

Abstract
Not only is gene regulation in polyoma interesting, but it has also proven to be highly informative and illustrative of a number of novel concepts in gene regulation. Of special interest and importance are the mechanisms by which this virus switches from the expression of early gene products to late gene products after the onset of viral DNA replication. This switch is mediated at least in part by changes in transcription elongation and polyadenylation in the late region, and by the formation and editing of dsRNA in the nucleus. In this review we will summarize the regulation of RNA synthesis and processing during polyoma infection, and will point out in particular those aspects that have been most novel.
Keywords: Polyoma, Gene Regulation, Antisense RNA, Editing, Polyadenylation, Review
2. INTRODUCTION
Murine polyoma virus lytically infects mouse cells in tissue culture. In wild mice it is not known to be pathogenic, but in baby mice and in other rodents it is highly tumorigenic, and efficiently transforms rat or hamster cells in culture (1). In cell culture, polyoma lytically infects mouse cells and oncogenically transforms rat cells. The polyoma genome is small and compact, and viral gene expression is carefully regulated during infection in order to maximize mature viral output and to minimize effective host antiviral responses. Because its genome is so small, slightly larger than 5000 base pairs, the expression and regulation of viral gene expression relies heavily on the host cell machinery. This small size allows ease of manipulation of the viral genome and makes polyoma a good model system for studying not only the molecular biology of cell transformation and tumorigenesis, but also mechanisms of regulation of eukaryotic gene expression. As we will point out below, there are a number of aspects of polyoma RNA processing that offer unique insights into several novel modes of mammalian gene control.
3. GENOME ORGANIZATION
The polyoma genome is a circular DNA molecule of about 5300 base pairs. The genome is divided into “early” and “late” regions, which are expressed and regulated differently as infection proceeds (2–5) (Figure 1A). The early and late transcription units extend in opposite directions around the circular genome from startsites near the unique, bidirectional origin of DNA replication (5, 6). Primary RNA products from the early transcription unit are alternatively spliced to yield three early mRNAs which code for the large T antigen (100 kDa), the middle T antigen (56 kDa) and the small T antigen (22 kDa). Large T binds to sequences in or near the DNA replication origin region (7–10) and is involved in the initiation of DNA replication, indirectly in the autoregulation of early-strand RNA levels (11–13) and indirectly in the activation of high levels of expression from the late promoter (13, 14). The other two early proteins are dispensable for lytic infection, but are important for cell transformation. Late primary transcripts accumulate after the onset of DNA replication and are also spliced in alternative ways to give mRNAs which code for the three viral structural proteins VP1, VP2 and VP3. Figure 2 shows the relevant features of the early and late regions. Note in particular that in the early region there are two alternative 5′ splice sites and two alternative 3′ splice sites, while in the late region there is a single 5′ splice site and three 3′ splice sites, one lying upstream of the 5′ splice site.
Figure 1.
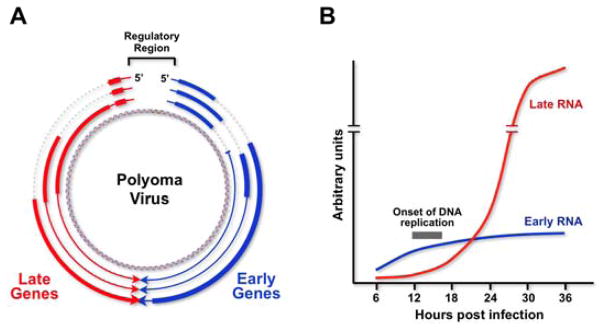
A. The circular polyoma genome expressed early and late mRNAs from opposite directions from promoters lying within an intergenic regulatory region that contains the origin of DNA replication. B. This is an idealized diagram of the temporal pattern of expression of early and late mRNAs throughout the virus life cycle. Before the onset of DNA replication, early mRNAs predominate. After DNA replication begins late RNA accumulation is dramatically enhanced, while early mRNA levels accumulate much more slowly. Since the early and late promoters are of similar strength, the regulation of relative levels of early and late mRNAs at both early and late times is posttranscriptional.
Figure 2.

Diagram of the intron-exon structures of early and late viral mRNAs. The open reading frames for early (blue) and late (pink) proteins are indicated. There are two 5′ splice sites and two 3′ splice sites in the early region. The majority of early region splicing acts to generate the three mRNAs shown. Note that middle T antigen mRNA uses a distinct 3′ splice site just downstream of that used for small T and large T mRNAs. In the late region, there are three 3′ splices sites and only a single 5′ splice site.
