

Abstract
Purpose of review
This review summarizes recent developments in the activity, regulation, and physiology of the ABCG5 ABCG8 (G5G8) transporter and the use of its xenobiotic substrates, phytosterols, as cholesterol lowering agents in the treatment of cardiovascular disease. Recent progress has significant implications for the role of G5G8 and its substrates in complications associated with features of the metabolic syndrome.
Recent findings
Recent reports expand the clinical presentation of sitosterolemia to include platelet and adrenal dysfunction. The G5G8 sterol transporter is critical to hepatobiliary excretion of cholesterol under nonpathological conditions and has been linked to the cholesterol gallstone susceptibility. Finally, the cardiovascular benefits of cholesterol lowering through the use of phytosterol supplements were offset by vascular dysfunction, suggesting that alternative strategies to reduced cholesterol absorption offer greater benefit.
Summary
Insulin resistance elevates G5G8 and increases susceptibility to cholesterol gallstones. However, this transporter is critical for the exclusion of phytosterols from the absorptive pathways in the intestine. Challenging the limits of this protective mechanism through phytosterol supplementation diminishes the cardioprotective benefits of cholesterol lowering in mouse models of cardiovascular disease.
Keywords: ABCG5, ABCG8, atherosclerosis, cholesterol, phytosterol
Introduction
The G5G8 transporter is the body’s primary defense against the accumulation of neutral sterols from the diet. It promotes the removal of sterols from the body by mediating the transport of cholesterol and noncholesterol sterols (phytosterols) from intestinal enterocytes into the gut lumen and from hepatocytes into the biliary space. This review discusses recent advances in the understanding of the regulation and function of this complex, the implications of ABCG5 and ABCG8 polymorphisms and phytosterols on cardiovascular disease and cholethiasis, and emerging concerns over the use of phytosterols as a therapeutic lifestyle change or ‘add-on’ therapy for cholesterol lowering.
Sitosterolemia
The discovery of ABCG5 and ABCG8 as the defective genes in sitosterolemia, the animal models of this disease, and the molecular and cellular biology of G5G8 regulation have been recently reviewed [1,2]. The accumulation of phytosterols, tendon xanthomas, and accelerated atherosclerosis were previously recognized symptoms of this disease. Although hematological abnormalities had been reported in some sitosterolemic patients, this was an inconsistent finding and had not been observed in mouse models of this disease. A recent report indicates that platelet numbers are reduced in mice lacking Abcg5 [3•]. Compared with wild-type, the spleens of Abcg5−/− mice are enlarged, harbor an increased number of megakaryocytes that display disrupted membranes, and produce enlarged platelets that are depleted of granules. Interestingly, the megakaryocytes contain multivesicular bodies and have reduced mRNA levels of the liver X receptor (LXR) target genes, ABCA1 and ABCG1, and normal levels of hydroxymethylglutaryl CoA reductase (HMGR). This is in striking contrast to observations in the adrenal gland in which cholesterol synthesis is reduced, efflux pathways are increased, and cholesterol esters are depleted. Although adrenal dysfunction was not observed in G5G8-deficient mice, a recent report indicates that adrenal insufficiency and short stature were observed in sitosterolemic patients [4]. The mother of these patients also presented with ovarian failure, suggesting that phytosterol accumulation may broadly disrupt steroidogenic tissues.
The disruptions of cholesterol homeostatic mechanisms by plant sterols appear to be both phytosterol-specific and cell-type-specific. Consequently, the dietary phytosterol profile likely influences the clinical presentation of sitosterolemia. Fortunately, phytosterols use a common pathway for absorption in the intestinal enterocytes. A recent study on a mouse model of sitosterolemia [5] demonstrates that the absence of NPC1L1 prevents the accumulation of plant sterols, confirming previous reports that inhibition of NPC1L1 is a viable treatment strategy for sitosterolemia. A 2-year continuation of a clinical trial testing the use of ezetimibe for the treatment of sitosterolemic patients demonstrated that this drug effectively reduced plasma phytosterols from baseline levels for at least 2 years [6•].
