h to explore whether prolonged exposure would achieve improved efficacy.
Patients and Methods
Patient Selection
Eligibility criteria included men or women ≥18 years of age with Eastern Cooperative Oncology Group (ECOG) performance status 0, 1, or 2 and histological or cytological confirmation of relapsed or refractory acute leukemias, CML, high-risk MDS, or advanced myelofibrosis. Patients were excluded if they had inadequate liver function, impaired renal function, received radio- or chemotherapy or investigational anti-cancer treatment within 14 days or had unresolved CTCAE grade 2 or greater toxicity from previous anti-cancer therapy, evidence of severe or uncontrolled systemic conditions or current unstable or uncompensated respiratory or cardiac conditions, active and uncontrolled central nervous system disease, left ventricular ejection fraction (LVEF) <50% (criterion added by amendment), ischemic heart disease or myocardial infarction or unstable cardiac disease within 3 months of study entry, prior infection with human immunodeficiency virus (HIV) or hepatitis B or C viruses, or major surgery within 28 days before the start of AT9283 infusion.
All patients provided written informed consent. The study was approved by the independent ethics committee for each trial center and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice (GCP).
Study Design
This was a phase 1, open-label, dose-escalation study to identify the MTD, as well as to assess the preliminary safety, tolerability, and efficacy, of AT9283. The MTD was identified based on the incidence of dose-limiting toxicities (DLTs) during the first cycle of treatment. During the initial part of the study, AT9283 was administered as a 72-hour continuous infusion, and doses were escalated according to a standard 3+3 design. The interval between treatment cycles could be increased up to six weeks in the presence of unresolved toxicity. Safety information from the first cycle of treatment was evaluated for dose escalation decisions by the Safety Monitoring Committee (SMC).
In the second part of the study, the duration of AT9283 infusion (starting at 40 mg/m2/day) was increased sequentially in 24h increments to four (160 mg/m2/96h) and five (200 mg/m2/120h) days to define the MTD of longer infusions.
Toxicity Criteria
Adverse events (AEs) were evaluated throughout the study according to CTCAE Version 3.0. A DLT was defined as any one of the following: (1) CTCAE grade 3 or 4 nausea, vomiting, or diarrhea despite maximum prophylactic and supportive care, (2) any other CTCAE grade 3 or 4 nonhematological toxicity prolonged for ≥7 days that was considered to be clinically significant and causally related to AT9283, (3) pancytopenia with a hypocellular bone marrow (≤5% cellularity) and no evidence of leukemia, lasting longer than 42 days; where, in the investigator’s opinion, it was likely that administration of AT9283 was causally linked with the toxicity or observed effect.
Evaluations
Safety was additionally evaluated based on physical examinations, hematology and clinical chemistry profiles, electrocardiogram, measurements of LVEF, blood pressure and heart rate. AEs were recorded from the first dose of study medication until up to 30 days after the last dose or 30 days after withdrawal from treatment.
Efficacy was assessed as objective remission rate (complete and partial) and duration of remissions. To assess change in blasts, bone marrow samples were collected within 7 days before initial dosing, within 5 days before Cycle 2 dosing, and thereafter as clinically indicated. Western blot analysis was performed on PBMCs extracted from patient blood samples, which were collected immediately before dosing, and at different time points up to Day 8 after the start of the infusion in Cycle 1 and Cycle 2 using antibodies directed against anti-phospho-histone H3(Ser10) (pHH3), pHH3(Ser28) Histone H3 (HH3), pSTAT5(Tyr694), pHH3(Ser10), Beta-Actin, Phospho-Crkl(Tyr207) and total Crkl antibodies (all from Cell Signalling Technology, Beverly, MA).
Blood samples for pharmacokinetic (PK) analysis were collected during Cycle 1 at the following times: predose, 1, 5, 8, and 22 (±2) hours after the start of the first 24-hr infusion; 8 (±2) and 22 (±2) hours after the start of the second 24-hr infusion; and 5, 15, 30, and 45 minutes, and 1, 1.5, 2, 3, 4, 6, 8 (±2), and 24(±2) hr after the end of the last infusion.
