

Abstract
Calcium signaling in astrocytes couples changes in brain activity to regional alterations in cerebral blood flow (CBF) by eliciting vasoconstriction or vasodilation of adjacent arterioles1-7. However, the mechanism for how these disparate astrocyte influences provide appropriate changes in cerebral vascular tone within a cellular environment that has dynamic metabolic requirements remains unclear. The regulation of CBF has recently been shown to be tightly coupled to the lactate/pyruvate ratio and thus the NADH/NAD+ ratio in animals8 and humans9,10. We tested the impact of metabolic changes on the regulation of cerebral blood vessel diameter by astrocytes, by manipulating tissue oxygenation which changes dynamically with brain activity11-14. Using two-photon Ca2+ imaging and uncaging as well as intrinsic nicotinamide adenine dinucleotide (NADH) imaging of single cells as a measure of redox state, we show that the ability of astrocytes to induce vasodilations over vasoconstrictions critically relies on the metabolic state of the tissue. When O2 availability is lowered and astrocyte [Ca2+]i is elevated, astrocyte glycolysis and lactate release are maximized. Extracellular lactate contributes to prostaglandin E2 (PGE2) accumulation by hindering its transporter-mediated uptake, subsequently causing vasodilation. These data reveal the role of metabolic substrates in regulating CBF and provide a mechanism for differential astrocyte control over cerebrovascular diameter during different states of brain activation.
We first examined whether changing PO2 consistently determined whether vasoconstrictions or vasodilations were evoked by astrocyte Ca2+ transients. The mGluR agonist t-ACPD (100 μM) potently increased [Ca2+]i (240.0 ± 19.1 %, n = 14, P < 0.01, Fig. 1 a) in astrocytes1,4,5 and differential responses were observed in the adjacent cerebral blood vessels whereby vasodilation (107.0 ± 0.8 % (100% = control diameter), P < 0.01, n = 23, Fig. 1 a, d) occurred in low O2 but in high O2 arterioles constricted1 (85.4 ± 4.0 %, P < 0.01, n = 10, Fig. 1 d). We next used two-photon photolysis of the Ca2+ cage DMNPE-415 to examine the effect of low O2 levels on the impact of directly evoked astrocyte Ca2+ signals on adjacent blood vessels. Uncaging DMNPE-4 triggered Ca2+ waves that spread to multiple endfeet circumscribing arterioles (Fig. 1 b, c). In contrast to previous results in high PO2 where Ca2+ transients in astrocyte endfeet always caused constriction1, in low O2 endfeet Ca2+ (232.2 ± 8.5 %, n = 17, P < 0.01) caused arteriole dilations (107.4 ± 1.0 %, P < 0.01, n = 17, Fig. 1 b, c, d) that were repeatable (Fig. 1 b). The Ca2+ change preceded the enlargement of lumen diameter (Fig. 1 b, c). These data suggest PO2 dictates the polarity of the change in arteriole diameter caused by either mGluR activity or an elevation in endfoot Ca2+ and that the dilation process can be initiated solely by astrocyte activation (see Supplemental Fig. 1 for PO2 measurements). Ca2+ activates cPLA2 triggering arachidonic acid (AA) formation that is either converted to 20-HETE1 in (SMCs) causing vasoconstriction or is converted to the vasodilator PGE2 in astrocytes via cyclooxygenase (COX) enzymes4,7. We confirmed COX expression in astrocytes and their endfeet (Supplemental Fig. 2). The COX inhibitor indomethacin (100 μM) blocked vasodilations caused by t-ACPD (98.4 ± 0.8 %, P > 0.05, n = 9, Fig. 1 d) and by caged Ca2+ photolysis (101.0 ± 0.5 %, P > 0.05, n = 6, Fig. 1 d) in low O2. Application of the COX product PGE2 (1 μM) elicited vasodilation (109.4 ± 2.7 %, P < 0.05, n = 4, Fig. 1 d), confirming COX activation and the generation of PGE2 is an important signaling molecule in astrocyte-mediated vasodilations in low O24,7.
Fig. 1.