Enzymology of the G5G8 pump
Each of the G-subfamily members (G1, G2, G4, G5G8) mediates the transport of native or modified sterols from cells. Unlike other family members, the pace of discovery for the regulation of G5G8 function has been severely hampered by the inability to develop reliable cell-based assays to measure G5G8 activity. Efforts to establish such assays in hepatocytes and polarized cells of intestinal origin were unsuccessful despite robust expression and successful targeting of G5G8 to the cell surface in a variety of cell types (unpublished observation). Some progress on this front was made using a canine gallbladder epithelial cell line and a lentoviral expression system encoding recombinant G5 and G8 [7]. In these studies, G5G8 mediated cholesterol efflux was greatest to bileacid micelles composed of tauroursodeoxycholate and phosphatidylcholine. G5G8 did not mediate efflux of cholesterol to Apo A-I or high-density lipoprotein (HDL), indicating that the molecular mechanism by which G5G8 promotes sterol transport is functionally distinct from that of other members of the G-subfamily as well as ABCA1. Wang et al. [8] examined the kinetics of G5G8-mediated sterol transport from donor liposomes using native complexes purified from mouse liver and reconstituted in proteoliposomes. The rates of cholesterol transport (~0.6 μmol cholesterol per mg per min) and ATP hydrolysis (~0.4 μmol per mg per min) are in close agreement, consistent with a 1 : 1 stoichiometry for ATP hydrolysis and cholesterol transport. These studies also demonstrated that the native complex is stereospecific, functions as a dimer, and is modestly inhibited by its substrates. The functional significance of this last point is not clear. The mechanism by which G5G8 promotes cholesterol efflux is not known. Previous investigators have suggested that G5G8 functions as a ‘flippase’, ‘floppase’, or perhaps lowers the activation energy required for cholesterol extraction from the canalicular membrane. Although circumstantial, a mechanism within the membrane is consistent with studies of reconstituted G5G8 in which transbilayer transport of cholesterol could not be demonstrated [8].
Posttranslational regulation of the G5G8 transporter remains largely unexplored. A recent report indicates that the G5 subunit is palmitoylated, but mutation of this site to prevent fatty acylation did not prevent complex formation, stability, trafficking to the cell surface, or its ability to mediate cholesterol excretion into bile [8]. Conversely, deletion of the C-terminal tail of G8, but not G5, prevented trafficking of the complex to the apical surface in cultured hepatocytes and biliary cholesterol excretion in G5G8-deficient mice. However, truncation of G8 was also associated with a decrease in the presence of the mature, glycosylated proteins, suggesting that the reduced activity may merely be due to a failure in the formation and folding of the heterodimer within the endoplasmic reticulum. Consequently, the functional significance of palmitoylation and the C-terminal cytoplasmic domains on G5G8 regulation remains unclear.
Physiology: excretion and absorption
Under physiological conditions, G5G8 is the principal mechanism for hepatobiliary transport of cholesterol. G5G8-independent transport of cholesterol has been observed in a mouse model of progressive familial intrahepatic cholestasis that harbors a disease causing mutation in Atp8b1 [9]. ATP8B1 (familial intrahepatic cholestasis gene 1, FIC1) is a phosphatidylserine flippase that confines this phospholipid to the cytofacial leaflet of the apical membrane and is critical for maintaining resistance of canalicular membranes to detergent extraction by bile acids. These results are consistent with the findings of previous studies in which G5G8-independent excretion of cholesterol is associated with a loss of canalicular membrane integrity and the appearance of canalicular membrane proteins in bile. However, it is likely that other mechanisms beyond hepatobiliary disease can contribute to G5G8-independent biliary excretion of cholesterol. Using HDL labeled with [14C]-cholesterol and [3H]-sitostanol, mice lacking G8 retained 25–35% of biliary cholesterol excretion [10]. This was in stark contrast to sitostanol, which accumulated in liver, but was not detectible in hepatic bile. These studies underscore that whereas G5G8-independent mechanisms may contribute to hepatobiliary transport of cholesterol, the transport of sitostanol and perhaps other phytosterols and stanols is exclusively dependent on G5G8. Similarly, G5G8 is more critical for the exclusion of noncholesterol sterols from absorption into the lymph. Whereas the absorption of cholesterol is increased by 15–20% in the absence of G8, the absorption of stanols is elevated six-fold [10]. This raises modest concern over measurements of cholesterol absorption by the dual-isotope method in which sitostanol is used as a nonabsorbable standard as fluctuations in G5G8 may allow for modest sitostanol absorption.