Results
Between September 2006 and April 2009, 48 patients received AT9283 (32 patients [8 dose cohorts] in the first part of the study, and 16 patients [12 who received a 96h infusion and 4 who received a 120h infusion] in the second part of the study). Most patients (75%) had a diagnosis of AML, and 38% (n=18) received more than one cycle of treatment (Table 2). Most patients received treatment as prescribed. Retreatment in subsequent cycles was started when evidence of peripheral blood count recovery was observed. Only 5 of 48 patients had a dose interrupted or an incomplete infusion. Twenty-seven patients (56%) were withdrawn from the study due to disease progression. Seven patients (15%) were withdrawn after treatment due to AEs, and 1 patient (2%) was withdrawn due to safety reasons after an AE of cardiomyopathy. Four patients (8%) withdrew consent and 9 patients (19%) withdrew for other reasons.
Table 2.
Baseline patient demographics and characteristics and AT9283 treatment cycles
| Characteristic | AT9283 dose (mg/m2) | All (N=48) | ||
|---|---|---|---|---|
| Part A (72h infusion) | Part B | |||
| 9 – 486 (n=32) | 160a (n=12) | 200b (n=4) | ||
| Median age (y, range) | 61.5 (22–86) | 61.5 (31–78) | 57.5 (53–68) | 61 (22–86) |
| Male, n (%) | 16 (50) | 8 (67) | 3 (75) | 27 (56) |
| Diagnosis, n (%) | ||||
| ALL | 1 (33) | 0 | 0 | 1 (2) |
| AML | 24 (75) | 9 (75) | 3 (75) | 36 (75) |
| CML | 2 (6) | 0 | 0 | 2 (4) |
| MMM (I) | 1 (3) | 2 (17) | 1 (25) | 4 (8) |
| MMM (II) | 4 (13) | 1 (8) | 0 | 5 (10) |
| Number of prior lines of therapy, n (%) | ||||
| 1 | 3 (10) c | 0 | 0 | 3 (7) c |
| 2 | 2 (6) c | 0 | 0 | 2 (4) c |
| ≥3 | 26 (84) c | 10 (100) c | 4 (100) | 40 (89) c |
| Best response to most recent therapy, n (%) | ||||
| - Complete remission | 2 (6) c | 0 | 0 | 2 (4) c |
| - Disease progression | 8 (26) c | 3 (30) c | 4 (100) | 15 (33) c |
| - Disease transformation | 1 (3) c | 0 | 0 | 1 (2) c |
| - Failure | 15 (48) c | 3 (30) | 0 | 18 (40) c |
| - Relapse after complete or partial remission | 2 (6) c | 0 | 0 | 2 (4) c |
| - Stable disease | 2 (6) c | 1 (10) c | 0 | 3 (7) c |
| - Unknown | 1 (3) c | 3 (30) c | 0 | 4 (9) c |
| Previous chemotherapy or leukemia, n (%) | ||||
| Yes | 4 (13) | 2 (17) | 1 (25) | 7 (15) |
| No | 28 (88) | 10 (83) | 3 (75) | 41 (85) |
| Number of AT9283 cycles received, n (%) | ||||
| ≥1 | 32 (100) | 12 (100) | 4 (100) | 48 (100) |
| ≥2 | 15 (47) | 3 (25) | 0 | 18 (38) |
| ≥3 | 4 (13) | 1 (8) | 0 | 5 (10) |
| ≥4 | 1 (3) | 0 | 0 | 1 (2) |
| ≥5 | 1 (3) | 0 | 0 | 1 (2) |
| ≥6 | 1 (3) | 0 | 0 | 1 (2) |
96 hr infusion (includes 2 cohorts)
120 hr infusion
Percentage calculated based on available data (5 of 6 patients in the 324 mg/m2 group [i.e., 31 of 32 patients in Part A] and 10 of 12 patients in the 160 mg/m2 group)
Efficacy and PD
Approximately one-third of patients with relapsed/refractory AML had a ≥38% reduction in bone marrow blasts after AT9283 treatment and received 2–3 cycles of drug. Reduction of leukemic blasts in the bone marrow was not accompanied by significant hematological reconstitution, so no objective or partial remissions were observed. Both patients with accelerated phase CML who received AT9283 showed evidence of benefit, which in 1 patient was manifest by a cytogenetic response; that patient completed 6 cycles of treatment. Both patients had received previous treatment with imatinib and dasatinib for CML. Although treatment with AT9283 led to transient reductions in hepatosplenomegaly, there was no sustained evidence of treatment benefit in patients with myeloproliferative disease.
