

Abstract
The relationships between GWAS-identified and replicated genetic variants associated to Alzheimer’s disease (AD) risk and disease progression or therapeutic responses in AD patients are almost unexplored. 701 AD patients with at least three different cognitive evaluations and genotypic information for APOE and six GWAS-significant SNPs were selected for this study. Mean differences in GDS and MMSE were evaluated using non-parametric tests, GLM and mixed models for repeated measurements. Each chart was also reviewed for evidence of treatment with any cholinesterase inhibitor (AChEI), memantine or both. Relationships between therapeutic protocols, genetic markers and progression were explored using stratified analysis looking for specific effects on progression in each therapeutic category separately. Neither calculation rendered a Bonferroni-corrected statistically significant difference in any genetic marker. Mixed model results suggested differences in the average point in MMSE test for patients carrying PICALM GA or AA genotype compared to GG carriers at the end of the follow up (MMSE mean difference= −0.57 C.I.95%[−1.145−0.009], p=0.047). This observations remained unaltered after covariate adjustments although did not achieve predefined multiple testing significance threshold. PICALM SNP also displayed a significant effect protecting against rapid progression during pharmacogenetics assays although it observed effect displayed heterogeneity among AD therapeutic protocols (p=0.039). None of studied genetic markers was convincingly linked to AD progression or drug response. However, by using different statistical approaches, PICALM rs3851179 marker displayed consistent but weak effects on disease progression phenotypes.
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia. It is expected that AD prevalence will be quadrupled by 2040, reaching a worldwide number of 81.1 million affected individuals (Ballard, et al., 2011). In spite of the knowledge that genetic factors may account for about 60–80% of AD susceptibility (Wingo, et al., 2012), until very recently the only genetic factor almost universally associated to non-hereditary or sporadic AD risk was the APOE haplotype ɛ4 (Corder, et al., 1993, Strittmatter, et al., 1993). Furthermore, APOE ɛ4 effect on AD age-at onset (AAO) and mild cognitive impairment conversion rate are also well known (Aggarwal, et al., 2005, Locke, et al., 1995). In contrast, APOE locus involvement on AD progression or its pharmacogenetics effects on AD therapies has been largely debated and disputed (Schmidt, et al., 2011). Although still controversial, this last observation might imply that APOE could be involved mainly in human susceptibility to AD and not in the disease progression, its prognosis or AD drug’s effectiveness which could depend on completely different set of genetic and exogenous factors.
Genome wide association studies (GWAS) are revolutionizing the genetic knowledge of AD. Of note, the discovery of novel risk factors associated to AD is still underway and it is suspected that discovered markers are just the tip of a genomic iceberg containing several hundreds or even thousands of very low penetrance alleles weakly linked to the disease risk (Roses, 1998). Currently, GWAS, together with extensive meta-analyses of multiple independent studies, have elevated up to ten the genetic markers with an uncontroversial link to AD risk (Antunez, et al., 2011a, Harold, et al., 2009, Hollingworth, et al., 2011, Lambert, et al., 2009, Naj, et al., 2011, Seshadri, et al., 2010). These novel loci are dispersed in the entire genome and its mechanisms of action in AD pathogenesis are mostly unknown.
The relationships between uncontroversial GWAS-isolated genetic SNPs associated to AD risk and disease progression or therapeutic responses in AD patients are almost unexplored to date. Of Note, recent studies using follow up data obtained from AD patients suggested that PICALM and CLU variants could be associated with cognitive decline in AD as measured by change in Clinical Dementia Rating-sum of boxes (CDR-SB) score from the baseline. However, obtained findings did not pass multiple-test correction (Hu, et al., 2011).
In the present study we systematically analyzed the clinical effect of seven GWAS-isolated genetic SNPs. To our knowledge none of studied genetic markers was previously analyzed in relation with AD progression or drug response in our population. We have selected only SNPs that have been previously isolated as GWAS-significant for AD risk (Harold, et al., 2009, Lambert, et al., 2009, Seshadri, et al., 2010) and that have been corroborated in our population as risk or protective factors for AD (Antunez, et al., 2011a, Antunez, et al., 2011b, Ramirez-Lorca, et al., 2009, Seshadri, et al., 2010). Only CR1 rs3818361 SNP displayed a weaker effect on AD risk in Spain compared to other European studies although its effect direction (risk allele) was the same as originally reported in the French population (Lambert, et al., 2009). The rest of markers were statistically significant in our series during risk analysis and displayed identical effects on AD susceptibility in terms of magnitude and direction of their effect when compared with other European populations.