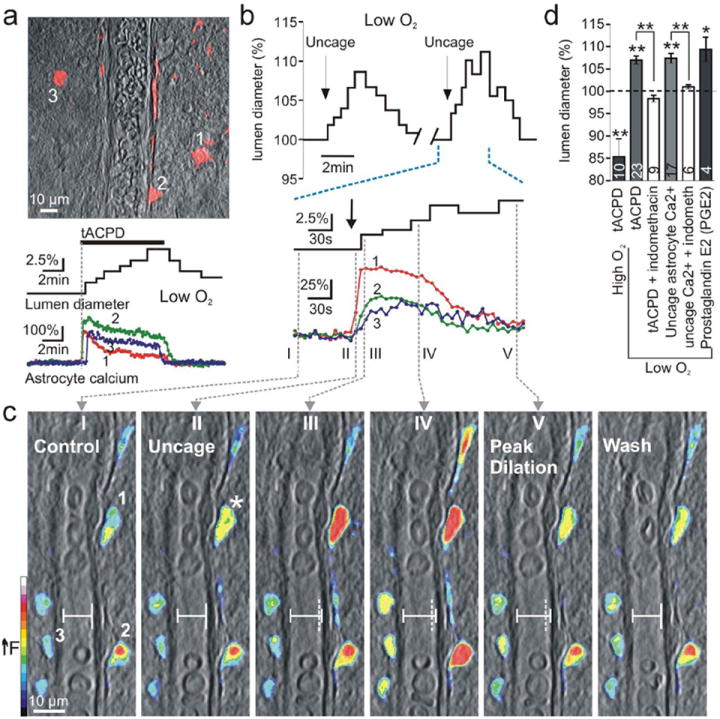
Lowering O2 converts astrocyte-mediated vasoconstrictions to vasodilations. (a) Top: astrocytes (red) loaded with Ca2+ indicator surround arteriole. Bottom: astrocyte Ca2+ signals occur coincident with dilation caused by the mGluR agonist t-ACPD in low O2. (b) Top: uncaging astrocyte Ca2+ (indicated by arrow) causes vasodilation in low O2 and is repeatable. Bottom: expanded time scale shows the Ca2+ signal in endfoot 1 (shown in c I) precedes the lumen diameter increase. (c) Overlay of vessel and pseudo colored endfoot Ca2+ changes. Images I-V correspond to times in b. I: control state and regions of interest: endfeet 1-3. Image II: endfoot 1 shows first Ca2+ rise (star) before lumen diameter starts to increase. Vertical dotted line (III-V) indicates previous position of vessel wall. (d) Summary data.
Next we examined how changing tissue PO2 could modify whether astrocyte Ca2+ transients induce constrictions versus dilations. We examined the possibility that a lower PO2 elevates anaerobic metabolism, increasing external lactate which subsequently enhances external PGE2 concentrations. Once produced, PGE2 passively diffuses into the extracellular space16 and its action on smooth muscle cells is terminated when prostaglandin transporters (PGTs) take up PGE2 by an exchange of intracellular lactate17. Due to the influence of a lactate concentration gradient on PGT efficacy, we tested the hypothesis that higher levels of extracellular lactate reduce the ability of PGT to uptake PGE217 from the extracellular space, thereby increasing PGE2 concentrations resulting in vasodilation. Such a role for lactate is consistent with the observations that extracellular lactate levels increase immediately during brain activity18 and that lactate is known to enhance activity dependent increases in blood flow8. We found using immuohistochemistry that PGT is widely expressed in brain grey matter in both astrocyte endfeet (Fig. 2 a), and neurons (Fig. 2 b), indicating dendrites as well as endfeet take up prostaglandins. Consistent with a positive correlation between external lactate and PGE2, we found that low O2 enhanced lactate release from brain slices (low: 114.2 ± 9.1 μM, n = 6; high: 41.9 ± 5.6 μM, n = 6, P < 0.01, Fig. 2 c). Also, significantly greater levels of PGE2 were observed in low O2 when PGE2 production was triggered by mGluR activation, (low: 136.1 ± 10.2 pg/ml, n = 6; high: 91.9 ± 12.6 pg/ml, n = 4, P < 0.01, Fig. 2 d). Furthermore, addition of lactate (1 mM) enhanced the PGE2 level (control: 40.5 ± 3.3 pg/ml; + lactate: 58.0 ± 3.1 pg/ml, n = 5, P < 0.01) and increased arteriole diameter (107.5 ± 1.0 %, P < 0.01, n = 15, Fig. 2 e, f, g). Dilations induced by lactate were blocked by indomethacin (100.4 ± 0.4 %, P > 0.05, n = 11, Fig. 2 g) indicating they were mediated by PGE2. These data indicate that in low O2 the higher level of extracellular lactate raises external levels of PGE2. This is consistent with lactate reducing PGE2 uptake through PGT.
Fig. 2.