Inhibition of kinase signalling was observed in peripheral mononuclear cells extracted from patients during and after AT9283 infusion. Sample quality precluded comprehensive analysis, including comparisons among dose regimens. In two of the three patients who provided informative samples at the MTD (108 mg/m2/day; 324 mg/m2/72h), rapid inhibition of phosphorylation of the aurora kinase B substrate pHH3 was observed after initiation of the AT9283 infusion. In both patients (Figure 1) inhibition was achieved near the end of the infusion and maintained for up to 8 days following treatment. Similar data was observed for some of the other signalling molecules, including JAK2 and ABL kinases (data not shown). Similar effects were observed in patients in a lower dose group; in two of the three patients in the 3 mg/m2/day (9 mg/m2/72h) dose group, inhibition of pHH3 phosphorylation was observed (no detectable levels were observed in the third patient) (Figure 1).
Figure 1.

Mononuclear cells were isolated from the blood of three patients before, during and after infusion of AT9283, Cycle 1. Samples were prepared for western blotting with the indicated antibodies to assess inhibition of signalling pathways by AT9283. Patients A and B received the MTD (108 mg/m2/day over 72h). Patient A received 1 cycle of treatment and therefore had no follow-up for response (% blasts). Patient B received 2 cycles of treatment and a 48% reduction in bone marrow blasts (62% at baseline v 32% at follow-up). Patient C received 3 cycles of treatment with the lowest dose (3 mg/m2/day (9 mg/m2/72h) and had a 69% reduction in bone marrow blasts (29% at baseline v 9% at follow-up). A number of pathways were analyzed in all patients including pERK and pSTAT, however, the phosphoSTAT5 and phosphoCrkl signals were not activated and measurable for Patient C in contrast to Patients A and B.
Safety and tolerability
The AT9283 dose of 108 mg/m2/day (324 mg/m2/72h) was determined to be the MTD of AT9283 for a 72-hour infusion. DLTs were myocardial infarction, hypertension, cardiomyopathy, tumor lysis syndrome, pneumonia, and multiorgan failure. Two patients experienced severe tumor lysis syndrome necessitating dialysis and intensive care (one receiving 36 mg/m2/72h and one receiving 200 mg/m2/120h). Although the resulting renal impairment was reversible, patients experienced subsequent infectious complications. Two patients developed a severe drop in left ventricular ejection fraction (LVEF) (to 20–25%) during the third day of AT9283 infusion (one receiving 144 mg/m2/72h and one receiving 160 mg/m2/96h), resulting in pulmonary edema necessitating respiratory support and diuretics; ventricular function recovered both patients. Neither patient had pretreatment measurement of LVEF. The role of previous anthracycline therapy in the etiology of these events was unclear, although both patients had prior anthracycline therapy and other significant pre-existing cardiac conditions including cardiomyopathy that may be contributory. A high incidence of cardiac tachyarrythmias (15%), especially atrial fibrillation, was seen; these often occurred during severe infections, particularly pneumonia, and electrolyte disturbance but not during AT9283 infusion, so the relationship between these events and AT9283 treatment was unclear.
All 48 patients had at least one AE. The overall incidence of AEs was high, as expected in patients with heavily pre-treated relapsed/refractory AML receiving escalating doses of a cytotoxic. The AEs with the highest incidence overall were gastrointestinal disorders (75%), including nausea (29%), diarrhea (29%), constipation (21%), stomatitis (19%) and vomiting (19%). Other AEs with high incidence were pneumonia (29%), fatigue (27%), febrile neutropenia (25%), peripheral edema (25%), headache (21%), and epistaxis (17%). The AEs assessed by the investigator as possibly, probably, or definitely related to AT9283 with the highest incidence were nausea (15%), stomatitis (13%), and alopecia (8%) (Table 3). Overall toxicity was dose dependent with a clear increase in the number and severity of AEs at higher doses; incidence was highest in those who received 216 mg/m2/72hrs (72 mg/m2/day) (100%) and lowest in those who received 36 mg/m2/72hrs (12 mg/m2/day) (29%).