4. TEMPORAL REGULATION OF VIRAL GENE EXPRESSION
Gene expression during lytic infection of permissive mouse cells proceeds in a well defined temporally regulated manner (2, 15, 16) (see Figure 1B). Immediately after infection, RNA from the early transcription unit (E-RNA) begins to accumulate; however, RNA from the late transcription unit (L-RNA) accumulates more slowly. At 12 hours after infection, the early-late RNA ratio is about 4 to 1 (2, 15, 17) and in the presence of DNA replication inhibitors, the ratio is 10 to 1 or higher. At 12–15 hours post-infection, viral DNA replication commences and L-RNA begins to accumulate rapidly while E-RNA accumulates at a slower rate. In fact, the absolute amount of E-RNA in the cell is similar at 12 hr and 24 hr post infection (2, 15, 17). Thus there is a dramatic change in the relative abundances of E- and L-RNA; by 24 hours post-infection, the early to late RNA ratio is as low as 1 to 50 (2, 15, 17). This early-late “switch” is dependent on viral DNA replication; if replication is inhibited, E-RNA accumulates to abnormally high levels with minimal accumulation of L-RNA (11, 12, 17–19). It has been commonly accepted in the field for many years that the early-late switch is the result of T antigen repression of the early promoter, coupled with a transactivation of the late promoter (1). In fact, there is little experimental support for this notion, and it is not true. This temporally regulated switch is not controlled mainly at the level of transcription initiation, but results from changes in transcription elongation and/or RNA stability (13, 17, 20, 21). Work from our laboratory has shown clearly that late RNA accumulation is regulated post-transcriptionally, by what appears to be a novel RNA titration event (21), while early RNA levels are regulated by nuclear antisense RNA, which results in extensive editing of early-strand RNA molecules at late times in infection (see below) (21).
5. THE EARLY PROMOTER
The polyoma early promoter resembles other known viral and cellular promoters, and the regulation of early gene expression has been extensively studied. About 30 nt before the startsite there is a TATA box sequence and far upstream lies a 240 bp enhancer region which consists of two basic enhancer elements, A and B, both required for wild type levels of early transcription (22–25). These elements can function independently to stimulate early transcription, with differing cell specificities (23). Specific sequences in the enhancer region that are required for early promoter function have been revealed by deletion analysis (23, 24, 26–31). Interestingly, wild type polyoma cannot grow in embryonic mouse cells, owing to a deficiency in essential transcription factors in undifferentiated cells.
However, mutations in the enhancer B region can allow viral expression in these cells (32–37).
6. EARLY RNA SPLICING
The mechanism by which early mRNAs are produced by alternative splicing is interesting and unusual. In particular, the introns for mRNAs for both small T and middle T antigen are very short (62 nt for the middle T intron and only 48 nt for the small T intron) (Figure 3A). Further, the branchpoint for middle T lies 18 nt upstream of its 3′ splice site, while one prominent (mapped in vitro) branchpoint for small T is at the same position, lying only 4 nt upstream of the small T 3′ splice site (38). This distance between branchpoint and 3′ splice site is far shorter than that found in other systems. Curiously, and by a mechanism that remains unclear, there is essential cooperation between these two splice sites, both in vitro (38) and in vivo. Thus, remarkably, when we mutated either the middle T 3′ splice site dinucleotide AG to AA (mutant Py808A) (39) or the middle T branchpoint from G to G (Py791G), the splicing of both small and middle T antigen was completely blocked (Figure 3B, C). At the same time, however, large T antigen mRNA was still efficiently spliced, even though it uses the same 3′ splice site as small T. Rather than being a mere curiosity, this system may prove of interest in future studies on the mechanisms of regulation of spliceosome assembly on closely spaced, alternative 3′ splice sites.
Figure 3.

An interesting cooperation between two closely spaced 3′ splice sites is important for splicing of small T and middle T antigen mRNAs. A. The sequence in the early region spanning the small T and middle T introns. The conserved dinucleotides at the intron borders are indicated in red and green, and the mapped branchpoints are shown by the colored dots. The mutations used are indicated. B. A riboprobe spanning the early introns was prepared and used in RNAse protection assays to monitor the splicing at each of the early splice sites. Note the predicted sizes of the protected bands that result from each mRNA exon. C. Cytoplasmic RNA was isolated 24 hours after transfection with wild type polyoma genomic DNA, or that of the two indicated mutants, and subjected to RNAse protection analysis. Note that each of the mutations completely abolishes the splicing of both small T and middle T mRNAs.
7. THE LATE PROMOTER
Late transcription startsites are heterogeneous; there are at least 15 startsites, within a 96 bp region, from nt 5077–5170 (40–45). More than 90% of these are in a 25 bp region, just upstream of the late leader exon. Almost every purine in this region can be used; pyrimidines are not used. Several labs, including ours, have studied the late promoter (14, 46–51). While there is some evidence that the early and late promoters may share common elements (47–51), both we (14, 46) and others (47) have reported similar results indicating that the major contributing element to late promoter function lies in the enhancer A region. We have shown (L. Rapp and G. Carmichael, unpublished) that our minimal late promoter consists of an initiator element (Inr) (52) that specifies the major late startsite, and which is not GC-rich as are Inrs of the housekeeping gene type (53), or homologous to the terminal deoxynucleotidyl transferase type (54). It binds the zinc finger protein Miz-1,which also binds the cellular c-myc protein (55). Although late promoter activity increases in the presence of large T antigen, there is no specific effect on the late promoter, and much of the increase in late-strand transcription may be the consequence of template amplification through DNA replication, or replication-induced template conformation (14, 20).