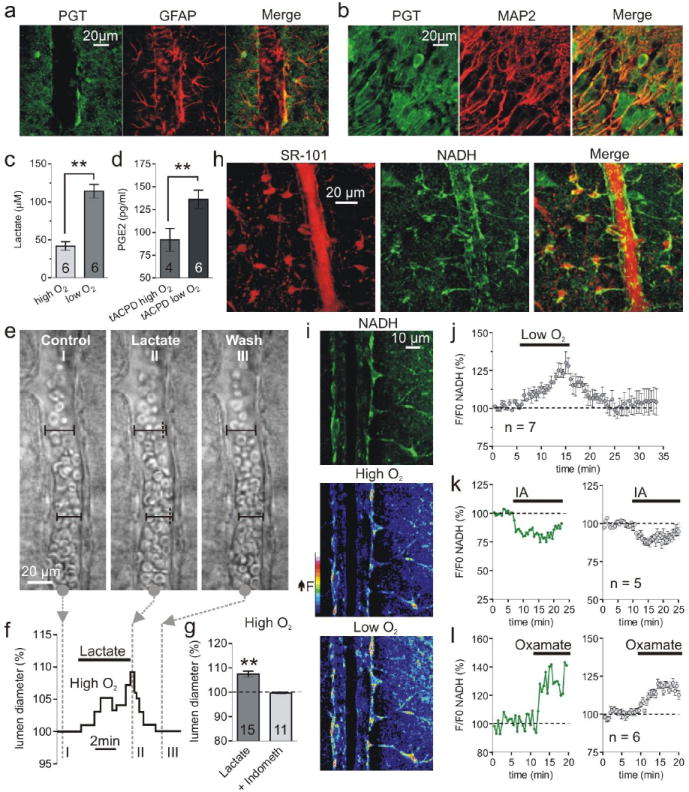
Low O2 facilitates lactate and PGE2 release and enhances astrocyte glycolysis. (a and b) Immunohistochemistry showing the astrocyte marker GFAP and the neuron marker MAP2 colocalize with PGT labeling. (c) Lactate release is elevated in low O2. (d) t-APCD increases PGE2 release most in low O2. (e and f) Lactate dilates an arteriole. Images I-III correspond to time points in f, which shows the lumen diameter increase. (g) Summary showing percent dilation by lactate and block by indomethacin. (h) Colocalization of the astrocyte maker SR-101 (left stack) and the NADH signal (middle stack) from perivascular glia somas and endfeet. (i) Top: arteriole NADH fluorescence from a single image plane showing astrocyte compartments, SMCs/endothelial cells and empty lumen of the blood vessel. Middle and Lower: pseudo colored image of NADH fluorescence in high and low O2. (j) Summary of NADH changes in astrocyte compartments caused by low O2. (k and l) Astrocyte NADH in response to glycolysis inhibition with iodoacetate (IA) (k) and LDH inhibition with Oxamate (l). Left: single experiment; right: summary.
We next examined the role of glycolysis in lactate production by imaging the intrinsic fluorescence of the metabolic electron carrier NADH in astrocytes. Two-photon excitation of NADH provides a sensitive, subcellular measure of both oxidative metabolism (punctate NADH fluorescence from mitochondria) and glycolytic metabolism (diffuse NADH fluorescence from the cytosol)19. We observed, as previously reported, that astrocytes (stained with SR-10120) showed bright intracellularly diffuse NADH fluorescence in the soma and endfeet (Fig. 2 h, i). Stimulating glycolysis with low O2 increased the astrocyte NADH signal (124.6 ± 1.4 %, n = 7, P < 0.01, Fig. 2 i, j) and inhibiting glycolysis with iodoacetate (IA, 200 μM) reduced basal NADH fluorescence (87.8 ± 2.0 %, n = 5, P < 0.05, Fig. 2 k). Furthermore, inhibiting lactate dehydrogenase (LDH) (which converts pyruvate and NADH to lactate and NAD+; see Fig. 4 e diagram) with oxamate (2.5 mM) increased the NADH signal (119.3 ± 2.0 %, n = 6, P < 0.01, Fig. 2 l). These data support the conclusion that NADH is generated to a significant extent in astrocytes as a consequence of glycolytic metabolism—likely originating from glycogen breakdown21—which can be augmented by reducing O2.
Fig. 4.