Table 3.
AEs assessed by the investigator as related to study treatment (possibly, probably, or definitely) reported in ≥4% of patients
| System Organ Class / AE | n (%) patients | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AT9283 dose (mg/m2) | Total | ||||||||||
| Part A (72 hr infusion) | Part B | ||||||||||
| 9 (n=3) | 18 (n=3) | 36 (n=7) | 72 (n=3) | 144 (n=4) | 216 (n=3) | 324 (n=6) | 486 (n=3) | 160 a (n=12) | 200 b (n=4) | (N=48) | |
| Blood & lymphatic system disorders | |||||||||||
| Febrile neutropenia | 0 | 0 | 0 | 0 | 0 | 3 (100) | 0 | 0 | 0 | 0 | 3 (6) |
| Cardiac disorders | |||||||||||
| Cardiomyopathy | 0 | 0 | 0 | 0 | 1 (25) | 0 | 0 | 0 | 1 (8) | 0 | 2 (4) |
| Myocardial infarction | 0 | 1 (33) | 0 | 0 | 0 | 0 | 0 | 1 (33) | 0 | 0 | 2 (4) |
| Gastrointestinal disorders | |||||||||||
| Diarrhoea | 0 | 0 | 0 | 0 | 0 | 1 (33) | 0 | 1 (33) | 1 (8) | 0 | 3 (6) |
| Nausea | 1 (33) | 0 | 0 | 0 | 0 | 3 (100) | 2 (33) | 1 (33) | 0 | 0 | 7 (15) |
| Stomatitis | 0 | 0 | 0 | 1 (33) | 1 (25) | 3 (100) | 0 | 0 | 0 | 1 (25) | 6 (13) |
| Vomiting | 1 (33) | 0 | 0 | 0 | 0 | 1 (33) | 0 | 1 (33) | 0 | 0 | 3 (6) |
| Neoplasms benign, malignant and unspecified | |||||||||||
| Tumour lysis syndrome | 0 | 0 | 1 (14) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (25) | 2 (4) |
| Skin and subcutaneous tissue disorders | |||||||||||
| Alopecia | 0 | 0 | 0 | 0 | 0 | 3 (100) | 1 (17) | 0 | 0 | 0 | 4 (8) |
| Vascular disorders | |||||||||||
| Hypertension | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17) | 0 | 2 (17) | 0 | 3 (6) |
96 hr infusion (includes 2 cohorts)
120 hr infusion
There were 13 deaths (27%), the most frequent cause of which was infection. In most cases, infection was considered secondary to immunosuppression from the underlying leukemia rather than related to AT9283 treatment. The 486 mg/m2/72hr (162 mg/m2/day) regimen was determined to be unacceptably toxic, as all 3 patients who received this dose died shortly after completing their first infusion; 2 of these deaths (caused by myocardial infarction and sepsis/progressive leukemia) were considered related to treatment. At other dose levels, two other deaths were considered related to AT9283 treatment: in one patient, who received 18 mg/m2/72h AT9283, death occurred approximately 3 weeks after the second cycle of AT9283 and was caused by cardiopulmonary arrest and respiratory failure; and in the other patient, who received 144 mg/m2/72h AT9283, death occurred approximately 1 week after the first cycle of AT9283 and was attributed to respiratory failure and refractory AML.
Pharmacokinetics
In Cycle 1, mean maximum plasma concentrations (Cmax) occurred at Tmax between 41 and 62 hours and appeared independent of the dose (Table 4, Figure 2). Exposure to AT9283 (as measured by Cmax and AUC0-inf) generally increased with increasing dose, although it was slightly greater than proportional to the increase in dose. PK parameters exhibited a similar pattern in Cycle 2. With extended dosing, exposure to AT9283 was similar between the 2 infusion lengths (96 and 120 hours).