8. LATE RNA SPLICING AND EXPORT
Late viral gene expression has a number of unusual features that have turned out to be useful for helping to unravel fundamental aspects of RNA synthesis, processing, regulation, and mRNA transport from the nucleus. Late-strand pre-mRNA molecules are processed into mature mRNAs using a highly unusual pathway that involves inefficient polyadenylation and ordered splice site selection from precursors containing tandemly repeated introns and exons. Unlike early primary transcripts, late nuclear pre-mRNAs are heterogeneous in size, the result of inefficient transcription termination and polyadenylation, and range from about 2.5 Kb to over 60 Kb in length (40, 41, 56–59). Most late pre-mRNAs are not polyadenylated (41). Further, most late RNA sequences never leave the nucleus as they are removed during mRNA splicing, and are subsequently degraded (57, 60).
Mature late mRNA molecules have polyadenylated 3′-ends which map very close to those of the early RNAs (4, 18). At their 5′-ends, late messages contain tandem repeats of the 57-base noncoding “late leader” sequence, which appears only once in the viral genome. In multigenomic late-strand transcripts, the repeats of this exon are spliced to one another, removing genome-length introns before leader-to-body splicing generates the final VP1, VP2 or VP3 messages (Figure 4). Thus, leader-leader splicing is part of an alternative splicing pathway that is unique in the field: during RNA processing, precise alternative splice site choices are made, but between identical splice sites! For example, if late transcription proceeds around the genome five times, there will be five copies of the VP1 3′ splice site in the transcript, but only the one nearest to the site of polyadenylation will be selected for splicing, and the others, though identical, will be skipped. Experimental analysis of this phenomenon revealed that in polyoma splicing requires the concomitant selection of both ends of exons, even if one of them is a poly(A) site (61, 62).
Figure 4.

Processing of late pre-mRNAs. Late pre-mRNAs contain tandem copies of the 57-nucleotide noncoding late leader exon (L). In multigenomic late-strand transcripts, these exons are efficiently spliced to one another, leading to the further stabilization and processing of late mRNAs. In the example shown here, transcription has proceeded around the genome one and a half times. The two leader exons splice to one another. Only the 3′-terminal VP1 exon (closest to the site of eventual polyadenylation) is selected for splicing, while the first one is ignored.
What is the function of the late leader exon, and does its multiplicity at the 5′-ends of late mRNAs confer any advantage to the virus? Since the leader exon contains two regions having strong complementarity to the 3′-end of mouse 18S rRNA, the possibility existed that it might play a role in facilitating ribosome loading and translation efficiency. However, replacing the leader sequence with a variety of other inserts did not affect the translation of viral late proteins (63) and there is no evidence that the leader sequence, per se, has any role other than to participate in the late pre-mRNA splicing described above (64–66).
How are the relative abundances of late mRNAs regulated? Polyoma virus late pre-mRNAs contain a single 5′ splice site and two message body 3′ splice sites (Figure 2), which are not used at equal frequencies. Owing to alternative splicing, about 5% of all late mRNAs encode VP2 (no message body splice chosen), about 15% encode VP3 (promoter-proximal 3′ splice site chosen), and about 80% encode VP1 (promoter-distal 3′ splice site chosen). Interestingly, splice site strength does not appear to determine the ratio of spliced products. Constructs containing duplicated or rearranged 3′ splice sites and sequences throughout the late region indicated that the 3′ splice closest to the polyadenylation site (the shortest terminal exon) is always used preferentially. Thus, in polyoma, late splicing choices appear to be determined largely not by sequence, but rather by the relative position of the message body 3′-splice sites (67).
In contrast to mVP1 and mVP3, mVP2 messages have no body splice, and a fraction of them (representing only about 5% of all late mRNAs) are completely unspliced. This balance between unspliced and alternatively spliced products is reminiscent of the situation found in retroviruses, which must produce both spliced and unspliced messages. Interestingly, although as many as 50% of all mVP2 RNAs are unspliced, many of these mRNAs are nevertheless exported to the cytoplasm. This is of interest because splicing is generally a prerequisite for mRNA export, unless specific cis-acting sequences in the unspliced mRNAs (often found in retroviral genomes) override retention. Examination of the intracellular distribution of late viral messages revealed, however, that mVP2 molecules are exported less efficiently than mVP1 and mVP3, in which the 5′ splice site has been removed by splicing (68). Point mutations and deletion analyses demonstrated that the efficiency of mVP2 export is inversely correlated with the strength of the 5′ splice site and that unused 3′ splice sites present in the message have little or no effect on export (68). These results suggested that the unused 5′ splice is a key player in mVP2 export. Further, results comparing spliced and unspliced forms of mVP2 molecules indicated that the process of splicing does not enhance nuclear export. Since mVP2 and some of its mutant forms can accumulate in the cytoplasm in the absence of splicing, it was proposed that in the polyoma virus system, removal of splicing machinery from mRNAs may be required, but that splicing itself is not essential (68).
9. REGULATION OF THE EARLY-LATE SWITCH
Contrary to the straightforward notion that the early-late switch is controlled primarily at the level of transcription initiation, the regulation has turned out to be much more complex, and consequently much more interesting. Both reporter assays (14, 46) and nuclear run-on analyses (17) revealed that the early and late promoters are of similar strength, and do not appear to be differentially regulated in response to viral DNA replication or early proteins such as large T antigen. Thus, at both early and late times in infection, transcription initiates at similar rates from both promoters. However, at early times late-strand RNAs fail to accumulate efficiently, and at late times early-strand RNAs appear to accumulate to much lower levels than late-strand RNAs. The result is that before viral DNA replication commences early mRNAs outnumber late mRNAs by a factor of 5 or more, while at late times in infection late mRNAs can accumulate to levels 20–50 times more abundant than early mRNAs.