Materials & Methods
Subjects
1. Patient evaluation (diagnostics and follow up)
AD cases represent patients seen at a single recruiting center: The Memory Clinic of Fundació ACE, Institut Català de Neurociències Aplicades. Fundació ACE is one of the two reference Alzheimer centers for a population of 550.000 inhabitants living in central Barcelona. All subjects in this area of influence live at a distance of less than 30 minutes from Fundació ACE. Patients are referred for evaluation of cognitive impairment by their primary care physicians or primary care neurologist. Seven hundred and one Alzheimer’s disease patients with at least three different cognitive evaluations (basal plus two follow up examinations) were selected for this study. Follow-up diagnoses were made with full knowledge of prior classification, and prior neurobehavioral data. The diagnosis and follow-up evaluations of patients were made following standard criteria (McKhann, et al., 1984, Neary, et al., 1998, Petersen, et al., 1999, Winblad, et al., 2004).
All subjects receive a thorough structured neurological evaluation including history, examination, Mini-mental state examination (MMSE), Blessed dementia rating scale, Neuropsychiatric Inventory questionnaire (NPI-Q), Tinnetti scale for gait and balance, as well as GDS scoring. Family members or caregivers are interviewed by a social worker. A neuropsychological evaluation was administered to all patients including tests sensitive for attention, verbal learning and memory, language, visual gnosis, praxis and executive functions. Tests included: Temporal, Spatial and Personal Orientation, Digit spans (forwards and backwards), Block Design, and Similarities subtests of Wechsler Adult Intelligence Scale-III (WAIS-III); The Word List Learning from the Wechsler Memory Scale (WMS-III); The 15-item abbreviated Boston Naming Test; Poppelreuter’s Test, and Luria’s Clocks Test; Ideomotor and Imitation praxis; the Automatic Inhibition subtest of the Syndrom-Kurtz Test (SKT); Phonemic Verbal Fluency (words with ‘p’ in one minute), Semantic Verbal Fluency (‘animals’ in one minute) and the Spanish version of the Clock Test”. Patients have neuroimaging (mostly CT) and complete blood workup (including vitamin B12, folate and TSH) performed. SPECT imaging would be solicited in cases with unclear differential diagnosis. A daily diagnostic conference is held with the participation of six neurologists, four neuropsychologists and two social workers. Diagnosis of dementia and type of dementia are established by consensus according to DSM-IV criteria for dementia, and NINCDS-ADRDA criteria for possible or probable AD(McKhann, et al., 1984).
2. Pharmacotherapy categories
Each chart was reviewed for evidence of treatment with any cholinesterase inhibitor (AChEI), memantine or both. The patients were classified into four usage groups: those who never used AChEIs or Memantine during the entire course of the study, those who were taking AChEIs as monotherapy (irrespective of the AChEI drug employed), those who were taking a combined therapy (Memantine plus AChEI), and those who were taking Memantine as monotherapy. The decision to treat with an AChEI, memantine or both was made at the neurologist’s discretion and depends on clinical situation of each patient.
3. Follow up measurements
MMSE and Global Deterioration Score (GDS) variation during follow up were selected as target variables to evaluate disease progression in this study. We constructed four different variables based on MMSE and GDS values and their timing of administration, i.e.: at the time of AD diagnosis (basal) and at the last available follow up data point for both scales. Variables were defined as: 1) “MMSE decay”= Basal MMSE score minus Last Follow up MMSE score; 2) “GDS grow” =Last Follow up GDS minus Basal GDS; 3) MMSE rate=MMSE decay/follow up time expressed in years; and 4) GDS rate= GDS grow/follow up time expressed in years).
4. Rapid progression definition
We established the cut-off to define rapid progression according to Cortes et al. (Cortes, et al., 2008). Individuals with MMSE score point decrease per year (MMSE rate) higher than 4.5 were considered rapid progressors. Following this criterion 14.3% of AD patients displayed a rapidly progressive AD phenotype. Using this criterion we found 601 normal progressors and 100 rapid progressors.
5. Ethical issues
Written informed consents were obtained from all individuals included in this study. The referral center ethics committees and NeoCodex have approved this research protocol that is in compliance with national legislation and the Code of Ethical Principles for Medical Research Involving Human Subjects of the World Medical Association.