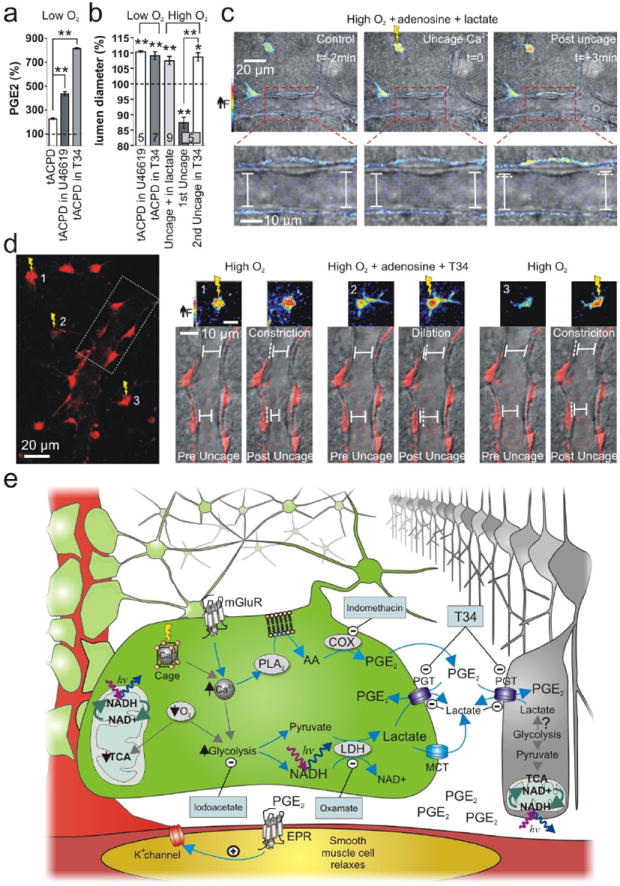
Raising PGE2 levels by inhibiting PGT changes the polarity of astrocyte-mediating vasomotion. (a) PGE2 levels were further elevated by mGluR activation in low O2 when PGT was inhibited by U46619 or T34. (b) Summary data of vasomotion during PGT manipulation. (c) Uncaging Ca2+ in high O2 causes vasodilation in exogenous lactate. Top panels show the astrocyte Ca2+ signal change (pseudo colour) from uncaging. Lower panels show close up of vessel lumen. (d) In high O2, astrocyte-mediated vasoconstriction is converted to vasodilation during PGT blockade. Left: astrocytes and endfeet circumscribing an arteriole. Ca2+ is uncaged in 3 astrocytes separated in time; box indicates the vessel region examined on the right. Right: vasomotions corresponding to the separate uncaging events. Small pseudo colour images show the Ca2+ signal change from uncaging in each astrocyte. Lower images of the vessel and endfeet (red) show that the vasomotion switches polarity when PGTs are blocked. (e) Diagram of the supported model.
Recent two-photon NADH imaging demonstrated an increase in astrocyte glycolysis subsequent to the onset of neuronal activity19. We hypothesized that mGluR activation further enhances glycolysis in astrocytes in low O2, promoting greater extracellular lactate levels that cause vasodilation. Consistent with this idea, t-ACPD enhanced extracellular lactate, which was greatest in low O2 (low: 186.7 ± 11.2 μM, n = 6; high: 98.6 ± 10.2 μM, n = 7, P < 0.01, Fig. 3 f). The astrocytes were a site of increased glycolysis because t-ACPD triggered an increase in cytosolic NADH (128.7 ± 4.1 %, n = 7, P < 0.01, Fig. 3 a, b, c, d, e) coincident with lumen widening (108.5 ± 0.7 %, n = 5 of 7, P < 0.01, Fig. 3 b, c). In contrast, peak NADH signals from perivascular neurons were not as great and were qualitatively different, showing distinct punctate mitochondrial fluorescence (Supplemental Fig. 3). These data signify a change in anaerobic metabolism may be important for astrocyte-mediated vasomotion in low O2.
Fig. 3.