9.1. Downregulation of early RNAs at late times in infection via late-strand antisense
When late RNAs accumulate, late-strand transcription termination is inefficient (Figure 4). This gives rise to giant primary transcripts that include intronic late-strand sequences that are antisense to early-strand transcripts. Mutations that destabilize these naturally-occurring nuclear antisense RNAs (splice site mutations in the late region) always have the phenotype of overexpression of early RNAs, as one would expect if early RNAs were in fact regulated by nuclear antisense RNA (13). In addition, if late RNAs are overexpressed, early-strand RNAs are repressed even more. This natural regulation can be quantitatively mimicked using nuclear antisense expression vectors (13, 21). If the polyadenylation signal is replaced with a hammerhead ribozyme sequence then RNA polymerase II transcripts are not exported from the nucleus, and accumulate there (69). Such molecules can act as specific and effective antisense inhibitors of gene expression. Nuclear antisense RNA leads to double strand RNA (dsRNA) formation in the nucleus. These dsRNA molecules serve as substrates for promiscuous adenosine-to-inosine editing by the ADAR enzyme at late times in infection (70). Editing of long dsRNAs results in up to about 50% editing of all A’s to I’s within these molecules, and editing occurs on both strands (71). Promiscuously edited RNAs are quantitatively retained in the nucleus and therefore are not translated into mutant proteins in the cytoplasm (70, 72). Thus, RNA editing dramatically reduces the amount of cytoplasmic translatable early-strand mRNAs at late times. On the other hand, edited late-strand sequences lie within a large intron which is removed and degraded in the nucleus, so that editing does not directly impact late-strand mRNAs.
9.2. Activation of late RNA accumulation
Why do late mRNAs accumulate to high levels only after the onset of viral DNA replication? Nonreplicating genomes express only very low levels of late-strand transcripts, while early-strand RNAs accumulate normally (13). However, late genes from nonreplicating genomes can be turned on if a replicating polyoma genome is introduced into the same cell (13). By systematically altering the replicating genome and looking at the effects of mutations on its ability to activate late gene expression from the non-replicating genome, it was demonstrated that late genes are not activated by large, middle or small T antigens, late virion structural proteins or even replicating DNA molecules (13). Rather, late mRNA accumulation appeared to be most strongly affected by late transcripts themselves! Thus, there is something about late transcripts that, in concert with the onset of viral DNA replication, can lead to the activation of late genes from a nonreplicating genome in the same cells. If a mutation that destabilizes late RNAs in the nucleus was introduced into an otherwise wild type genome, then that genome could not transactivate the late genes from the nonreplicating genome, even though DNA replication was normal for both and early gene expression (including T antigen expression) was efficient (13). Thus, the mechanism of activation of late gene expression was unexpected, and quite different from the incorrect but commonly assumed mechanism of late promoter transactivation by large T antigen (which, in fact, is based on very little, if any, direct experimental data).
In order to obtain more molecular details about this novel regulation of late RNA accumulation by late-strand RNA, a number of additional late region deletion and mutation constructs were made, and sequences responsible for activating late RNA accumulation were narrowed down to the late polyadenylation signal, or to the region containing it (73). Close inspection of this region of the genome revealed an interesting feature: the early and late polyadenylation signals overlap one another (Figure 5). Thus, at all times in infection, there is the potential for the early-strand and late-strand transcripts to anneal at their 3′-ends, leading to a duplex region of at least 45 base pairs. Could this overlap lead to dsRNA formation and ADAR editing of the poly(A) signals? If so, this would provide the basis for a model that can account for one of the major keys to the early-late switch. Further, this model would represent a novel way in which gene expression can be regulated in mammalian cells - regulation of polyadenylation by dsRNA formation. Our current working model is presented in Figure 6. Before the onset of viral DNA replication, transcripts from both the early and late promoters are produced. From these, early mRNAs are made and accumulate. However, late-strand primary transcripts that contain only a single leader exon splice inefficiently, and are rapidly degraded in the nucleus (66). This may be related to the fact that the VP1 and VP3 3′ splice sites are weak (67). Thus, since leader-leader splicing is important for late mRNA accumulation, late-strand RNAs are turned over rapidly in the nucleus (Figure 6A). As DNA replication commences, something changes in the nature of viral RNA processing. While we do not yet know the precise nature of this molecular trigger, all available data are consistent with the notion that it involves promoting the annealing of the 3′-ends of early-strand and late-strand RNAs. This in turn could lead to the editing of at least some of them. Since polyadenylation and transcription termination are intimately linked, poly(A) site editing (or perhaps just dsRNA formation) would lead to transcription readthrough, as is observed. On the late strand, this would generate multigenomic transcripts that would allow leader-leader splicing and late mRNA accumulation. On the early strand, transcription readthrough would lead to transcripts that cannot be productively processed and which would therefore most likely be degraded rapidly in the nucleus (Figure 6B). Further, in this model dsRNA and/or editing and polyadenylation/transcription termination would be in competition with each other. Since editing would occur before polyadenylation only some of the time, there would be a given probability of each occurring and this would lead to the heterogeneity observed in the late-strand primary transcripts in the nucleus. This new model makes a number of predictions that have been tested and which are being reported elsewhere (74). One of these is that knockdown of ADAR editing activity in mouse cells should interfere with the viral life cycle, which is observed. Another, and critical, prediction is that mutant viruses in which the 3′-ends of early and late strand transcripts can not overlap should be defective for the early-late switch, even when all other known regulatory signals are present and unchanged. This is also the case. In conclusion, studies of the polyoma virus life cycle have revealed a number of very new and interesting insights not only into how a small DNA virus can effectively use the host RNA processing machinery to regulate its temporal pattern of gene expression, but also into novel underlying mechanisms of regulation of mammalian gene expression.