Genotyping
We extracted DNA using Magnapure technology (Roche Diagnostics, Mannheim, Germany). SNPs were selected on the basis of GWAS significance during meta-analyses (Antunez, et al., 2011a, Harold, et al., 2009, Hollingworth, et al., 2011, Lambert, et al., 2009, Naj, et al., 2011, Seshadri, et al., 2010) and positive validation in the Spanish population (Antunez, et al., 2011a, Antunez, et al., 2011b, Seshadri, et al., 2010). Genotypes for selected SNPs in APOE (rs429358 (SNP 112) and rs7412 (SNP 158)), CLU (rs11136000), PICALM (rs3851179), CR1 (rs3818361), BIN1 (rs744373), EXOC3L2 (rs597668) and MS4A gene cluster (rs1562990) were generated during case-control analyses previously reported by us. Consequently, all genotyping protocols have been described (Antunez, et al., 2011a, Antunez, et al., 2011b, Ramirez-Lorca, et al., 2009, Seshadri, et al., 2010). Of note, we selected genotypes from 701 AD patients having clinical follow-up data available. We obtained 4156 analyzable genotypes in 701 individuals. Calculated genotype conversion rate for this study reached 98.9%. Genotype distribution, minor allele frequency and Hardy-Weinberg equilibrium were also calculated for each SNP marker (supplementary table 1). A slightly Hardy-Weinberg deviation was observed for rs3818361 (CR1) (p=0.01, Pearson’s goodness-of-fit chi-square).
Statistical analysis
a. Basic statistics of Single Nucleotide Polymorphisms
For statistical analysis of genotype distribution and test for deviation of Hardy–Weinberg equilibrium (HWE) we employed tests adapted from Sasieni (1997 (Sasieni, 1997)). These calculations were performed on the online resource facility at the Institute for Human Genetics, Munich, Germany (http://ihg.gsf.de).
b. Studies for modeling AD progression and pharmacogenetics
Due to the exploratory nature of our research, we decided to analyze the effect of SNP markers in disease progression in multiple ways by employing different measurements, assuming pre-fixed end-points or analyzing its interaction with important covariates and therapeutic protocols. The basic idea was to look for consistent effects on the phenotype irrespective of statistic methodologies or covariates employed.
Having this number of phenotypes to evaluate, we decided to analyze a single genetic model to diminish multiple testing problems. We selected the dominant model for the minor allele which is a well-recognized biological model and also provides insights on recessive effects of the major allele. Of note, risk or protective effects previously described for selected SNPs are also referred to same alleles. Because observed MAF in most of selected markers was relatively low (MAF<0.3), selected model may also capture the allelic and trend (additive) models due to the lack of independence between different models (Sasieni, 1997).
Mean differences in GDS (grow and rate) and MMSE (decay and rate) between genotypic groups were evaluated using a non-parametric test (Mann-Whitney U-Test) assuming a dominant model for each SNP maker. To discard a random co-linearity of SNP genotypes with well-established variables affecting disease progression and to demonstrate independence for nominal effects, we further re-calculate genotype estimates using a general linear model (GLM). GLM was employed to adjust observed genotype effects on MMSE and GDS metrics by basal cognition (Baseline MMSE), age at diagnostic, gender, education, presence of depression, pharmacotherapy category and follow up time (expressed in number of days to follow up). Selected covariates are classic factors usually affecting to AD progression.
Mixed models were also employed to analyze longitudinally the effect of SNPs in disease progression. Specifically, the associations between loci and cognitive functions (GDS and MMSE) were evaluated with linear mixed models for three repeated measurements (basal evaluation plus two follow ups). All mixed models included time, genotype and its interaction term with time. Genotype effects on GDS and MMSE dynamics were initially calculated using genotype, follow-up time and interaction term as fixed factors without any additional adjustment. Covariate adjustments were performed only for significant signals by adding the same set of covariates employed for GLM modeling (i.e.: Baseline MMSE, age at diagnostic, gender, education, presence of depression, pharmacotherapy category and follow up time).
The coefficient for time represents the mean cognitive decline during the study. Coefficients for the main genotype effect could be interpreted as the average point difference in cognitive tests (MMSE or GDS) associated with each genotypic group (presence of the minor allele, dominant model) compared with the reference group (individuals with absence of the minor allele). The coefficients of the genotype-by-time interaction terms can be interpreted as the average annual difference in slopes (rate of changes in MMSE or GDS scores) between genotypic groups.
c. Pharmacogenetics
We completed the exploration of available data looking for interactions between selected SNPs and therapeutic protocols. The basic idea was to classify the patients into rapid and normal progressors and stratified them by therapeutic groups. The general characteristics of both groups (rapid and normal progressors) are described in supplementary table 2. Relationships between therapeutic protocols and SNPs were assessed in the simplest way using Mantel-Haenszel stratified analysis looking for specific effects of genotypes on rapid progressors rate in each therapeutic categories separately (no treatments, AChEIs only, AChEIS plus memantine and memantine only). Interactions between selected genotypes and therapeutic protocols were the measured using the Breslow-Day test with three degrees of freedom (d.f). Independence of observed interactions with common factors affecting rapid progression was assessed using conventional binary logistic regression-based analysis by employing rapid progression (rapid versus normal progressors) as dependent variable and genotype, therapeutic protocol and interaction term (genotype*therapeutic protocol) as independent variables. Covariate adjustments were performed as previously described for GLM and mixed models (for details see above).