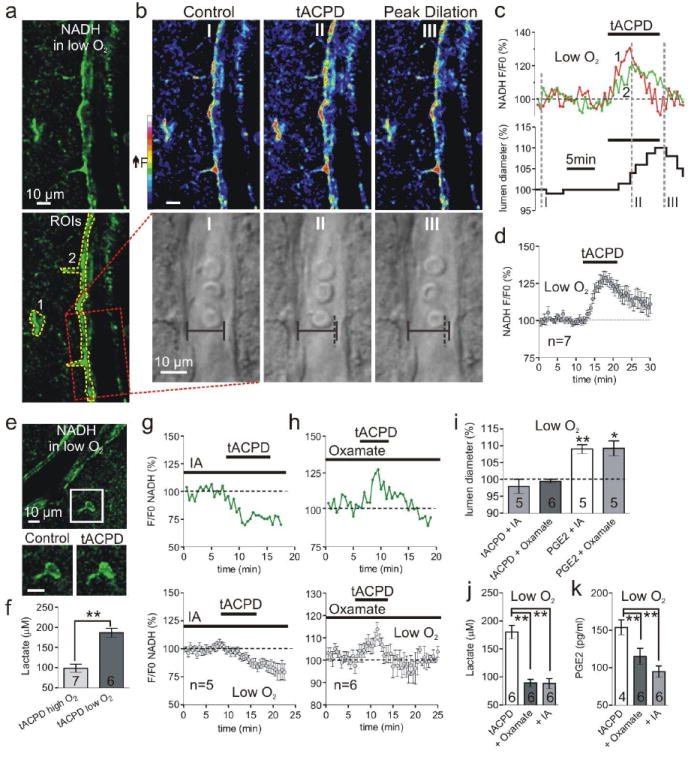
Glycolysis and lactate release is critical for astrocyte-mediated dilations. (a) Arteriole and astrocyte NADH image in low O2; bottom shows ROIs for astrocyte compartments (1 and 2) and vessel (box). (b) Pseudo color NADH (top) and diameter (bottom) changes caused by t-ACPD at time points I-III in c. Vertical dotted line (II and III) indicates previous position of vessel wall. (c) Astrocyte NADH (top) and lumen diameter (bottom) in response to t-ACPD; same experiment as a and b. (d) Summary of NADH increase from mGluR activation in low O2. (e) Top: NADH image of an arteriole and astrocyte. Lower: soma close-up showing t-ACPD causes a diffuse increase in NADH. (f) t-ACPD increases extracellular lactate most in low O2. (g) t-ACPD decreases astrocyte NADH during glycolysis inhibition; top: single experiment; bottom: summary. (h) mGluR activation increases astrocyte NADH during LDH inhibition; top: single experiment; bottom: summary. (i) Summary showing t-ACPD fails to dilate vessels in oxamate or IA and PGE2 rescues vasodilation in these compounds. (j and k) The increase in lactate (j) and PGE2 (k) caused by t-ACPD is significantly less in oxamate and IA.
Recent functional imaging data from human subjects reveals a relationship between a rise in the lactate/pyruvate and thus NADH/NAD+ ratios and an increase in CBF9, but how this enhanced glycolytic state is a critical step translating information on the level of brain activity into vasodilation of arterioles is not known. Our data demonstrate that lactate, astrocyte NADH and PGE2 are maximally increased in low O2 when mGluRs are activated and that exogenous lactate can mediate prostaglandin dependent vasodilation. Therefore, we tested the hypothesis that glycolysis and thus lactate production is essential for prostaglandin dependent astrocyte-mediated vasodilations. We used two separate pharmacological treatments to limit the lactate released by mGluR activity in low O2. First, in the presence of IA to block the source of lactate, t-ACPD instead decreased astrocyte NADH (81 ± 0.4 %, n = 5, P < 0.01, Fig. 3 g) and failed to dilate vessels (97.8 ± 0.2 %, n = 5, P > 0.05, Fig. 3 i). Second, in the presence of oxamate to curtail lactate formation, t-ACPD still increased astrocyte NADH (113.8 ± 3.1 %, n = 6, P < 0.05, Fig. 3 h)—an expected outcome as oxamate acts downstream of NADH production (see Fig. 4 c diagram)—yet vasodilations no longer occurred (99.0 ± 0.1 %, n = 6, P > 0.05, Fig. 3 i). We then confirmed the lack of vasomotion observed during these treatments was associated with a significant reduction in lactate and PGE2 release when mGluRs were activated (Lactate release in t-ACPD: 180.2 ± 11.9 μM, n = 6; + IA: 88.2 ± 8.8 μM, n = 6, P < 0.01; + Oxamate: 89.1 ± 6.6 μM, n = 6, P < 0.01, Fig. 3 j)(PGE2 release in t-ACPD: 154.0 ± 10.1 pg/ml, n = 4; + IA: 115.4 ± 10.6 pg/ml, n = 6, P < 0.01; + Oxamate: 94.6 ± 7.6 pg/ml, n = 6, P < 0.01, Fig. 3 k). Finally, because PGE2 is the final effector molecule on SMCs (downstream of astrocyte glycolysis), we could rescue vasodilation by applying PGE2 in the presence of either oxamate (109.2 ± 2.2 %, P < 005, n = 5, Fig. 3 i) or IA (109.0 ± 1.3 %, P < 0.01, n = 5, Fig. 3 i). These data demonstrate that inhibition of glycolysis or LDH limits extracellular lactate and PGE2 accumulation in response to t-ACPD in low O2, preventing astrocyte mediated vasodilations.