Although the absorption of cholesterol and noncholesterol sterols uses a common NPC1L1, ezetimibe sensitive pathway, other factors clearly contribute to the relative absorption of individual phytosterols. Although the absorption of plant sterols is universally increased in the absence of G5G8, differences in relative absorption persist. Using the stroke prone spontaneously hypertensive rat that harbors a mutation in ABCG5, lymphatic absorption of plant sterols following intralipid gavage varied by 20-fold with campesterol being the most highly absorbed and stigmasterol being the least [11]. These differences generally correlated with the extent of esterification, which is consistent with previous reports indicating a role for ACAT2 in the selectivity of sterol absorption.
An emerging body of literature suggests a substantial role of the intestine in cholesterol elimination from the body. This was first evidenced in mdr2 (ABCB4)-deficient mice, which have disrupted hepatobiliary cholesterol excretion, yet normal fecal neutral sterol output. Using isolated, perfused intestinal segments, van der Velde et al. [12•] demonstrated not only evidence for such a pathway, but also increases in intestinal cholesterol excretion following high-fat feeding. A role for G5G8 would seem likely, but G5 and G8 mRNAs were not upregulated with high-fat diet. However, anecdotal evidence is supportive of this notion, as this pathway appears to be more prominent in mice than humans and the relative abundance of mRNAs for G5G8 is greater in the intestine when compared with the liver in mice, whereas the opposite is true in humans.
Regulation of G5G8 abundance
Activation of the LXR pathway in response to cholesterol feeding promotes reduced cholesterol absorption and increased biliary excretion. Although mice lacking G5G8 do not accumulate excessive levels of plasma and tissue cholesterol, this transporter is required for both LXR-mediated fecal neutral sterol elimination and reduced cholesterol absorption [10,13]. However, an upregulation of G5G8 mRNA is not essential for increased biliary output of cholesterol. Mice lacking LXR-α failed to upregulate G5G8 in response to cholesterol feeding, but biliary cholesterol excretion was similar to that in wild-type mice, which showed a four to five-fold increase in mRNA [14].
Although the regulation of G5G8 expression by LXR is well established, more recent findings indicate that the transcriptional regulation of G5G8 is considerably more complex (Fig. 1). First, hepatocyte nuclear factor (HNF) 4-α along with GATA-binding protein 4 (GATA4) and GATA6 were shown to act synergistically to increase expression of G5G8 [15]. Consistent with coregulation of G5 and G8 subunit expression, these transcription factors promote gene expression in both directions from the ABCG5 ABCG8 intergenic promoter. Further, these authors speculated that the limited tissue distribution of G5G8 to liver and intestine was dependent on colocalization ofHNF 4-α with GATA4 or GATA6 in these tissues.
Figure 1. Regulatory elements within the ABCG5 ABCG8 intergenic promoter.
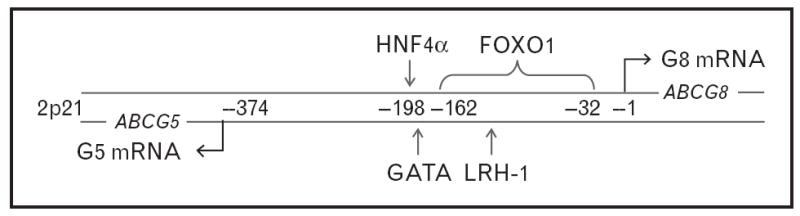
ABCG5 and ABCG8 are located on 2p21, are separated by 374 base pairs, and are encoded on opposite strands of the DNA. Numbers between the strands represent base pairs from the G8 transcriptional start site. The locations of the regulatory elements within the intergenic promoter are indicated by arrows. LXR and thyroid hormone also regulate G5 and G8 mRNA, but regulatory elements for these have not been identified. FOXO, forkhead box O1A; HNF, hepatocyte nuclear factor.
An emerging body of literature supports a link between energy metabolism, hepatic G5G8 levels, and biliary cholesterol elimination. Hypophesectomized rats demonstrate pronounced reduction in hepatic G5G8 that is accompanied by decreased biliary elimination of cholesterol and fecal neutral sterol excretion and increased cholesterol absorption [16]. Replacement of thyroid hormone restored G5G8 mRNAs and increased fecal neutral sterols. The mechanism by which thyroid hormone regulates G5G8 mRNA is not known. Likewise, hepatic insulin resistance through tissue-specific deletion of insulin receptor resulted in upregulation of G5G8 and increased biliary cholesterol excretion [17••]. The loss in hepatic insulin signaling results in disinhibition of forkhead box O1A (FOXO1) transcription factor by insulin. A FOXO1 response element was identified in the human ABCG5 ABCG8 intergenic promoter and adenoviral expression of a constitutively active FOXO1 increased mRNA for G5 and G8 in a cultured rat hepatocytes. Of considerable clinical significance, the increased G5G8 expression in insulin-resistant liver resulted in increased susceptibility to cholesterol gallstones, thereby linking insulin resistance to the increase in gallbladder disease (Fig. 2).