All Statistical analyses were carried out using the Statistical Package for the Social Sciences Software (SPSS 15.0, Evanston, IL, USA). Study wise or global significance for this study was established at p<0.00102 assuming independence for all statistical test performed (49 tests).
Results
To explore the role of candidate SNPs in disease progression we constructed four different variables (GDS grow and rate; MMSE decay and rate). We analyzed the effect of SNPs in disease progression but neither unadjusted nor adjusted calculations rendered a Bonferroni-corrected statistically significant difference in candidate genotypes or APOE (p<0.00102 for 49 tests, table 1). For almost all SNPs the lack of association was the rule among analyzed end-points. In spite of nominal association obtained for APOE genotype in GDS grow (p=0.048) and for CR1 genotype in GDS rate (p=0.043), both associations did not persist after covariate analyses and multiple testing adjustment (table 1).
Table 1.
Effect of SNP markers on Alzheimer’s disease progression measurements.
| Loci | SNP | GDS grow | GDS rate | MMSE decay | MMSE rate | ||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | Sd | Mean | Sd | Mean | Sd | Mean | Sd | ||
| APOE (diplotypes based on rs429358 and rs7412 SNPs) | |||||||||
| ɛ4 negative | 1.14** | 1.06 | 0.37 | 0.47 | 7.2 | 6.4 | 2.44 | 2.72 | |
| ɛ4 positive | 1.30** | 1.08 | 0.37 | 0.41 | 7.7 | 7 | 2.16 | 2.75 | |
| BIN1 rs744373 | |||||||||
| TT | 1.17 | 1.09 | 0.36 | 0.46 | 7.15 | 6.8 | 2.2* | 2.96 | |
| TC+CC | 1.26 | 1.06 | 0.38 | 0.42 | 7.77 | 6.4 | 2.4* | 2.5 | |
| CLU rs11136000 | |||||||||
| AA | 1.22 | 1.03 | 0.36 | 0.42 | 7.6 | 2.3 | 2.33 | 2.79 | |
| AC+CC | 1.20 | 1.1 | 0.38 | 0.45 | 7.4 | 2.3 | 2.3 | 2.71 | |
| PICALM rs3851179 | |||||||||
| GG | 1.27 | 1.09 | 0.38 | 0.45 | 7.9 | 6.9 | 2.38 | 2.67 | |
| GA+AA | 1.16 | 1.06 | 0.37 | 0.43 | 7.08 | 6.4 | 2.29 | 2.84 | |
| EXOC3L2 rs597668 | |||||||||
| TT | 1.18* | 1.09 | 0.37 | 0.44 | 7.5 | 6.8 | 2.35 | 2.9 | |
| TC+CC | 1.34* | 1.05 | 0.39 | 0.47 | 7.6 | 6.4 | 2.22 | 2.3 | |
| CR1 rs3818361 | |||||||||
| CC | 1.19 | 1.1 | 0.36** | 0.48 | 7.4 | 6.8 | 2.25 | 2.9 | |
| CT+TT | 1.27 | 1.04 | 0.39** | 0.39 | 7.6 | 6.3 | 2.44 | 2.3 | |
| MS4A rs1562990 | |||||||||
| AA | 1.22 | 1.04 | 0.36 | 0.42 | 7.6 | 6.9 | 2.33 | 2.8 | |
| AC+CC | 1.20 | 1.1 | 0.38 | 0.46 | 7.4 | 6.5 | 2.31 | 2.7 | |
Note: GDS grow and MMSE decay metrics correspond to mean score points on each genotype category. GDS and MMSE rates are expressed as mean loss or gain of points per year respectively.
Trend toward association for mean difference (diff) using U-test or GLM: 0.1>p>0.05.
nominal statistical significance for diff using U-test or GLM: p<0.05. P-values observed on each analysis is summarized supplementary table 2.
In contrast, a weak, but non-significant trend for BIN1 and PICALM genotypes was observed in the four cognitive metrics examined (table 1; supplementary table 3). Accordingly, results obtained using linear mixed models for the main genotype effects of PICALM and BIN1 displayed the same trend towards association with p values below 0.05 with or without covariate adjustment. Observed effect size and direction were also consistent with U-tests and GLM results (supplementary table 3 and 4).