As a final test of our hypothesis we pharmacologically targeted PGTs to manipulate extracellular levels of PGE2 and examined the effects on vasomotion. First, blockade of PGTs with U4661922 or TGBz T3416 resulted in elevated levels of PGE2 when mGluRs were activated in low O2 (tACPD: 227.5 ± 7.2 %, tACPD + U46619: 435.6 ± 16.9, P < 0.01 to tACPD alone, n = 5; tACPD + T34: 816.4 ± 6.0, P < 0.01 to tACPD alone, n = 4, Fig. 4 a) and did not interfere with low O2 astrocyte-mediated dilations (U46619: 110.4 ± 0.3 %, P < 0.01, n = 5; T34: 109.1 ± 1.3 %, P < 0.01, n = 7, Fig. 4 b). Second, we hypothesized that adding exogenous lactate in high O2 would enable astrocyte-mediated vasomotion to change polarity by attenuating PGE2 uptake. However, because there is nothing preventing the initiation of vasoconstriction in high O2 from the action of AA in SMCs, we blocked constriction at the level of SMCs by adding adenosine23. Extracellular adenosine is elevated in a reduced oxygen environment24 and is an additional modulator of vasoconstriction23. In the presence of adenosine (100 μM), uncaging Ca2+ within astrocytes in high O2 produced no vasomotion (99.6 ± 0.9 %, P > 0.05, n = 4, Supplemental Fig. 4). However, when lactate (100 μM - 1 mM) was included to mitigate PGE2 clearance in addition to adenosine, Ca2+ uncaging now resulted in dilation (107.5 ± 1.4 %, P < 0.01, n = 9, Fig. 4 b, c), despite equivalent endfeet Ca2+ signals compared to adenosine alone (adenosine: F/F0 = 160.4 ± 6.1; adenosine + lactate: F/F0 = 159.6 ± 5.1, P > 0.05, Supplemental Fig. 4). From this, we hypothesized that PGT blockade would have a similar result by raising the extracellular PGE2 level. Indeed, in high O2 we found that in the same vessel, the first uncaging event caused constriction (87.4 ± 1.8 %, P < 0.01, Fig. 4 b, d) but the second uncaging event during PGT inhibition with T34 caused dilation (108.7 ± 1.4 %, P < 0.05, n = 5, Fig. 4 b, d). These data indicate that the ability of astrocytes to induce dilation over constriction critically relies on reduced efficacy of PGE2 uptake under conditions where lactate release is enhanced.
Here we show that when the glycolytic state of the brain is enhanced by lower O2 levels, astrocyte-mediated vasoconstrictions convert to vasodilations because the elevated extracellular lactate causes PGE2 accumulation which is maximized during mGluR activation. Flow through blood vessels is proportional to vessel radius to the 4th power; therefore a ~9 % change in arteriole diameter reported here equates to a ~45 % increase in CBF. This change is consistent with astrocyte-mediated vasodilations observed in vivo7 and can more than account for regional CBF changes measured by PET25 and with two-photon microscopy26 when the brain is physiologically activated. These data add an important and until now unrecognized mechanism in the bidirectional control of cerebral vessels by astrocytes. Previous studies implicated different populations of GABA neurons27 and tissue nitric oxide levels2. The metabolic control of these antagonistic pathways by astrocytes implies that brain blood flow would reach a homeostatic level depending on metabolic activity. Under quiescent periods when O2 is not being rapidly consumed, astrocyte Ca2+ signals induce constrictor tone, keeping CBF at an appropriate level. At the onset of neural activity there is a drop in PO2 and rise in extracellular lactate which promotes astrocyte mediated dilation. Manipulating this homeostatic balance may be a therapeutic avenue for treating the inappropriate declines in CBF that occurs in some dementias and after stroke.