Figure 2. Relationships among obesity, insulin resistance, endoplasmic reticulum stress, and leptin on the abundance of G5G8 and cholethiasis.
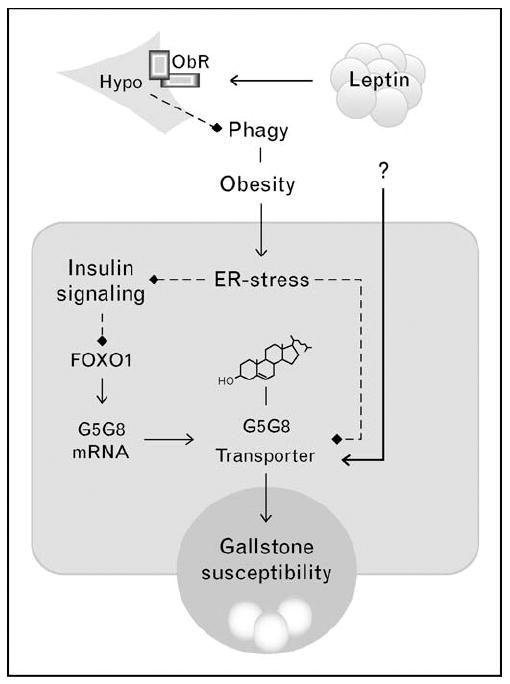
Solid lines with arrows indicate positive regulation. Dashed lines with diamonds indicate negative regulation. Insulin resistance increase G5G8 expression via disinhibition of FOXO1. Despite obesity and insulin resistance, G5G8 is reduced in leptin-deficient mice. The mechanism by which leptin increases G5G8 is not known. ER, endoplasmic reticulum; FOXO1, forkhead box O1A.
Conversely, the abundance of G5G8 protein, but not mRNA, is reduced in mice lacking a functional leptin axis [18]. Leptin replacement restored G5G8, suggesting a role for this hormone in regulating the abundance of G5G8. G5G8 can also be restored by the small molecule chaperone, tauroursodeoxycholate, suggesting a role for endoplasmic reticulum stress and the unfolded protein response (UPR). A rapidly expanding body of literature links endoplasmic reticulum stress to a number of complications associated with obesity and diabetes, including insulin resistance [19]. In this regard, insulin resistance, and endoplasmic reticulum stress appear to have opposing effects on the abundance of the G5G8 sterol transporter and biliary cholesterol elimination. In the absence of leptin signaling, reductions in G5G8 prevail. The mechanism by which leptin preserves G5G8 is not known.
G5G8 and metabolic syndrome
Cholesterol synthesis is generally greater than cholesterol absorption in obese and type II diabetic patients [20,21]. Conversely, type I diabetes is associated with greater cholesterol absorption than synthesis. This was confirmed in a rat model of type I diabetes and was associated with a decrease in G5G8 mRNAs and an increase in phytosterol absorption [22]. The extent to which changes in hepatic or intestinal G5G8 expression contribute to these differences is not clear. In a model of intestinal lipid absorption, high glucose was shown to increase cholesterol absorption as well as NPC1L1, CD36, and ABCA1 expression, but had no discernable effect on G8 mRNA. Interestingly, polymorphisms in ABCG5 were associated with changes in cholesterol metabolism that favor synthesis following weight loss [23]. These results are consistent with the notion that G5G8 plays a pivotal role in determining whether individuals are predominantly synthesizers versus absorbers of cholesterol and may therefore influence the response to lipid-lowering statin therapy as suggested by early studies of G5G8.
Although seldom life-threatening, cholethiasis, a common and serious complication, is associated with obesity and insulin resistance. In contrast to cardiovascular disease, the literature has been far more consistent with respect to polymorphisms in ABCG8 being associated with cholesterol gallstones. The D19H polymorphism has been positively associated with the risk of cholesterol gallstones in three independent studies using multiple ethnicities [24-26]. In a few studies, plasma levels of plant sterols were inversely related to BMI and markers of insulin resistance [27]. However, phytosterol supplementation had no beneficial effect on the development of obesity and insulin resistance in a high-fat diet model of obesity in mice [28].