Table 4.
Cycle 1 Mean plasma PK parameters for AT9283
| Plasma PK parameter | AT9283 dose (mg/m2) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Part A (72 hr infusion) | Part B | |||||||||
| 9 (n=3) | 18 (n=3) | 36 (n=7) | 72 (n=3) | 144 (n=4) | 216 (n=3) | 324 (n=6) | 486 (n=3) | 160 a (n=12) | 200 b (n=4) | |
| Cmax (ng/mL) | 7.35 | 18.1 | 41.1 | 131 | 229 | 228 | 447 | 1280 | 164 | 184 |
| Tmax (h) | 47.2 | 62.0 | 53.2 | 46.7 | 54.7 | 41.3 | 53.8 | 51.0 | 59.3 | 69.0 |
| AUC0-inf (h.ng/mL) | 465 | 1070 | 2860 | 16300 | 14300 | 13000 | 28000 | ND | 13700 | 19100 |
| Cl (mL/min/kg) | 9.52 | 7.17 | 6.03 | 1.84 | 4.27 | 7.13 | 5.36 | ND | 5.61 | 4.58 |
96 hr infusion (includes 2 cohorts)
120 hr infusion
Figure 2.
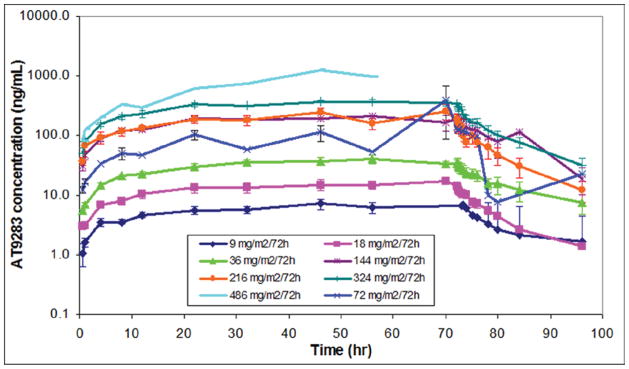
Cycle 1 mean plasma concentration profiles after IV infusion of doses from 9 to 486 mg/m2/72h
Discussion
This was primarily a safety and tolerability study. Analysis of the dose-escalation identified the MTD to be 108 mg/m2/day for a 72h infusion (324 mg/m2/72h) and 40mg/m2/day for a 96h infusion (160 mg/m2/96h). Data from patients enrolled at the MTDs suggest that AT9283 can be administered with an acceptable safety profile. The pattern of toxicity observed was consistent with previous reports of other cytotoxic anticancer therapies and included reversible dose-related myelosuppression, gastrointestinal disturbance, and alopecia. A high incidence of cardiac tachyarrythmias occurred (often during severe infections and electrolyte disturbance but not during AT9283 infusion), and two patients experienced severe cardiomyopathy.
PK analysis confirmed that exposure to AT9283 was generally proportional to dose, and pharmacologically active levels of AT9283 were identified in the plasma of patients treated at well tolerated doses. Target inhibition was sustained to Day 8 after dosing (the only follow-up time point).
One-third of patients with relapsed/refractory AML had significantly reduced bone marrow blasts after treatment and received multiple cycles of AT9283. Reduction in bone marrow blasts was not accompanied by hematological reconstitution and no objective remissions were achieved. Both patients with accelerated phase CML who received AT9283 treatment had a hematological response, but no treatment benefit was evident in any of the patients with myeloproliferative disease in this study.
Despite the initial promise of aurora kinase inhibition for treatment of patients with leukemia,6,7,9 recent findings also point to new independent mechanisms of resistance to therapy,15 and the clinical utility of this group of agents, at least as monotherapy, remains unproven.
Conclusion
The MTD for AT9283 was determined to be 108 mg/m2/day for a 72-hour infusion (324 mg/m2/72hr) and 40mg/m2/day for a 96-hour infusion (160 mg/m2/96hr). Toxicity of AT9283 was consistent with other cytotoxic anticancer therapies, although cardiac tachyarrythmias and severe reversible cardiomyopathy were also observed, thus further studies of AT9283 should include close cardiovascular monitoring. Biological activity observed with AT9283 therapy was consistent with aurora kinase inhibition, including evidence of inhibition of aurora kinase B during the infusion. Treatment was associated with a reduction in leukemic blasts in a proportion of patients with relapsed/refractory AML. This activity, however, was not sustained and did not lead to objective clinical responses in the patients studied.