Figure 5.
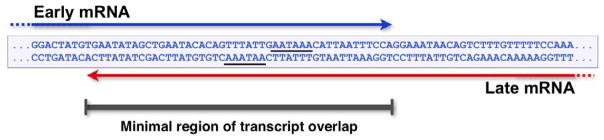
The polyoma early and late polyadenylation signals overlap, with the potential for early-strand and late-strand transcripts to anneal with one another over a region of at least 45 nucleotides. The AAUAAA hexanucleotide sequences important for cleavage and polyadenylation are underlined.
Figure 6.
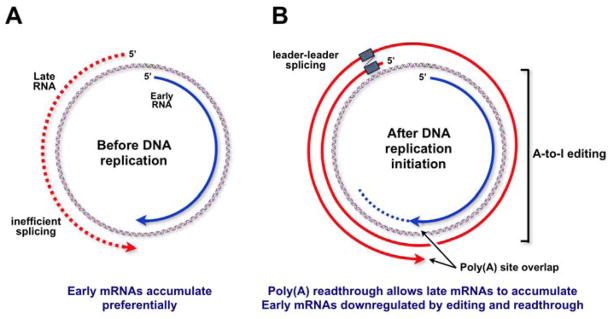
Our working model for the early-late switch. See text for details. A. At early times, both strands are transcribed. However, late-strand RNAs are not efficiently spliced and are rapidly degraded in the nucleus. B. After the onset of viral DNA replication, late polyadenylation/transcription termination becomes inefficient, perhaps owing to transcript overlap and A-to-I editing. This leads to multigenomic primary transcripts which are efficiently spliced, leading to mature late messages. In addition, late pre-mRNAs contain intronic sequences complementary to early-strand RNAs, and this nuclear antisense RNA may downregulate early-strand gene expression via A-to-I editing.
Acknowledgments
We thank N. Barrett for help with some of the work shown in Figure 3. This work was supported by grant CA04382 from the National Cancer Institute.
References
- 1.Tooze J, editor. DNA Tumour Viruses. 2. Cold Spring Harbor Laboratory, Cold Spring Harbor; New York: 1980. Molecular Biology of Tumour Viruses. [Google Scholar]
- 2.Kamen R, Lindstrom DM, Shure H, Old RW. Virus-specific RNA in cells productively infected or transformed by polyoma virus. Cold Spring Harb Symp Quant Biol. 1975;39:187–198. doi: 10.1101/sqb.1974.039.01.025. [DOI] [PubMed] [Google Scholar]
- 3.Kamen R, Favaloro J, Parker J, Treisman R, Lania L, Fried M, Mellor A. A comparison of polyomavirus transcription in productively infected mouse cells and transformed rodent cell lines. Cold Spring Harb Symp Quant Biol. 1980;44:189. doi: 10.1101/sqb.1980.044.01.009. [DOI] [PubMed] [Google Scholar]
- 4.Kamen R, Favaloro J, Parker J. Topography of the three late mRNA’s of polyoma virus which encode the virion proteins. J Virol. 1980;33:637–651. doi: 10.1128/jvi.33.2.637-651.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Griffin B, Fried M. Amplification of a specific region of the polyoma virus genome. Nature. 1975;256:175–179. doi: 10.1038/256175a0. [DOI] [PubMed] [Google Scholar]
- 6.Crawford L, Robbins A, Nicklin P. Location of the origin and terminus of replication in polyoma virus DNA. J Gen Virol. 1974;25:133–138. doi: 10.1099/0022-1317-25-1-133. [DOI] [PubMed] [Google Scholar]
- 7.Gaudray P, Tyndall C, Kamen R, Cuzin F. The high affinity binding site on polyoma virus DNA for the viral large-T protein. Nucleic Acids Res. 1981;9:5697–5710. doi: 10.1093/nar/9.21.5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pomerantz BJ, Mueller CR, Hassell JA. Polyomavirus large T antigen binds independently to multiple, unique regions on the viral genome. J Virol. 1983;47:600–610. doi: 10.1128/jvi.47.3.600-610.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dilworth S, Cowie A, Kamen R, Griffin B. DNA binding activity of polyoma virus large tumor antigen. Proc Natl Acad Sci USA. 1984;81:1941–1945. doi: 10.1073/pnas.81.7.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cowie A, Kamen R. Multiple binding sites for polyomavirus large T antigen within regulatory sequences of polyomavirus DNA. J Virol. 1984;52:750–760. doi: 10.1128/jvi.52.3.750-760.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cogen B. Virus-specific early RNA in 3T6 cells infected by a tsA mutant of polyoma virus. Virology. 1978;85:222–230. doi: 10.1016/0042-6822(78)90426-9. [DOI] [PubMed] [Google Scholar]
- 12.Farmerie WG, Folk WR. Regulation of polyomavirus transcription by large tumor antigen. Proc Natl Acad Sci USA. 1984;81:6919–6923. doi: 10.1073/pnas.81.22.6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Z, Carmichael GG. Polyoma virus early late switch - regulation of late RNA accumulation by DNA replication. Proc Natl Acad Sci USA. 1993;90(18):8494–8498. doi: 10.1073/pnas.90.18.8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cahill KB, Roome AJ, Carmichael GG. Replication-dependent transactivation of the polyomavirus late promoter. J Virol. 1990;64:992–1001. doi: 10.1128/jvi.64.3.992-1001.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piper P. Polyoma virus transcription early during productive infection of mouse 3T6 cells. J Mol Biol. 1979;131:399–407. doi: 10.1016/0022-2836(79)90083-4. [DOI] [PubMed] [Google Scholar]