Phytosterols, G5G8, and cardiovascular disease
The history of the development and use of phytosterols and phytostanols as cholesterol-lowering agents, the use of plasma phytosterols as biomarkers of cardiovascular disease, and the concern about their accumulation in individuals consuming gram quantities of these supplements have been reviewed [29,30]. Recent studies [31-35] extend previous observations that phytosterols and phytostanols reduce cholesterol and increase phytosterols and phytostanols in plasma. The use of plant sterols and stanols as a therapeutic lifestyle change in coordination with pharmacotherapy to achieve cholesterollowering goals is currently recommended by the National Cholesterol Education Program Adult Treatment Panel III Guidelines [36].
However, other studies have raised concerns over the use of phytosterols. Phytosterols accumulate in vascular lesions and this increases with phytosterol supplementation [37,38••]. Among the commonly consumed phytosterols, campesterol preferentially accumulates in both plasma and tissues of mice [38••,39]. Although plant sterols and ezetimibe decreased plasma cholesterol to a similar extent, ezetimibe produced a greater protective effect on atherosclerotic lesion area [38••]. An important finding of these experiments is that plant sterols impaired endothelial function and increased cerebral lesion area in a middle cerebral artery occlusion model of stroke in mice. These results indicate that the cardiovascular benefits of cholesterol lowering are not fully realized with the use of plant sterols, perhaps due to deleterious effects on the endothelium.
Beyond this single study, the extent to which phytosterols induce deleterious phenotypes in the cardiovascular system is difficult to assess because virtually all of the research on the effects of phytosterols has been conducted in vitro and often uses concentrations observed in sitosterolemic patients. Within the ‘physiological’ range, sitosterol was shown to decrease viability of human abdominal aortic endothelial cells [40]. Sitosterol induces growth arrest and apoptosis in two cancer cell lines [41]. At lower concentrations, sitosterol disrupted ceramide metabolism and sensitized cells to tamoxifen suppression of cell proliferation [42].
Although studies consistently demonstrate a relationship between cholesterol absorption, plasma plant sterols, and genetic variants of ABCG5 and ABCG8, there is no consensus on the predictive value of plasma phytosterols for cardiovascular disease. A recent report in a heterozygous familial hypercholesterolemia cohort indicates that the D19H polymorphism in the ABCG8 gene is associated with increased risk of coronary heart disease [43]. In this same cohort, the presence of both D19H and T400K increased risk of coronary heart disease and cardiovascular disease. These observations demonstrate a fundamental disconnect between the influence of genetic variants of ABCG5 and ABCG8 on plasma levels of plant sterols and their influence on cardiovascular disease risk. A similar disconnect exists for the efficacy of phytosterols on cholesterol lowering. Although the T400K polymorphism in ABCG8 influences the cholesterol-lowering effect of phytosterol supplementation, efficacy is independent of baseline plant sterol levels as well as cholesterol intake [31,34,35]. These discrepancies suggest that dietary intake of phytosterols may have a greater impact on plasma plant sterol levels than ABCG5 and ABCG8 polymorphisms, thereby undermining the predictive value of plasma phytosterol levels for cardiovascular risk.
Conclusion
G5G8 functions as a xenobiotic transporter as well as a regulator of endogenous cholesterol homeostasis. G5G8 is a major route for the elimination of excess cholesterol from the body and mediates excretion in both the liver and intestine. Disruptions in insulin signaling lead to increased G5G8 and contribute to cholesterol gallstone formation. Conversely, reductions in G5G8 lead to the accumulation of neutral sterols in plasma and tissues. Although the coordinated efforts of G5G8 and ACAT2 proficiently protect the body from phytosterols, challenging the limits of this protective mechanism for modest, but reproducible reductions in plasma cholesterol, may not confer the expected reduction in cardiovascular risk in hypercholesterolemic patients.
Acknowledgments
The authors thank Todd Porter for review of the manuscript and helpful discussions. This work was supported by the American Heart Association, Scientist Development Grant (053025N).
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
-
•
of special interest
-
••
of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (p. 196).