Clinical Practice Points.
Aurora kinases are key regulators of mitosis and have roles in centrosome function, mitotic spindle formation, chromosome segregation and cytokinesis. Overexpression of Aurora kinases A and B has been linked to genetic instability and cancer, due to dysregulation of the process of cell division.
AT9283 is a small molecule inhibitor of Aurora kinases A and B, c-ABL, JAK2, and other kinases that may be targets for therapeutic intervention in leukemia. AT9283 was evaluated in a phase I, openlabel, dose-escalation study of patients with relapsed/refractory leukemias.
The MTD for AT9283 was determined to be 324 mg/m2/72h. AT9283 tolerability was strongly dose dependent with reversible myelosuppression predominating at lower doses and events such as cardiovascular toxicities manifesting at higher doses. DLTs were myocardial infarction, hypertension, cardiomyopathy, tumor lysis syndrome, pneumonia and multiorgan failure. Bone marrow blasts decreased ≥38% after AT9283 treatment in approximately one-third of patients with relapsed/refractory AML, but no objective responses were observed, despite rapid inhibition of phosphorylation of aurora kinase B substrate pHH3 after initiation of the AT9283 infusion in clinical samples. Two patients with accelerated phase CML showed evidence of benefit, accompanied by a cytogenetic response in one patient who completed 6 cycles of treatment. Exposure to AT9283 was generally dose proportional.
Anti-leukemic activity was insufficient to warrant further exploration of AT9283 as monotherapy in this patient population. Clinical trials with AT9283 are ongoing in alternative patient populations.
Acknowledgments
We thank the patients involved in this study and the staff who participated in their care. We acknowledge Sam Lewis for technical assistance and Laurie Haynes for editorial assistance funded by Astex.
Funding Sources: This study was sponsored by Astex Pharmaceuticals, Inc.
Footnotes
Conflict of Interest:
- James Foran: funding for clinical study by Astex Pharmaceuticals Inc.
- Farhad Ravandi: funding for clinical study by Astex Pharmaceuticals Inc.
- William Wierda: funding for clinical study by Astex Pharmaceuticals Inc.
- Guillermo Garcia-Manero: funding for clinical study by Astex Pharmaceuticals Inc.
- Srdan Verstovsek: funding for clinical study by Astex Pharmaceuticals Inc.
- Tapan Kadia: funding for clinical study by Astex Pharmaceuticals Inc.
- Jan Burger: funding for clinical study by Astex Pharmaceuticals Inc.
- Gautham Borthakur: funding for clinical study by Astex Pharmaceuticals Inc.
- Jorge Cortes: funding for clinical study by Astex Pharmaceuticals Inc.
- Hagop Kantarjian: funding for clinical study by Astex Pharmaceuticals Inc.
- Dr. Yule, Dr. Langford, Dr. Lyons, and Dr. Ayrton are employees of or contractors for AstexPharmaceuticals, Inc. Victoria Lock was previously employed by Astex Pharmaceuticals, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
James Foran, University of Alabama at Birmingham (UAB) Comprehensive Cancer Center, Birmingham, Alabama.
Farhad Ravandi, Anderson Cancer Center, Houston, Texas.
William Wierda, Anderson Cancer Center, Houston, Texas.
Guillermo Garcia-Manero, Anderson Cancer Center, Houston, Texas.
Srdan Verstovsek, Anderson Cancer Center, Houston, Texas.
Tapan Kadia, Anderson Cancer Center, Houston, Texas.
Jan Burger, Anderson Cancer Center, Houston, Texas.
Murray Yule, Astex Pharmaceuticals, Inc., Cambridge, United Kingdom.
Gillian Langford, Astex Pharmaceuticals, Inc., Cambridge, United Kingdom.
John Lyons, Astex Pharmaceuticals, Inc., Cambridge, United Kingdom.