- 16.Beard P, Acheson NH, Maxwell IH. Strand-specific transcription of polyoma virus DNA early in productive infection and in transformed cells. J Virol. 1976;17:20–26. doi: 10.1128/jvi.17.1.20-26.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hyde-DeRuyscher R, Carmichael GG. Polyomavirus early-late switch is not regulated at the level of transcription initiation and is associated with changes in RNA processing. Proc Natl Acad Sci USA. 1988;85:8993–8997. doi: 10.1073/pnas.85.23.8993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heiser WC, Eckhart W. Polyoma virus early and late mRNAs in productively infected mouse 3T6 cells. J Virol. 1982;44:175–188. doi: 10.1128/jvi.44.1.175-188.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamen R, Jat P, Treisman R, Favaloro J, Folk WR. 5′ Termini of polyoma virus early region transcripts synthesized in vivo by wild-type virus and viable deletion mutants. J Mol Biol. 1982;159:189–224. doi: 10.1016/0022-2836(82)90493-4. [DOI] [PubMed] [Google Scholar]
- 20.Hyde-DeRuyscher RP, Carmichael GG. Polyomavirus late pre-mRNA processing: DNA-replication-associated changes in leader exon multiplicity suggest a role for leader-to-leader splicing in the early-late switch. J Virol. 1990;64:5823–5832. doi: 10.1128/jvi.64.12.5823-5832.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Z, Batt DB, Carmichael GG. Targeted nuclear antisense RNA mimics natural antisense-induced degradation of polyoma virus early RNA. Proc Natl Acad Sci USA. 1994;91(10):4258–62. doi: 10.1073/pnas.91.10.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Villiers J, Schaffner W. A small segment of polyoma virus DNA enhances the expression of a cloned beta-globin gene over a distance of 1400 base pairs. Nucleic Acids Res. 1981;9:6251–6264. doi: 10.1093/nar/9.23.6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herbomel P, Bourachot B, Yaniv M. Two distinct enhancers with different cell specificities coexist in the regulatory region of polyoma. Cell. 1984;39:653–62. doi: 10.1016/0092-8674(84)90472-0. [DOI] [PubMed] [Google Scholar]
- 24.Mueller CR, Mes-Masson AM, Bouvier M, Hassell JA. Location of sequences in polyomavirus DNA that are required for early gene expression in vivo and in vitro. Mol Cell Biol. 1984;4:2594–2609. doi: 10.1128/mcb.4.12.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Velcich A, Kern FG, Basilico C, Ziff EB. Adenovirus E1a proteins repress expression from polyomavirus early and late promoters. Mol Cell Biol. 1986;6:4019–4025. doi: 10.1128/mcb.6.11.4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dailey L, Basilico C. Sequences in the polyomavirus DNA regulatory region involved in viral DNA replication and early gene expression. J Virol. 1985;54:739–749. doi: 10.1128/jvi.54.3.739-749.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luthman H, Nilsson MG, Magnusson G. Noncontiguous segments of the polyoma genome required in cis for DNA replication. J Mol Biol. 1982;161:533–550. doi: 10.1016/0022-2836(82)90406-5. [DOI] [PubMed] [Google Scholar]
- 28.Mueller CR, Muller WJ, Hassell JA. The polyomavirus enhancer comprises multiple functional elements. J Virol. 1988;62:1667–1678. doi: 10.1128/jvi.62.5.1667-1678.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mueller CR, Dufort D, Hassell JA. Multiple subelements within the polyomavirus enhancer function synergistically to activate DNA replication. Mol Cell Biol. 1988;8:5000–5015. doi: 10.1128/mcb.8.11.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tyndall C, LaMantia G, Thacker C, Kamen R. A region of the polyoma virus genome between the replication origin and late protein coding sequences is required in cis for both early gene expression and viral DNA replication. Nucleic Acids Res. 1981;9:6231–6250. doi: 10.1093/nar/9.23.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veldman G, Lupton S, Kamen R. Polyomavirus enhancer contains multiple redundant sequence elements that activate both DNA replication and gene expression. Mol Cell Biol. 1985;5:649–658. doi: 10.1128/mcb.5.4.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ariizumi K, Ariga H. New class of polyomavirus mutant that can persist as free copies in F9 embryonal carcinoma cells. Mol Cell Biol. 1986;6:3920–3927. doi: 10.1128/mcb.6.11.3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin ME, Yang XY, Folk WR. Expression of a 91-kilodalton PEA3-binding protein is down-regulated during differentiation of F9 embryonal carcinoma cells. Mol Cell Biol. 1992;12(5):2213–2221. doi: 10.1128/mcb.12.5.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nothias JY, Weinmann R, Blangy D, Melin F. Analysis of transcription factors binding to the duplicated PEA1 and PEA3 sites that are required for polyomavirus mutant expression in PCC4 embryonic carcinoma cells. J Virol. 