- 1.Kidambi S, Patel SB. Sitosterolaemia: pathophysiology, clinical presentation and laboratory diagnosis. J Clin Pathol. 2008;61:588–594. doi: 10.1136/jcp.2007.049775. [DOI] [PubMed] [Google Scholar]
- 2.Kidambi S, Patel SB. Cholesterol and noncholesterol sterol transporters: ABCG5, ABCG8 and NPC1L1– a review. Xenobiotica. 2008;38:1119–1139. doi: 10.1080/00498250802007930. [DOI] [PubMed] [Google Scholar]
- 3•.Kruit JK, Drayer AL, Bloks VW, et al. Plant sterols cause macrothrombocytopenia in a mouse model of sitosterolemia. J Biol Chem. 2008;283:6281–6287. doi: 10.1074/jbc.M706689200. This study provides a mechanism for platelet dysfunction sitosterolemia. [DOI] [PubMed] [Google Scholar]
- 4.Mushtaq T, Wales JK, Wright NP. Adrenal insufficiency in phytosterolaemia. Eur J Endocrinol. 2007;157(Suppl 1):S61–S65. doi: 10.1530/EJE-07-0222. [DOI] [PubMed] [Google Scholar]
- 5.Tang W, Ma Y, Jia L, et al. Genetic inactivation of NPC1L1 protects against sitosterolemia in mice lacking ABCG5/ABCG8. J Lipid Res. 2009;50:293–300. doi: 10.1194/jlr.M800439-JLR200. [DOI] [PubMed] [Google Scholar]
- 6•.Lutjohann D, von Bergmann K, Sirah W, et al. Long-term efficacy and safety of ezetimibe 10mg in patients with homozygous sitosterolemia: a 2-year, openlabel extension study. Int J Clin Pract. 2008;62:1499–1510. doi: 10.1111/j.1742-1241.2008.01841.x. This study demonstrates that long-term treatment of sitosterolemia with ezetimibe is effective and safe. [DOI] [PubMed] [Google Scholar]
- 7.Vrins C, Vink E, Vandenberghe KE, et al. The sterol transporting heterodimer ABCG5/ABCG8 requires bile salts to mediate cholesterol efflux. FEBS Lett. 2007;581:4616–4620. doi: 10.1016/j.febslet.2007.08.052. [DOI] [PubMed] [Google Scholar]
- 8.Wang J, Zhang DW, Lei Y, et al. Purification and reconstitution of sterol transfer by native mouse ABCG5 and ABCG8. Biochemistry. 2008;47:5194–5204. doi: 10.1021/bi800292v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groen A, Kunne C, Jongsma G, et al. Abcg5/8 independent biliary cholesterol excretion in Atp8b1-deficient mice. Gastroenterology. 2008;134:2091–2100. doi: 10.1053/j.gastro.2008.02.097. [DOI] [PubMed] [Google Scholar]
- 10.Wang HH, Patel SB, Carey MC, Wang DQ. Quantifying anomalous intestinal sterol uptake, lymphatic transport, and biliary secretion in Abcg8(−/−) mice. Hepatology (Baltimore, Md) 2007;45:998–1006. doi: 10.1002/hep.21579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamada T, Egashira N, Nishizono S, et al. Lymphatic absorption and deposition of various plant sterols in stroke-prone spontaneously hypertensive rats, a strain having a mutation in ATP binding cassette transporter G5. Lipids. 2007;42:241–248. doi: 10.1007/s11745-006-3015-3. [DOI] [PubMed] [Google Scholar]
- 12•.van der Velde AE, Vrins CL, van den Oever K, et al. Regulation of direct transintestinal cholesterol excretion in mice. Am J Physiol. 2008;295:G203–G208. doi: 10.1152/ajpgi.90231.2008. This study demonstrates an active role for the intestine in the removal of cholesterol from the body. [DOI] [PubMed] [Google Scholar]
- 13.Calpe-Berdiel L, Rotllan N, Fievet C, et al. Liver X receptor-mediated activation of reverse cholesterol transport from macrophages to feces in vivo requires ABCG5/G8. J Lipid Res. 2008;49:1904–1911. doi: 10.1194/jlr.M700470-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.van der Veen JN, Havinga R, Bloks VW, et al. Cholesterol feeding strongly reduces hepatic VLDL-triglyceride production in mice lacking the liver X receptor alpha. J Lipid Res. 2007;48:337–347. doi: 10.1194/jlr.M600170-JLR200. [DOI] [PubMed] [Google Scholar]