John Ayrton, Astex Pharmaceuticals, Inc., Cambridge, United Kingdom.
Victoria Lock, Astex Pharmaceuticals, Inc., Cambridge, United Kingdom.
Gautham Borthakur, Anderson Cancer Center, Houston, Texas.
Jorge Cortes, Anderson Cancer Center, Houston, Texas.
Hagop Kantarjian, Anderson Cancer Center, Houston, Texas.
References
- 1.Carmena M, Earnshaw WC. The cellular geography of Aurora Kinases. Nat Rev Mol Cell Biol. 2003;4:842–854. doi: 10.1038/nrm1245. [DOI] [PubMed] [Google Scholar]
- 2.Sen S, Zhou H, White RA. A putative serine/threonine kinase encoding gene BTAK on chromosome 20q13 is amplified and overexpressed in human breast cancer cell lines. Oncogene. 1997;14:2195–200. doi: 10.1038/sj.onc.1201065. [DOI] [PubMed] [Google Scholar]
- 3.Bischoff JR, Anderson L, Zhu Y, et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998;17:3052–3065. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katayama H, Ota T, Jisaki F, et al. Mitotic kinase expression and colorectal cancer progression. J Natl Cancer Inst. 1999;91:1160–2. doi: 10.1093/jnci/91.13.1160. [DOI] [PubMed] [Google Scholar]
- 5.Kamada K, Yamada Y, Hirao T, et al. Amplification/overexpression of Aurora-A in human gastric carcinoma: Potential role in differentiated type gastric carcinogenesis. Oncol Rep. 2004;12:593–9. [PubMed] [Google Scholar]
- 6.Farag SS. The potential role of Aurora kinase inhibitors in haematological malignancies. Br J Haematol. 2011;155:561–79. doi: 10.1111/j.1365-2141.2011.08898.x. published online October 8, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moore AS, Blagg J, Linardopoulos S, Pearson AD. Aurora kinase inhibitors: novel small molecules with promising activity in acute myeloid and Philadelphia-positive leukemias. Leukemia. 2010;24:671–8. doi: 10.1038/leu.2010.15. published online February 11, 2010. [DOI] [PubMed] [Google Scholar]
- 8.Hartsink-Segers SA, Zwaan CM, Exalto C, et al. Aurora kinases in childhood acute leukemia: the promise of aurora B as therapeutic target. Leukaemia. 2013;27:560–8. doi: 10.1038/leu.2012.256. published online September 3, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Löwenberg B, Muus P, Ossenkoppele G, et al. Phase 1/2 study to assess the safety, efficacy, and pharmacokinetics of barasertib (AZD1152) in patients with advanced acute myeloid leukemia. Blood. 2011;118:6030–6. doi: 10.1182/blood-2011-07-366930. published online October 5, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carter TA, Wodicka LM, Shah NP, et al. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. PNAS. 2005;102:11011–11016. doi: 10.1073/pnas.0504952102. published online July 26, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodall J, Squires MS, Lock V, et al. AT9283, a multi-targeted kinase inhibitor, has potent activity in AML cell lines and patient samples. Poster 1613, American Society of Hematology (ASH) annual meeting; 2008. [Google Scholar]
- 12.Barosi G, Poletto V, Massa M, et al. JAK2 V617F genotype is a strong determinant of blast transformation in primary myelofibrosis. PLoS One. doi: 10.1371/journal.pone.0059791. Published online March 26, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hubbeling HG, Frank DM, Hexner EO. Myelofibrosis 2012: it’s complicated. Ther Adv Hematol. 2012;3:131–46. doi: 10.1177/2040620712437754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quintás-Cardama A, Verstovsek S. Molecular pathways: Jak/STAT pathway: mutations, inhibitors, and resistance. Clin Cancer Res. 2013;19:1933–40. doi: 10.1158/1078-0432.CCR-12-0284. published online February 13, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Failes TW, Mitic G, Abdel-Halim H, et al. Evolution of resistance to Aurora kinase B inhibitors in leukaemia cells. PLoS One. doi: 10.1371/journal.pone.0030734. Published online February 16, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]