1993;67(6):3036–3047. doi: 10.1128/jvi.67.6.3036-3047.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Satake M, Furukawa K, Ito Y. Biological activities of oligonucleotides spanning the F9 point mutation within the enhancer region of polyomavirus DNA. J Virol. 1988;62:970–977. doi: 10.1128/jvi.62.3.970-977.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tseng RW, Fujimura FK. Multiple domains in the polyomavirus B enhancer are required for productive infection of F9 embryonal carcinoma cells. J Virol. 1988;62:2890–2895. doi: 10.1128/jvi.62.8.2890-2895.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tseng RW, Williams T, Fujimura FK. Unique requirement for the PyF441 mutation for polyomavirus infection of F9 embryonal carcinoma cells. J Virol. 1988;62:2896–2902. doi: 10.1128/jvi.62.8.2896-2902.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ge H, Noble J, Colgan J, Manley JL. Polyoma virus small tumor antigen pre-messenger RNA splicing requires cooperation between 2 3′ splice sites. Proc Natl Acad Sci USA. 1990;87:3338–3342. doi: 10.1073/pnas.87.9.3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang TJ, Carmichael GG, Benjamin TL. A polyoma mutant encoding small but not middle T antigen demonstrates uncoupling of cell surface and cytoskeletal changes associated with cell transformation. Mol Cell Biol. 1984;4:2774–2783. doi: 10.1128/mcb.4.12.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Treisman R. Characterization of polyoma late mRNA leader sequences by molecular cloning and DNA sequence analysis. Nucleic Acids Res. 1980;8:4867–4888. doi: 10.1093/nar/8.21.4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Treisman R, Kamen R. Structure of polyoma virus late nuclear RNA. J Mol Biol. 1981;148:273–301. doi: 10.1016/0022-2836(81)90539-8. [DOI] [PubMed] [Google Scholar]
- 42.Fenton RG, Basilico C. Viral gene expression in polyomavirus-transformed cells and their cured revertants. J Virol. 1981;40:150–163. doi: 10.1128/jvi.40.1.150-163.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flavell AJ, Cowie A, Legon S, Kamen R. Multiple 5′-terminal cap structures in late polyoma virus RNA. Cell. 1979;16:357–372. doi: 10.1016/0092-8674(79)90012-6. [DOI] [PubMed] [Google Scholar]
- 44.Flavell A, Cowie A, Arrand JR, Kamen R. Localization of three major capped 5′ ends of polyoma virus late mRNA’s within a single tetranucleotide sequence in the viral genome. J Virol. 1980;33:902–908. doi: 10.1128/jvi.33.2.902-908.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cowie A, Tyndall C, Kamen R. Sequences at the capped 5′-ends of polyoma virus late region mRNAs. An example of extreme terminal heterogeneity. Nucleic Acids Res. 1981;9:6305–6322. doi: 10.1093/nar/9.23.6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cahill KB, Carmichael GG. Deletion analysis of the polyomavirus late promoter: Evidence for both positive and negative elements in the absence of early proteins. J Virol. 1989;63:3634–3642. doi: 10.1128/jvi.63.9.3634-3642.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bourachot B, Yaniv M, Herbomel P. Control elements situated downstream of the major transcriptional start site are sufficient for highly efficient polyomavirus late transcription. J Virol. 1989;63(6):2567–77. doi: 10.1128/jvi.63.6.2567-2577.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kern FG, Delli-Bovi P, Pellegrini S, Basilico C. Regulation of gene expression from the polyoma late promoter. In: Aloni Y, editor. Molecular Aspects of Papovaviruses. Martinus Nijhoff; Boston: 1987. [Google Scholar]
- 49.Kern FG, Pellegrini S, Cowie A, Basilico C. Regulation of polyomavirus late promoter activity by viral early proteins. J Virol. 1986;60:275–285. doi: 10.1128/jvi.60.1.275-285.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kern FG, Pellegrini S, Basilico C. Cis and transacting factors regulating gene expression from the polyomavirus late promoter. Cancer Cells. 1986;4:115–124. [Google Scholar]
- 51.Kern FG, Basilico C. Transcription from the polyoma late promoter in cells stably transformed by chimeric plasmids. Mol Cell Biol. 1985;5:797–807. doi: 10.1128/mcb.5.4.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smale ST, Baltimore D. The “initiator” as a transcription control element. Cell. 1989;57:103–113. doi: 10.1016/0092-8674(89)90176-1. [DOI] [PubMed] [Google Scholar]