- 15.Sumi K, Tanaka T, Uchida A, et al. Cooperative interaction between hepatocyte nuclear factor 4{alpha} and GATA transcription factors regulates ATPbinding cassette sterol transporters ABCG5 and ABCG8. Mol Cell Biol. 2007;27:4248–4260. doi: 10.1128/MCB.01894-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galman C, Bonde Y, Matasconi M, et al. Dramatically increased intestinal absorption of cholesterol following hypophysectomy is normalized by thyroid hormone. Gastroenterology. 2008;134:1127–1136. doi: 10.1053/j.gastro.2008.01.032. [DOI] [PubMed] [Google Scholar]
- 17••.Biddinger SB, Haas JT, Yu BB, et al. Hepatic insulin resistance directly promotes formation of cholesterol gallstones. Nat Med. 2008;14:778–782. doi: 10.1038/nm1785. This study provides a mechanistic link between hepatic insulin resistance and the risk of cholethiasis. This has substantial clinical implications for the regulation of G5G8 as well as bile acid synthesis in the development of cholesterol gallstones in obese and diabetic patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sabeva NS, Rouse EJ, Graf GA. Defects in the leptin axis reduce abundance of the ABCG5-ABCG8 sterol transporter in liver. J Biol Chem. 2007;282:22397–22405. doi: 10.1074/jbc.M702236200. [DOI] [PubMed] [Google Scholar]
- 19.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29:42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- 20.Gylling H, Hallikainen M, Kolehmainen M, et al. Cholesterol synthesis prevails over absorption in metabolic syndrome. Transl Res. 2007;149:310–316. doi: 10.1016/j.trsl.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 21.Smahelova A, Hyspler R, Haas T. Relation of cholesterol metabolism and noncholesterol sterols to insulin resistance. Physiol Res. 2007;56:749–755. doi: 10.33549/physiolres.931065. [DOI] [PubMed] [Google Scholar]
- 22.Scoggan KA, Gruber H, Chen Q, et al. Increased incorporation of dietary plant sterols and cholesterol correlates with decreased expression of hepatic and intestinal Abcg5 and Abcg8 in diabetic BB rats. J Nutr Biochem. 2009;20:177–186. doi: 10.1016/j.jnutbio.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 23.Santosa S, Demonty I, Lichtenstein AH, et al. Single nucleotide polymorphisms in ABCG5 and ABCG8 are associated with changes in cholesterol metabolism during weight loss. J Lipid Res. 2007;48:2607–2613. doi: 10.1194/jlr.M600452-JLR200. [DOI] [PubMed] [Google Scholar]
- 24.Buch S, Schafmayer C, Volzke H, et al. A genome-wide association scan identifies the hepatic cholesterol transporter ABCG8 as a susceptibility factor for human gallstone disease. Nat Genet. 2007;39:995–999. doi: 10.1038/ng2101. [DOI] [PubMed] [Google Scholar]
- 25.Kuo KK, Shin SJ, Chen ZC, et al. Significant association of ABCG5 604Q and ABCG8 D19H polymorphisms with gallstone disease. Br J Surg. 2008;95:1005–1011. doi: 10.1002/bjs.6178. [DOI] [PubMed] [Google Scholar]
- 26.Grunhage F, Acalovschi M, Tirziu S, et al. Increased gallstone risk in humans conferred by common variant of hepatic ATP-binding cassette transporter for cholesterol. Hepatology (Baltimore, Md) 2007;46:793–801. doi: 10.1002/hep.21847. [DOI] [PubMed] [Google Scholar]
- 27.Chen ZC, Shin SJ, Kuo KK, et al. Significant association of ABCG8:D19H gene polymorphism with hypercholesterolemia and insulin resistance. J Hum Genet. 2008;53:757–763. doi: 10.1007/s10038-008-0310-2. [DOI] [PubMed] [Google Scholar]
- 28.Calpe-Berdiel L, Escola-Gil JC, Rotllan N, Blanco-Vaca F. Phytosterols do not change susceptibility to obesity, insulin resistance, and diabetes induced by a high-fat diet in mice. Metabolism. 2008;57:1497–1501. doi: 10.1016/j.metabol.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 29.John S, Sorokin AV, Thompson PD. Phytosterols and vascular disease. Curr Opin Lipidol. 2007;18:35–40. doi: 10.1097/MOL.0b013e328011e9e3. [DOI] [PubMed] [Google Scholar]