The effect of candidate genotypes using rapidly progressive AD definition was also examined (Cortes, et al., 2008, Schmidt, et al., 2011). APOE and, PICALM genotypes displayed nominal interaction with therapy on rapidly progressing phenotype. Only PICALM resisted covariation. Interaction between PICALM and therapeutic protocol on rapid disease progression suggested the existence of effect heterogeneity among therapeutic categories (p=0.039 for Breslow-day test) and resisted multivariate adjustments p=0.019 (table 2). PICALM genotype effect separated by therapeutic protocol is represented in figure 1.
Table 2.
Pharmacogenetics effect of candidate SNPs.
| Loci | SNP | AD Progression | OR (Mantel-Haenszel)* | Breslow-Day Test | Logistic Regression | ||||
|---|---|---|---|---|---|---|---|---|---|
| Rapid | Normal | Common OR | C.I.95% | Chi-square (3df) | p-value | Mean Effect (p-value) | I (p-value) | ||
| APOE rs429358; rs7412 | |||||||||
| ɛ4− | 63 | 323 | 0.685 | 0.44–1.06 | 7.88 | 0.048 | 0.56 | 0.54 | |
| ɛ4+ | 37 | 277 | |||||||
| MS4A rs1562990 | |||||||||
| AA | 35 | 222 | 1.078 | 0.69–1.68 | 2.85 | 0.414 | n.a. | ||
| AC+CC | 65 | 378 | |||||||
| CLU rs11136000 | |||||||||
| CC | 36 | 240 | 1.185 | 0.76–1.85 | 1.908 | 0.592 | n.a. | ||
| CT+TT | 64 | 360 | |||||||
| PICALM rs3851179 | |||||||||
| GG | 54 | 275 | 0.739 | 0.48–1.13 | 8.364 | 0.039 | 0.016 | 0.019 | |
| GA+AA | 45 | 310 | |||||||
| CR1 rs3818361 | |||||||||
| CC | 66 | 404 | 1.047 | 0.66–1.65 | 1.018 | 0.797 | n.a. | ||
| CT+TT | 32 | 189 | |||||||
| BIN1 rs744373 | |||||||||
| TT | 46 | 297 | 1.174 | 0.77–1.79 | 5.691 | 0.132 | n.a. | ||
| TC+CC | 54 | 295 | |||||||
| EXOC3L2 rs597668 | |||||||||
| TT | 74 | 430 | 0.86 | 0.52–1.43 | 2.668 | 0.446 | n.a. | ||
| TC+CC | 23 | 156 | |||||||
Note:OR was calculated using Mantel Haenszel method. OR estimates were calculated separately according to therapeutic categories (untreated, AChEIs only, Memantine plus AChEIS and Memantine only). Once obtained each, a common OR is calculated using MH stratified analysis. The effect estimate is valid only when Breslow-Day interaction test is negative. I: genotype*therapeutics interaction term included in regression-based analyses. A significant p-value confirms SNP effect heterogeneity between therapeutic groups.
Fig 1.
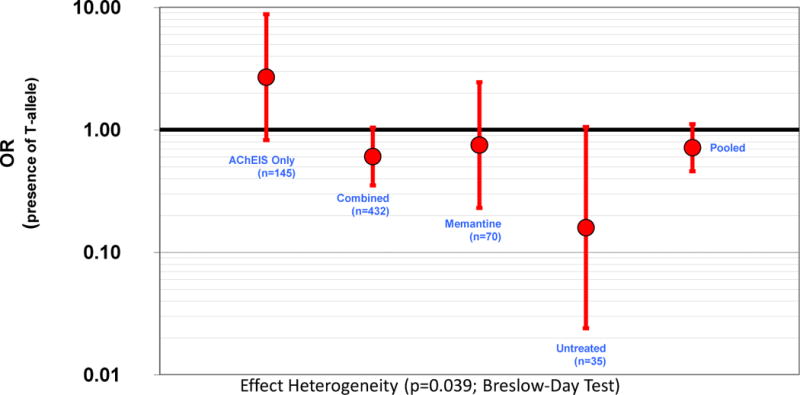
PICALM rs3851179 effect on AD rapid progression phenotype. The patients have been splited in four therapeutic categories
Discussion
The genetics of AD is being disentangled using agnostic genome-wide research methods (Bertram, 2011). However, genetic research using identified SNPs on AD prognosis or pharmacogenetics is still pending. Six out of seven SNPs analyzed in this study are considered uncontroversial GWAS-significant AD loci (Antunez, et al., 2011a, Corder, et al., 1993, Harold, et al., 2009, Lambert, et al., 2009, Seshadri, et al., 2010, Strittmatter, et al., 1993). In contrast, EXOC3L2 SNP effect on AD risk is still disputed (Naj, et al., 2011, Seshadri, et al., 2010). EXOC3L2 is a controversial signal and it could be considered an APOE proxy rather than a true independent AD signal. Both loci (APOE and EXOC3L2) are displaying negative results which in turn could be interpreted as consistent due to its physical proximity and weak LD.