- 53.Sehgal A, Patil N, Chao M. A constitutive promoter directs expression of the nerve growth factor receptor gene. Mol Cell Biol. 1988;8:3160–3167. doi: 10.1128/mcb.8.8.3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Landau NR, John TPS, Weissman IL, Wolf SC, Silverstone AE, Baltimore D. Cloning of terminal transferase cDNA by antibody screening. Proc Natl Acad Sci USA. 1984;81:5836–5840. doi: 10.1073/pnas.81.18.5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peukert K, Staller P, Schneider A, Carmichael G, Hänel F, Eilers M. An alternate pathway for gene regulation by Myc. EMBO J. 1997;16(18):5672–5686. doi: 10.1093/emboj/16.18.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Acheson N, Buetti E, Scherrer K, Weil R. Transcription of the polyoma virus genome: Synthesis and cleavage of giant late polyoma specific RNA. Proc Natl Acad Sci USA. 1971;68:2231–2235. doi: 10.1073/pnas.68.9.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Acheson N. Transcription during productive infection with polyoma virus and SV40. Cell. 1976;8:1–12. doi: 10.1016/0092-8674(76)90179-3. [DOI] [PubMed] [Google Scholar]
- 58.Acheson NH. Polyoma giant RNAs contain tandem repeats of the nucleotide sequence of the entire viral genome. Proc Natl Acad Sci USA. 1978;75:4754–4758. doi: 10.1073/pnas.75.10.4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Birg F, Favaloro J, Kamen R. Analysis of polyoma viral nuclear RNA by miniblot hybridization. Proc Natl Acad Sci USA. 1977;74:3138–3142. doi: 10.1073/pnas.74.8.3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Acheson N. Kinetics and efficiency of polyadenylation of late polyomavirus nuclear RNA: Generation of oligomeric polyadenylated RNAs and their processing into mRNA. Mol Cell Biol. 1984;4:722–729. doi: 10.1128/mcb.4.4.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luo Y, Carmichael GG. Splice site choice in a complex transcription unit containing multiple inefficient polyadenylation signals. Mol Cell Biol. 1991;11(10):5291–5300. doi: 10.1128/mcb.11.10.5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luo Y, Carmichael GG. Splice site skipping in polyomavirus late pre-mRNA processing. J Virol. 1991;65(12):6637–6644. doi: 10.1128/jvi.65.12.6637-6644.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rhee E, Carmichael GG. Translational efficiency of polyoma virus late messenger RNA molecules that differ in the sequence of their 5′-noncoding late leader exons. J Virol. 1989;63:432–435. doi: 10.1128/jvi.63.1.432-435.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adami GR, Carmichael GG. Polyomavirus late leader region serves and essential spacer function necessary for viability and late gene expression. J Virol. 1986;58:417–425. doi: 10.1128/jvi.58.2.417-425.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Adami GR, Carmichael GG. The length but not the sequence of the polyoma virus late leader exon is important for both late RNA splicing and stability. Nucleic Acids Res. 1987;15:2593–2610. doi: 10.1093/nar/15.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adami GR, Marlor CW, Barrett NL, Carmichael GG. Leader-to-leader splicing is required for the efficient production and accumulation of polyomavirus late mRNA’s. J Virol. 1989;63:85–93. doi: 10.1128/jvi.63.1.85-93.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Batt DB, Rapp LM, Carmichael GG. Splice site selection in polyomavirus late pre-mRNA processing. J Virol. 1994;68(3):1797–804. doi: 10.1128/jvi.68.3.1797-1804.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang Y, Carmichael GG. A suboptimal 5′ splice site is a cis-acting determinant of nuclear export of polyomavirus late mRNAs. Mol Cell Biol. 1996;16(11):6046–6054. doi: 10.1128/mcb.16.11.6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang Y, Carmichael GG. Role of polyadenylation in nucleocytoplasmic transport of mRNA. Mol Cell Biol. 1996;16(4):1534–1542. doi: 10.1128/mcb.16.4.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kumar M, Carmichael GG. Nuclear antisense RNA induces extensive adenosine modifications and nuclear retention of target transcripts. Proc Natl Acad Sci USA. 1997;94(8):3542–3547. doi: 10.1073/pnas.94.8.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang Z, Carmichael GG. The fate of dsRNA in the nucleus. A p54(nrb)-containing complex mediates the nuclear retention of promiscuously A-to-I edited RNAs. Cell. 2001;106(4):465–475. doi: 10.1016/s0092-8674(01)00466-4. [DOI] [PubMed] [Google Scholar]
- 73.Gu R, Zhang Z, Carmichael GG. How a small DNA virus uses dsRNA but not RNAi to regulate its life cycle. Cold Spring Harb Symp Quant Biol. 2006;71:293–9. doi: 10.1101/sqb.2006.71.017. [DOI] [PubMed] [Google Scholar]
- 74.Gu R, Zhang Z, DeCerbo J, Carmichael GG. 2008 submitted. [Google Scholar]