- 30.Thompson GR, Grundy SM. History and development of plant sterol and stanol esters for cholesterol-lowering purposes. Am J Cardiol. 2005;96(1A):3D–9D. doi: 10.1016/j.amjcard.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 31.Houweling AH, Vanstone CA, Trautwein EA, et al. Baseline plasma plant sterol concentrations do not predict changes in serum lipids, C-reactive protein (CRP) and plasma plant sterols following intake of a plant sterolenriched food. Eur J Clin Nutr. 2007 doi: 10.1038/sj.ejcn.1602969. http://www.nature.com/ejcn/journal/vaop/ncurrent/full/1602969a.html. [DOI] [PubMed]
- 32.Fransen HP, de Jong N, Wolfs M, et al. Customary use of plant sterol and plant stanol enriched margarine is associated with changes in serum plant sterol and stanol concentrations in humans. J Nutr. 2007;137:1301–1306. doi: 10.1093/jn/137.5.1301. [DOI] [PubMed] [Google Scholar]
- 33.de Jong A, Plat J, Lutjohann D, Mensink RP. Effects of long-term plant sterol or stanol ester consumption on lipid and lipoprotein metabolism in subjects on statin treatment. Br J Nutr. 2008;100:937–941. doi: 10.1017/s0007114508966113. [DOI] [PubMed] [Google Scholar]
- 34.Zhao HL, Houweling AH, Vanstone CA, et al. Genetic variation in ABC G5/G8 and NPC1L1 impact cholesterol response to plant sterols in hypercholesterolemic men. Lipids. 2008;43:1155–1164. doi: 10.1007/s11745-008-3241-y. [DOI] [PubMed] [Google Scholar]
- 35.Kassis AN, Vanstone CA, AbuMweis SS, Jones PJ. Efficacy of plant sterols is not influenced by dietary cholesterol intake in hypercholesterolemic individuals. Metabolism. 2008;57:339–346. doi: 10.1016/j.metabol.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 36.Grundy SM, Cleeman JI, Merz CN, et al. Implications of Recent Clinical Trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. Circulation. 2004;110:227–239. doi: 10.1161/01.CIR.0000133317.49796.0E. [DOI] [PubMed] [Google Scholar]
- 37.Helske S, Miettinen T, Gylling H, et al. Accumulation of cholesterol precursors and plant sterols in human stenotic aortic valves. J Lipid Res. 2008;49:1511–1518. doi: 10.1194/jlr.M800058-JLR200. [DOI] [PubMed] [Google Scholar]
- 38••.Weingartner O, Lutjohann D, Ji S, et al. Vascular effects of diet supplementation with plant sterols. J Am Coll Cardiol. 2008;51:1553–1561. doi: 10.1016/j.jacc.2007.09.074. This study demonstrates that phytosterol supplementation causes vascular dysfunction in mice. In addition, it demonstrates that inhibition of cholesterol absorption with ezetimibe is more effective at reducing atherosclerosis than phytosterols, despite similar effects on plasma cholesterol levels. [DOI] [PubMed] [Google Scholar]
- 39.Plat J, de Jong A, Volger OL, et al. Preferential campesterol incorporation into various tissues in apolipoprotein E*3-Leiden mice consuming plant sterols or stanols. Metabolism. 2008;57:1241–1247. doi: 10.1016/j.metabol.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 40.Rubis B, Paszel A, Kaczmarek M, et al. Beneficial or harmful influence of phytosterols on human cells? Br J Nutr. 2008;100:1183–1191. doi: 10.1017/S0007114508981423. [DOI] [PubMed] [Google Scholar]
- 41.Moon DO, Kim MO, Choi YH, Kim GY. beta-Sitosterol induces G2/M arrest, endoreduplication, and apoptosis through the Bcl-2 and PI3K/Akt signaling pathways. Cancer Lett. 2008;264:181–191. doi: 10.1016/j.canlet.2008.01.032. [DOI] [PubMed] [Google Scholar]
- 42.Awad AB, Barta SL, Fink CS, Bradford PG. beta-Sitosterol enhances tamoxifen effectiveness on breast cancer cells by affecting ceramide metabolism. Mol Nutr Food Res. 2008;52:419–426. doi: 10.1002/mnfr.200700222. [DOI] [PubMed] [Google Scholar]
- 43.Koeijvoets KC, van der Net JB, Dallinga-Thie GM, et al. ABCG8 gene polymorphisms, plasma cholesterol concentrations, and risk of cardiovascular disease in familial hypercholesterolemia. Atherosclerosis. 2008 doi: 10.1016/j.atherosclerosis.2008.09.018. [DOI] [PubMed] [Google Scholar]