We faced the research question of AD prognosis using a relatively large and homogeneous series of AD patients (n=701) from Fundació ACE (Barcelona, Spain). The use of homogeneous series for follow-up analyses has advantages in terms of diagnostic and treatment criteria homogeneity of studied individuals. However, independent replication of our observations will be also an essential step to corroborate observed findings in present study. Using Rapid progression analyses and linear mixed models we obtained more significant results compared to quantitative cross-sectional studies in two related cognitive scores (MMSE and GDS), however we consider interesting to examine involvement of genetic markers using multiple end-points and statistical approaches looking for consistent results.
Observed results suggested differences in the average point in MMSE test for patients carrying PICALM GA or AA genotype compared to GG carriers at the end of the follow up. Conversely, patients carrying minor allele for BIN1 (TC+CC) displayed the opposite effect. Both observations remained almost unaltered after covariate adjustments (supplementary table 4) and both also converged with mixed model observations. PICALM data also suggested that genotype protection against rapid progression may be effective using a combined therapy but not using cholinesterase inhibitors alone.
We are very cautious on these observations because observed effects are based on relatively small numbers and didn’t reach statistical significance after correction for multiple testing. Furthermore, our study has limitations. The lack of information in our series for other psychoactive treatments or the presence of psychotic symptoms could be the most important. Both factors have been related with disease progression and could distort observed associations. Moreover, tautological considerations have to take in mind when pre-established therapeutic categories are analyzed. In fact, each subcategory is strongly related to a specific clinical status (i.e. ACHEIS group is related to mild to moderate AD). Although we tried to control this phenomenon, named channeling effect, by using baseline MMSE as a covariate, we think that channeling could persists even after controlling for baseline cognitive status. Unfortunately, this is a general limitation of retrospective studies and it only can be corrected by increasing sample size and by analyzing each therapeutic category separately. Alternatively, prospective and controlled clinical trials might help to corroborate the effect of target SNPs on disease progression.
However, we think that the consistence of SNP effects among different analyses could be an interesting alternative to find out true genetic SNPs related to AD progression. This could be the case of PICALM rs3851179 SNP which has been associated with disease progression using linear mixed models and with rapid progression using stratified analyses in our series. Importantly, PICALM rs3851179 marker also resisted intense covariate adjustments. Taking into account our findings, we feel that, if exist, the role of this PICALM marker in disease progression or pharmacogenetics will be small. In accordance with this last suggestion, a weak effect for the same marker was recently detected in an independent study (Hu, et al., 2011). Among the studied markers tested in that data set, only one marker, PICALM (rs3851179), showed a trend towards association for genotype effects on the change in CDR-SB over time for AD subjects (Bonferroni adjusted p=0.08) whereas CLU marker was associated with CDR-SB slopes (Hu, et al., 2011). Interestingly, the result for PICALM seems concordant with our findings. However, differences in patient selection, genotype modeling and statistical methods prevented a direct comparison of both results. Taking together, PICALM rs3851179 would be the most promising result obtained to date although cannot consider our results as a replication of previous findings due to methodological differences among both studies.
It is important to reinforce the notion that none of the analysis surpassed pre-established multiple testing corrected significance threshold. Consequently, the main finding of this research is that none of studied genetic markers can be convincingly linked to AD progression or drug efficacy. We suspect that the real effects of GWAS AD markers in disease progression might be as small as those observed during risk analyses (Antunez, et al., 2011a, Harold, et al., 2009, Lambert, et al., 2009, Seshadri, et al., 2010). Power analysis supported this speculation (supplementary table 5). Our results indicated that this series has enough power to capture small effect sizes assuming an alpha of 0.05. As we expected, detectable differences between genotypic groups must be bigger if we assume an alpha corrected for multiple testing (P<0.001). However, minimum detectable effect sizes are relatively small on average even assuming an alpha=0.001. Evidently, below calculated thresholds genuine effects could exist and they cannot be safely detected using available sample size. Such putative small differences, if truly exist, would have little impact on clinical management of patients. Further research by increasing sample size or using independent validating series will be necessary to corroborate or discard current observations. In addition we believe that GWAS analysis in well controlled samples collected with the goal of studying disease progression in treated populations would be able to isolate stronger genetic factors with a high impact on disease progression and pharmacogenetics. This information will be critical for clinical trials designs, to improve prognosis and therapeutic management of AD patients.