Methods Summary
Brain Slices and Imaging
Hippocampal and neocortical slices were prepared from juvenile (p 16-21), male, Sprague-Dawley rats. Care and use of animals was approved by the University of British Columbia Animal Care and Use Committee. ACSF bubbled with 95 % O2, 5 % CO2 (standard for brain slice experimentation), was defined as a high O2 treatment and ACSF bubbled with 20 % O2, 5 % CO2, balanced N2, was defined as a low O2 treatment. For astrocyte dye loading, slices were transferred to a 3 ml ACSF well containing the Ca2+ cage DMNPE-4/AM and/or the Ca2+ indicator rhod-2/AM dissolved in DMSO (final DMSO concentration 0.2 %) for 1.5 hours. A two-photon laser-scanning microscope (Zeiss LSM510-Axioskop-2 fitted with a 40X-W/0.80 numerical aperture objective lens) directly coupled to a Chameleon Ultra tunable ultrafast laser (~100-fs pulses and 76MHz, Coherent) provided excitation of rhod-2 intrinsic NADH fluorescence and was used for uncaging using two photon photolysis. Arterioles were identified with IR-DIC optics and vessel diameter changes were imaged by acquiring the transmitted laser light. Quantification of lumen diameter, NADH and Ca2+ changes were performed with Zeiss LSM (version 3.2) software and ImageJ.
Lactate and PGE2 Measurements
The assay used for measuring PGE2 release was Specific Parameter PGE2 ELISA kits (R&D systems). Extracellular lactate levels were measured using a Lactate Assay Kit (Biomedical Research Service Centre, SUNY Buffalo).
Full Methods and associated references can be found below in the supplemental materials section.
Supplementary Material
Acknowledgments
We thank Drs. T. Murphy, Tony Phillips and Yu Tian Wang for reading an earlier version of the manuscript. T34 was a gift from the lab of Victor L. Schuster from the Departments of Medicine and Physiology and Biophysics, Albert Einstein College of Medicine. Supported by an operating Grant from the Canadian Institutes of Health Research. BAM is a Canada Research Chair and a Michael Smith Foundation for Health Research (MSFHR) Distinguished Scholar. GRJG is supported by fellowships from the Alberta Heritage Foundation for Medical Research and MSFHR. HBC is supported by postdoctoral fellowships from Wilms Foundation and the Heart and Stroke Foundation of BC and the Yukon.
Footnotes
Author Contributions G.R.J.G. and B.A.M. designed the imaging experiments and wrote the manuscript. G.R.J.G. performed the imaging experiments and analysis and took slice PO2 measurements. H.B.C., G.R.J.G. and B.A.M. designed the lactate and PGE2 experiments. H.B.C. and B.A.M. designed the immunohistochemistry experiments. H.B.C. performed the lactate and PGE2 measurements and analysis and the immunohistochemistry. G.C.R.E-D designed and synthesized the calcium cage. All authors helped edit the manuscript.
References
- 1.Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431(7005):195. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- 2.Metea MR, Newman EA. Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J Neurosci. 2006;26(11):2862. doi: 10.1523/JNEUROSCI.4048-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chuquet J, Hollender L, Nimchinsky EA. High-resolution in vivo imaging of the neurovascular unit during spreading depression. J Neurosci. 2007;27(15):4036. doi: 10.1523/JNEUROSCI.0721-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zonta M, Angulo MC, Gobbo S, et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nature neuroscience. 2003;6(1):43. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]
- 5.Filosa JA, Bonev AD, Nelson MT. Calcium dynamics in cortical astrocytes and arterioles during neurovascular coupling. Circulation research. 2004;95(10):e73. doi: 10.1161/01.RES.0000148636.60732.2e. [DOI] [PubMed] [Google Scholar]
- 6.Filosa JA, Bonev AD, Straub SV, et al. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nature neuroscience. 2006;9(11):1397. doi: 10.1038/nn1779. [DOI] [PubMed] [Google Scholar]