Supplementary Material
Acknowledgments
We would like to thank patients and controls who participated in this project. We are indebted to Trinitat Port-Carbó and her family who are supporting Fundació ACE research programs. This work has been partially funded by the Ministerio de Educación y Ciencia (PCT-010000-2007-18), (DEX-580000-2008-4), (Gobierno de España), Corporación Tecnológica de Andalucía (08/211) and Agencia IDEA (841318)(Consejería de Innovación, Junta de Andalucía).
Bibliography
- Aggarwal NT, Wilson RS, Beck TL, Bienias JL, Berry-Kravis E, Bennett DA. The apolipoprotein E epsilon4 allele and incident Alzheimer’s disease in persons with mild cognitive impairment. Neurocase. 2005;11(1):3–7. doi: 10.1080/13554790490903038. [DOI] [PubMed] [Google Scholar]
- Antunez C, Boada M, Gonzalez-Perez A, Gayan J, Ramirez-Lorca R, Marin J, Hernandez I, Moreno-Rey C, Moron FJ, Lopez-Arrieta J, Mauleon A, Rosende-Roca M, Noguera-Perea F, Legaz-Garcia A, Vivancos-Moreau L, Velasco J, Carrasco JM, Alegret M, Antequera-Torres M, Manzanares S, Romo A, Blanca I, Ruiz S, Espinosa A, Castano S, Garcia B, Martinez-Herrada B, Vinyes G, Lafuente A, Becker JT, Galan JJ, Serrano-Rios M, Vazquez E, Tarraga L, Saez ME, Lopez OL, Real LM, Ruiz A. The membrane-spanning 4-domains, subfamily A (MS4A) gene cluster contains a common variant associated with Alzheimer’s disease. Genome Med. 2011a;3(5):33. doi: 10.1186/gm249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunez C, Boada M, Lopez-Arrieta J, Moreno-Rey C, Hernandez I, Marin J, Gayan J, Gonzalez-Perez A, Real LM, Alegret M, Tarraga L, Ramirez-Lorca R, Ruiz A. Genetic association of complement receptor 1 polymorphism rs3818361 in Alzheimer’s disease. Alzheimers Dement. 2011b;7(4):e124–9. doi: 10.1016/j.jalz.2011.05.2412. [DOI] [PubMed] [Google Scholar]
- Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer’s disease. Lancet. 2011;377(9770):1019–31. doi: 10.1016/S0140-6736(10)61349-9. [DOI] [PubMed] [Google Scholar]
- Bertram L. Alzheimer’s genetics in the GWAS era: a continuing story of ‘replications and refutations’. Curr Neurol Neurosci Rep. 2011;11(3):246–53. doi: 10.1007/s11910-011-0193-z. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Cortes F, Nourhashemi F, Guerin O, Cantet C, Gillette-Guyonnet S, Andrieu S, Ousset PJ, Vellas B. Prognosis of Alzheimer’s disease today: a two-year prospective study in 686 patients from the REAL-FR Study. Alzheimers Dement. 2008;4(1):22–9. doi: 10.1016/j.jalz.2007.10.018. [DOI] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1088–93. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Beaumont H, Warden D, Wilcock G, Love S, Kehoe PG, Hooper NM, Vardy ER, Hardy J, Mead S, Fox NC, Rossor M, Collinge J, Maier W, Jessen F, Ruther E, Schurmann B, Heun R, Kolsch H, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Gallacher J, Hull M, Rujescu D, Giegling I, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Pankratz VS, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Dickson DW, Graff-Radford NR, Petersen RC, van Duijn CM, Breteler MM, Ikram MA, DeStefano AL, Fitzpatrick AL, Lopez O, Launer LJ, Seshadri S, Berr C, Campion D, Epelbaum J, Dartigues JF, Tzourio C, Alperovitch A, Lathrop M, Feulner TM, Friedrich P, Riehle C, Krawczak M, Schreiber S, Mayhaus M, Nicolhaus S, Wagenpfeil S, Steinberg S, Stefansson H, Stefansson K, Snaedal J, Bjornsson S, Jonsson PV, Chouraki V, Genier-Boley B, Hiltunen M, Soininen H, Combarros O, Zelenika D, Delepine M, Bullido MJ, Pasquier F, Mateo I, Frank-Garcia A, Porcellini E, Hanon O, Coto E, Alvarez V, Bosco P, Siciliano G, Mancuso M, Panza F, Solfrizzi V, Nacmias B, Sorbi S, Bossu P, Piccardi P, Arosio B, Annoni G, Seripa D, Pilotto A, Scarpini E, Galimberti D, Brice A, Hannequin D, Licastro F, Jones L, Holmans PA, Jonsson T, Riemenschneider M, Morgan K, Younkin SG, Owen MJ, O’Donovan M, Amouyel P, Williams J. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43(5):429–35. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Pickering E, Liu YC, Hall S, Fournier H, Katz E, Dechairo B, John S, Van Eerdewegh P, Soares H. Meta-analysis for genome-wide association study identifies multiple variants at the BIN1 locus associated with late-onset Alzheimer’s disease. PLoS One. 2011;6(2):e16616. doi: 10.1371/journal.pone.0016616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fievet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossu P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanche H, Dartigues JF, Tzourio C, Gut I, Van Broeckhoven C, Alperovitch A, Lathrop M, Amouyel P. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1094–9. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Locke PA, Conneally PM, Tanzi RE, Gusella JF, Haines JL. Apolipoprotein E4 allele and Alzheimer disease: examination of allelic association and effect on age at onset in both early- and late-onset cases. Genet Epidemiol. 1995;12(1):83–92. doi: 10.1002/gepi.1370120108. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, DeCarli C, DeKosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43(5):436–41. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–54. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56(3):303–8. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- Ramirez-Lorca R, Boada M, Saez ME, Hernandez I, Mauleon A, Rosende-Roca M, Martinez-Lage P, Gutierrez M, Real LM, Lopez-Arrieta J, Gayan J, Antunez C, Gonzalez-Perez A, Tarraga L, Ruiz A. GAB2 gene does not modify the risk of Alzheimer’s disease in Spanish APOE 4 carriers. J Nutr Health Aging. 2009;13(3):214–9. doi: 10.1007/s12603-009-0061-6. [DOI] [PubMed] [Google Scholar]
- Roses AD. Apolipoprotein E and Alzheimer’s disease. The tip of the susceptibility iceberg. Ann N Y Acad Sci. 1998;855:738–43. doi: 10.1111/j.1749-6632.1998.tb10653.x. [DOI] [PubMed] [Google Scholar]
- Sasieni PD. From genotypes to genes: doubling the sample size. Biometrics. 1997;53(4):1253–61. [PubMed] [Google Scholar]
- Schmidt C, Wolff M, Weitz M, Bartlau T, Korth C, Zerr I. Rapidly progressive Alzheimer disease. Arch Neurol. 2011;68(9):1124–30. doi: 10.1001/archneurol.2011.189. [DOI] [PubMed] [Google Scholar]
- Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, Bis JC, Smith AV, Carassquillo MM, Lambert JC, Harold D, Schrijvers EM, Ramirez-Lorca R, Debette S, Longstreth WT, Jr, Janssens AC, Pankratz VS, Dartigues JF, Hollingworth P, Aspelund T, Hernandez I, Beiser A, Kuller LH, Koudstaal PJ, Dickson DW, Tzourio C, Abraham R, Antunez C, Du Y, Rotter JI, Aulchenko YS, Harris TB, Petersen RC, Berr C, Owen MJ, Lopez-Arrieta J, Varadarajan BN, Becker JT, Rivadeneira F, Nalls MA, Graff-Radford NR, Campion D, Auerbach S, Rice K, Hofman A, Jonsson PV, Schmidt H, Lathrop M, Mosley TH, Au R, Psaty BM, Uitterlinden AG, Farrer LA, Lumley T, Ruiz A, Williams J, Amouyel P, Younkin SG, Wolf PA, Launer LJ, Lopez OL, van Duijn CM, Breteler MM. Genome-wide analysis of genetic loci associated with Alzheimer disease. Jama. 2010;303(18):1832–40. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90(5):1977–81. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winblad B, Palmer K, Kivipelto M, Jelic V, Fratiglioni L, Wahlund LO, Nordberg A, Backman L, Albert M, Almkvist O, Arai H, Basun H, Blennow K, de Leon M, DeCarli C, Erkinjuntti T, Giacobini E, Graff C, Hardy J, Jack C, Jorm A, Ritchie K, van Duijn C, Visser P, Petersen RC. Mild cognitive impairment–beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J Intern Med. 2004;256(3):240–6. doi: 10.1111/j.1365-2796.2004.01380.x. [DOI] [PubMed] [Google Scholar]
- Wingo TS, Lah JJ, Levey AI, Cutler DJ. Autosomal recessive causes likely in early-onset Alzheimer disease. Arch Neurol. 2012;69(1):59–64. doi: 10.1001/archneurol.2011.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.