- 7.Takano T, Tian GF, Peng W, et al. Astrocyte-mediated control of cerebral blood flow. Nature neuroscience. 2006;9(2):260. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- 8.Ido Y, Chang K, Williamson JR. NADH augments blood flow in physiologically activated retina and visual cortex. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(2):653. doi: 10.1073/pnas.0307458100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mintun MA, Vlassenko AG, Rundle MM, et al. Increased lactate/pyruvate ratio augments blood flow in physiologically activated human brain. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(2):659. doi: 10.1073/pnas.0307457100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vlassenko AG, Rundle MM, Raichle ME, et al. Regulation of blood flow in activated human brain by cytosolic NADH/NAD+ ratio. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(6):1964. doi: 10.1073/pnas.0510632103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ances BM, Buerk DG, Greenberg JH, et al. Temporal dynamics of the partial pressure of brain tissue oxygen during functional forepaw stimulation in rats. Neuroscience letters. 2001;306(1-2):106. doi: 10.1016/s0304-3940(01)01868-7. [DOI] [PubMed] [Google Scholar]
- 12.Offenhauser N, Thomsen K, Caesar K, et al. Activity-induced tissue oxygenation changes in rat cerebellar cortex: interplay of postsynaptic activation and blood flow. The Journal of physiology. 2005;565(Pt 1):279. doi: 10.1113/jphysiol.2005.082776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vanzetta I, Grinvald A. Increased cortical oxidative metabolism due to sensory stimulation: implications for functional brain imaging. Science (New York, NY. 1999;286(5444):1555. doi: 10.1126/science.286.5444.1555. [DOI] [PubMed] [Google Scholar]
- 14.Devor A, Ulbert I, Dunn AK, et al. Coupling of the cortical hemodynamic response to cortical and thalamic neuronal activity. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(10):3822. doi: 10.1073/pnas.0407789102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ellis-Davies GC. Caged compounds: photorelease technology for control of cellular chemistry and physiology. Nature methods. 2007;4(8):619. doi: 10.1038/nmeth1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chi Y, Khersonsky SM, Chang YT, et al. Identification of a new class of prostaglandin transporter inhibitors and characterization of their biological effects on prostaglandin E2 transport. The Journal of pharmacology and experimental therapeutics. 2006;316(3):1346. doi: 10.1124/jpet.105.091975. [DOI] [PubMed] [Google Scholar]
- 17.Chan BS, Endo S, Kanai N, et al. Identification of lactate as a driving force for prostanoid transport by prostaglandin transporter PGT. Am J Physiol Renal Physiol. 2002;282(6):F1097. doi: 10.1152/ajprenal.00151.2001. [DOI] [PubMed] [Google Scholar]
- 18.Hu Y, Wilson GS. A temporary local energy pool coupled to neuronal activity: fluctuations of extracellular lactate levels in rat brain monitored with rapid-response enzyme-based sensor. Journal of neurochemistry. 1997;69(4):1484. doi: 10.1046/j.1471-4159.1997.69041484.x. [DOI] [PubMed] [Google Scholar]
- 19.Kasischke KA, Vishwasrao HD, Fisher PJ, et al. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science (New York, NY. 2004;305(5680):99. doi: 10.1126/science.1096485. [DOI] [PubMed] [Google Scholar]
- 20.Nimmerjahn A, Kirchhoff F, Kerr JN, et al. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nature methods. 2004;1(1):31. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- 21.Brown AM, Ransom BR. Astrocyte glycogen and brain energy metabolism. Glia. 2007;55(12):1263. doi: 10.1002/glia.20557. [DOI] [PubMed] [Google Scholar]
- 22.Pucci ML, Endo S, Nomura T, et al. Coordinate control of prostaglandin E2 synthesis and uptake by hyperosmolarity in renal medullary interstitial cells. Am J Physiol Renal Physiol. 2006;290(3):F641. doi: 10.1152/ajprenal.00426.2004. [DOI] [PubMed] [Google Scholar]
- 23.Bryan RM., Jr . In: Cerebral Blood Flow and Metabolism. Edvinson L, Krause DN, editors. Lippincott Williams and Wilkins; Philadelphia: 2002. p. 311. [Google Scholar]
- 24.Frenguelli BG, Llaudet E, Dale N. High-resolution real-time recording with microelectrode biosensors reveals novel aspects of adenosine release during hypoxia in rat hippocampal slices. Journal of neurochemistry. 2003;86(6):1506. doi: 10.1046/j.1471-4159.2003.01957.x. [DOI] [PubMed] [Google Scholar]
- 25.Fox PT, Raichle ME. Stimulus rate dependence of regional cerebral blood flow in human striate cortex, demonstrated by positron emission tomography. Journal of neurophysiology. 1984;51(5):1109. doi: 10.1152/jn.1984.51.5.1109. [DOI] [PubMed] [Google Scholar]
- 26.Chaigneau E, Tiret P, Lecoq J, et al. The relationship between blood flow and neuronal activity in the rodent olfactory bulb. J Neurosci. 2007;27(24):6452. doi: 10.1523/JNEUROSCI.3141-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cauli B, Tong XK, Rancillac A, et al. Cortical GABA interneurons in neurovascular coupling: relays for subcortical vasoactive pathways. J Neurosci. 2004;24(41):8940. doi: 10.1523/JNEUROSCI.3065-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.